Rare Stochastic Expression of O6-Methylguanine- DNA Methyltransferase (MGMT) in MGMT-Negative Melanoma Cells Determines Immediate Emergence of Drug-Resistant Populations upon Treatment with Temozolomide In Vitro and In Vivo


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Pharmacological Agents
2.2. Cell Lines
2.3. Colony Formation Assay
2.4. Generating Mass Cultures of TMZ-Treated Cells
2.5. Treatment with 5-Azacytidine
2.6. Immunoblots
2.7. MethyLight Analysis
2.8. Animal Model
2.9. Tissue Analysis
2.10. Statistical Analysis
3. Results
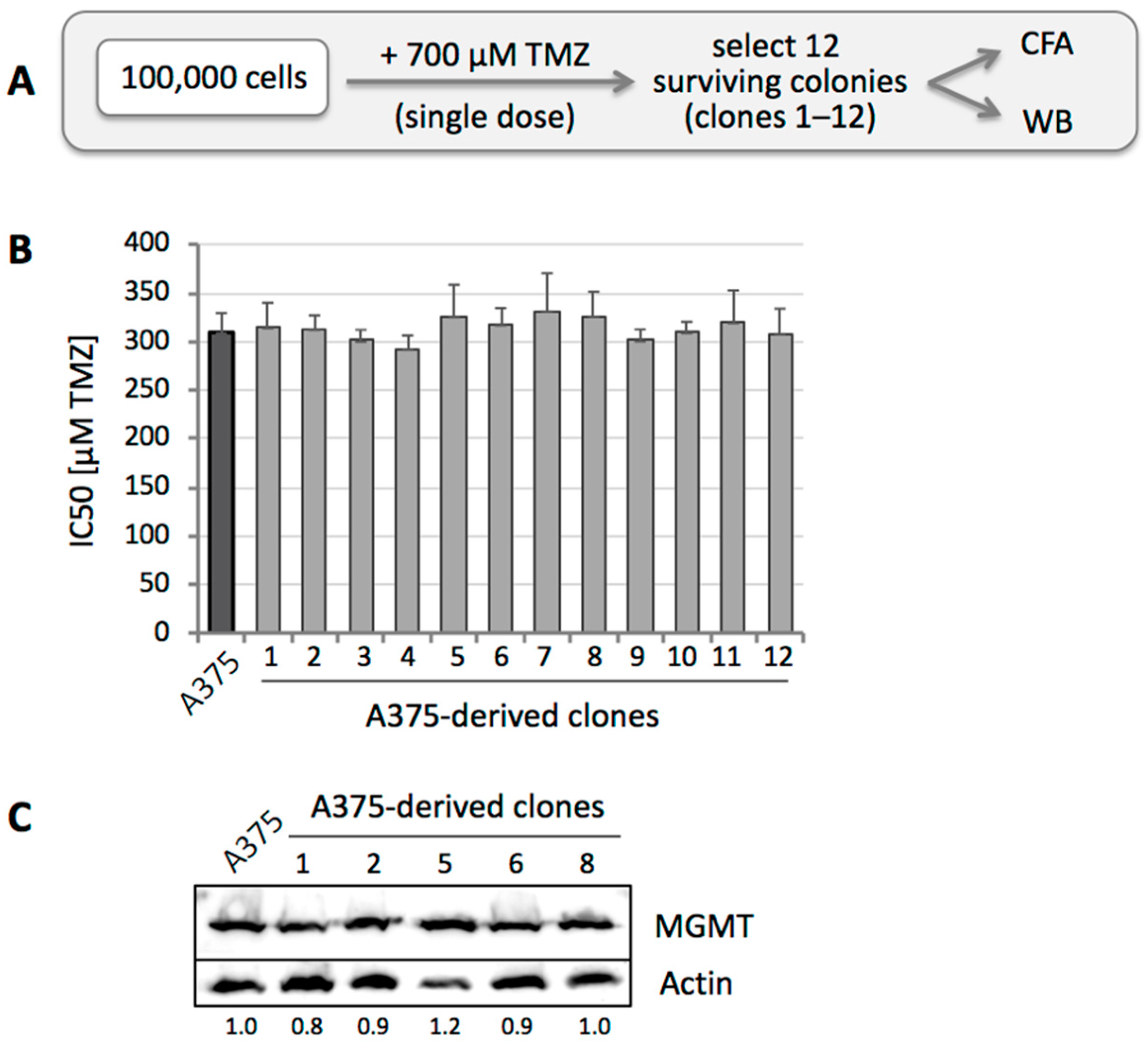
3.1. Sensitivity to TMZ Correlates with MGMT Protein Levels
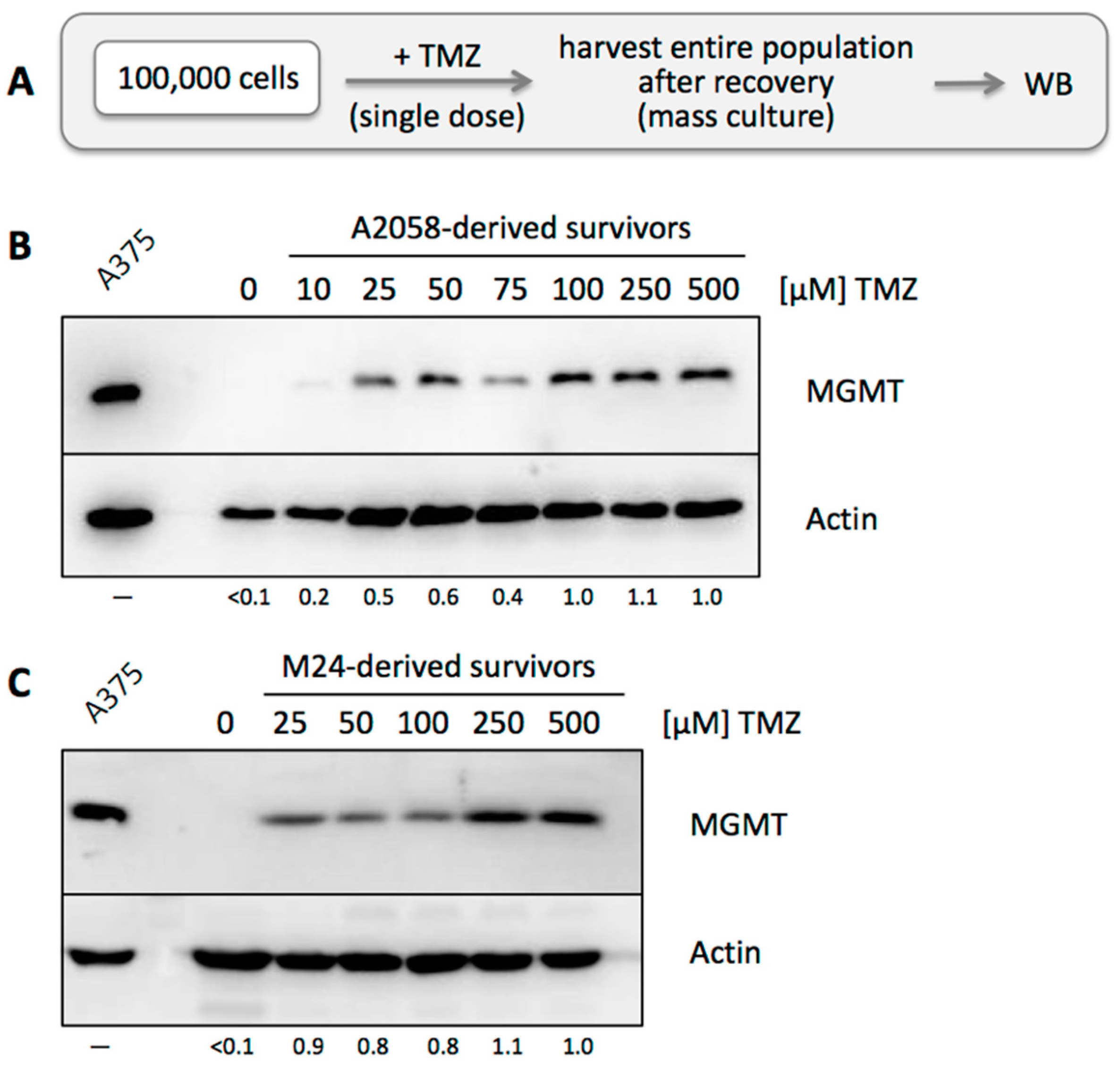
3.2. MGMT-Negative, but Not MGMT-Positive, Cell Populations Adjust to TMZ Treatment
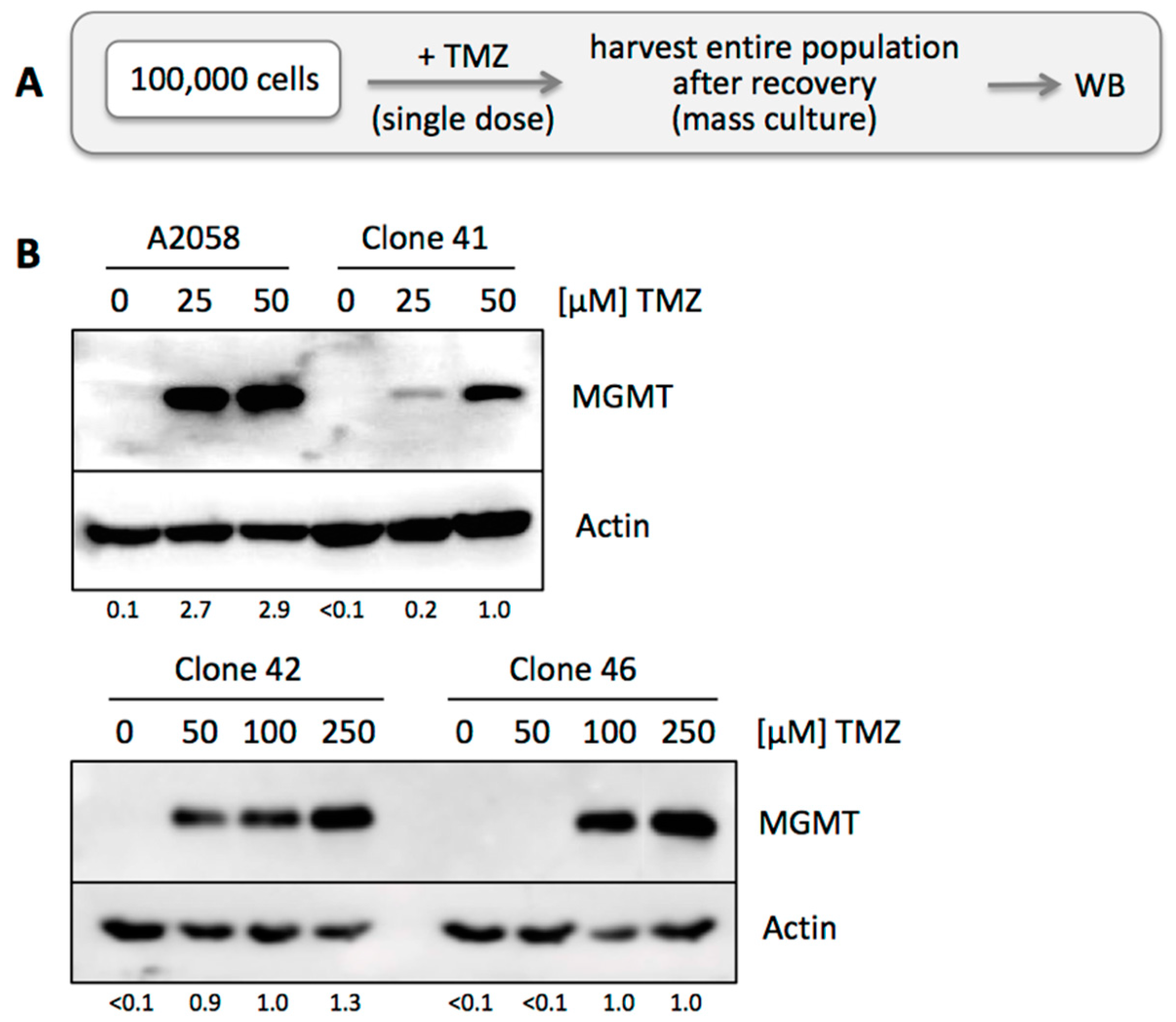
3.3. TMZ Resistance Emerges after Single Drug Treatment Even at Low Concentration
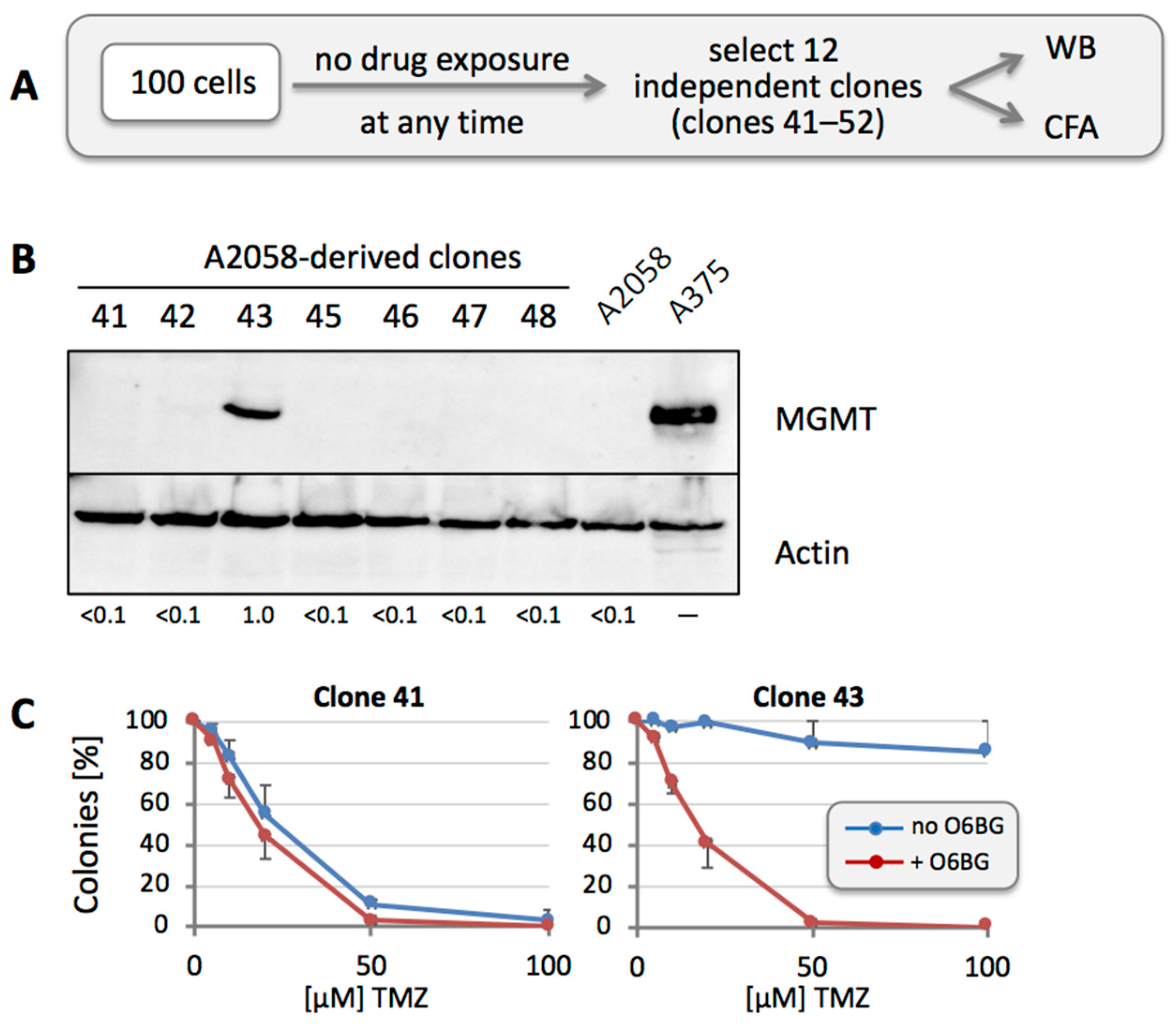
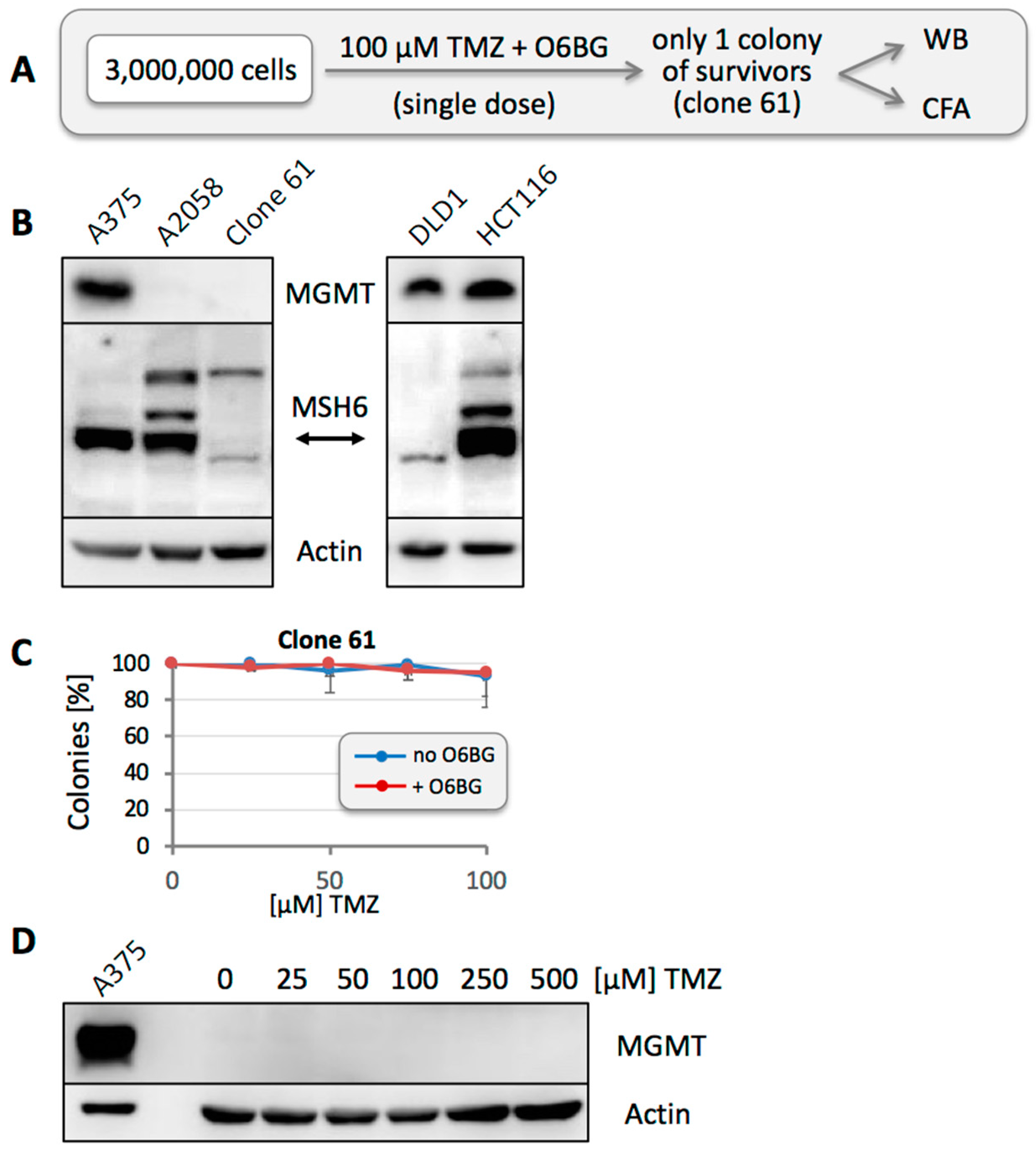
3.4. MGMT-Positive Cells Exist within an MGMT-Negative Population
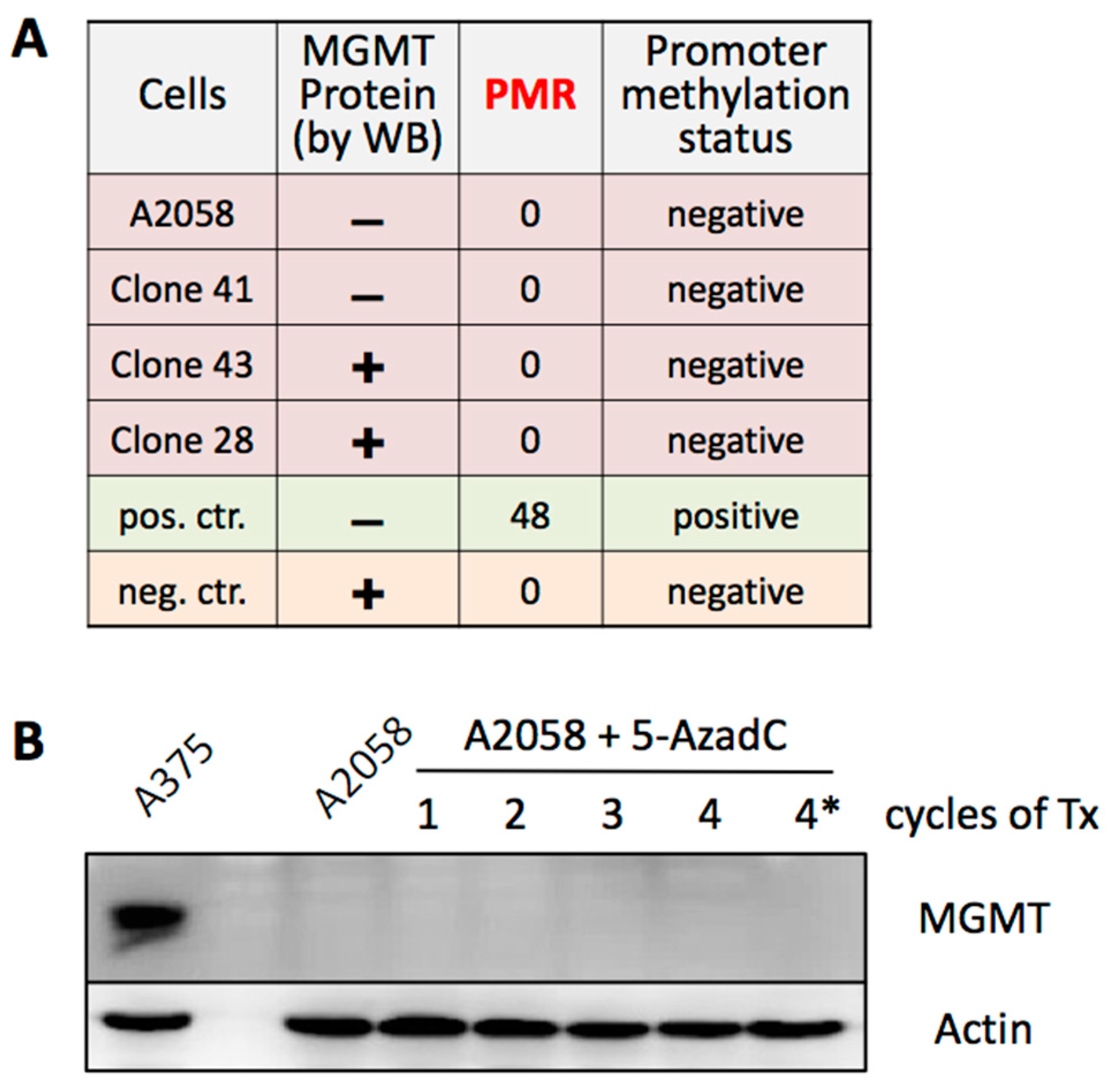
3.5. MGMT Expression Is Not Regulated by Promoter Methylation
3.6. Other Resistance Mechanisms Are Present, but Are Considerably Rarer
3.7. Repeated TMZ Treatment Accelerates the Emergence of Resistance
3.8. Emergence of MGMT-Driven TMZ Resistance Occurs In Vivo
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 5-AzaC | 5-Aza-deoxycytidine |
| CFA | Colony-formation assay |
| MGMT | O6-methylguanine-DNA methyltransferase |
| MMR | Mismatch repair |
| O6BG | O6-Benzylguanine |
| TMZ | Temozolomide |
| WB | Western blot |
References
- Oxnard, G.R. The cellular origins of drug resistance in cancer. Nat. Med. 2016, 22, 232–234. [Google Scholar] [CrossRef] [PubMed]
- Stewart, E.L.; Tsao, M.S. Characterizing the last holdouts. Oncotarget 2017, 8, 12542–12543. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.Y.; Wang, W.; Jhaveri, N.; Torres, S.; Tseng, J.; Leong, M.N.; Lee, D.J.; Goldkorn, A.; Xu, T.; Petasis, N.A.; et al. Perillyl alcohol for the treatment of temozolomide-resistant gliomas. Mol. Cancer Ther. 2012, 11, 2462–2472. [Google Scholar] [CrossRef] [PubMed]
- Happold, C.; Roth, P.; Wick, W.; Schmidt, N.; Florea, A.M.; Silginer, M.; Reifenberger, G.; Weller, M. Distinct molecular mechanisms of acquired resistance to temozolomide in glioblastoma cells. J. Neurochem. 2012, 122, 444–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.; Wong, T.S.; Zhang, X.Q.; Pu, J.K.; Lee, N.P.; Day, P.J.; Ng, G.K.; Lui, W.M.; Leung, G.K. Protein alterations associated with temozolomide resistance in subclones of human glioblastoma cell lines. J. Neurooncol. 2012, 107, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Velpula, K.K.; Guda, M.R.; Sahu, K.; Tuszynski, J.; Asuthkar, S.; Bach, S.E.; Lathia, J.D.; Tsung, A.J. Metabolic targeting of EGFRvIII/PDK1 axis in temozolomide resistant glioblastoma. Oncotarget 2017, 8, 35639–35655. [Google Scholar] [CrossRef] [PubMed]
- Lage, H.; Christmann, M.; Kern, M.A.; Dietel, M.; Pick, M.; Kaina, B.; Schadendorf, D. Expression of DNA repair proteins hMSH2, hMSH6, hMLH1, O6-methylguanine-DNA methyltransferase and N-methylpurine-DNA glycosylase in melanoma cells with acquired drug resistance. Int. J. Cancer 1999, 80, 744–750. [Google Scholar] [CrossRef]
- Brandes, A.A.; Franceschi, E.; Paccapelo, A.; Tallini, G.; De Biase, D.; Ghimenton, C.; Danieli, D.; Zunarelli, E.; Lanza, G.; Silini, E.M.; et al. Role of MGMT Methylation Status at Time of Diagnosis and Recurrence for Patients with Glioblastoma: Clinical Implications. Oncologist 2017, 22, 432–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felsberg, J.; Thon, N.; Eigenbrod, S.; Hentschel, B.; Sabel, M.C.; Westphal, M.; Schackert, G.; Kreth, F.W.; Pietsch, T.; Loffler, M.; et al. Promoter methylation and expression of MGMT and the DNA mismatch repair genes MLH1, MSH2, MSH6 and PMS2 in paired primary and recurrent glioblastomas. Int. J. Cancer 2011, 129, 659–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Calve, B.; Rynkowski, M.; Le Mercier, M.; Bruyere, C.; Lonez, C.; Gras, T.; Haibe-Kains, B.; Bontempi, G.; Decaestecker, C.; Ruysschaert, J.M.; et al. Long-term in vitro treatment of human glioblastoma cells with temozolomide increases resistance in vivo through up-regulation of GLUT transporter and aldo-keto reductase enzyme AKR1C expression. Neoplasia 2010, 12, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Stark, A.M.; Doukas, A.; Hugo, H.H.; Hedderich, J.; Hattermann, K.; Maximilian Mehdorn, H.; Held-Feindt, J. Expression of DNA mismatch repair proteins MLH1, MSH2, and MSH6 in recurrent glioblastoma. Neurol. Res. 2015, 37, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.; Smith, R.; Cahill, D.P.; Stephens, P.; Stevens, C.; Teague, J.; Greenman, C.; Edkins, S.; Bignell, G.; Davies, H.; et al. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res. 2006, 66, 3987–3991. [Google Scholar] [CrossRef] [PubMed]
- Yip, S.; Miao, J.; Cahill, D.P.; Iafrate, A.J.; Aldape, K.; Nutt, C.L.; Louis, D.N. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin. Cancer Res. 2009, 15, 4622–4629. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.Y.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 2014, 343, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Denny, B.J.; Wheelhouse, R.T.; Stevens, M.F.; Tsang, L.L.; Slack, J.A. NMR and molecular modeling investigation of the mechanism of activation of the antitumor drug temozolomide and its interaction with DNA. Biochemistry 1994, 33, 9045–9051. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.L.; Wheelhouse, R.T. The medicinal chemistry of imidazotetrazine prodrugs. Pharmaceuticals 2014, 7, 797–838. [Google Scholar] [CrossRef] [PubMed]
- Knizhnik, A.V.; Roos, W.P.; Nikolova, T.; Quiros, S.; Tomaszowski, K.H.; Christmann, M.; Kaina, B. Survival and death strategies in glioma cells: Autophagy, senescence and apoptosis triggered by a single type of temozolomide-induced DNA damage. PLoS ONE 2013, 8, e55665. [Google Scholar] [CrossRef] [PubMed]
- Christmann, M.; Verbeek, B.; Roos, W.P.; Kaina, B. O(6)-Methylguanine-DNA methyltransferase (MGMT) in normal tissues and tumors: Enzyme activity, promoter methylation and immunohistochemistry. Biochim. Biophys. Acta 2011, 1816, 179–190. [Google Scholar] [CrossRef] [PubMed]
- Kaina, B.; Christmann, M.; Naumann, S.; Roos, W.P. MGMT: Key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair 2007, 6, 1079–1099. [Google Scholar] [CrossRef] [PubMed]
- Pegg, A.E. Multifaceted roles of alkyltransferase and related proteins in DNA repair, DNA damage, resistance to chemotherapy, and research tools. Chem. Res. Toxicol. 2011, 24, 618–639. [Google Scholar] [CrossRef] [PubMed]
- Silber, J.R.; Bobola, M.S.; Blank, A.; Chamberlain, M.C. O(6)-methylguanine-DNA methyltransferase in glioma therapy: Promise and problems. Biochim. Biophys. Acta 2012, 1826, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Dolan, M.E.; Moschel, R.C.; Pegg, A.E. Depletion of mammalian O6-alkylguanine-DNA alkyltransferase activity by O6-benzylguanine provides a means to evaluate the role of this protein in protection against carcinogenic and therapeutic alkylating agents. Proc. Natl. Acad. Sci. USA 1990, 87, 5368–5372. [Google Scholar] [CrossRef] [PubMed]
- Kaina, B.; Margison, G.P.; Christmann, M. Targeting O(6)-methylguanine-DNA methyltransferase with specific inhibitors as a strategy in cancer therapy. Cell. Mol. Life Sci. 2010, 67, 3663–3681. [Google Scholar] [CrossRef] [PubMed]
- Pegg, A.E.; Wiest, L.; Mummert, C.; Stine, L.; Moschel, R.C.; Dolan, M.E. Use of antibodies to human O6-alkylguanine-DNA alkyltransferase to study the content of this protein in cells treated with O6-benzylguanine or N-methyl-N’-nitro-N-nitrosoguanidine. Carcinogenesis 1991, 12, 1679–1683. [Google Scholar] [CrossRef] [PubMed]
- Gladfelter, P.; Darwish, N.H.E.; Mousa, S.A. Current status and future direction in the management of malignant melanoma. Melanoma Res. 2017, 27, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Velho, T.R. Metastatic melanoma—A review of current and future drugs. Drugs Context 2012, 2012, 212242. [Google Scholar] [CrossRef] [PubMed]
- Karimkhani, C.; Gonzalez, R.; Dellavalle, R.P. A review of novel therapies for melanoma. Am. J. Clin. Dermatol. 2014, 15, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Quirbt, I.; Verma, S.; Petrella, T.; Bak, K.; Charette, M. Temozolomide for the treatment of metastatic melanoma. Curr. Oncol. 2007, 14, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Schwartz, G.K. Chemotherapy in the management of advanced cutaneous malignant melanoma. Clin. Dermatol. 2013, 31, 290–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Middleton, M.R.; Grob, J.J.; Aaronson, N.; Fierlbeck, G.; Tilgen, W.; Seiter, S.; Gore, M.; Aamdal, S.; Cebon, J.; Coates, A.; et al. Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J. Clin. Oncol. 2000, 18, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Augustine, C.K.; Yoo, J.S.; Potti, A.; Yoshimoto, Y.; Zipfel, P.A.; Friedman, H.S.; Nevins, J.R.; Ali-Osman, F.; Tyler, D.S. Genomic and molecular profiling predicts response to temozolomide in melanoma. Clin. Cancer Res. 2009, 15, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Qi, F.; Yin, Z.; Wang, G.; Zeng, S. Clinical and Prognostic Significance of O(6)-Methylguanine-DNA Methyltransferase Promoter Methylation in Patients with Melanoma: A Systematic Meta-Analysis. Ann. Dermatol. 2018, 30, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Ranson, M.; Hersey, P.; Thompson, D.; Beith, J.; McArthur, G.A.; Haydon, A.; Davis, I.D.; Kefford, R.F.; Mortimer, P.; Harris, P.A.; et al. Randomized trial of the combination of lomeguatrib and temozolomide compared with temozolomide alone in chemotherapy naive patients with metastatic cutaneous melanoma. J. Clin. Oncol. 2007, 25, 2540–2545. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.J.; Middleton, M.R.; McGown, G.; Thorncroft, M.; Ranson, M.; Hersey, P.; McArthur, G.; Davis, I.D.; Thomson, D.; Beith, J.; et al. O(6)-methylguanine-DNA methyltransferase depletion and DNA damage in patients with melanoma treated with temozolomide alone or with lomeguatrib. Br. J. Cancer 2009, 100, 1250–1256. [Google Scholar] [CrossRef] [PubMed]
- Middleton, M.R.; Kelly, J.; Thatcher, N.; Donnelly, D.J.; McElhinney, R.S.; McMurry, T.B.; McCormick, J.E.; Margison, G.P. O(6)-(4-bromothenyl)guanine improves the therapeutic index of temozolomide against A375M melanoma xenografts. Int. J. Cancer 2000, 85, 248–252. [Google Scholar] [CrossRef]
- Wedge, S.R.; Porteous, J.K.; Newlands, E.S. Effect of single and multiple administration of an O6-benzylguanine/temozolomide combination: An evaluation in a human melanoma xenograft model. Cancer Chemother. Pharmacol. 1997, 40, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Epstein, A.L.; Martin, A.O.; Kempson, R. Use of a newly established human cell line (SU-CCS-1) to demonstrate the relationship of clear cell sarcoma to malignant melanoma. Cancer Res. 1984, 44, 1265–1274. [Google Scholar] [PubMed]
- Henriet, P.; Zhong, Z.D.; Brooks, P.C.; Weinberg, K.I.; DeClerck, Y.A. Contact with fibrillar collagen inhibits melanoma cell proliferation by up-regulating p27KIP1. Proc. Natl. Acad. Sci. USA 2000, 97, 10026–10031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jazirehi, A.R.; Nazarian, R.; Torres-Collado, A.X.; Economou, J.S. Aberrant apoptotic machinery confers melanoma dual resistance to BRAF(V600E) inhibitor and immune effector cells: Immunosensitization by a histone deacetylase inhibitor. Am. J. Clin. Exp. Immunol. 2014, 3, 43–56. [Google Scholar] [PubMed]
- Chen, T.C.; Cho, H.Y.; Wang, W.; Barath, M.; Sharma, N.; Hofman, F.M.; Schönthal, A.H. A novel temozolomide-perillyl alcohol conjugate exhibits superior activity against breast cancer cells in vitro and intracranial triple-negative tumor growth in vivo. Mol. Cancer Ther. 2014, 13, 1181–1193. [Google Scholar] [CrossRef] [PubMed]
- Pyrko, P.; Soriano, N.; Kardosh, A.; Liu, Y.T.; Uddin, J.; Petasis, N.A.; Hofman, F.M.; Chen, C.S.; Chen, T.C.; Schönthal, A.H. Downregulation of survivin expression and concomitant induction of apoptosis by celecoxib and its non-cyclooxygenase-2-inhibitory analog, dimethyl-celecoxib (DMC), in tumor cells in vitro and in vivo. Mol. Cancer 2006, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Campan, M.; Weisenberger, D.J.; Trinh, B.; Laird, P.W. MethyLight. Methods Mol. Biol. 2009, 507, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Mullins, C.S.; Schneider, B.; Stockhammer, F.; Krohn, M.; Classen, C.F.; Linnebacher, M. Establishment and characterization of primary glioblastoma cell lines from fresh and frozen material: A detailed comparison. PLoS ONE 2013, 8, e71070. [Google Scholar] [CrossRef] [PubMed]
- Showalter, T.N.; Andrel, J.; Andrews, D.W.; Curran, W.J., Jr.; Daskalakis, C.; Werner-Wasik, M. Multifocal glioblastoma multiforme: Prognostic factors and patterns of progression. Int. J. Radiat. Oncol. Biol. Phys. 2007, 69, 820–824. [Google Scholar] [CrossRef] [PubMed]
- Diez, B.D.; Statkevich, P.; Zhu, Y.; Abutarif, M.A.; Xuan, F.; Kantesaria, B.; Cutler, D.; Cantillon, M.; Schwarz, M.; Pallotta, M.G.; et al. Evaluation of the exposure equivalence of oral versus intravenous temozolomide. Cancer Chemother. Pharmacol. 2010, 65, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Hammond, L.A.; Eckardt, J.R.; Baker, S.D.; Eckhardt, S.G.; Dugan, M.; Forral, K.; Reidenberg, P.; Statkevich, P.; Weiss, G.R.; Rinaldi, D.A.; et al. Phase I and pharmacokinetic study of temozolomide on a daily-for-5-days schedule in patients with advanced solid malignancies. J. Clin. Oncol. 1999, 17, 2604–2613. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Pei, C.; Li, Q.; Dong, T.; Dong, Y.; Xing, W.; Zhou, P.; Gong, Y.; Zhen, Z.; Gao, Y.; et al. Up-regulation of MSH6 is associated with temozolomide resistance in human glioblastoma. Biochem. Biophys. Res. Commun. 2018, 496, 1040–1046. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Weller, M.; van den Bent, M.; Sanson, M.; Weiler, M.; von Deimling, A.; Plass, C.; Hegi, M.; Platten, M.; Reifenberger, G. MGMT testing—The challenges for biomarker-based glioma treatment. Nat. Rev. Neurol. 2014, 10, 372–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christman, J.K. 5-Azacytidine and 5-aza-2′-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene 2002, 21, 5483–5495. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, K.; Mizoguchi, M.; Hata, N.; Murata, H.; Hatae, R.; Amano, T.; Nakamizo, A.; Sasaki, T. Complex DNA repair pathways as possible therapeutic targets to overcome temozolomide resistance in glioblastoma. Front. Oncol. 2012, 2, 186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Stevens, M.F.; Bradshaw, T.D. Temozolomide: Mechanisms of action, repair and resistance. Curr. Mol. Pharmacol. 2012, 5, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Boeckmann, L.; Nickel, A.C.; Kuschal, C.; Schaefer, A.; Thoms, K.M.; Schon, M.P.; Thomale, J.; Emmert, S. Temozolomide chemoresistance heterogeneity in melanoma with different treatment regimens: DNA damage accumulation contribution. Melanoma Res. 2011, 21, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Berghoff, A.S.; Hainfellner, J.A.; Marosi, C.; Preusser, M. Assessing MGMT methylation status and its current impact on treatment in glioblastoma. CNS Oncol. 2015, 4, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Thon, N.; Kreth, S.; Kreth, F.W. Personalized treatment strategies in glioblastoma: MGMT promoter methylation status. Onco Targets Ther. 2013, 6, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Hassel, J.C.; Sucker, A.; Edler, L.; Kurzen, H.; Moll, I.; Stresemann, C.; Spieth, K.; Mauch, C.; Rass, K.; Dummer, R.; et al. MGMT gene promoter methylation correlates with tolerance of temozolomide treatment in melanoma but not with clinical outcome. Br. J. Cancer 2010, 103, 820–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rietschel, P.; Wolchok, J.D.; Krown, S.; Gerst, S.; Jungbluth, A.A.; Busam, K.; Smith, K.; Orlow, I.; Panageas, K.; Chapman, P.B. Phase II study of extended-dose temozolomide in patients with melanoma. J. Clin. Oncol. 2008, 26, 2299–2304. [Google Scholar] [CrossRef] [PubMed]
- Kohonen-Corish, M.R.; Cooper, W.A.; Saab, J.; Thompson, J.F.; Trent, R.J.; Millward, M.J. Promoter hypermethylation of the O(6)-methylguanine DNA methyltransferase gene and microsatellite instability in metastatic melanoma. J. Investig. Dermatol. 2006, 126, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Rastetter, M.; Schagdarsurengin, U.; Lahtz, C.; Fiedler, E.; Marsch, W.; Dammann, R.; Helmbold, P. Frequent intra-tumoural heterogeneity of promoter hypermethylation in malignant melanoma. Histol. Histopathol. 2007, 22, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Parker, N.R.; Hudson, A.L.; Khong, P.; Parkinson, J.F.; Dwight, T.; Ikin, R.J.; Zhu, Y.; Cheng, Z.J.; Vafaee, F.; Chen, J.; et al. Intratumoral heterogeneity identified at the epigenetic, genetic and transcriptional level in glioblastoma. Sci. Rep. 2016, 6, 22477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, J.A.; Jiang, S.X.; Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; Rich, J.N.; Gururangan, S.; Friedman, A.H.; Bigner, D.D.; Sampson, J.H.; et al. Phase II trial of temozolomide plus o6-benzylguanine in adults with recurrent, temozolomide-resistant malignant glioma. J. Clin. Oncol. 2009, 27, 1262–1267. [Google Scholar] [CrossRef] [PubMed]
- Warren, K.E.; Gururangan, S.; Geyer, J.R.; McLendon, R.E.; Poussaint, T.Y.; Wallace, D.; Balis, F.M.; Berg, S.L.; Packer, R.J.; Goldman, S.; et al. A phase II study of O6-benzylguanine and temozolomide in pediatric patients with recurrent or progressive high-grade gliomas and brainstem gliomas: A Pediatric Brain Tumor Consortium study. J. Neurooncol. 2012, 106, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Cahill, D.P.; Levine, K.K.; Betensky, R.A.; Codd, P.J.; Romany, C.A.; Reavie, L.B.; Batchelor, T.T.; Futreal, P.A.; Stratton, M.R.; Curry, W.T.; et al. Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin. Cancer Res. 2007, 13, 2038–2045. [Google Scholar] [CrossRef] [PubMed]
- Alvino, E.; Passarelli, F.; Cannavo, E.; Fortes, C.; Mastroeni, S.; Caporali, S.; Jiricny, J.; Cappellini, G.C.; Scoppola, A.; Marchetti, P.; et al. High expression of the mismatch repair protein MSH6 is associated with poor patient survival in melanoma. Am. J. Clin. Pathol. 2014, 142, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Murphy, M.; O’Dwyer, P.J.; Berman, E.; Reed, K.; Gallo, J.M. Biochemical changes associated with a multidrug-resistant phenotype of a human glioma cell line with temozolomide-acquired resistance. Biochem. Pharmacol. 2002, 63, 1219–1228. [Google Scholar] [CrossRef]
- Germano, G.; Lamba, S.; Rospo, G.; Barault, L.; Magri, A.; Maione, F.; Russo, M.; Crisafulli, G.; Bartolini, A.; Lerda, G.; et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 2017, 552, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Campbell, B.B.; Light, N.; Fabrizio, D.; Zatzman, M.; Fuligni, F.; de Borja, R.; Davidson, S.; Edwards, M.; Elvin, J.A.; Hodel, K.P.; et al. Comprehensive Analysis of Hypermutation in Human Cancer. Cell 2017, 171, 1042–1056.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinnadurai, M.; McDonald, K.L. Immune checkpoint inhibition and its relationship with hypermutation phenoytype as a potential treatment for Glioblastoma. J. Neurooncol. 2017, 132, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, V.; Kaestner, V.; Mailankody, S. Cancer Drugs Approved Based on Biomarkers and Not Tumor Type-FDA Approval of Pembrolizumab for Mismatch Repair-Deficient Solid Cancers. JAMA Oncol. 2018, 4, 157–158. [Google Scholar] [CrossRef] [PubMed]











© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, T.C.; Chan, N.; Minea, R.O.; Hartman, H.; Hofman, F.M.; Schönthal, A.H. Rare Stochastic Expression of O6-Methylguanine- DNA Methyltransferase (MGMT) in MGMT-Negative Melanoma Cells Determines Immediate Emergence of Drug-Resistant Populations upon Treatment with Temozolomide In Vitro and In Vivo. Cancers 2018, 10, 362. https://doi.org/10.3390/cancers10100362
Chen TC, Chan N, Minea RO, Hartman H, Hofman FM, Schönthal AH. Rare Stochastic Expression of O6-Methylguanine- DNA Methyltransferase (MGMT) in MGMT-Negative Melanoma Cells Determines Immediate Emergence of Drug-Resistant Populations upon Treatment with Temozolomide In Vitro and In Vivo. Cancers. 2018; 10(10):362. https://doi.org/10.3390/cancers10100362
Chicago/Turabian StyleChen, Thomas C., Nymph Chan, Radu O. Minea, Hannah Hartman, Florence M. Hofman, and Axel H. Schönthal. 2018. "Rare Stochastic Expression of O6-Methylguanine- DNA Methyltransferase (MGMT) in MGMT-Negative Melanoma Cells Determines Immediate Emergence of Drug-Resistant Populations upon Treatment with Temozolomide In Vitro and In Vivo" Cancers 10, no. 10: 362. https://doi.org/10.3390/cancers10100362