
A Novel Zak Knockout Mouse with a Defective Ribotoxic Stress Response

 , ,
, , {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
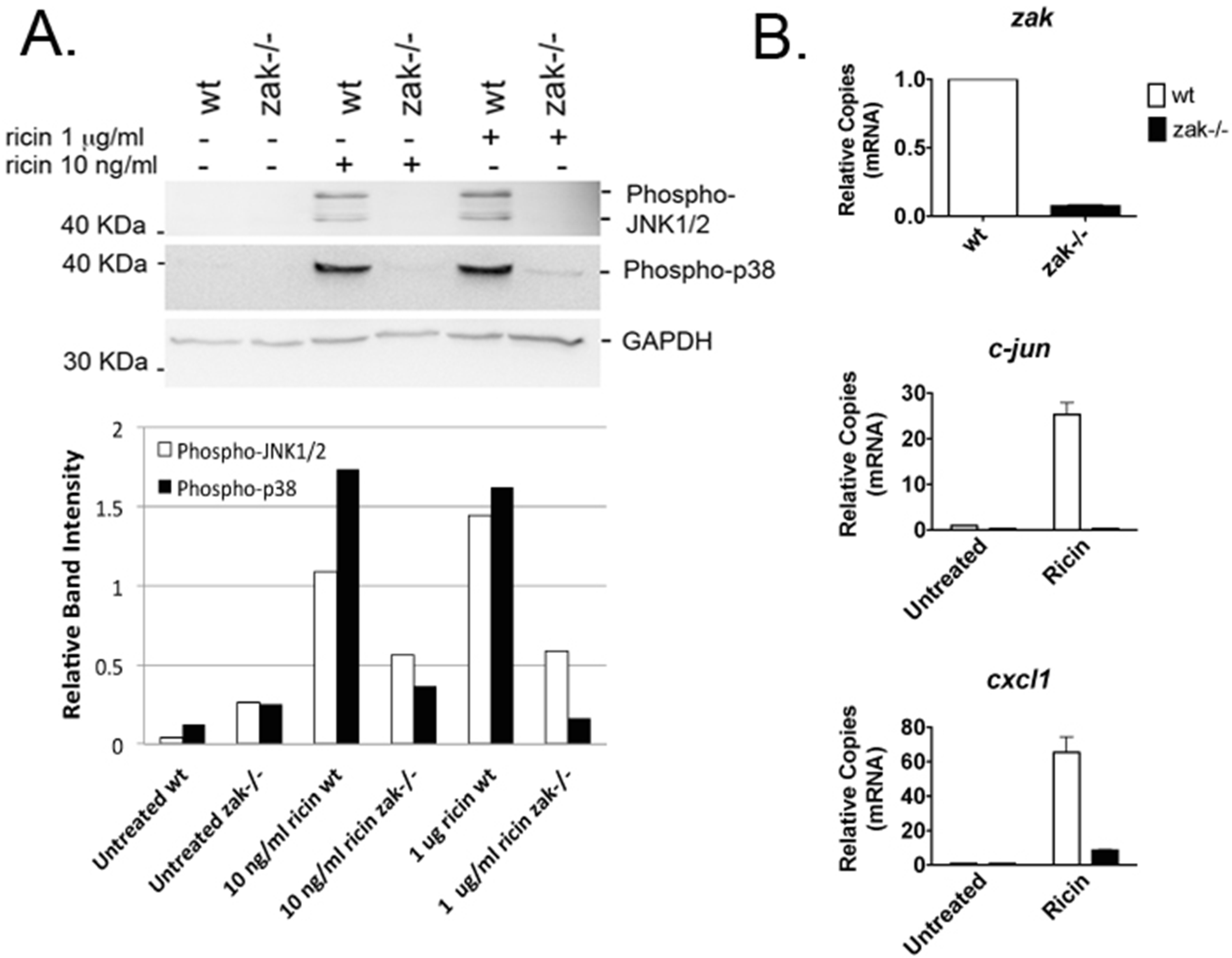
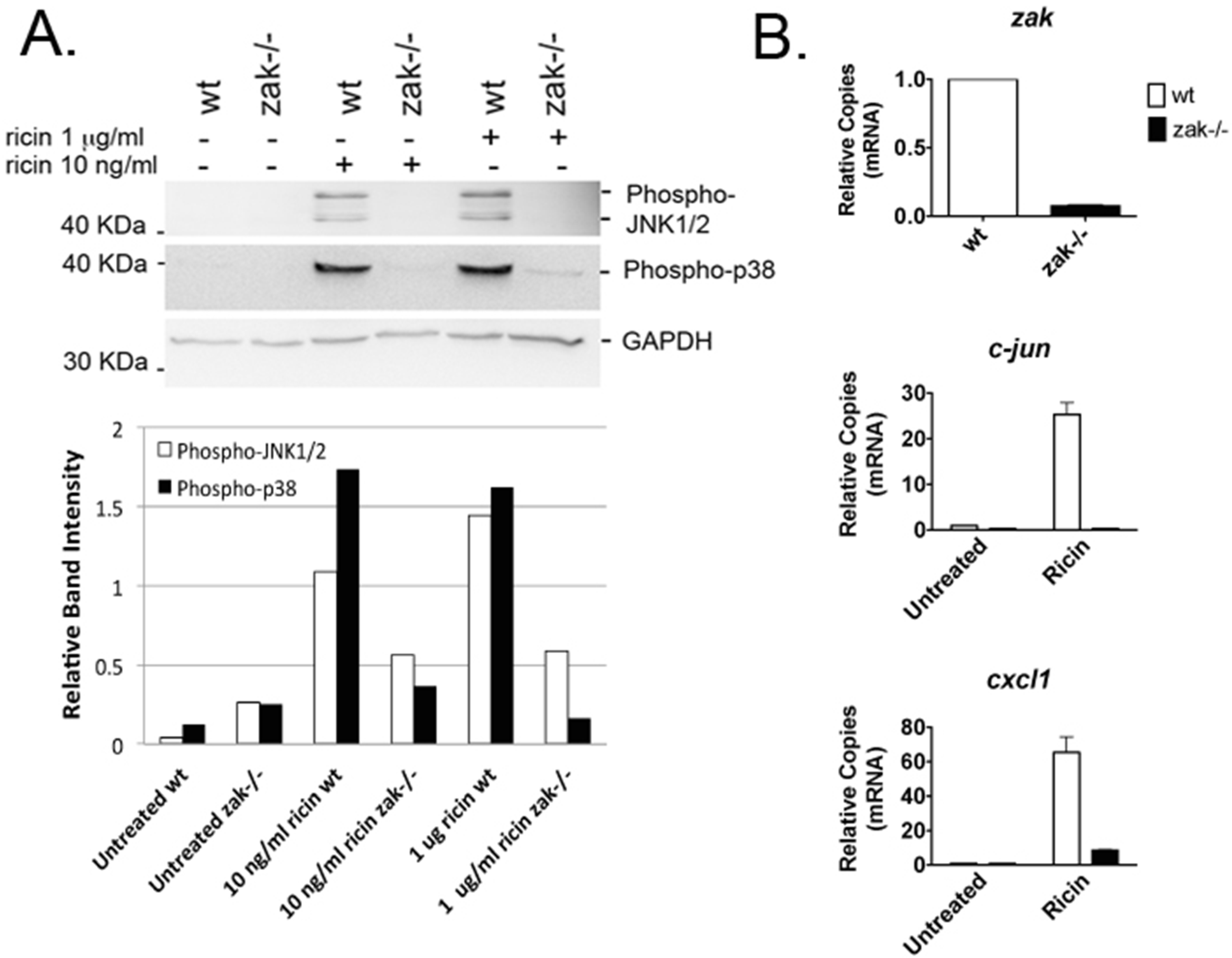
2.1. Bone Marrow–Derived Macrophages (BMDMs) from zak−/− Mice Have a Distinct Defect in JNK1/2 and p38 Activation and Associated Gene Expression Following Ricin Intoxication
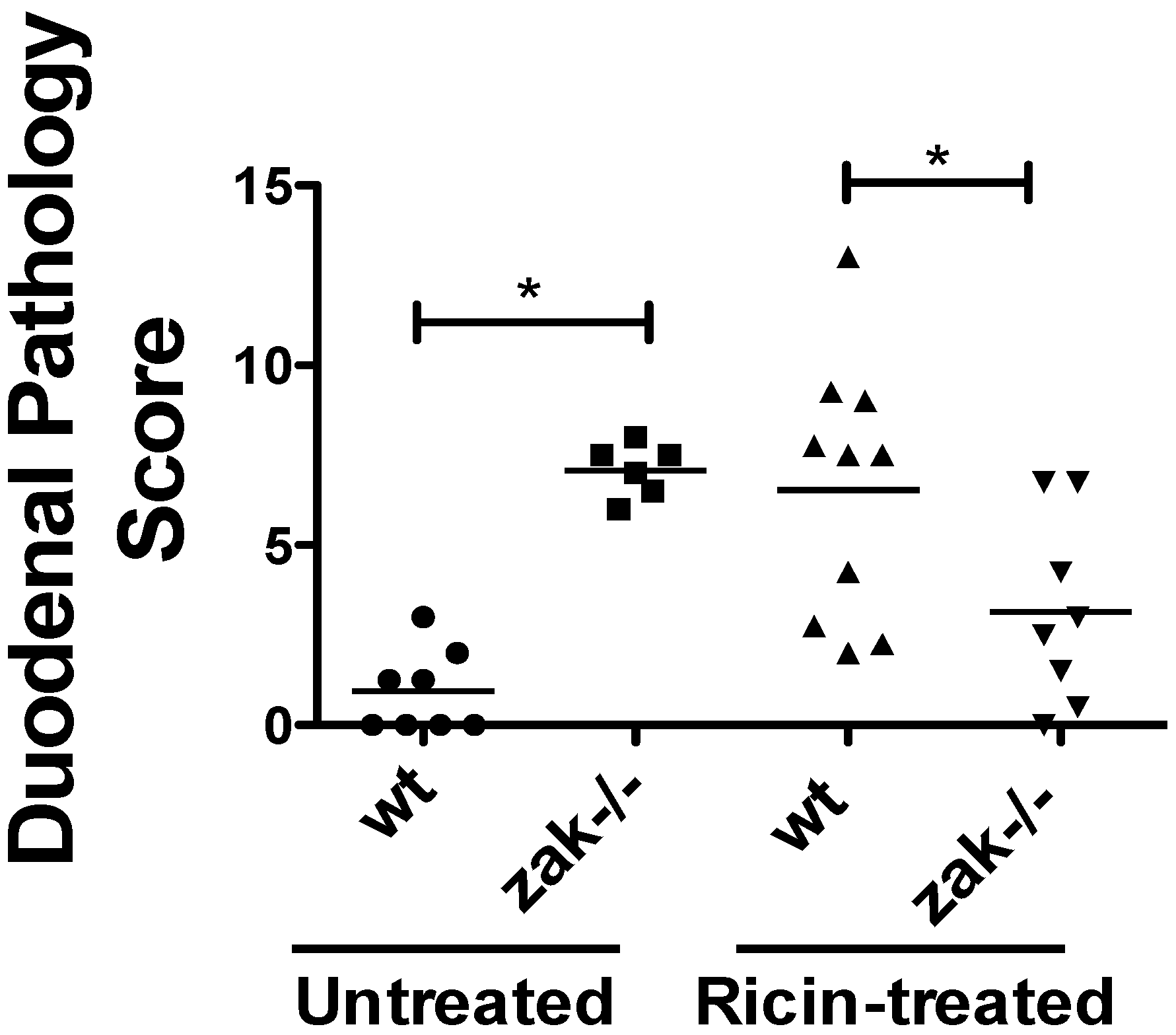
2.2. zak−/− Mice Demonstrate Decreased Epithelial Damage When Gavaged with Ricin
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Mice
5.2. Harvesting of Bone Marrow–Derived Macrophages (BMDMs)
5.3. Ricin
5.4. Preparation of Whole Cell Extracts, MAPK Immunoprecipitation Assays, and Western Blotting
5.5. Quantitative Real-Time PCR
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| MAPK | Mitogen activated protein kinase |
| MAP3K | Mitogen activated protein 3 kinase |
| BMDMs | Bone Marrow Derived Macrophages |
| RSR | Ribotoxic Stress Response |
| qRT-PCR | quantitative real-time polymerase chain reaction |
References
- Kent, K.J. Ricin. In Handbook of Bioterrorism and disaster Medicine; Springer Science: New York, NY, USA, 2006; pp. 143–146. [Google Scholar]
- Schmitt, E.; Shanker, T. Qaeda Tryng to Harness Toxin for Bombs, U.S. Officials Fear. The New York Times. 12 August 2011. Available online: http://www.nytimes.com/2011/08/13/world/middleeast/13terror.html?_r=0 (accessed on 31 August 2016).
- CDC. Investigation of a Ricin-Containing Envelope at a Postal Facility—South Carolina, 2003. MMWR 2003, 52, 1129–1131. [Google Scholar]
- Risen, J.; Van Natta, D., Jr. Plot to poison food of british troops is suspected. The New York Times. 24 January 2003. Available online: http://www.nytimes.com/2003/01/24/international/europe/24TERR.html (accessed on 31 August 2016).
- Pollack, A.F.D.A. Finds Traces of Poison in 2 Jars of Baby Food in California. The New York Times. 29 July 2004. Available online: http://www.nytimes.com/2004/07/29/us/fda-finds-traces-of-poison-in-2-jars-of-baby-food-in-california-no-one-is-harmed.html (accessed on 31 August 2016).
- CDC. Ricin in Senate Office Building (Washington, DC). Available online: https://stacks.cdc.gov/view/cdc/25095 (accessed on 8 August 2016).
- FBI. North Georgia Men Arrested, Charged in Plots to Purchase Explosives, Silencer and to Manufacture a Biological Toxin. Available online: http://archives.fbi.gov/archives/atlanta/press-releases/2011/north-georgia-men-arrested-charged-in-plots-to-purchase-explosives-silencer-and-to-manufacture-a-biological-toxin (accessed on 8 August 2016).
- Friess, S. In Accord, Ricin Owner Enters Pleas of Guilty. The New York Times. 5 August 2008. Available online: http://www.nytimes.com/2008/08/05/us/05ricin.html (accessed on 31 August 2016).
- Iordanov, M.S.; Pribnow, D.; Magun, J.L.; Dinh, T.H.; Pearson, J.A.; Chen, S.L.; Magun, B.E. Ribotoxic stress response: Activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the alpha-sarcin/ricin loop in the 28S rRNA. Mol. Cell. Biol. 1997, 17, 3373–3381. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Tsurugi, K. RNA N-glycosidase activity of ricin A-chain. Mechanism of action of the toxic lectin ricin on eukaryotic ribosomes. J. Biol. Chem. 1987, 262, 8128–8130. [Google Scholar] [PubMed]
- Endo, Y.; Mitsui, K.; Motizuki, M.; Tsurugi, K. The mechanism of action of ricin and related toxic lectins on eukaryotic ribosomes. The site and the characteristics of the modification in 28 S ribosomal RNA caused by the toxins. J. Biol. Chem. 1987, 262, 5908–5912. [Google Scholar] [PubMed]
- Jandhyala, D.M.; Thorpe, C.M.; Magun, B. Ricin and Shiga toxins: Effects on host cell signal transduction. Curr. Top. Microbiol. Immunol. 2012, 357, 41–65. [Google Scholar] [PubMed]
- Iordanov, M.S.; Pribnow, D.; Magun, J.L.; Dinh, T.H.; Pearson, J.A.; Magun, B.E. Ultraviolet radiation triggers the ribotoxic stress response in mammalian cells. J. Biol. Chem. 1998, 273, 15794–15803. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, C.M.; Hurley, B.P.; Lincicome, L.L.; Jacewicz, M.S.; Keusch, G.T.; Acheson, D.W. Shiga toxins stimulate secretion of interleukin-8 from intestinal epithelial cells. Infect. Immun. 1999, 67, 5985–5993. [Google Scholar]
- Smith, W.E.; Kane, A.V.; Campbell, S.T.; Acheson, D.W.; Cochran, B.H.; Thorpe, C.M. Shiga toxin 1 triggers a ribotoxic stress response leading to p38 and JNK activation and induction of apoptosis in intestinal epithelial cells. Infect. Immun. 2003, 71, 1497–1504. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, C.M.; Smith, W.E.; Hurley, B.P.; Acheson, D.W. Shiga toxins induce, superinduce, and stabilize a variety of C-X-C chemokine mRNAs in intestinal epithelial cells, resulting in increased chemokine expression. Infect. Immun. 2001, 69, 6140–6147. [Google Scholar] [CrossRef] [PubMed]
- Foster, G.H.; Tesh, V.L. Shiga toxin 1-induced activation of c-Jun NH(2)-terminal kinase and p38 in the human monocytic cell line THP-1: Possible involvement in the production of TNF-alpha. J. Leukoc. Biol. 2002, 71, 107–114. [Google Scholar] [PubMed]
- Cherla, R.P.; Lee, S.Y.; Mees, P.L.; Tesh, V.L. Shiga toxin 1-induced cytokine production is mediated by MAP kinase pathways and translation initiation factor eIF4E in the macrophage-like THP-1 cell line. J. Leukoc. Biol. 2006, 79, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Lee, M.S.; Cherla, R.P.; Tesh, V.L. Shiga toxin 1 induces apoptosis through the endoplasmic reticulum stress response in human monocytic cells. Cell. Microbiol. 2008, 10, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Korcheva, V.; Wong, J.; Corless, C.; Iordanov, M.; Magun, B. Administration of ricin induces a severe inflammatory response via nonredundant stimulation of ERK, JNK, and P38 MAPK and provides a mouse model of hemolytic uremic syndrome. Am. J. Pathol. 2005, 166, 323–339. [Google Scholar] [CrossRef]
- Korcheva, V.; Wong, J.; Lindauer, M.; Jacoby, D.B.; Iordanov, M.S.; Magun, B. Role of apoptotic signaling pathways in regulation of inflammatory responses to ricin in primary murine macrophages. Mol. Immunol. 2007, 44, 2761–2771. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Korcheva, V.; Jacoby, D.B.; Magun, B.E. Proinflammatory responses of human airway cells to ricin involve stress-activated protein kinases and NF-kappaB. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L1385–L1394. [Google Scholar] [CrossRef] [PubMed]
- Yoder, J.M.; Aslam, R.U.; Mantis, N.J. Evidence for widespread epithelial damage and coincident production of monocyte chemotactic protein 1 in a murine model of intestinal ricin intoxication. Infect. Immun. 2007, 75, 1745–1750. [Google Scholar] [CrossRef] [PubMed]
- Lindauer, M.L.; Wong, J.; Iwakura, Y.; Magun, B.E. Pulmonary inflammation triggered by ricin toxin requires macrophages and IL-1 signaling. J. Immunol. 2009, 183, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, I.; Adachi, M.; Nishida, E. Identification and characterization of a novel MAP kinase kinase kinase, MLTK. J. Biol. Chem. 2001, 276, 4276–4286. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Mader, M.M.; Toth, J.E.; Yu, X.; Jin, N.; Campbell, R.M.; Smallwood, J.K.; Christe, M.E.; Chatterjee, A.; Goodson, T., Jr.; et al. Complete inhibition of anisomycin and UV radiation but not cytokine induced JNK and p38 activation by an aryl-substituted dihydropyrrolopyrazole quinoline and mixed lineage kinase 7 small interfering RNA. J. Biol. Chem. 2005, 280, 19298–19305. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.H.; Zhang, J.B.; Dang, Z.; Li, X.; Zhou, T.; Liu, J.; Wang, D.S.; Song, W.J.; Dou, K.F. Long non-coding RNA URHC regulates cell proliferation and apoptosis via ZAK through the ERK/MAPK signaling pathway in hepatocellular carcinoma. Int. J. Biol. Sci. 2014, 10, 664–676. [Google Scholar] [CrossRef] [PubMed]
- Gross, E.A.; Callow, M.G.; Waldbaum, L.; Thomas, S.; Ruggieri, R. MRK, a mixed lineage kinase-related molecule that plays a role in gamma-radiation-induced cell cycle arrest. J. Biol. Chem. 2002, 277, 13873–13882. [Google Scholar] [CrossRef] [PubMed]
- Jandhyala, D.M.; Ahluwalia, A.; Obrig, T.; Thorpe, C.M. ZAK: A MAP3Kinase that transduces Shiga toxin- and ricin-induced proinflammatory cytokine expression. Cell. Microbiol. 2008, 10, 1468–1477. [Google Scholar] [CrossRef] [PubMed]
- Kyriakis, J.M.; Avruch, J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: A 10-year update. Physiol. Rev. 2012, 92, 689–737. [Google Scholar] [CrossRef] [PubMed]
- Tesh, V.L. The induction of apoptosis by Shiga toxins and ricin. Curr. Top. Microbiol. Immunol. 2012, 357, 137–178. [Google Scholar] [PubMed]
- Flora, A.D.; Teel, L.D.; Smith, M.A.; Sinclair, J.F.; Melton-Celsa, A.R.; O’Brien, A.D. Ricin crosses polarized human intestinal cells and intestines of ricin-gavaged mice without evident damage and then disseminates to mouse kidneys. PLoS ONE 2013, 8, e69706. [Google Scholar] [CrossRef] [PubMed]
- Audi, J.; Belson, M.; Patel, M.; Schier, J.; Osterloh, J. Ricin poisoning: A comprehensive review. JAMA 2005, 294, 2342–2351. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.L.; David, J.; Griffiths, G.D. Retrospective identification of ricin in animal tissues following administration by pulmonary and oral routes. Toxicology 2006, 223, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Tosti, E.; Waldbaum, L.; Warshaw, G.; Gross, E.A.; Ruggieri, R. The stress kinase MRK contributes to regulation of DNA damage checkpoints through a p38gamma-independent pathway. J. Biol. Chem. 2004, 279, 47652–47660. [Google Scholar] [CrossRef] [PubMed]
- Rey, C.; Faustin, B.; Mahouche, I.; Ruggieri, R.; Brulard, C.; Ichas, F.; Soubeyran, I.; Lartigue, L.; De Giorgi, F. The MAP3K ZAK, a novel modulator of ERK-dependent migration, is upregulated in colorectal cancer. Oncogene 2016, 35, 3190–3200. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Smith, L.B.; Magun, E.A.; Engstrom, T.; Kelley-Howard, K.; Jandhyala, D.M.; Thorpe, C.M.; Magun, B.E.; Wood, L.J. Small molecule kinase inhibitors block the ZAK-dependent inflammatory effects of doxorubicin. Cancer Biol. Ther. 2013, 14, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.R.; Islam, Z.; Pestka, J.J. Rapid, sequential activation of mitogen-activated protein kinases and transcription factors precedes proinflammatory cytokine mRNA expression in spleens of mice exposed to the trichothecene vomitoxin. Toxicol. Sci. 2003, 72, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.R.; Islam, Z.; Pestka, J.J. Induction of competing apoptotic and survival signaling pathways in the macrophage by the ribotoxic trichothecene deoxynivalenol. Toxicol. Sci. 2005, 87, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; McCleland, M.; Stawiski, E.W.; Gnad, F.; Mayba, O.; Haverty, P.M.; Durinck, S.; Chen, Y.J.; Klijn, C.; Jhunjhunwala, S.; et al. Integrated exome and transcriptome sequencing reveals ZAK isoform usage in gastric cancer. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Korkina, O.; Dong, Z.; Marullo, A.; Warshaw, G.; Symons, M.; Ruggieri, R. The MLK-related kinase (MRK) is a novel RhoC effector that mediates lysophosphatidic acid (LPA)-stimulated tumor cell invasion. J. Biol. Chem. 2013, 288, 5364–5373. [Google Scholar] [CrossRef] [PubMed]
- Bloem, L.J.; Pickard, T.R.; Acton, S.; Donoghue, M.; Beavis, R.C.; Knierman, M.D.; Wang, X. Tissue distribution and functional expression of a cDNA encoding a novel mixed lineage kinase. J. Mol. Cell. Cardiol. 2001, 33, 1739–1750. [Google Scholar] [CrossRef] [PubMed]
- Vin, H.; Ojeda, S.S.; Ching, G.; Leung, M.L.; Chitsazzadeh, V.; Dwyer, D.W.; Adelmann, C.H.; Restrepo, M.; Richards, K.N.; Stewart, L.R.; et al. BRAF inhibitors suppress apoptosis through off-target inhibition of JNK signaling. Elife 2013, 2, e00969. [Google Scholar] [CrossRef] [PubMed]
- Pothoulakis, C.; Castagliuolo, I.; LaMont, J.T.; Jaffer, A.; O'Keane, J.C.; Snider, R.M.; Leeman, S.E. CP-96,345, a substance P antagonist, inhibits rat intestinal responses to Clostridium difficile toxin A but not cholera toxin. Proc. Natl. Acad. Sci. USA 1994, 91, 947–951. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jandhyala, D.M.; Wong, J.; Mantis, N.J.; Magun, B.E.; Leong, J.M.; Thorpe, C.M. A Novel Zak Knockout Mouse with a Defective Ribotoxic Stress Response. Toxins 2016, 8, 259. https://doi.org/10.3390/toxins8090259
Jandhyala DM, Wong J, Mantis NJ, Magun BE, Leong JM, Thorpe CM. A Novel Zak Knockout Mouse with a Defective Ribotoxic Stress Response. Toxins. 2016; 8(9):259. https://doi.org/10.3390/toxins8090259
Chicago/Turabian StyleJandhyala, Dakshina M., John Wong, Nicholas J. Mantis, Bruce E. Magun, John M. Leong, and Cheleste M. Thorpe. 2016. "A Novel Zak Knockout Mouse with a Defective Ribotoxic Stress Response" Toxins 8, no. 9: 259. https://doi.org/10.3390/toxins8090259
APA StyleJandhyala, D. M., Wong, J., Mantis, N. J., Magun, B. E., Leong, J. M., & Thorpe, C. M. (2016). A Novel Zak Knockout Mouse with a Defective Ribotoxic Stress Response. Toxins, 8(9), 259. https://doi.org/10.3390/toxins8090259