Partial Reconstruction of the Ergot Alkaloid Pathway by Heterologous Gene Expression in Aspergillus nidulans

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
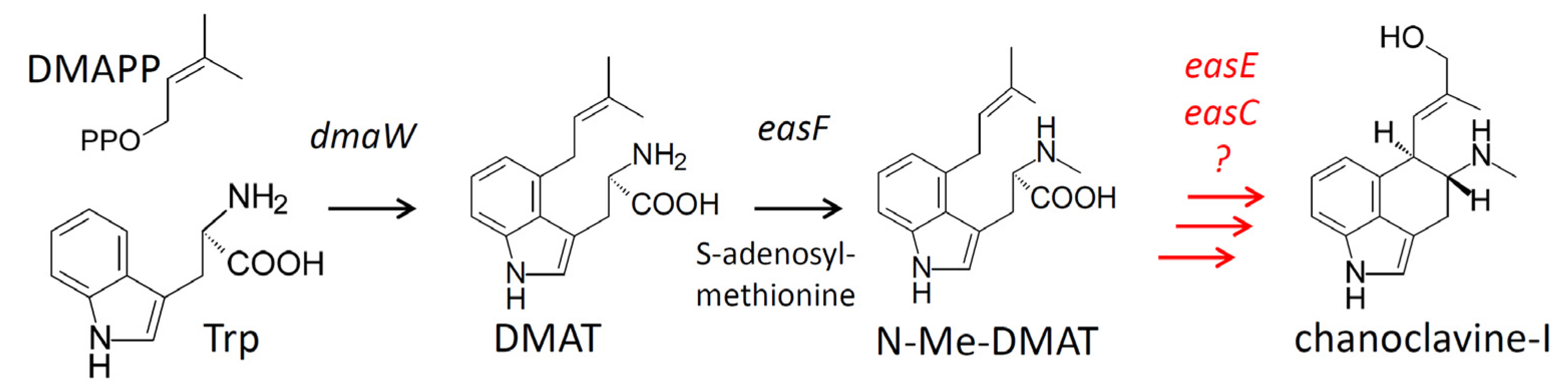
:1. Introduction
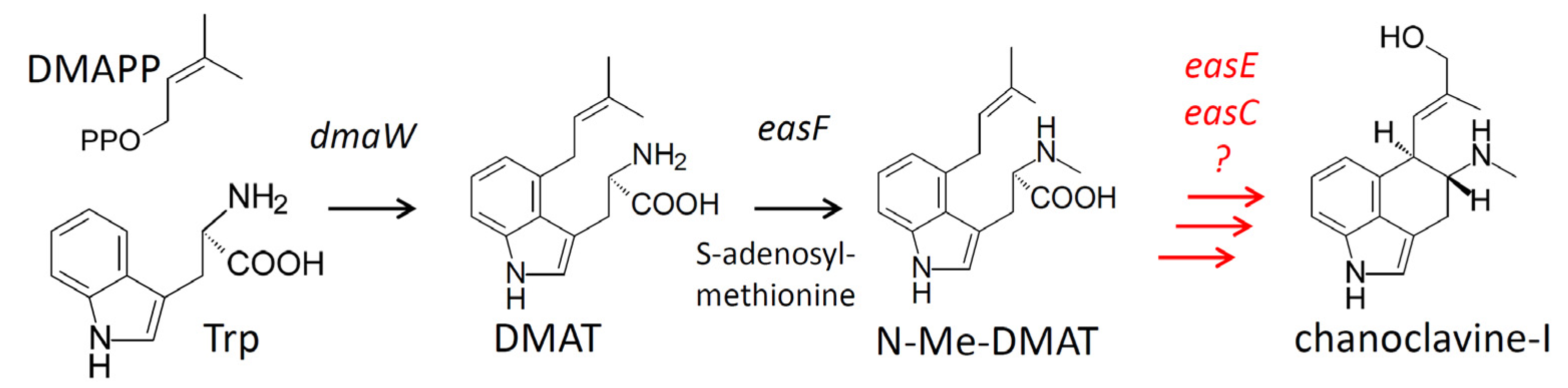
2. Results
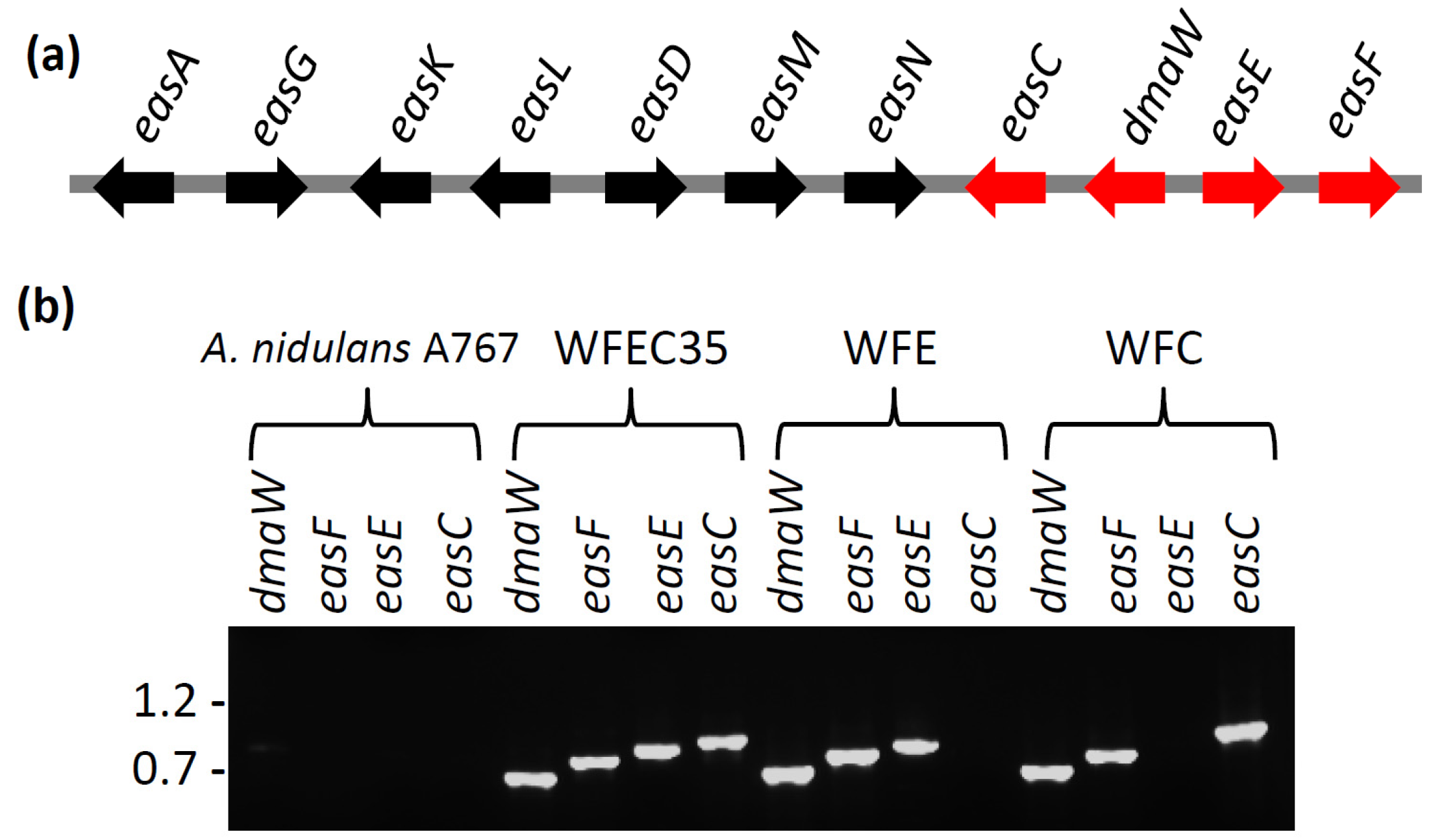
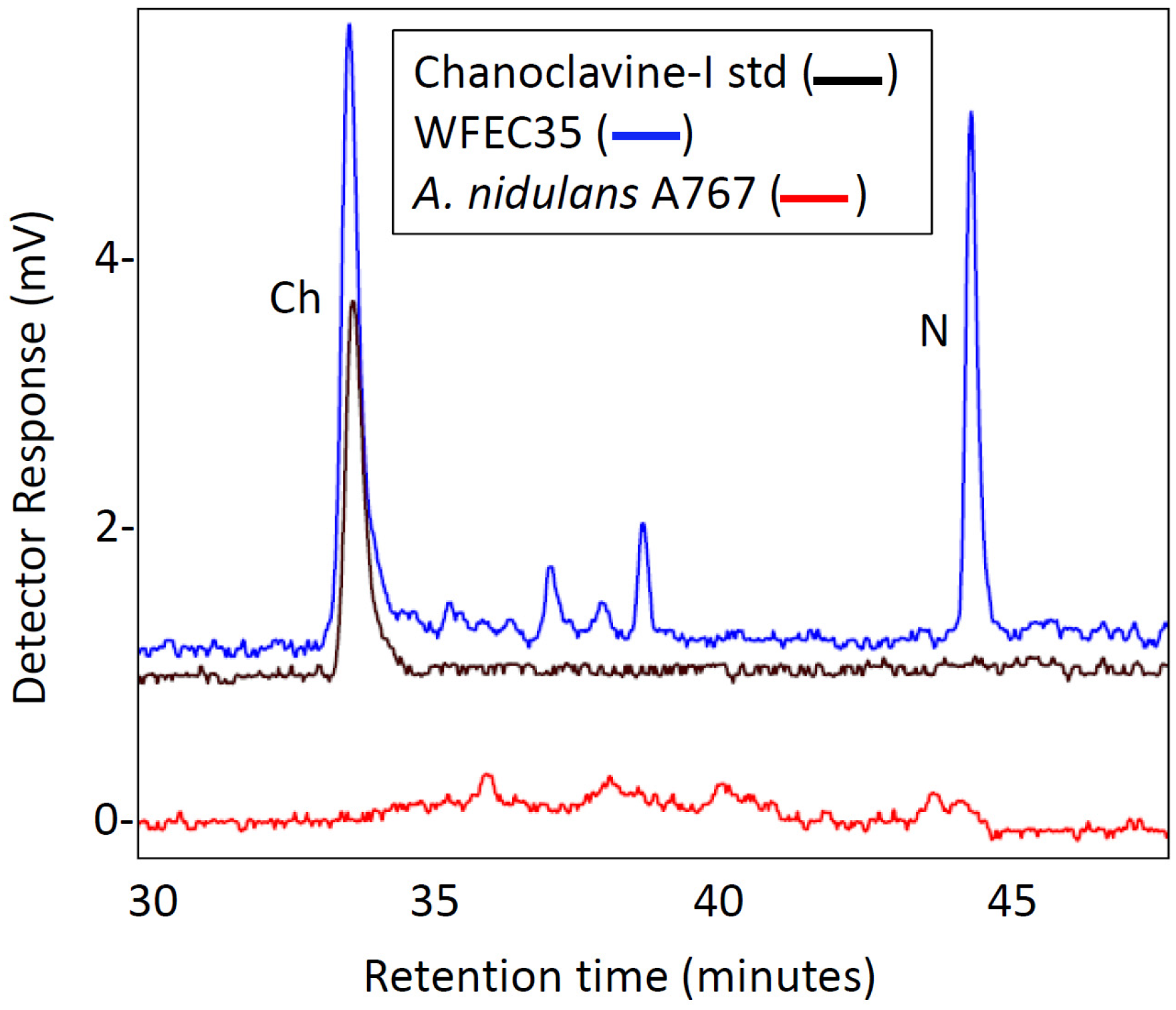
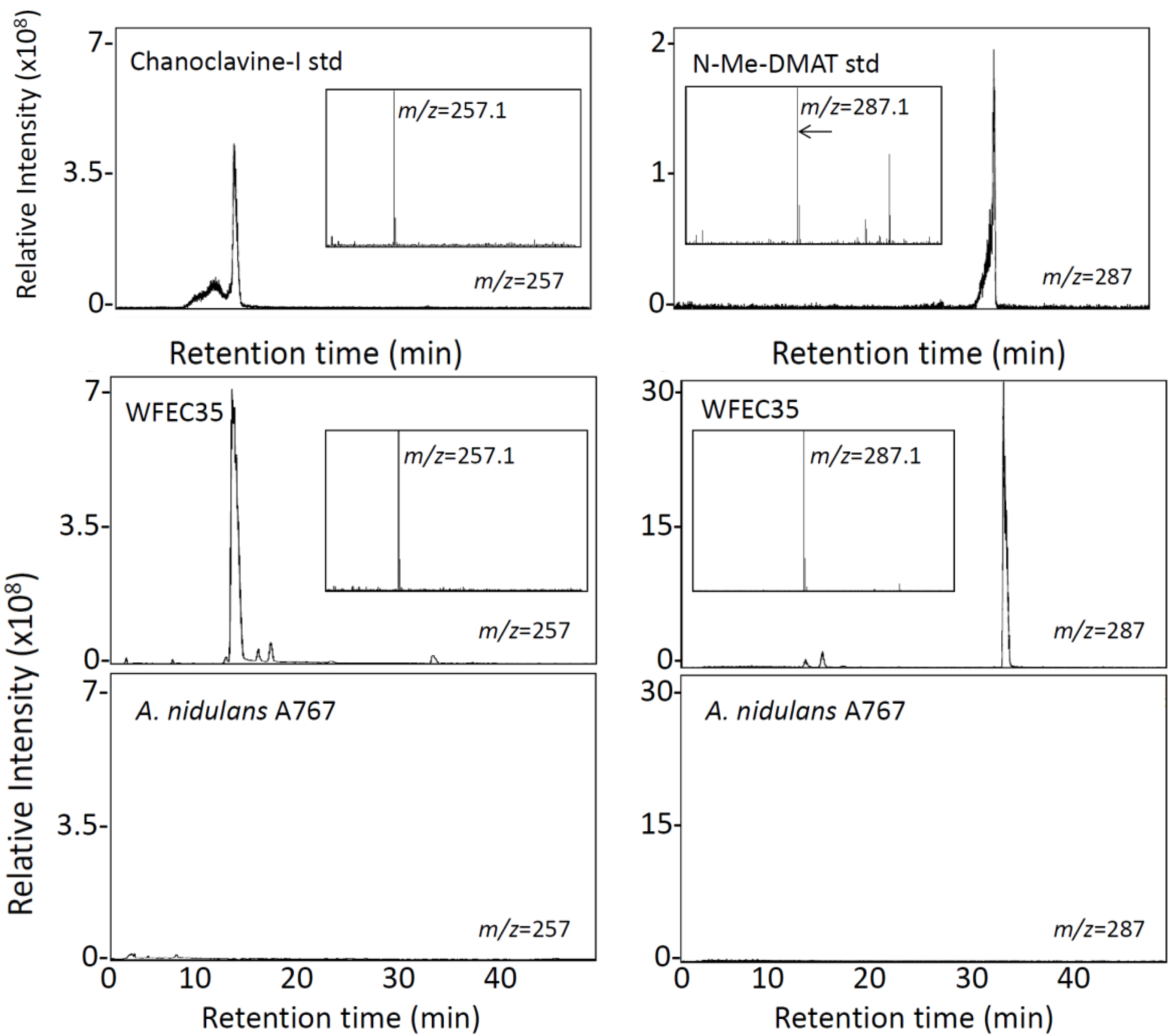
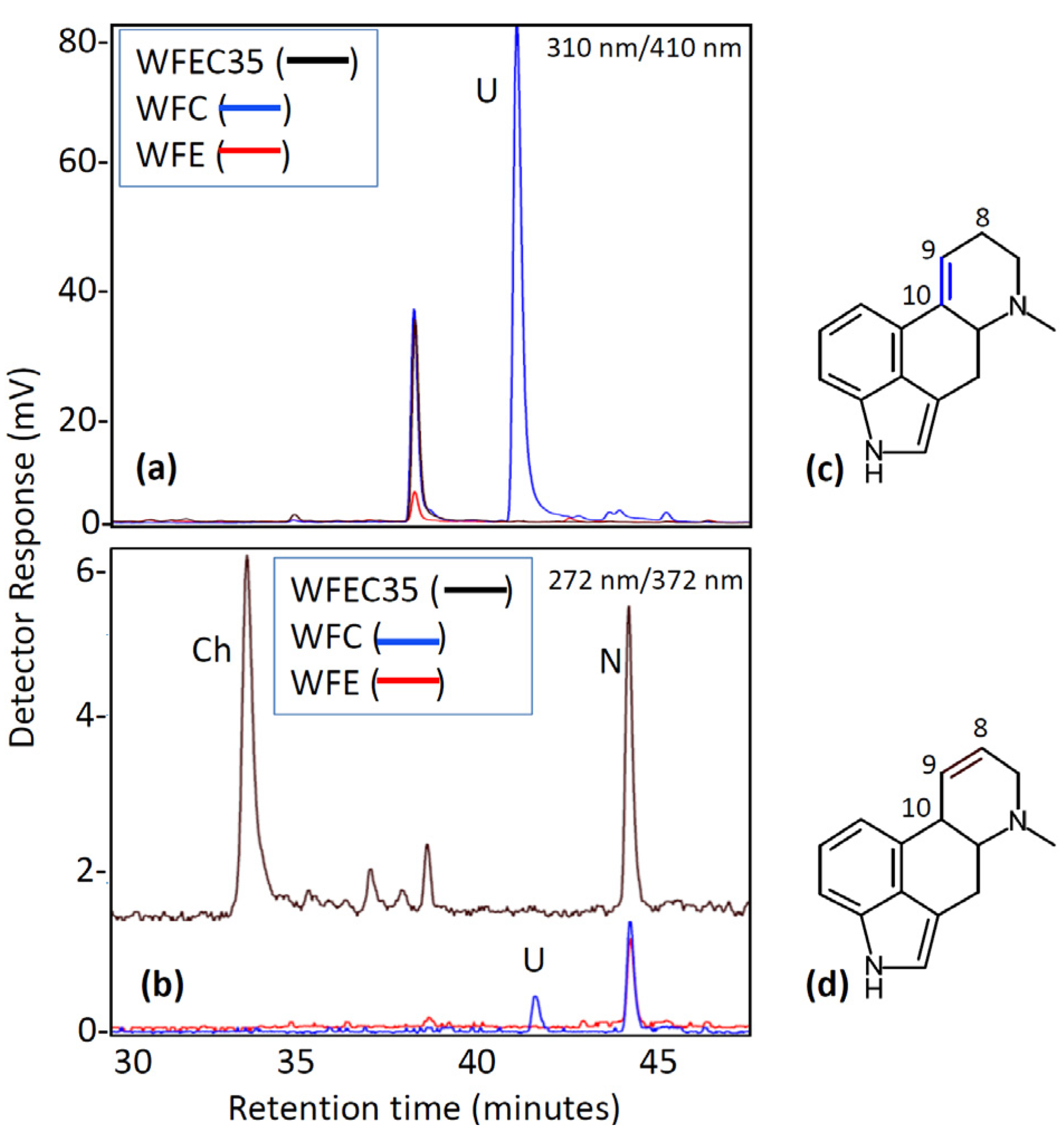
2.1. Engineering of Chanoclavine-I Producing Strains of A. nidulans
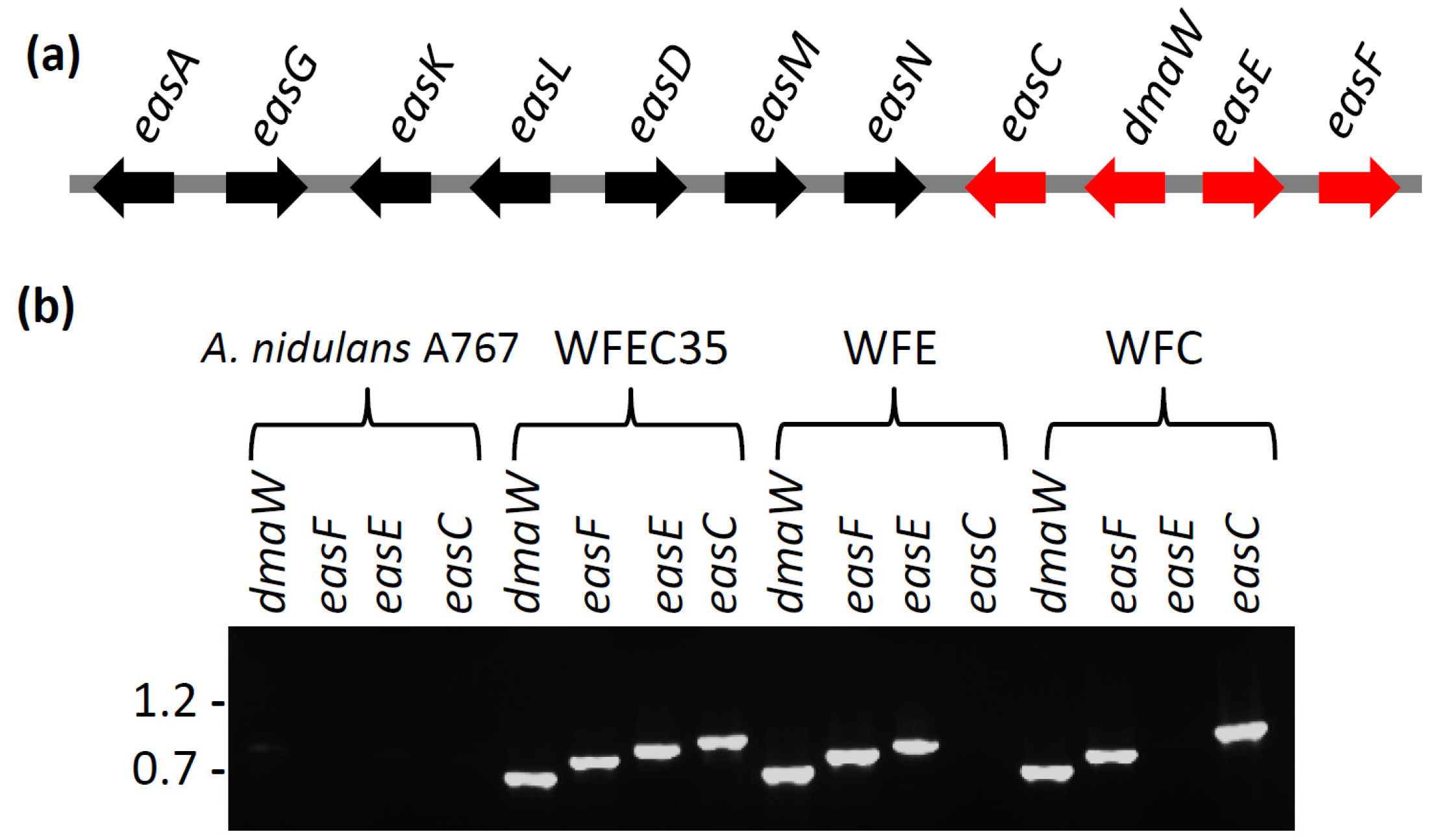

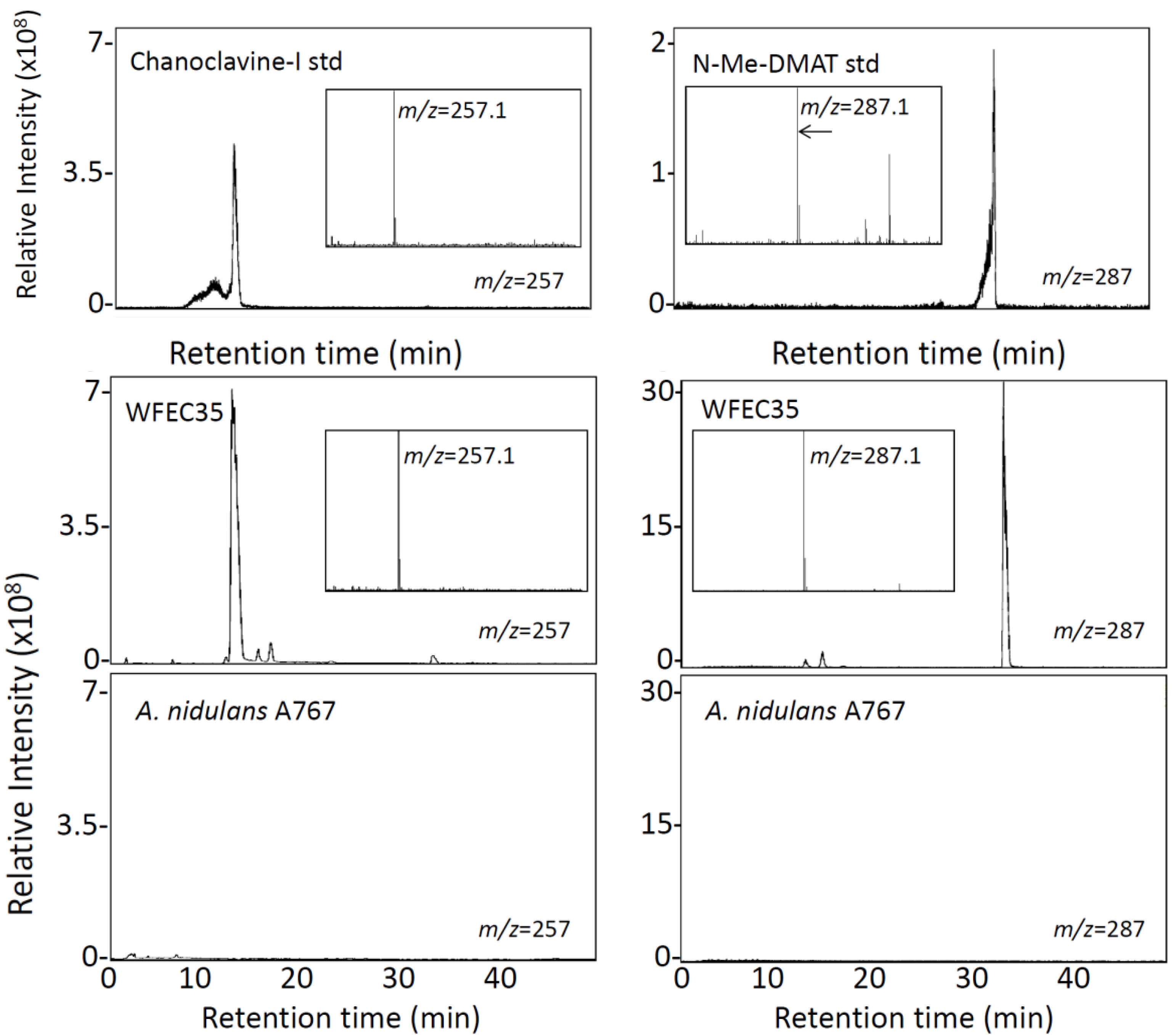
2.2. Engineering of A. nidulans Strains Expressing Subsets of Early Pathway Genes

3. Discussion
4. Experimental Section
4.1. Fungal Strains and Growth Conditions
4.2. DNA Manipulations
4.3. Fungal Transformation and Screening
4.4. HPLC Analyses
4.5. LC-MS Analyses
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Lorenz, N.; Haarmann, T.; Pažoutová, S.; Jung, M.; Tudzynski, P. The ergot alkaloid gene cluster: Functional analyses and evolutionary aspects. Phytochemistry 2009, 70, 1822–1832. [Google Scholar] [CrossRef]
- Panaccione, D.G. Ergot Alkaloids. In The Mycota, Industrial Applications, 2nd; Hofrichter, M., Ed.; Springer-Verlag: Berlin, Germany, 2010; Volume X, pp. 195–214. [Google Scholar]
- Wallwey, C.; Li, S.-M. Ergot alkaloids: Structure diversity, biosynthetic gene clusters and functional proof of biosynthetic genes. Nat. Prod. Rep. 2011, 28, 496–510. [Google Scholar] [CrossRef]
- Schardl, C.L.; Young, C.A.; Hesse, U.; Amyotte, S.G.; Andreeva, K.; Calie, P.J.; Fleetwood, D.J.; Haws, D.C.; Moore, N.; Oeser, B.; et al. Plant-symbiotic fungi as chemical engineers: Multi-genome analysis of the Clavicipitaceae reveals dynamics of alkaloid loci. PLoS Genet. 2013, in press. [Google Scholar]
- Kozikowski, A.P.; Chen, C.; Wu, J.-P.; Shibuya, M.; Kim, C.-G.; Floss, H.G. Probing ergot alkaloid biosynthesis: Intermediates in the formation of ring C. J. Am. Chem. Soc. 1993, 115, 2482–2488. [Google Scholar]
- Wang, J.; Machado, C.; Panaccione, D.G.; Tsai, H.-F.; Schardl, C.L. The determinant step in ergot alkaloid biosynthesis by an endophyte of perennial ryegrass. Fungal Genet. Biol. 2004, 41, 189–198. [Google Scholar] [CrossRef]
- Rigbers, O.; Li, S.-M. Ergot alkaloid biosynthesis in Aspergillus fumigatus: Overproduction and biochemical characterization of a 4-dimethylallyltryptophan N-methyltransferase. J. Biol. Chem. 2008, 283, 26859–26868. [Google Scholar] [CrossRef]
- Lorenz, N.; Olšovská, J.; Šulc, M.; Tudzynski, P. Alkaloid cluster gene ccsA of the ergot fungus Claviceps purpurea encodes chanoclavine I synthase, a flavin adenine dinucleotide-containing oxidoreductase mediating the transformation of N-methyl-dimethylallyltryptophan to chanoclavine I. Appl. Environ. Microbiol. 2010, 76, 1822–1830. [Google Scholar] [CrossRef]
- Goetz, K.E.; Coyle, C.M.; Cheng, J.Z.; O’Connor, S.E.; Panaccione, D.G. Ergot cluster-encoded catalase is required for synthesis of chanoclavine-I in Aspergillus fumigatus. Curr. Genet. 2011, 57, 201–211. [Google Scholar] [CrossRef]
- Unsöld, I.A.; Li, S.-M. Overproduction, purification and characterization of FgaPT2, a dimethylallyltryptophan synthase from Aspergillus fumigatu. Microbiology 2005, 151, 1499–1505. [Google Scholar] [CrossRef]
- Coyle, C.M.; Panaccione, D.G. An ergot alkaloid biosynthesis gene and clustered hypothetical genes from Aspergillus fumigatus. Appl. Environ. Microbiol. 2005, 71, 3112–3118. [Google Scholar] [CrossRef]
- Latgé, J.-P. Aspergillus fumigatus and aspergillosis. Clin. Microbiol. Rev. 1999, 12, 310–350. [Google Scholar]
- Coyle, C.M.; Kenaley, S.C.; Rittenour, W.R.; Panaccione, D.G. Association of ergot alkaloids with conidiation in Aspergillus fumigatus. Mycologia 2007, 99, 804–811. [Google Scholar] [CrossRef]
- Twumasi-Boateng, K.; Yu, Y.; Chen, D.; Gravelat, F.N.; Nierman, W.C.; Sheppard, D.C. Transcriptional profiling identifies a role for BrlA in the response to nitrogen depletion and for StuA in the regulation of secondary metabolite clusters in Aspergillus fumigatus. Eukaryot. Cell 2009, 8, 104–115. [Google Scholar] [CrossRef]
- Panaccione, D.G.; Ryan, K.L.; Schardl, C.L.; Florea, S. Analysis and modification of ergot alkaloid profiles in fungi. Method. Enzymol. 2012, 515, 267–290. [Google Scholar] [CrossRef]
- Chiang, Y.M.; Chang, S.L.; Oakley, B.R.; Wang, C.C.C. Recent advances in awakening silent biosynthetic gene clusters and linking orphan clusters to natural products in microorganisms. Curr. Opin. Chem. Biol. 2011, 15, 137–143. [Google Scholar] [CrossRef]
- Hunter, S.H.; Provasoli, L.; Schatz, A.; Haskins, C.P. Some approaches to the study of the role of metals in the metabolism of microorganisms. Am. Philos. Soc. Proc. 1950, 94, 152–170. [Google Scholar]
- Richards, E.; Reichardt, M.; Rogers, S. Preparation of genomic DNA from plant tissue. Curr. Protoc. Mol. Biol. 2001, 27, 2.3.1–2.3.7. [Google Scholar]
- Coyle, C.M.; Cheng, J.Z.; O’Connor, S.E.; Panaccione, D.G. An old yellow enzyme gene controls the branch point between Aspergillus fumigatus and Claviceps purpurea ergot alkaloid pathways. Appl. Environ. Microbiol. 2010, 76, 3898–3903. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ryan, K.L.; Moore, C.T.; Panaccione, D.G. Partial Reconstruction of the Ergot Alkaloid Pathway by Heterologous Gene Expression in Aspergillus nidulans. Toxins 2013, 5, 445-455. https://doi.org/10.3390/toxins5020445
Ryan KL, Moore CT, Panaccione DG. Partial Reconstruction of the Ergot Alkaloid Pathway by Heterologous Gene Expression in Aspergillus nidulans. Toxins. 2013; 5(2):445-455. https://doi.org/10.3390/toxins5020445
Chicago/Turabian StyleRyan, Katy L., Christopher T. Moore, and Daniel G. Panaccione. 2013. "Partial Reconstruction of the Ergot Alkaloid Pathway by Heterologous Gene Expression in Aspergillus nidulans" Toxins 5, no. 2: 445-455. https://doi.org/10.3390/toxins5020445