Evolution Stings: The Origin and Diversification of Scorpion Toxin Peptide Scaffolds





Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Toxin Type | Generalised Bioactivity | Representative Key References |
| Cytotoxin “bradykinin potentiating peptide family” | Potent cytotoxins leading to cell haemolysis and death. Documented antimicrobial activity is a side effect due to generalised cell-killing. C-terminal coil region contributes additional activity of bradykinin potentiation. | [25,26,27] |
| Cytotoxin “NDBP 5 linear peptide family” | Potent cytotoxins leading to cell haemolysis and death. Documented antimicrobial activity is a side effect due to generalised cell-killing. | [28,29,30,31] |
| Cytoxin–“Short cationic antimicrobial peptide family” | Potent cytotoxins leading to cell haemolysis and death. Documented antimicrobial activity is a side effect due to generalised cell-killing. | [30,32,33,34] |
| Anionic | uncharacterised | [27,32,35,36,37,38] |
| Glycine-rich | uncharacterised | [27] |
| CSαβ | ||
| CSαβ ‘alpha’ | Prevent inactivation by binding to sodium channel receptor site 3 | [13,39,40,41,42,43,44,45] |
| CSαβ ‘beta’ | Promote activation by binding to sodium channel receptor site 4 | |
| CSαβ ‘lipo’ | Lipolysis | |
| CSαβ ‘chlorotoxin’ | Chloride channel | |
| CSαβ ‘short-chain’ | Potassium channel | |
| CSαβ ‘long-chain’ | Potassium channel and antimicrobial | |
| CSαβ ‘scorpine’ | Potassium channel and antimicrobial | |
| ICK/DDH | ||
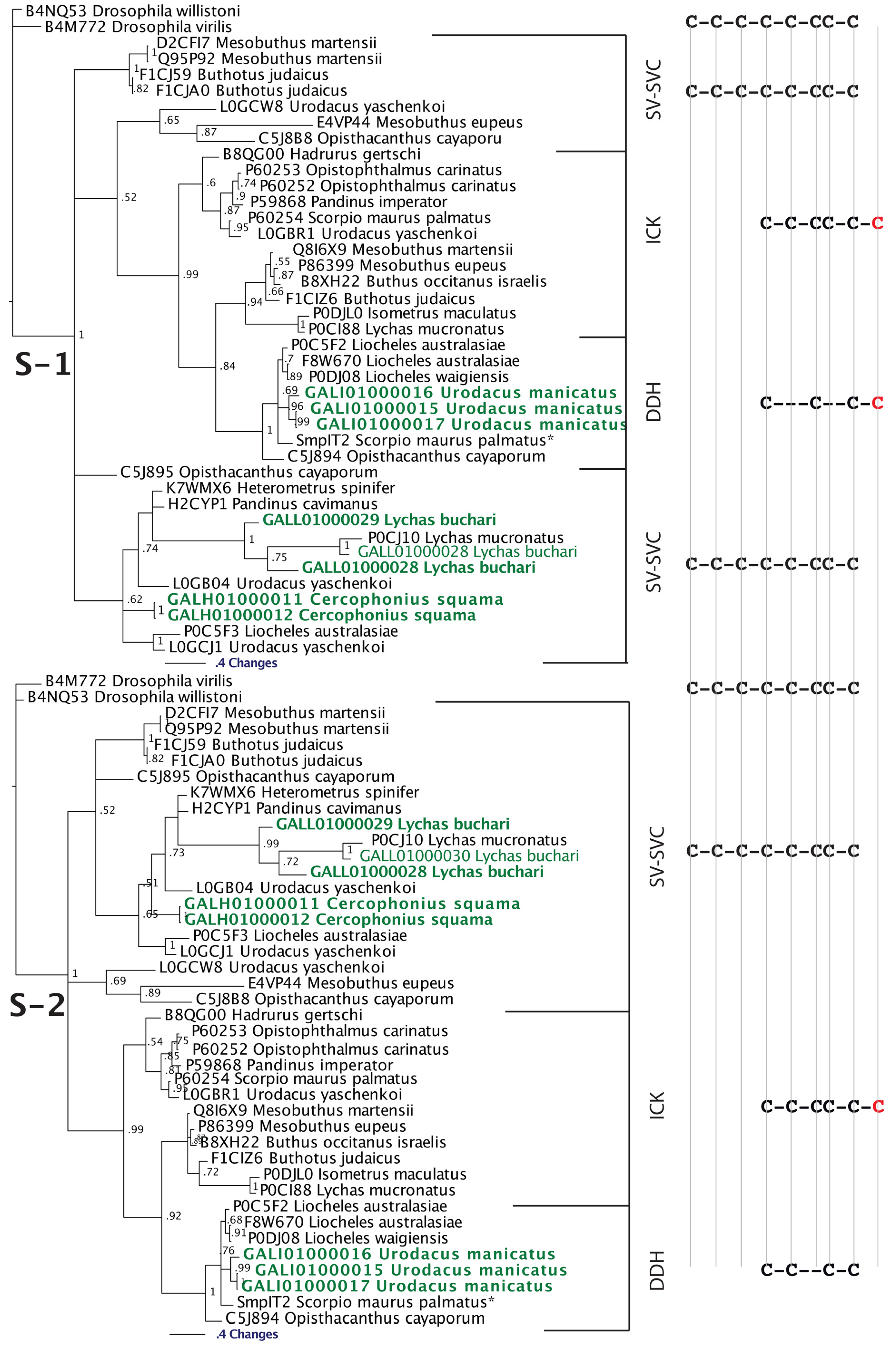
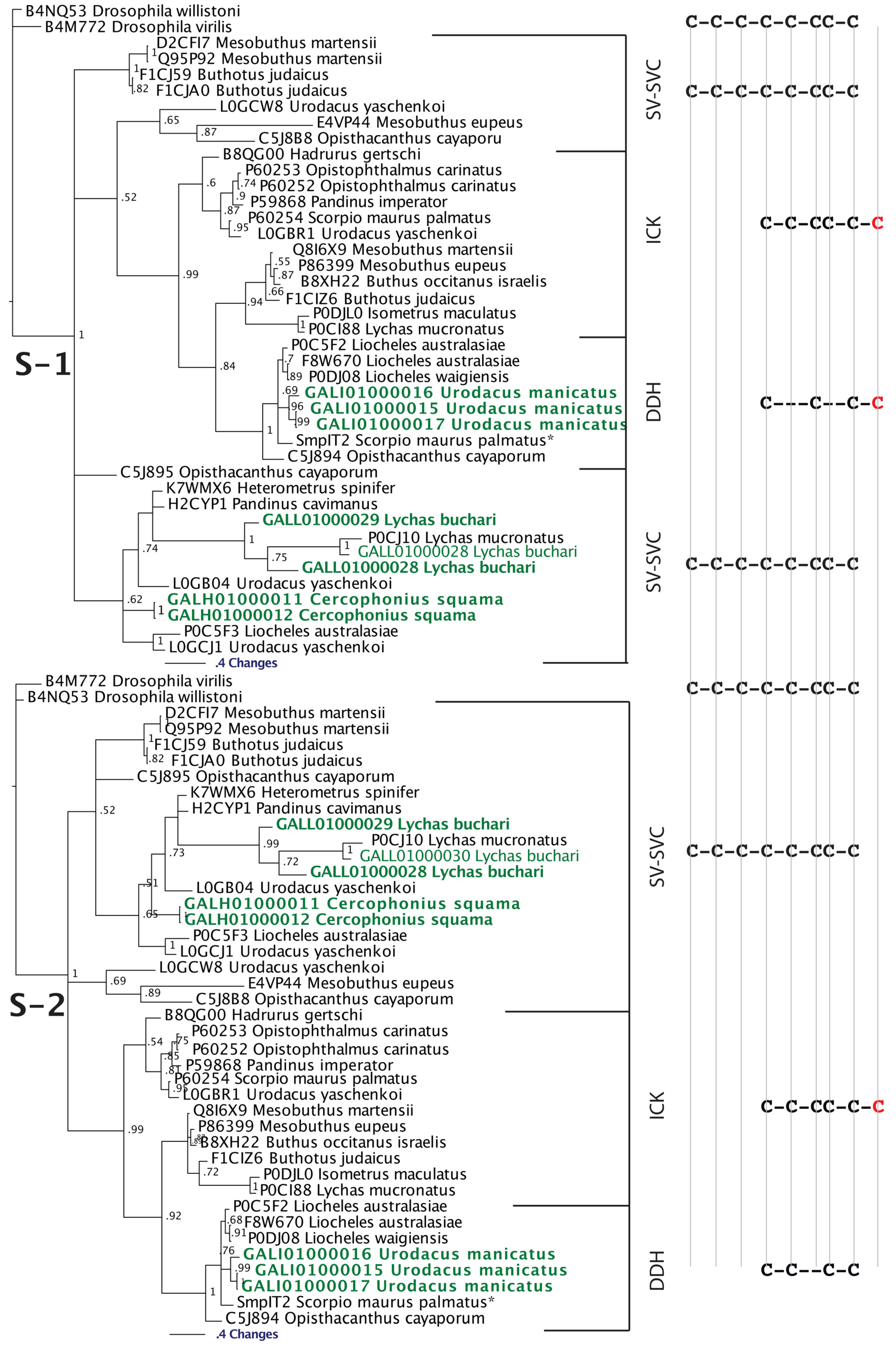
| SV-SVC | Functionally uncharacterised | [27,46,47,48] |
| ICK | Strong agonist of ryanodine receptors (calcium release channels). Induces voltage- and concentration-dependent subconductance states in both skeletal (RYR1 and RYR3) and cardiac (RYR2) ryanodine receptors by binding to a single, cytosolically accessible site different from the ryanodine binding site. Enhances calcium release. A derivative (GU187948) inhibits Shaker K+ channels | [27,42,49,50,51,52,53,54,55,56,57,58,59,60,61] |
| DDH | Types such as P60252 from Opistophthalmus carinatus, P59868 from Pandinus imperator and B8QG00 from Hadrurus gertschi potently and reversibly modify channel gating behavior of the type 1 ryanodine receptor (RYR1) by inducing prominent subconductance behavior. Binds a different site as ryanodine. Others with different cysteine frameworks such as P0DJ08 Liocheles waigiensis are insect-selective toxins. Provokes a dose-dependent contractile paralysis in crickets and blowfly larvae, followed by death. | [48,62,63,64,65,66] |
2. Results and Discussion
2.1. Transcriptomics of Scorpion Venom-Glands Highlights the True Diversity of Scorpion Venom-Arsenal
| Species | Australobuthus xerlomnion | Cercophonius squama | Isometroides vescus | Lychas buchari | Urodacus manicatus | |
|---|---|---|---|---|---|---|
| Toxin Type | ||||||
| Cytotoxin “bradykinin potentiating peptide family” | X | X | ||||
| Cytotoxin “NDBP 5 linear peptide family” | X | X | ||||
| Cytotoxin–“Short cationic antimicrobial peptide family | X | X | ||||
| Anionic | X | X | X | X | ||
| CSαβ | ||||||
| CSαβ ‘alpha’ NaV | ||||||
| CSαβ-‘beta’ NaV | X | |||||
| CSαβ ‘lipo’ NaV | X | X | X | |||
| CSαβ ‘chlorotoxin’ | ||||||
| CSαβ-‘short-chain’ KV | X | X | ||||
| CSαβ-‘long-chain’ KV | X | X | X | X | X | |
| CSαβ ‘scorpine’ KV | X | X | ||||
| Glycine | X | |||||
| ICK/DDH | ||||||
| ICK | X | |||||
| DDH | X | |||||
| SV-SVC | X | |||||
2.2. Implications for the Origin and Evolution of Scorpion Toxin Scaffolds
2.2.1. Apotypic (Derived) NaV-CSα/β Scaffolds

2.2.2. SV-SVCs: Putative Plesiotypic ICK-DDH Scaffold
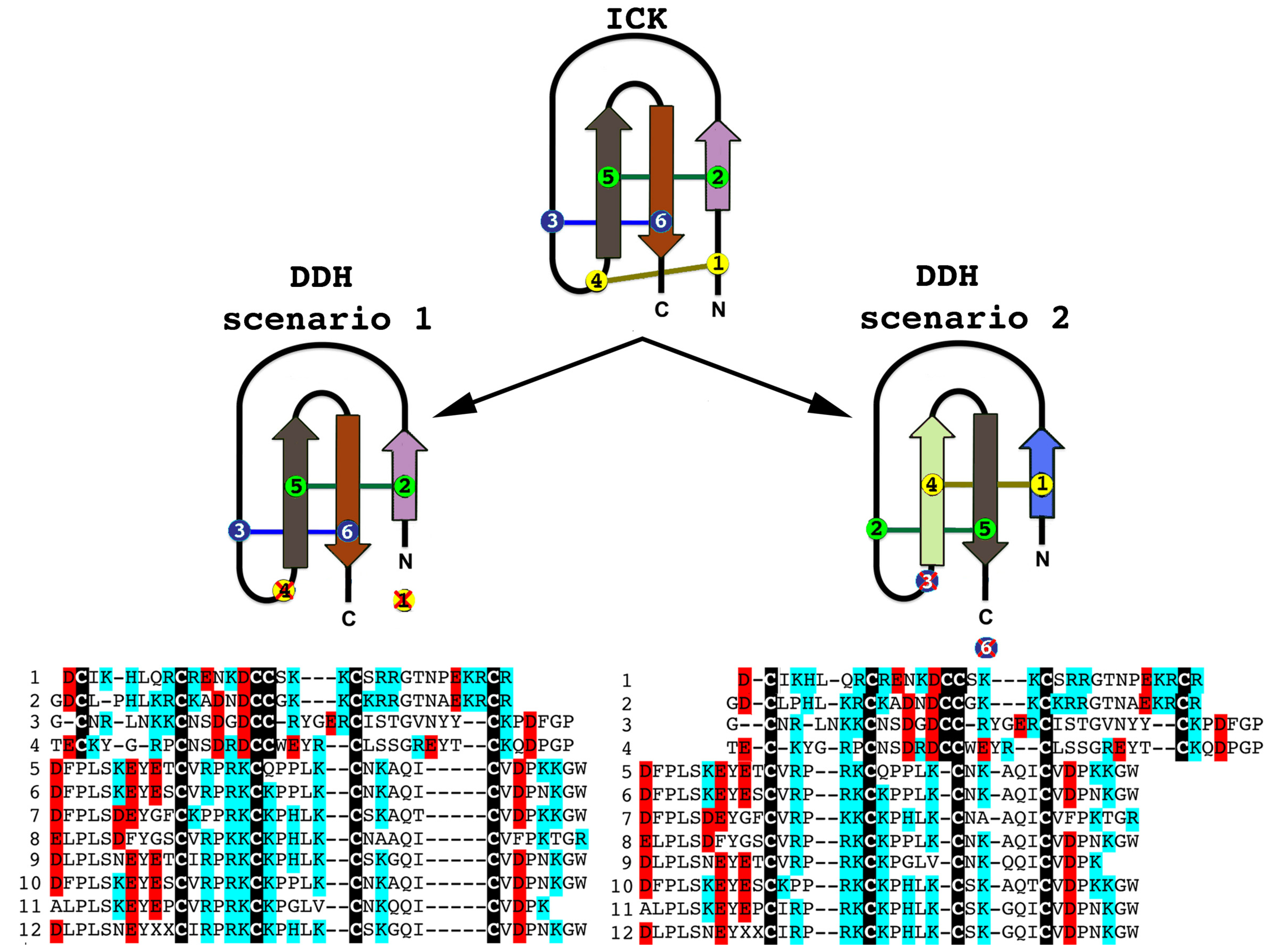
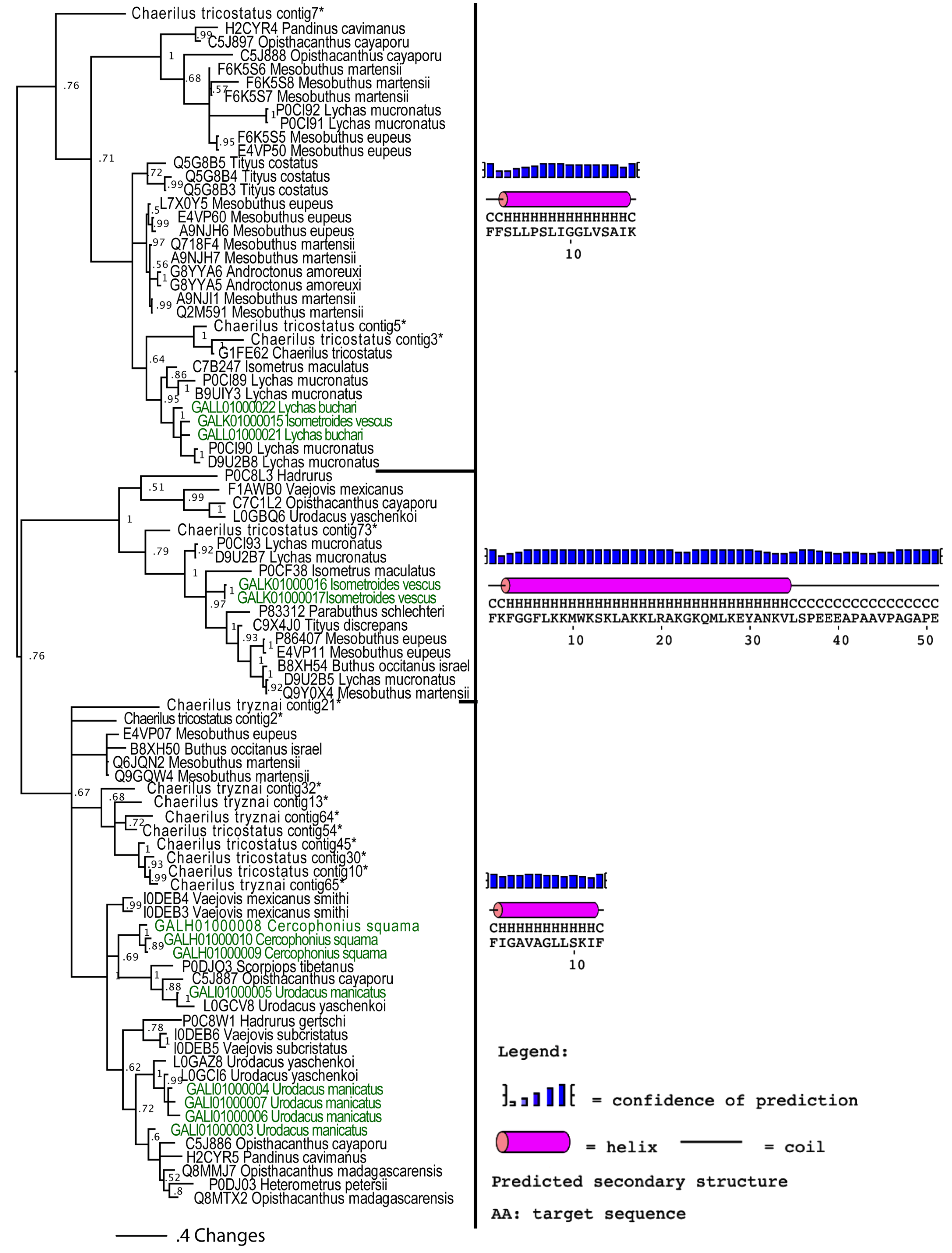
2.2.3. Putative Common Origin of Cytotoxic Peptides

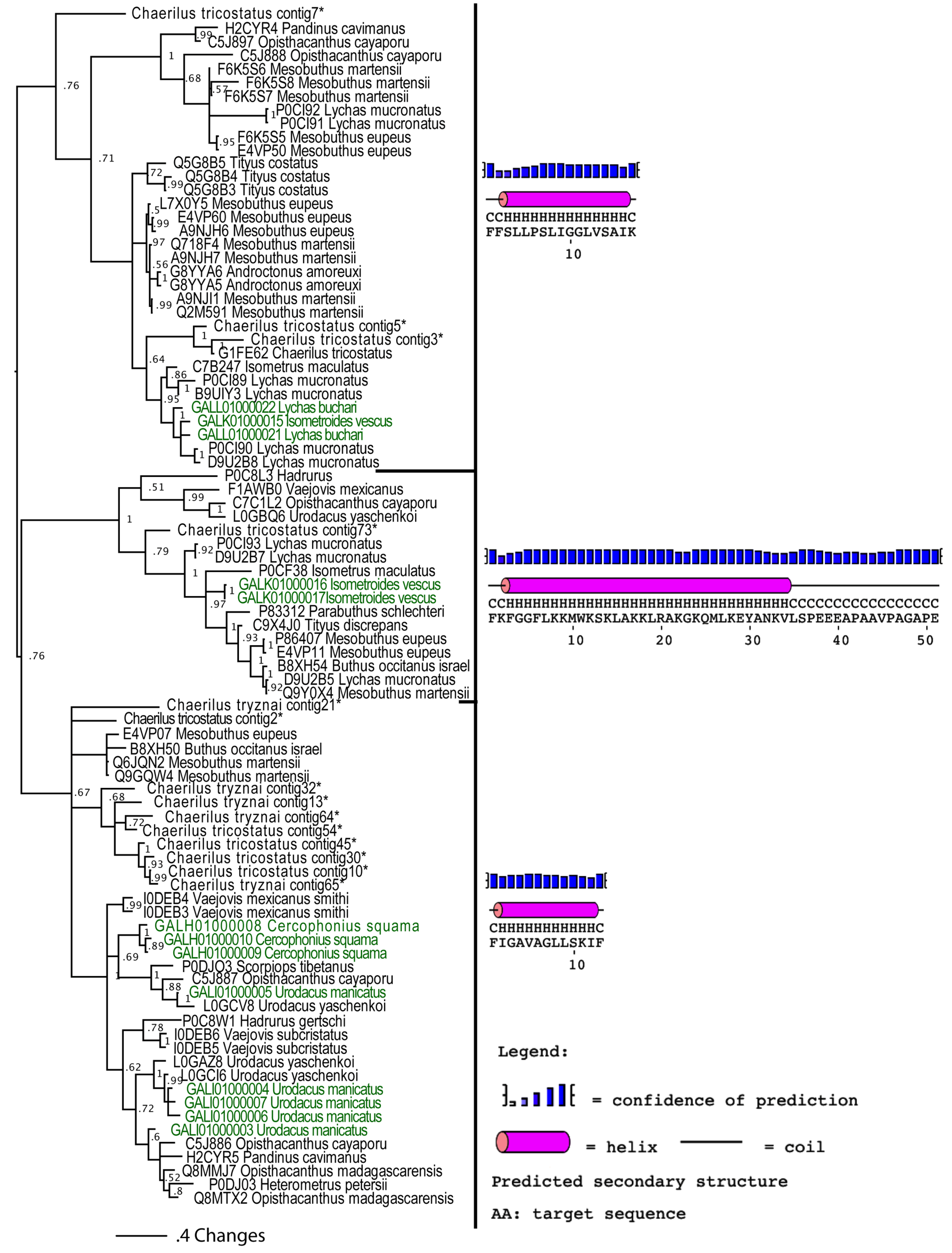
2.2.4. Unravelling the Dynamic Molecular Evolution of Scorpion Toxins
| FUBARa | MEMEb | BSRc | PAMLd | ||
|---|---|---|---|---|---|
| M8 | M2a | ||||
| Plesiotypic NaV-CSα/β | ω > 1e : 2
ω < 1f : 11 ω > 1e : 0 ω < 1f : 20 | 5 | 4 | 6 | 5 |
| (3 + 3) | (3 + 2) | ||||
| 1.63 | 1.76 | ||||
| Lipolytic NaV-CSα/β | 2 | 1 | 2 | 5 | |
| (2 + 0) | (2 + 3) | ||||
| 0.76 | 1.28 | ||||
| α-NaV-CSα/β | ω > 1e : 5
ω < 1f : 33 | 19 | 14 | 5 | 5 |
| (0 + 5) | (1 + 4) | ||||
| 0.54 | 0.8 | ||||
| β-NaV-CSα/β | ω > 1e : 0
ω < 1f : 37 | 18 | 16 | 4 | 8 |
| (2 + 2) | (6 + 2) | ||||
| 0.53 | 1.03 | ||||
| Long-KV-CSα/β | ω > 1e : 0
ω < 1f : 57 | 10 | 2 | 0 | 0 |
| 0.29 | 0.9 | ||||
| Short-KV-CSα/β | ω > 1e : 1
ω < 1f : 28 | 4 | 1 | 0 | 5 |
| (3 + 2) | |||||
| 0.4 | 1.02 | ||||
| ClV-CSα/β | ω > 1e : 0
ω < 1f : 10 | 5 | 2 | 2 | 0 |
| (0 + 2) | |||||
| 0.6 | 0.62 | ||||
| FUBARa | MEMEb | BSRc | PAMLd | ||
|---|---|---|---|---|---|
| M8 | M2a | ||||
| SV-SVC | ω > 1e : 2
ω < 1f : 32 | 3 | 0 | 0 | 0 |
| 0.34 | 0.68 | ||||
| ICK | ω > 1e : 0
ω < 1f : 12 | 0 | 0 | 0 | 0 |
| 0.34 | 0.49 | ||||
| DDH | ω > 1e : 0
ω < 1f : 6 | 1 | 0 | 0 | 0 |
| 0.32 | 0.32 | ||||
| AMP | ω > 1e : 1
ω < 1f : 34 | 2 | 0 | 1 | 0 |
| (0 + 1) | |||||
| 0.33 | 0.34 | ||||
| Linear | ω > 1e : 0
ω < 1f : 45 | 3 | 0 | 0 | 0 |
| 0.27 | 0.42 | ||||
| Bradykinin | ω > 1e : 0
ω < 1f : 36 | 2 | 0 | 0 | 0 |
| 0.2 | 0.22 | ||||
| Anionic | ω > 1e : 0
ω < 1f : 33 | 0 | 0 | 0 | 0 |
| 0.22 | 0.27 | ||||
| Glycine-rich | ω > 1e : 0
ω < 1f : 22 | 1 | 1 | 0 | 0 |
| 0.14 | 0.35 | ||||

2.2.5. Focal Mutagenesis Shapes the Molecular Evolution of Scorpion CSα/β Toxin Families
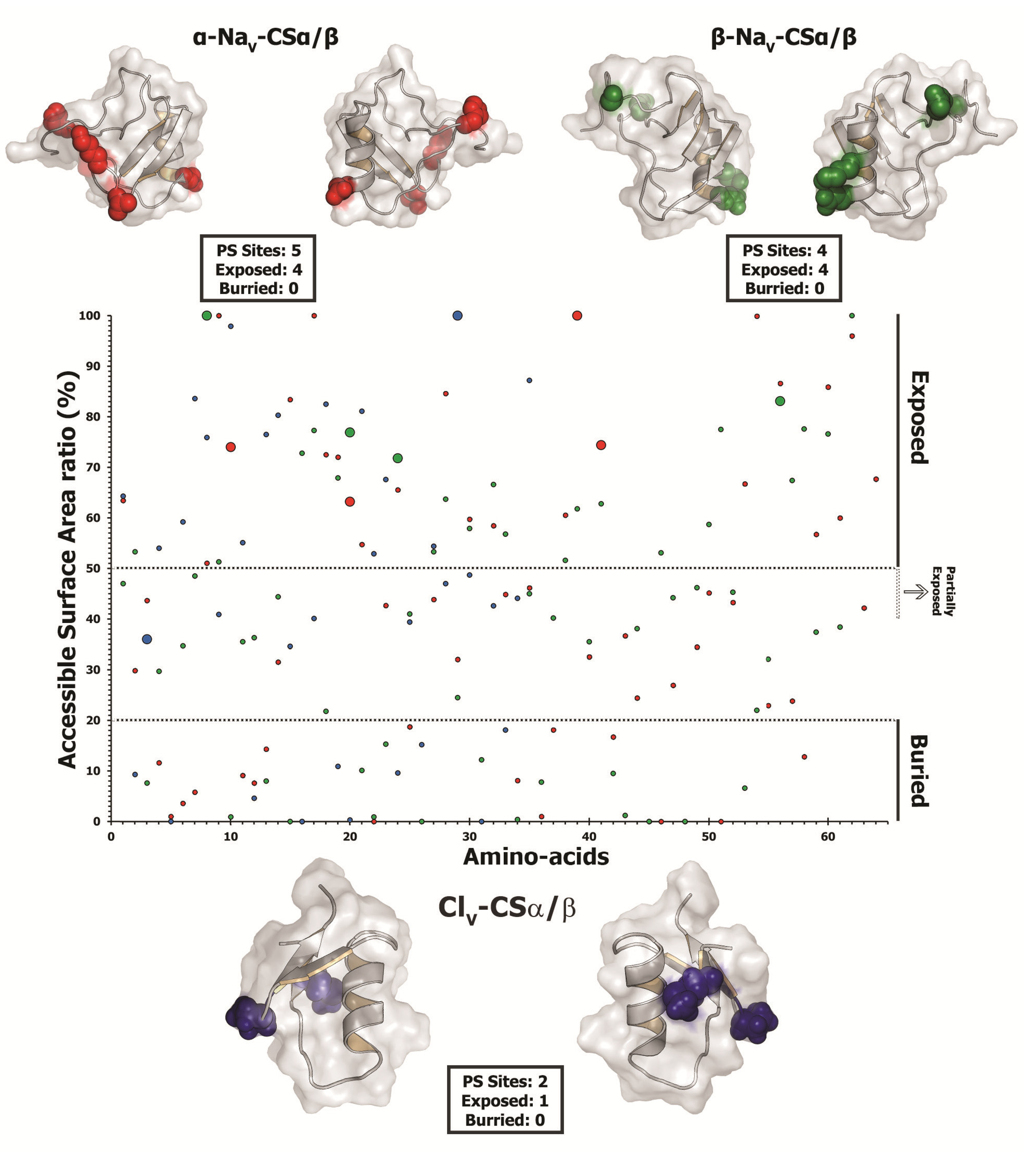
| Site | CodeML | TreeSAAP | ASA(%) | |||
|---|---|---|---|---|---|---|
| Codon | AA | M2a a | M8 b | Property c | Magnitude d | |
| Long 3CC | ||||||
| 33 | D | 5.858 ± 1.374 | 4.775 ± 1.168 | Pb, αC | 7, 8 | |
| (0.997)** | (0.998)** | |||||
| 42 | N | 5.752 ± 1.531 | 4.726 ± 1.233 | V° | 6 | |
| (0.975)* | (0.986)* | |||||
| 47 | K | 5.827 ± 1.427 | 4.761 ± 1.189 | V° | 6 | |
| (0.990)** | (0.995)** | |||||
| 51 | D | 5.703 ± 1.587 | 4.707 ± 1.253 | V° | 6 | |
| (0.966)* | (0.982)* | |||||
| 65 | Y | 5.870 ± 1.356 | 4.78 ± 1.162 | Pb, V°, αC, ESM | 7, 6, 6, 6 | |
| (0.999)** | (0.999)** | |||||
| 75 | D | 5.580 ± 1.740 | 4.643 ± 1.331 | Pb | 7 | |
| −0.941 | (0.966)* | |||||
| Lipolysis Activating | ||||||
| 61 | H | 4.678 ± 0.900 | 3.042 ± 0.857 | - | - | - |
| (1.000)** | (0.997)** | |||||
| 81 | S | 4.672 ± 0.914 | 3.026 ± 0.867 | αC | 6 | - |
| (0.998)** | (0.991)** | |||||
| Alpha NaScTx | ||||||
| 29 | E | 2.427 ± 0.324 | 1.471 ± 0.136 | pHi, El , Mv, αC, ESM | 8, 8, 7, 7, 6 | 74 |
| (0.952)* | (0.950)* | Exposed | ||||
| 40 | E | 2.463 ± 0.233 | 1.473 ± 0.133 | pHi, El, Mv, αC , ESM | 8, 8, 7, 7, 6 | 63.2 |
| (0.976)* | (0.955)* | Exposed | ||||
| 59 | G | 2.464 ± 0.232 | 1.478 ± 0.121 | Mv, ESM | 7, 6 | 100 |
| (0.977)* | (0.962)* | Exposed | ||||
| 61 | K | 2.491 ± 0.122 | 1.492 ± 0.083 | El , Mv, ESM | 8, 7, 6 | 74.4 |
| (0.994)** | (0.985)* | Exposed | ||||
| 85 | R | 2.439 ± 0.297 | 1.472 ± 0.135 | Mv | 7, 6 | |
| (0.960)* | (0.952)* | |||||
| Beta NaScTx | ||||||
| 27 | S | 2.500 ± 0.001 | 1.498 ± 0.035 | Pa, K°, pK! | 6, 6, 8 | 100 |
| (1.0)** | (0.996)** | Exposed | ||||
| 39 | E | 2.500 ± 0.005 | 1.487 ± 0.084 | Pa, K° | 6, 8 | 76.9 |
| (1.0)** | (0.978)* | Exposed | ||||
| 43 | K | 2.500 ± 0.001 | 1.497 ± 0.043 | Pa, K° | 6, 8 | 71.8 |
| (1.0)** | (0.994)** | Exposed | ||||
| 72 | L | 2.500 ± 0.003 | 1.493 ± 0.062 | K°, pK!, F | 8, 8, 8 | 83.1 |
| (1.0)** | (0.988)* | Exposed | ||||
| Chloride ion-channel Toxin | ||||||
| 27 | P | 1.833 ± 0.844 | 1.521 ± 0.223 | αC, Ra | 6, 7 | 36 |
| -0.814 | (0.984)* | NA | ||||
| 53 | Y | 1.792 ± 0.963 | 1.508 ± 0.244 | Ra | 7 | 100 |
| −0.737 | (0.966)* | Exposed | ||||
3. Experimental Section
3.1. Specimens
3.2. Transcriptome Library Construction
3.3. Sequence Retrieval and Alignment
3.4. Phylogenetics
3.5. Test for Recombination
3.6. Selection Analyses
3.7. Structural Analyses
4. Conclusion
Acknowledgements
Conflicts of Interest
References
- Sunagar, K.; Johnson, W.E.; O’Brien, S.J.; Vasconcelos, V.; Antunes, A. Evolution of CRISPs associated with toxicoferan-reptilian venom and mammalian reproduction. Mol. Biol. Evol. 2012, 29, 1807–1822. [Google Scholar] [CrossRef]
- Brust, A.; Sunagar, K.; Undheim, E.A.B.; Vetter, I.; Yang, D.C.; Casewell, N.R.; Jackson, T.N.W.; Koludarov, I.; Alewood, P.F.; Hodgson, W.C.; Lewis, R.J.; King, G.F.; Antunes, A.; Hendrikx, I.; Fry, B.G. Differential evolution and neofunctionalization of snake venom metalloprotease domains. Mol. Cell. Proteomics 2013, 12, 651–663. [Google Scholar] [CrossRef]
- Casewell, N.R.; Wagstaff, S.C.; Harrison, R.A.; Renjifo, C.; Wuster, W. Domain loss facilitates accelerated evolution and neofunctionalization of duplicate snake venom metalloproteinase toxin genes. Mol. Biol. Evol. 2011, 28, 2637–2649. [Google Scholar] [CrossRef]
- Fry, B.G.; Wüster, W.; Kini, R.M.; Brusic, V.; Khan, A.; Venkataraman, D.; Rooney, A.P. Molecular evolution and phylogeny of elapid snake venom three-finger toxins. J. Mol. Evol. 2003, 57, 110–129. [Google Scholar] [CrossRef]
- Fry, B.G.; Scheib, H.; Weerd, L. Van Der; Young, B.; Mcnaughtan, J.; Ramjan, S.F.R.; Vidal, N.; Poelmann, R.E.; Norman, J.A. Evolution of an Arsenal. Mol. Cell. Proteomics 2008, 14–18. [Google Scholar]
- Kordis, D.; Gubensek, F. Adaptive evolution of animal toxin multigene families. Gene 2000, 261, 43–52. [Google Scholar] [CrossRef]
- Puillandre, N.; Watkins, M.; Olivera, B.M. Evolution of Conus peptide genes: duplication and positive selection in the A-superfamily. J. Mol. Evol. 2010, 70, 190–202. [Google Scholar] [CrossRef]
- Weinberger, H.; Moran, Y.; Gordon, D.; Turkov, M.; Kahn, R.; Gurevitz, M. Positions under positive selection--key for selectivity and potency of scorpion alpha-toxins. Mol. Biol. Evol. 2010, 27, 1025–1034. [Google Scholar] [CrossRef]
- Duda, T.F.; Lee, T. Ecological Release and Venom Evolution of a Predatory Marine Snail at Easter Island. PLoS One 2009, 4, 7. [Google Scholar]
- Barlow, A.; Pook, C.E.; Harrison, R.A.; Wüster, W. Coevolution of diet and prey-specific venom activity supports the role of selection in snake venom evolution. Proc. Biol. Sci. 2009, 276, 2443–2449. [Google Scholar] [CrossRef]
- Daltry, J.C.; Wüster, W.; Thorpe, R.S. Diet and snake venom evolution. Nature 1996, 379, 537–540. [Google Scholar] [CrossRef]
- Low, D.H.W.; Sunagar, K.; Undheim, E.A.B.; Ali, S.A.; Alagon, A.C.; Ruder, T.; Jackson, T.N.W.; Pineda Gonzalez, S.; King, G.F.; Jones, A.; Antunes, A.; Fry, B.G. Dracula’s children: Molecular evolution of vampire bat venom. J. Proteomics 2013, 89, 95–111. [Google Scholar] [CrossRef]
- Rodríguez de la Vega, R.C.; Possani, L.D.; Vidal, N. Scorpion peptides. In Handbook of Biologically Active Peptides, 2nd ed.; Kastin, A.J., Ed.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 423–429. [Google Scholar]
- Dunlop, J.A.; Selden, P.A. Calibrating the chelicerate clock: a paleontological reply to Jeyaprakash and Hoy. Exp. Appl. Acarol. 2009, 48, 183–197. [Google Scholar] [CrossRef]
- Bryson, R.W.; Riddle, B.R.; Graham, M.R.; Smith, B.T.; Prendini, L. As Old as the hills: montane scorpions in Southwestern North America reveal ancient associations between biotic diversification and landscape history. PLoS One 2013, 8, e52822. [Google Scholar]
- Yamashita, T.; Rhoads, D.D. Species Delimitation and Morphological Divergence in the Scorpion Centruroides vittatus (Say, 1821): Insights from Phylogeography. PLoS One 2013, 8, e68282. [Google Scholar] [CrossRef]
- Zeh, D.W. The Biology of Scorpions. Gary A. Polis, Ed. Stanford University Press, Stanford, CA, 1990. xxvi, 587 pp., illus. $85. Science 1990, 249, 1176–1177. [Google Scholar]
- Gantenbein, B.; Largiadèr, C.R. The phylogeographic importance of the Strait of Gibraltar as a gene flow barrier in terrestrial arthropods: a case study with the scorpion Buthus occitanus as model organism. Mol. Phylogenet. Evol. 2003, 28, 119–130. [Google Scholar] [CrossRef]
- Polis, G.A. The Biology of Scorpions; Stanford University Press: Stanford, CA, USA, 1990. [Google Scholar]
- Possani, L.D.; Merino, E.; Corona, M.; Bolivar, F.; Becerril, B. Peptides and genes coding for scorpion toxins that affect ion-channels. Biochimie 2001, 82, 861–868. [Google Scholar]
- Catterall, W.A.; Cestèle, S.; Yarov-Yarovoy, V.; Yu, F.H.; Konoki, K.; Scheuer, T. Voltage-gated ion channels and gating modifier toxins. Toxicon 2007, 49, 124–141. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez De La Vega, R.C.; Schwartz, E.F.; Possani, L.D. Mining on scorpion venom biodiversity. Toxicon 2010, 56, 1155–1161. [Google Scholar] [CrossRef]
- Klint, J.K.; Senff, S.; Rupasinghe, D.B.; Er, S.Y.; Herzig, V.; Nicholson, G.M.; King, G.F. Spider-venom peptides that target voltage-gated sodium channels: Pharmacological tools and potential therapeutic leads. Toxicon 2012, 60, 478–491. [Google Scholar] [CrossRef]
- Quintero-Hernández, V.; Jiménez-Vargas, J.M.; Gurrola, G.B.; Valdivia, H.H.; Possani, L.D. Scorpion venom components that affect ion-channels function. Toxicon 2013. [Google Scholar] [CrossRef]
- Miyashita, M.; Sakai, A.; Matsushita, N.; Hanai, Y.; Nakagawa, Y.; Miyagawa, H. A novel amphipathic linear peptide with both insect toxicity and antimicrobial activity from the venom of the scorpion Isometrus maculatus. Biosci. Biotechnol. Biochem. 2010, 74, 364–369. [Google Scholar] [CrossRef]
- Zeng, X.-C.; Wang, S.; Nie, Y.; Zhang, L.; Luo, X. Characterization of BmKbpp, a multifunctional peptide from the Chinese scorpion Mesobuthus martensii Karsch: gaining insight into a new mechanism for the functional diversification of scorpion venom peptides. Peptides 2012, 33, 44–51. [Google Scholar] [CrossRef]
- Ruiming, Z.; Yibao, M.; Yawen, H.; Zhiyong, D.; Yingliang, W.; Zhijian, C.; Wenxin, L. Comparative venom gland transcriptome analysis of the scorpion Lychas mucronatus reveals intraspecific toxic gene diversity and new venomous components. BMC Genomics 2010, 11, 452. [Google Scholar] [CrossRef]
- Cao, L.; Li, Z.; Zhang, R.; Wu, Y.; Li, W.; Cao, Z. StCT2, a new antibacterial peptide characterized from the venom of the scorpion Scorpiops tibetanus. Peptides 2012, 36, 213–220. [Google Scholar] [CrossRef]
- Ramírez-Carreto, S.; Quintero-Hernández, V.; Jiménez-Vargas, J.M.; Corzo, G.; Possani, L.D.; Becerril, B.; Ortiz, E. Gene cloning and functional characterization of four novel antimicrobial-like peptides from scorpions of the family Vaejovidae. Peptides 2012, 34, 290–295. [Google Scholar] [CrossRef]
- Almaaytah, A.; Zhou, M.; Wang, L.; Chen, T.; Walker, B.; Shaw, C. Antimicrobial/cytolytic peptides from the venom of the North African scorpion, Androctonus amoreuxi: Biochemical and functional characterization of natural peptides and a single site-substituted analog. Peptides 2012, 35, 291–299. [Google Scholar] [CrossRef]
- Yan, R.; Zhao, Z.; He, Y.; Wu, L.; Cai, D.; Hong, W.; Wu, Y.; Cao, Z.; Zheng, C.; Li, W. A new natural α-helical peptide from the venom of the scorpion Heterometrus petersii kills HCV. Peptides 2011, 32, 11–19. [Google Scholar] [CrossRef]
- Zeng, X.-C.; Wang, S.-X.; Zhu, Y.; Zhu, S.-Y.; Li, W.-X. Identification and functional characterization of novel scorpion venom peptides with no disulfide bridge from Buthus martensii Karsch. Peptides 2004, 25, 143–150. [Google Scholar]
- Dai, C.; Ma, Y.; Zhao, Z.; Zhao, R.; Wang, Q.; Wu, Y.; Cao, Z.; Li, W. Mucroporin, the first cationic host defense peptide from the venom of Lychas mucronatus. Antimicrob. Agents Chemother. 2008, 52, 3967–3972. [Google Scholar] [CrossRef]
- Fan, Z.; Cao, L.; He, Y.; Hu, J.; Di, Z.; Wu, Y.; Li, W.; Cao, Z. Ctriporin, a New Anti-Methicillin-Resistant Staphylococcus aureus Peptide from the Venom of the Scorpion Chaerilus tricostatus. Antimicrob. Agents Chemother. 2011, 55, 5220–5229. [Google Scholar] [CrossRef]
- Diego-García, E.; Peigneur, S.; Clynen, E.; Marien, T.; Czech, L.; Schoofs, L.; Tytgat, J. Molecular diversity of the telson and venom components from Pandinus cavimanus (Scorpionidae Latreille 1802): Transcriptome, venomics and function. Proteomics 2012, 12, 313–328. [Google Scholar] [CrossRef]
- Luo, F.; Zeng, X.-C.; Hahin, R.; Cao, Z.-J.; Liu, H.; Li, W.-X. Genomic organization of four novel nondisulfide-bridged peptides from scorpion Mesobuthus martensii Karsch: gaining insight into evolutionary mechanism. Peptides 2005, 26, 2427–2433. [Google Scholar] [CrossRef]
- Diego-García, E.; Batista, C.V.F.; García-Gómez, B.I.; Lucas, S.; Candido, D.M.; Gómez-Lagunas, F.; Possani, L.D. The Brazilian scorpion Tityus costatus Karsch: genes, peptides and function. Toxicon 2005, 45, 273–283. [Google Scholar] [CrossRef]
- D’Suze, G.; Schwartz, E.F.; García-Gómez, B.I.; Sevcik, C.; Possani, L.D. Molecular cloning and nucleotide sequence analysis of genes from a cDNA library of the scorpion Tityus discrepans. Biochimie 2009, 91, 1010–1019. [Google Scholar] [CrossRef]
- Rodríguez De La Vega, R.C.; Possani, L.D. Overview of scorpion toxins specific for Na+ channels and related peptides: biodiversity, structure-function relationships and evolution. Toxicon 2005, 46, 831–844. [Google Scholar] [CrossRef]
- Del Río-Portilla, F.; Hernández-Marín, E.; Pimienta, G.; Coronas, F.V.; Zamudio, F.Z.; Rodríguez de la Vega, R.C.; Wanke, E.; Possani, L.D. NMR solution structure of Cn12, a novel peptide from the Mexican scorpion Centruroides noxius with a typical β-toxin sequence but with α-like physiological activity. Eur. J. Biochem. 2004, 271, 2504–2516. [Google Scholar] [CrossRef]
- Céard, B.; Martin-Eauclaire, M.-F.; Bougis, P.E. Evidence for a position-specific deletion as an evolutionary link between long- and short-chain scorpion toxins. FEBS Lett. 2001, 494, 246–248. [Google Scholar] [CrossRef]
- Zeng, X.-C.; Luo, F.; Li, W.-X. Molecular dissection of venom from Chinese scorpion Mesobuthus martensii: identification and characterization of four novel disulfide-bridged venom peptides. Peptides 2006, 27, 1745–1754. [Google Scholar] [CrossRef]
- Pedraza Escalona, M.; Possani, L.D. Scorpion beta-toxins and voltage-gated sodium channels: interactions and effects. Front. Biosci. Landmark 2013, 18, 572–587. [Google Scholar] [CrossRef]
- Diego-García, E.; Schwartz, E.F.; D’Suze, G.; González, S.A.R.; Batista, C.V.F.; García, B.I.; De La Vega, R.C.R.; Possani, L.D. Wide phylogenetic distribution of Scorpine and long-chain beta-KTx-like peptides in scorpion venoms: identification of “orphan” components. Peptides 2007, 28, 31–37. [Google Scholar] [CrossRef]
- Diego-García, E.; Abdel-Mottaleb, Y.; Schwartz, E.F.; De La Vega, R.C.R.; Tytgat, J.; Possani, L.D. Cytolytic and K+ channel blocking activities of beta-KTx and scorpine-like peptides purified from scorpion venoms. Cell. Mol. Life Sci. 2008, 65, 187–200. [Google Scholar] [CrossRef]
- Miyashita, M.; Otsuki, J.; Hanai, Y.; Nakagawa, Y.; Miyagawa, H. Characterization of peptide components in the venom of the scorpion Liocheles australasiae (Hemiscorpiidae). Toxicon 2007, 50, 428–437. [Google Scholar] [CrossRef]
- Zeng, X.-C.; Nie, Y.; Luo, X.; Wu, S.; Shi, W.; Zhang, L.; Liu, Y.; Cao, H.; Yang, Y.; Zhou, J. Molecular and bioinformatical characterization of a novel superfamily of cysteine-rich peptides from arthropods. Peptides 2013, 41, 45–58. [Google Scholar] [CrossRef]
- Silva, E.C.N.; Camargos, T.S.; Maranhão, A.Q.; Silva-Pereira, I.; Silva, L.P.; Possani, L.D.; Schwartz, E.F. Cloning and characterization of cDNA sequences encoding for new venom peptides of the Brazilian scorpion Opisthacanthus cayaporum. Toxicon 2009, 54, 252–261. [Google Scholar] [CrossRef]
- Gao, B.; Harvey, P.P.J.; Craik, D.D.J.; Ronjat, M.; De Waard, M.; Zhu, S. Functional evolution of scorpion venom peptides with an inhibitor cystine knot fold. Biosci. Rep. 2013, 33. [Google Scholar] [CrossRef]
- Schwartz, E.F.; Capes, E.M.; Diego-García, E.; Zamudio, F.Z.; Fuentes, O.; Possani, L.D.; Valdivia, H.H. Characterization of hadrucalcin, a peptide from Hadrurus gertschi scorpion venom with pharmacological activity on ryanodine receptors. Br. J. Pharmacol. 2009, 157, 392–403. [Google Scholar] [CrossRef]
- Capes, E.M.; Schwartz, E.; Diego-Garcia, E.; Zamudio, F.; Possani, L.D.; Valdivia, H.H. Hadrucalcin, a novel member of the Calcin scorpion toxin family, rapidly penetrates cellular membranes to bind ryanodine receptors and alter calcium release. J. Biophys. 2008, 94, 2631–2631. [Google Scholar] [CrossRef]
- Zhu, S.; Darbon, H.; Dyason, K.; Verdonck, F.; Tytgat, J. Evolutionary origin of inhibitor cystine knot peptides. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2003, 17, 1765–1767. [Google Scholar]
- Zamudio, F.Z.; Gurrola, G.B.; Arévalo, C.; Sreekumar, R.; Walker, J.W.; Valdivia, H.H.; Possani, L.D. Primary structure and synthesis of Imperatoxin A (IpTx(a)), a peptide activator of Ca2+ release channels/ryanodine receptors. FEBS Lett. 1997, 405, 385–389. [Google Scholar] [CrossRef]
- Valdivia, H.H.; Kirby, M.S.; Lederer, W.J.; Coronado, R. Scorpion toxins targeted against the sarcoplasmic reticulum Ca(2+)-release channel of skeletal and cardiac muscle. Proc. Natl. Acad. Sci. USA 1992, 89, 12185–12189. [Google Scholar] [CrossRef]
- Tripathy, A.; Resch, W.; Le Xu; Valdivia, H.H.; Meissner, G. Imperatoxin A Induces Subconductance States in Ca2+ Release Channels (Ryanodine Receptors) of Cardiac and Skeletal Muscle. J. Gen. Physiol. 1998, 111, 679–690. [Google Scholar] [CrossRef]
- Nabhani, T.; Zhu, X.; Simeoni, I.; Sorrentino, V.; Valdivia, H.H.; García, J. Imperatoxin a enhances Ca(2+) release in developing skeletal muscle containing ryanodine receptor type 3. Biophys. J. 2002, 82, 1319–1328. [Google Scholar] [CrossRef]
- Lee, C.W.; Lee, E.H.; Takeuchi, K.; Takahashi, H.; Shimada, I.; Sato, K.; Shin, S.Y.; Kim, D.H.; Kim, J. Il Molecular basis of the high-affinity activation of type 1 ryanodine receptors by imperatoxin A. Biochem. J. 2004, 377, 385–394. [Google Scholar] [CrossRef]
- Fajloun, Z.; Kharrat, R.; Chen, L.; Lecomte, C.; Di Luccio, E.; Bichet, D.; El Ayeb, M.; Rochat, H.; Allen, P.D.; Pessah, I.N.; De Waard, M.; Sabatier, J.M. Chemical synthesis and characterization of maurocalcine, a scorpion toxin that activates Ca(2+) release channel/ryanodine receptors. FEBS Lett. 2000, 469, 179–185. [Google Scholar] [CrossRef]
- Mosbah, A.; Kharrat, R.; Fajloun, Z.; Renisio, J.G.; Blanc, E.; Sabatier, J.M.; El Ayeb, M.; Darbon, H. A new fold in the scorpion toxin family, associated with an activity on a ryanodine-sensitive calcium channel. Proteins 2000, 40, 436–442. [Google Scholar] [CrossRef]
- Chen, Z.; Hu, Y.; Han, S.; Yin, S.; He, Y.; Wu, Y.; Cao, Z.; Li, W. ImKTx1, a new Kv1.3 channel blocker with a unique primary structure. J. Biochem. Mol. Toxicol. 2010, 25, 244–251. [Google Scholar]
- Zhijian, C.; Yun, X.; Chao, D.; Shunyi, Z.; Shijin, Y.; Yingliang, W.; Wenxin, L. Cloning and characterization of a novel calcium channel toxin-like gene BmCa1 from Chinese scorpion Mesobuthus martensii Karsch. Peptides 2006, 27, 1235–1240. [Google Scholar]
- Matsushita, N.; Miyashita, M.; Sakai, A.; Nakagawa, Y.; Miyagawa, H. Purification and characterization of a novel short-chain insecticidal toxin with two disulfide bridges from the venom of the scorpion Liocheles australasiae. Toxicon 2007, 50, 861–867. [Google Scholar] [CrossRef]
- Smith, J.J.; Hill, J.M.; Little, M.J.; Nicholson, G.M.; King, G.F.; Alewood, P.F. Unique scorpion toxin with a putative ancestral fold provides insight into evolution of the inhibitor cystine knot motif. Proc. Natl. Acad. Sci.USA 2011, 108, 10478–10483. [Google Scholar] [CrossRef]
- Smith, J.J.; Vetter, I.; Lewis, R.J.; Peigneur, S.; Tytgat, J.; Lam, A.; Gallant, E.M.; Beard, N.A.; Alewood, P.F.; Dulhunty, A.F. Multiple actions of φ-LITX-Lw1a on ryanodine receptors reveal a functional link between scorpion DDH and ICK toxins. Proc. Natl. Acad. Sci.USA 2013, 110, 8906–8911. [Google Scholar] [CrossRef]
- Horita, S.; Matsushita, N.; Kawachi, T.; Ayabe, R.; Miyashita, M.; Miyakawa, T.; Nakagawa, Y.; Nagata, K.; Miyagawa, H.; Tanokura, M. Solution structure of a short-chain insecticidal toxin LaIT1 from the venom of scorpion Liocheles australasiae. Biochem. Biophys. Res. Commun. 2011, 411, 738–744. [Google Scholar]
- Lazarovici, P.; Yanai, P.; Pelhate, M.; Zlotkin, E. Insect toxic component from the venom of a chactoid scorpion, Scorpio maurus palmatus (Scorpionidae). J. Biol. Chem. 1982, 257, 8397–8404. [Google Scholar]
- Luna-Ramírez, K.; Quintero-Hernández, V.; Vargas-Jaimes, L.; Batista, C.V.F.; Winkel, K.D.; Possani, L.D. Characterization of the venom from the Australian scorpion Urodacus yaschenkoi: Molecular mass analysis of components, cDNA sequences and peptides with antimicrobial activity. Toxicon 2013, 63, 44–54. [Google Scholar] [CrossRef]
- Froy, O.; Gurevitz, M. New insight on scorpion divergence inferred from comparative analysis of toxin structure, pharmacology and distribution. Toxicon 2003, 42, 549–555. [Google Scholar] [CrossRef]
- Inceoglu, A.B.; Hayashida, Y.; Lango, J.; Ishida, A.T.; Hammock, B.D. A single charged surface residue modifies the activity of ikitoxin, a beta-type Na+ channel toxin from Parabuthus transvaalicus. Eur. J. Biochem. 2002, 269, 5369–5376. [Google Scholar] [CrossRef]
- Bosmans, F.; Tytgat, J. Voltage-gated sodium channel modulation by scorpion alpha-toxins. Toxicon 2007, 49, 142–58. [Google Scholar] [CrossRef]
- He, Y.; Zhao, R.; Di, Z.; Li, Z.; Xu, X.; Hong, W.; Wu, Y.; Zhao, H.; Li, W.; Cao, Z. Molecular diversity of Chaerilidae venom peptides reveals the dynamic evolution of scorpion venom components from Buthidae to non-Buthidae. J. Proteomics 2013, 89C, 1–14. [Google Scholar]
- Soudani, N.; Gharbi-Chihi, J.; Srairi-Abid, N.; Yazidi, C.M.-E.; Planells, R.; Margotat, A.; Torresani, J.; El Ayeb, M. Isolation and molecular characterization of LVP1 lipolysis activating peptide from scorpion Buthus occitanus tunetanus. Biochim. Biophys. Acta 2005, 1747, 47–56. [Google Scholar] [CrossRef]
- Wang, X.; Connor, M.; Smith, R.; Maciejewski, M.W.; Howden, M.E. H.; Nicholson, G.M.; Christie, M.J.; King, G.F. Discovery and characterization of a family of insecticidal neurotoxins with a rare vicinal disulfide bridge. Nat. Struct. Mol. Biol. 2000, 7, 505–513. [Google Scholar] [CrossRef]
- Fry, B.G.; Roelants, K.; Winter, K.; Hodgson, W.C.; Griesman, L.; Kwok, H.F.; Scanlon, D.; Karas, J.; Shaw, C.; Wong, L.; Norman, J.A. Novel venom proteins produced by differential domain-expression strategies in beaded lizards and gila monsters (genus Heloderma). Mol. Biol. Evol. 2010, 27, 395–407. [Google Scholar] [CrossRef]
- Verdonck, F.; Bosteels, S.; Desmet, J.; Moerman, L.; Noppe, W.; Willems, J.; Tytgat, J.; Van der Walt, J. A novel class of pore-forming peptides in the venom of parabuthus schlechteri Purcell (scorpions: buthidae). Cimbebasia 2000, 16, 247–260. [Google Scholar]
- Moerman, L.; Bosteels, S.; Noppe, W.; Willems, J.; Clynen, E.; Schoofs, L.; Thevissen, K.; Tytgat, J.; Van Eldere, J.; Van Der Walt, J.; Verdonck, F. Antibacterial and antifungal properties of α-helical, cationic peptides in the venom of scorpions from southern Africa. Eur. J. Biochem. 2002, 269, 4799–4810. [Google Scholar] [CrossRef]
- Willems, J.; Noppe, W.; Moerman, L.; Van Der Walt, J.; Verdonck, F. Cationic peptides from scorpion venom can stimulate and inhibit polymorphonuclear granulocytes. Toxicon 2002, 40, 1679–1683. [Google Scholar] [CrossRef]
- Moerman, L.; Verdonck, F.; Willems, J.; Tytgat, J.; Bosteels, S. Antimicrobial peptides from scorpion venom induce Ca(2+) signaling in HL-60 cells. Biochem. Biophys. Res. Commun. 2003, 311, 90–97. [Google Scholar] [CrossRef]
- Willems, J.; Moerman, L.; Bosteels, S.; Bruyneel, E.; Ryniers, F.; Verdonck, F. Parabutoporin--an antibiotic peptide from scorpion venom--can both induce activation and inhibition of granulocyte cell functions. Peptides 2004, 25, 1079–1084. [Google Scholar] [CrossRef]
- Roelants, K.; Fry, B.G.; Ye, L.; Norman, J.A.; Stijlemans, B.; Kok, P.; Clynen, E.; Schoofs, L.; Cornelis, P.; Bossuyt, F. Evolutionary genomics of an amphibian defense peptide arsenal. PLoS Genet. 2013, in press. [Google Scholar]
- Sunagar, K.; Fry, B.G.; Jackson, T.; Casewell, N.R.; Undheim, E.A.B.; Vidal, N.; Ali, S.; King, G.F.; Vasudevan, K.; Vasconcelos, V.; Antunes, A. Molecular evolution of vertebrate neurotrophins: Co-option of the highly conserved nerve growth factor gene into the advanced snake venom arsenal. PLoS One 2013, 8, e81827. [Google Scholar] [CrossRef]
- Girish, K.S.; Jagadeesha, D.K.; Rajeev, K.B.; Kemparaju, K. Snake venom hyaluronidase: an evidence for isoforms and extracellular matrix degradation. Mol Cell Biochem 2002, 240, 105–110. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Yang, Z.; Nielsen, R. Synonymous and nonsynonymous rate variation in nuclear genes of mammals. J. Mol. Evol. 1998, 46, 409–418. [Google Scholar] [CrossRef]
- Anisimova, M.; Bielawski, J.P.; Yang, Z. Accuracy and power of Bayes prediction of amino acid sites under positive selection. Mol. Biol. Evol. 2002, 19, 950–958. [Google Scholar] [CrossRef]
- Reboiro-Jato, D.; Reboiro-Jato, M.; Fdez-Riverola, F.; Vieira, C.P.; Fonseca, N.A.; Vieira, J. ADOPS--Automatic Detection Of Positively Selected Sites. J. Integr. Bioinform. 2012, 9, 200. [Google Scholar]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: a fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Frost, S.D.W.; Pond, S.L.K. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef]
- Woolley, S.; Johnson, J.; Smith, M.J.; Crandall, K.A.; McClellan, D.A. TreeSAAP: selection on amino acid properties using phylogenetic trees. Bioinformatics 2003, 19, 671–672. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Murrell, B.; Fourment, M.; Frost, S.D.W.; Delport, W.; Scheffler, K. A random effects branch-site model for detecting episodic diversifying selection. Mol. Biol. Evol. 2011, 28, 3033–3043. [Google Scholar] [CrossRef]
- Smith, J.J.; Jones, A.; Alewood, P.F. Mass landscapes of seven scorpion species: The first analyses of Australian species with 1,5-DAN matrix. J. Venom Res. 2012, 3, 7–14. [Google Scholar]
- Chen, Z.-Y.; Hu, Y.-T.; Yang, W.-S.; He, Y.-W.; Feng, J.; Wang, B.; Zhao, R.-M.; Ding, J.-P.; Cao, Z.-J.; Li, W.-X.; Wu, Y.-L. Hg1, novel peptide inhibitor specific for Kv1.3 channels from first scorpion Kunitz-type potassium channel toxin family. J. Biol. Chem. 2012, 287, 13813–13821. [Google Scholar] [CrossRef]
- Kini, R.M.; Chan, Y.M. Accelerated evolution and molecular surface of venom phospholipase A2 enzymes. J. Mol. Evol. 1999, 48, 125–132. [Google Scholar]
- Ruder, T.; Sunagar, K.; Undheim, E.A.B.; Ali, S.A.; Wai, T.-C.; Low, D.H.W.; Jackson, T.N.W.; King, G.F.; Antunes, A.; Fry, B.G. Molecular phylogeny and evolution of the proteins encoded by coleoid (cuttlefish, octopus, and squid) posterior venom glands. J. Mol. Evol. 2013, 76, 192–204. [Google Scholar] [CrossRef]
- Sunagar, K.; Jackson, T.; Undheim, E.; Ali, S.; Antunes, A.; Fry, B. Three-Fingered RAVERs: Rapid Accumulation of Variations in Exposed Residues of Snake Venom Toxins. Toxins 2013, 5, 2172–2208. [Google Scholar] [CrossRef]
- Kozminsky-Atias, A.; Zilberberg, N. Molding the business end of neurotoxins by diversifying evolution. FASEB J. 2012, 26, 576–586. [Google Scholar] [CrossRef]
- Tian, C.; Yuan, Y.; Zhu, S. Positively selected sites of scorpion depressant toxins: possible roles in toxin functional divergence. Toxicon 2008, 51, 555–562. [Google Scholar] [CrossRef]
- Zhu, S.; Bosmans, F.; Tytgat, J. Adaptive evolution of scorpion sodium channel toxins. J. Mol. Evol. 2004, 58, 145–153. [Google Scholar] [CrossRef]
- Nisani, Z.; Boskovic, D.S.; Dunbar, S.G.; Kelln, W.; Hayes, W.K. Investigating the chemical profile of regenerated scorpion (Parabuthus transvaalicus) venom in relation to metabolic cost and toxicity. Toxicon 2012, 60, 315–323. [Google Scholar] [CrossRef]
- Delpietro, H.A.; Russo, R.G. Acquired resistance to saliva anticoagulants by prey previously fed upon by vampire bats (Desmodus rotundus): Evidence for immune response. J. Mamm. 2009, 90, 1132–1138. [Google Scholar] [CrossRef]
- Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Williams, T.D.; Nagaraj, S.H.; Nueda, M.J.; Robles, M.; Talon, M.; Dopazo, J.; Conesa, A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef]
- Gotz, S.; Arnold, R.; Sebastian-Leon, P.; Martin-Rodriguez, S.; Tischler, P.; Jehl, M.A.; Dopazo, J.; Rattei, T.; Conesa, A. B2G-FAR, a species-centered GO annotation repository. Bioinformatics 2011, 27, 919–924. [Google Scholar] [CrossRef]
- Chairman, V.; Kusk, K. CLC Main Workbench, version 6.7.1; CLC bio: Aarhus, Denmark, 2011. [Google Scholar]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D. W.; Pond, S.L. K. Automated phylogenetic detection of recombination using a genetic algorithm. Mol. Biol. Evol. 2006, 23, 1891–1901. [Google Scholar] [CrossRef]
- Scheffler, K.; Martin, D.P.; Seoighe, C. Robust inference of positive selection from recombining coding sequences. Bioinformatics 2006, 22, 2493–2499. [Google Scholar] [CrossRef]
- Goldman, N.; Yang, Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol. Biol. Evol. 1994, 11, 725–736. [Google Scholar]
- Yang, Z. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol. Biol. Evol. 1998, 15, 568–573. [Google Scholar] [CrossRef]
- Nielsen, R.; Yang, Z. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 1998, 148, 929–936. [Google Scholar]
- Yang, Z.; Wong, W.S. W.; Nielsen, R. Bayes empirical Bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [CrossRef]
- Pond, S.L.K.; Frost, S.D. W.; Muse, S. V HyPhy: hypothesis testing using phylogenies. Bioinformatics 2005, 21, 676–679. [Google Scholar] [CrossRef]
- Pond, S.L.K.; Scheffler, K.; Gravenor, M.B.; Poon, A.F.Y.; Frost, S.D.W. Evolutionary fingerprinting of genes. Mol. Biol. Evol. 2010, 27, 520–536. [Google Scholar] [CrossRef]
- Kelley, L.A.; Sternberg, M.J.E. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 2009, 4, 363–371. [Google Scholar] [CrossRef]
- DeLano, W.L. The PyMOL Molecular Graphics System, version 1.5; Schrödinger, LLC: Camberley, UK, 2002. [Google Scholar]
- Armon, A.; Graur, D.; Ben-Tal, N. ConSurf: an algorithmic tool for the identification of functional regions in proteins by surface mapping of phylogenetic information. J. Mol. Biol. 2001, 307, 447–463. [Google Scholar] [CrossRef]
- Fraczkiewicz, R.; Braun, W. Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J. Comput. Chem. 1998, 19, 319–333. [Google Scholar] [CrossRef]
Supplementary Files
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sunagar, K.; Undheim, E.A.B.; Chan, A.H.C.; Koludarov, I.; Muñoz-Gómez, S.A.; Antunes, A.; Fry, B.G. Evolution Stings: The Origin and Diversification of Scorpion Toxin Peptide Scaffolds. Toxins 2013, 5, 2456-2487. https://doi.org/10.3390/toxins5122456
Sunagar K, Undheim EAB, Chan AHC, Koludarov I, Muñoz-Gómez SA, Antunes A, Fry BG. Evolution Stings: The Origin and Diversification of Scorpion Toxin Peptide Scaffolds. Toxins. 2013; 5(12):2456-2487. https://doi.org/10.3390/toxins5122456
Chicago/Turabian StyleSunagar, Kartik, Eivind A. B. Undheim, Angelo H. C. Chan, Ivan Koludarov, Sergio A. Muñoz-Gómez, Agostinho Antunes, and Bryan G. Fry. 2013. "Evolution Stings: The Origin and Diversification of Scorpion Toxin Peptide Scaffolds" Toxins 5, no. 12: 2456-2487. https://doi.org/10.3390/toxins5122456