Effect of Thioridazine on Erythrocytes

 , ,
, , 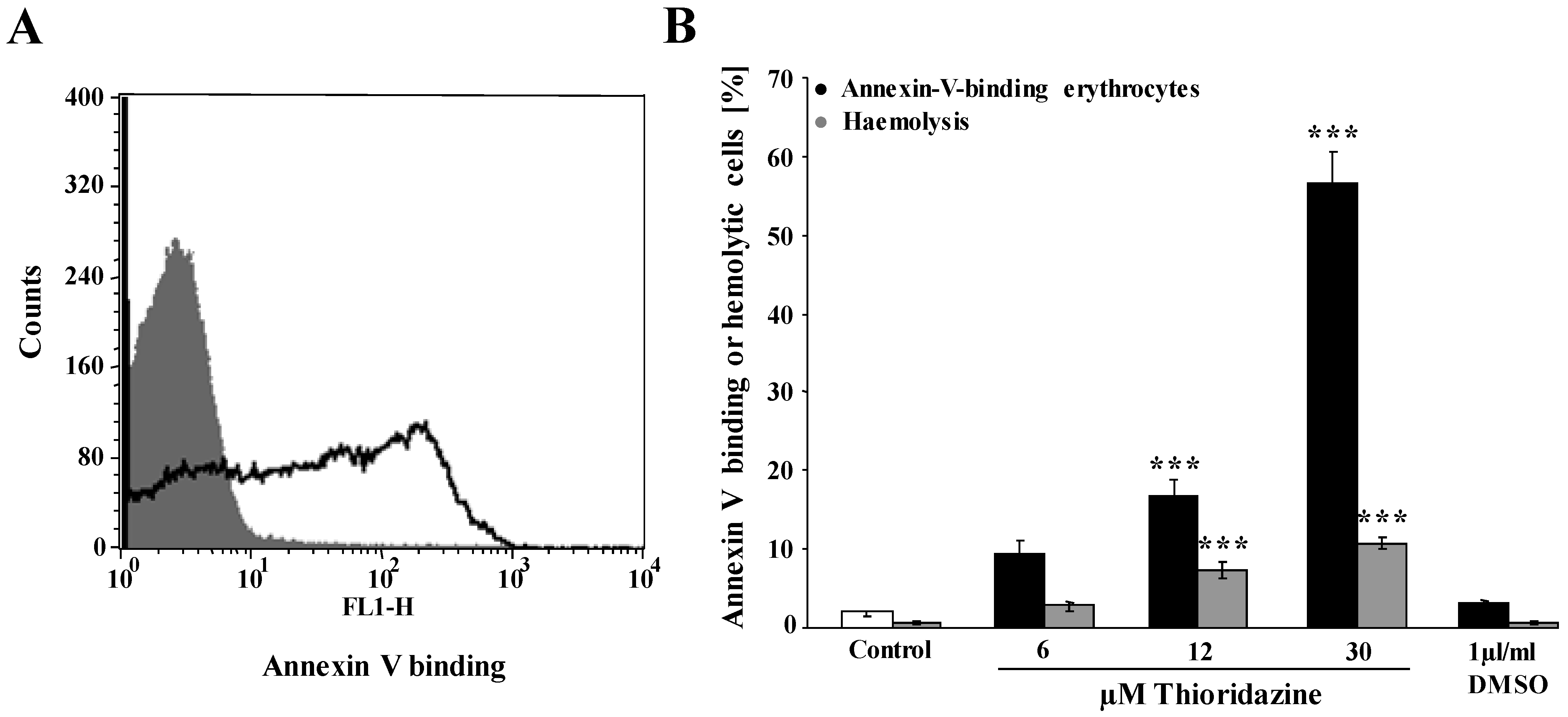{kind=link}
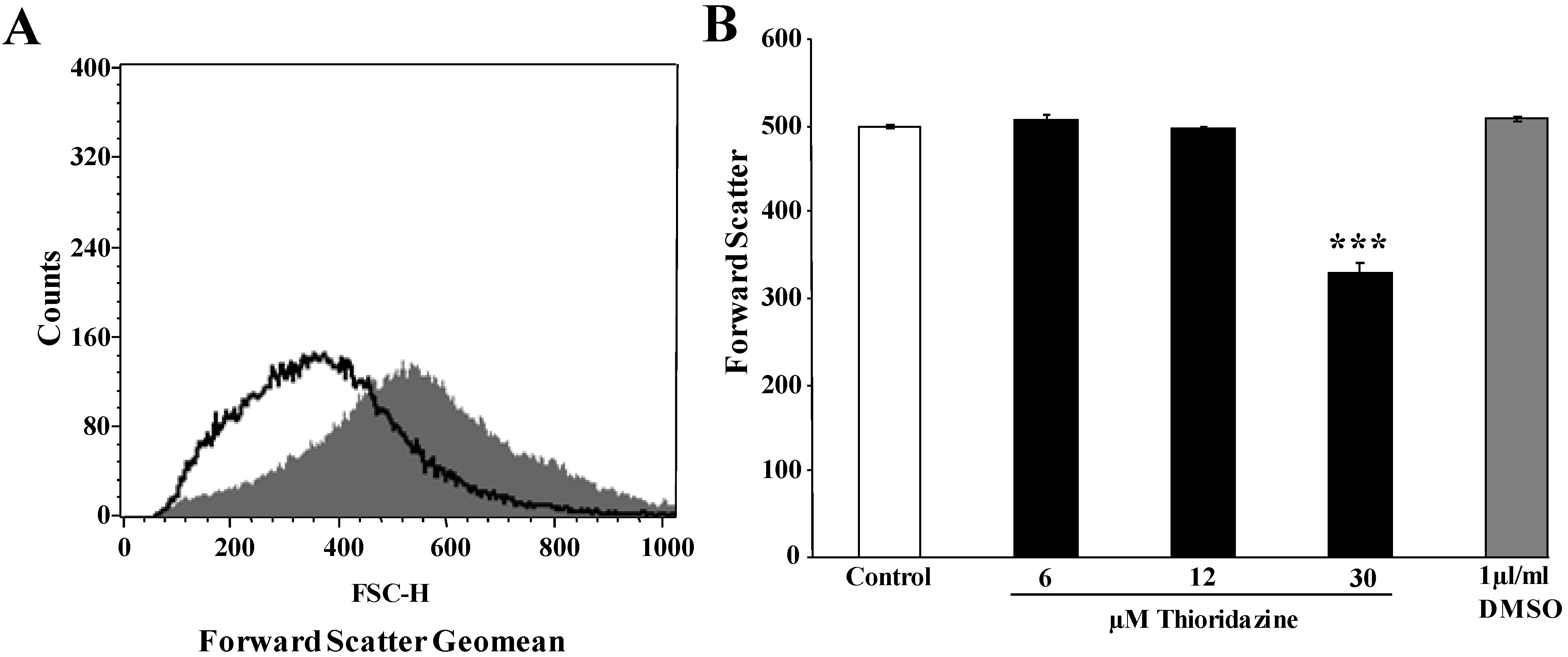{kind=link}
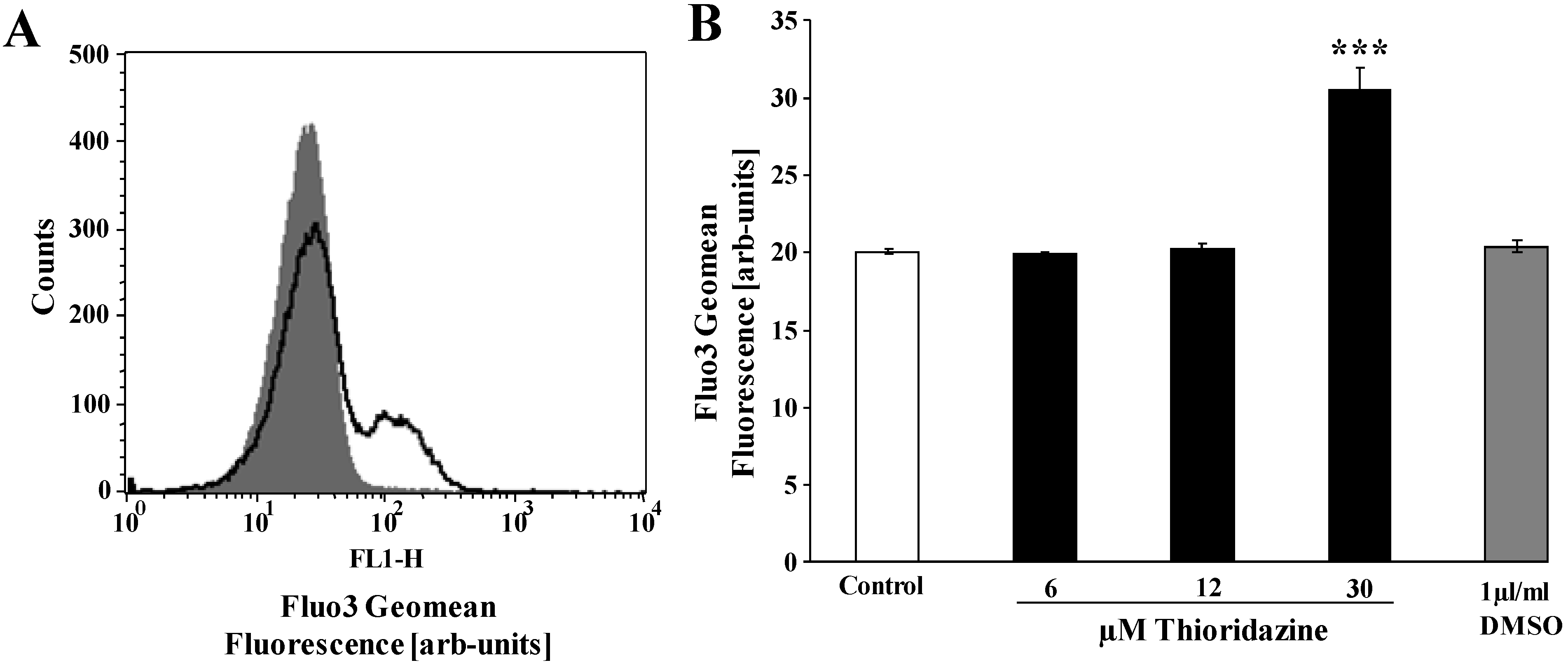{kind=link}
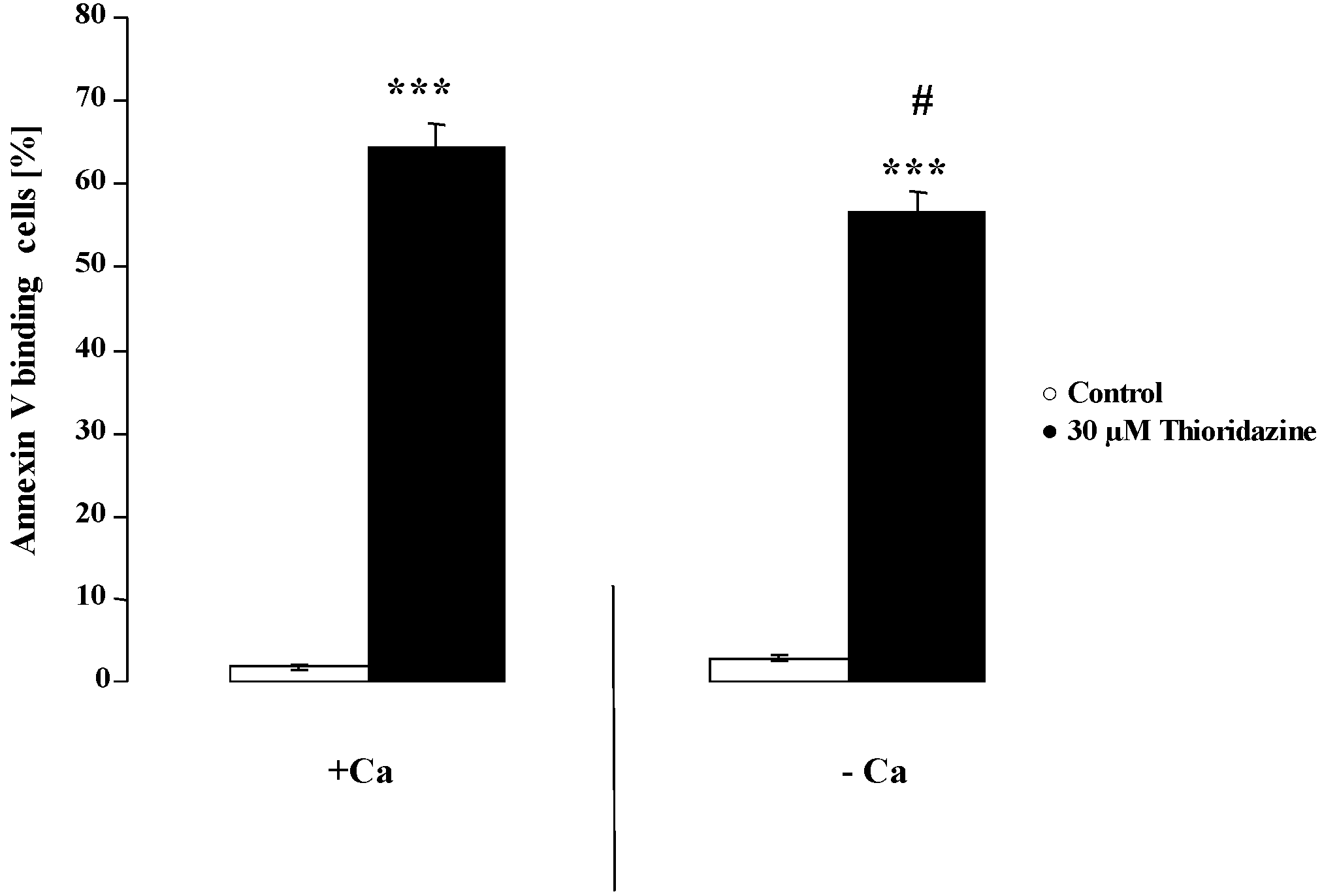{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
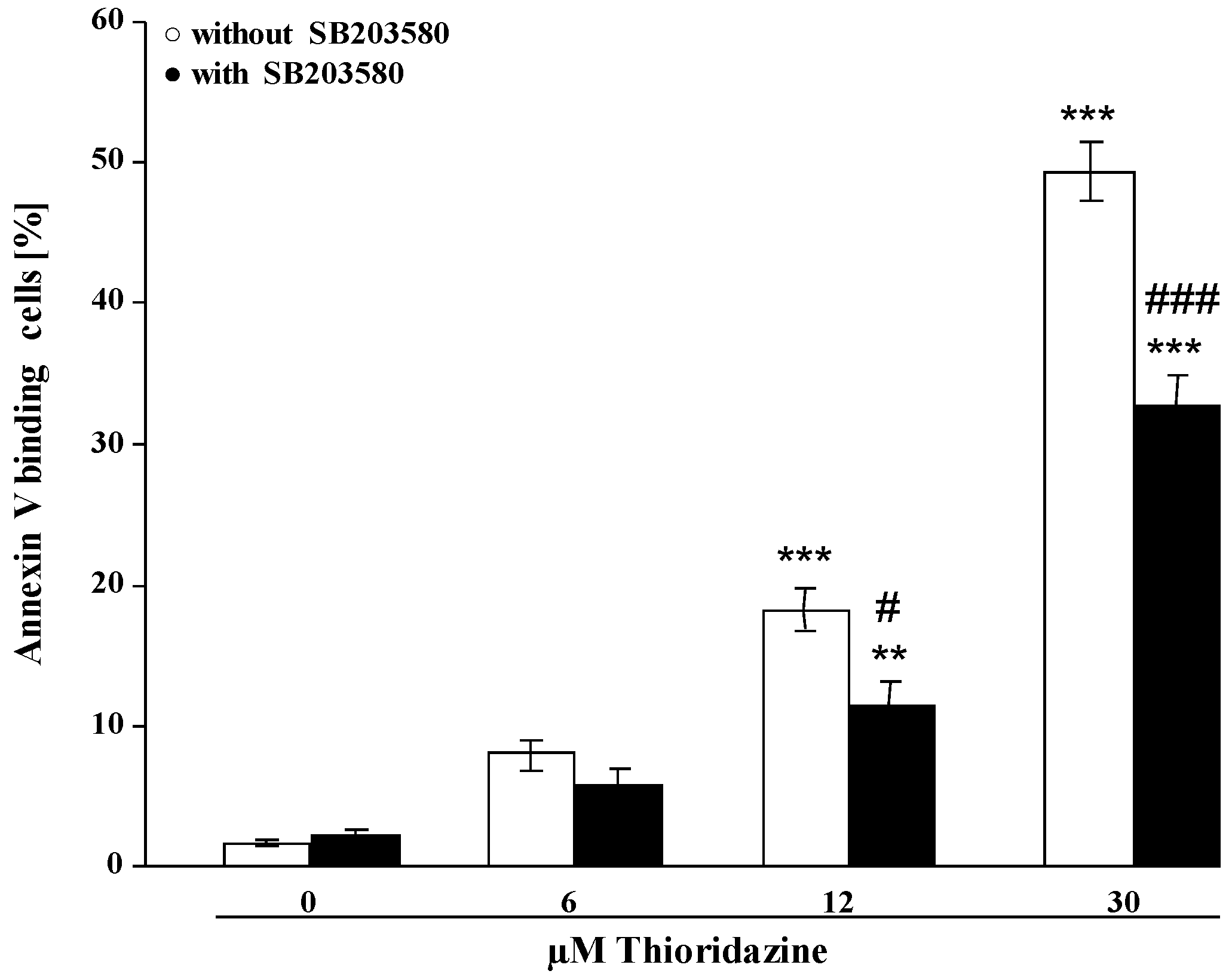
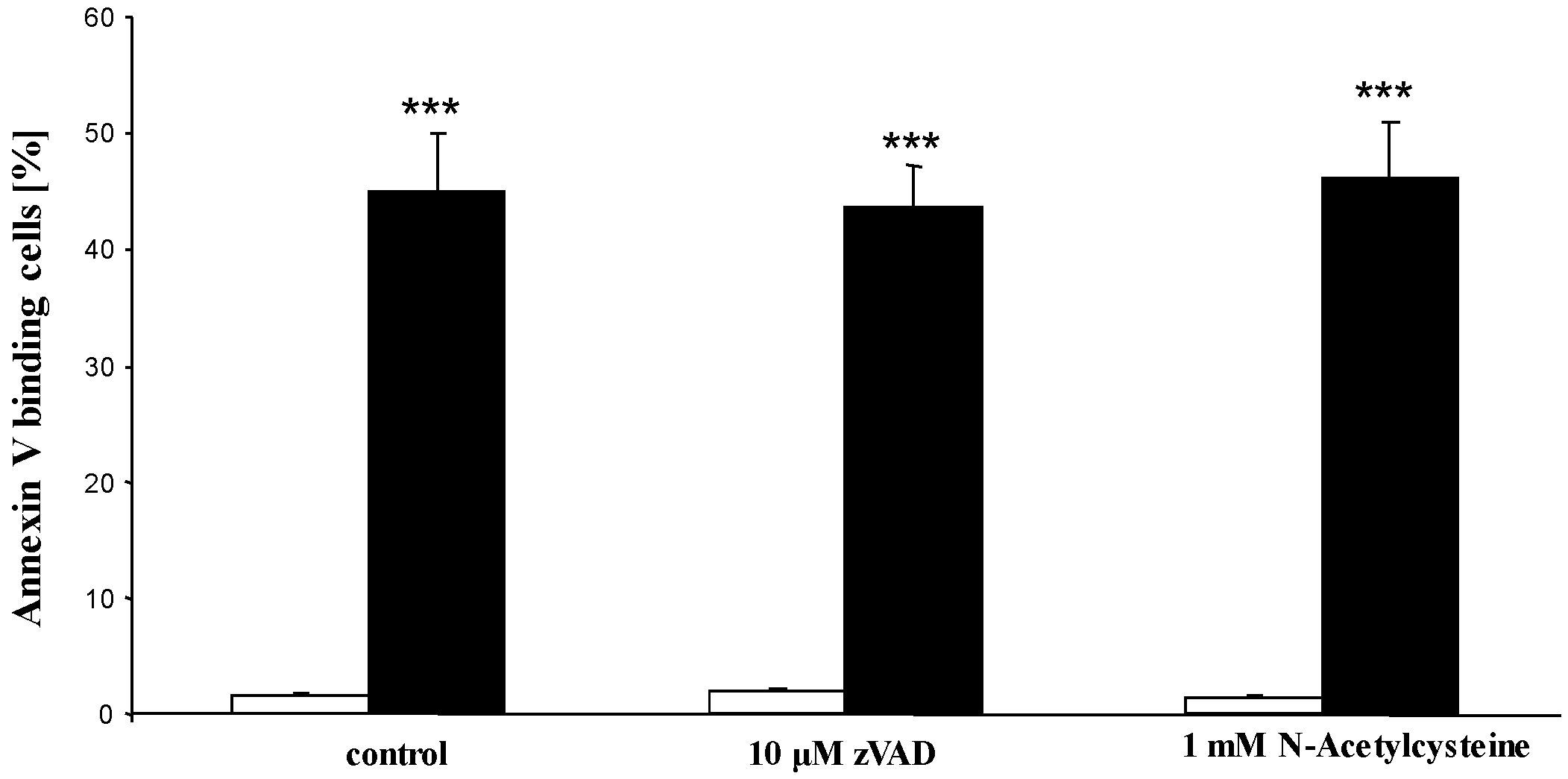
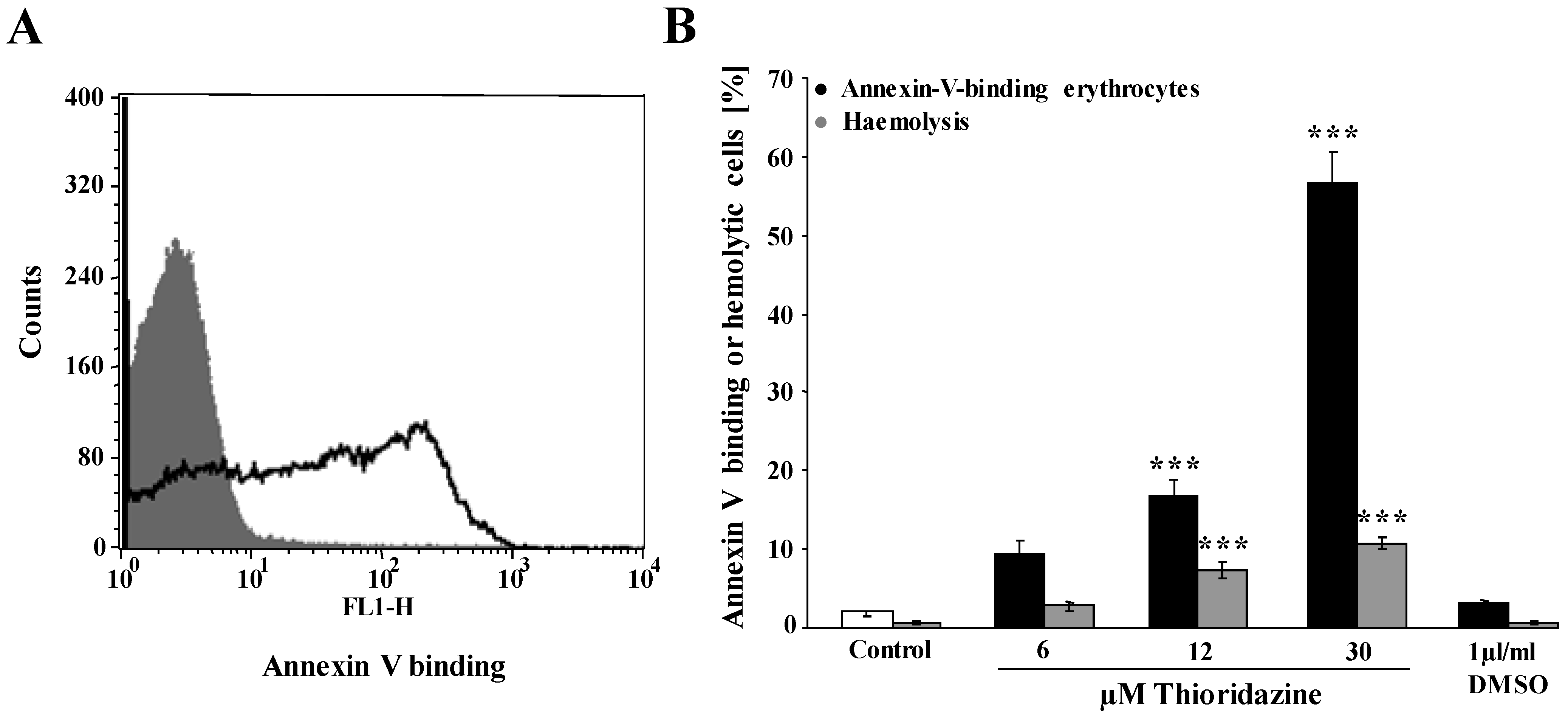
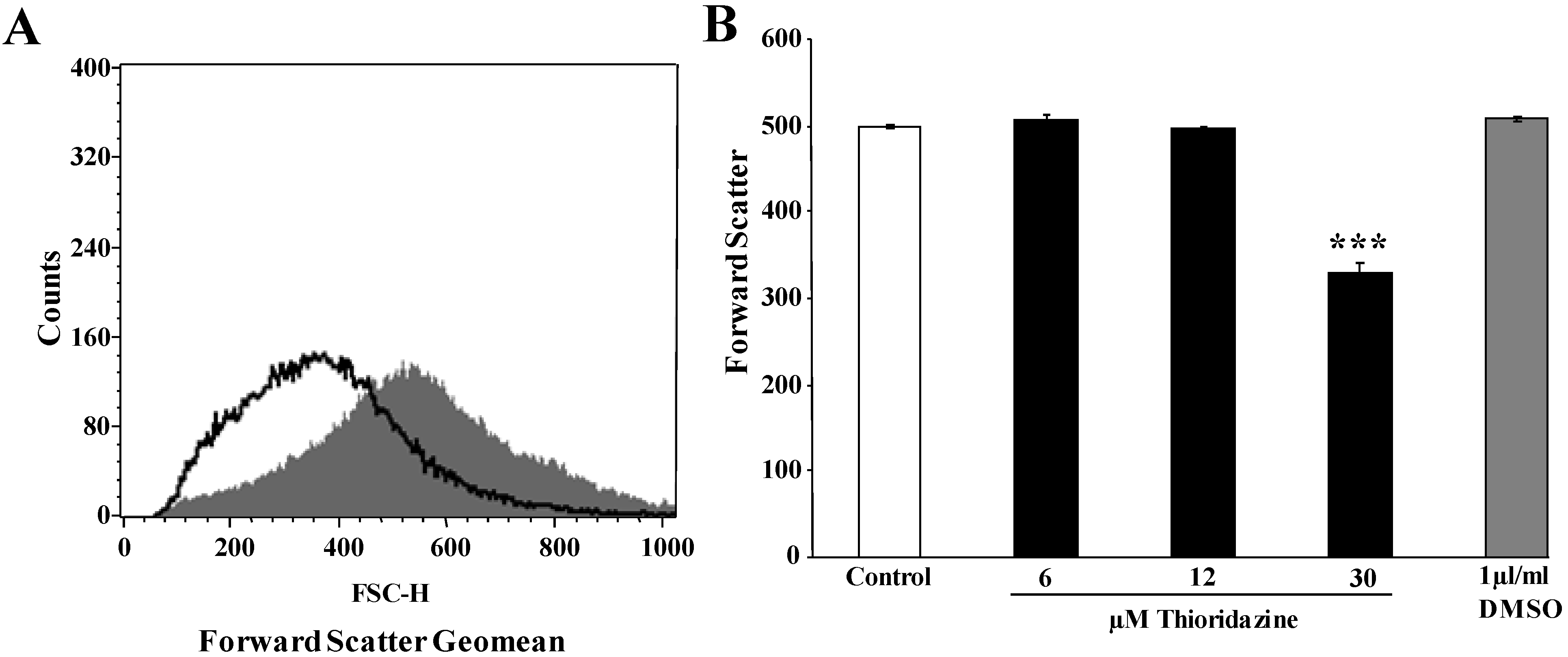
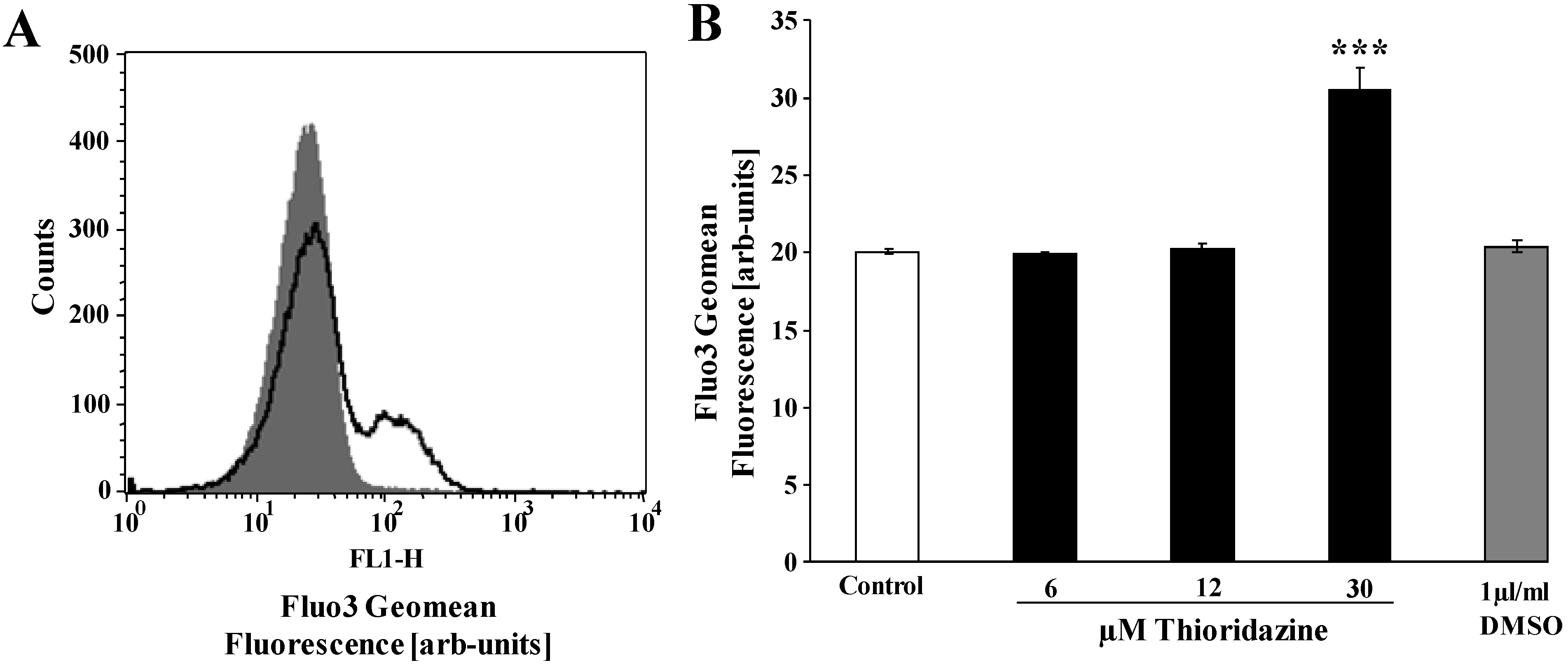
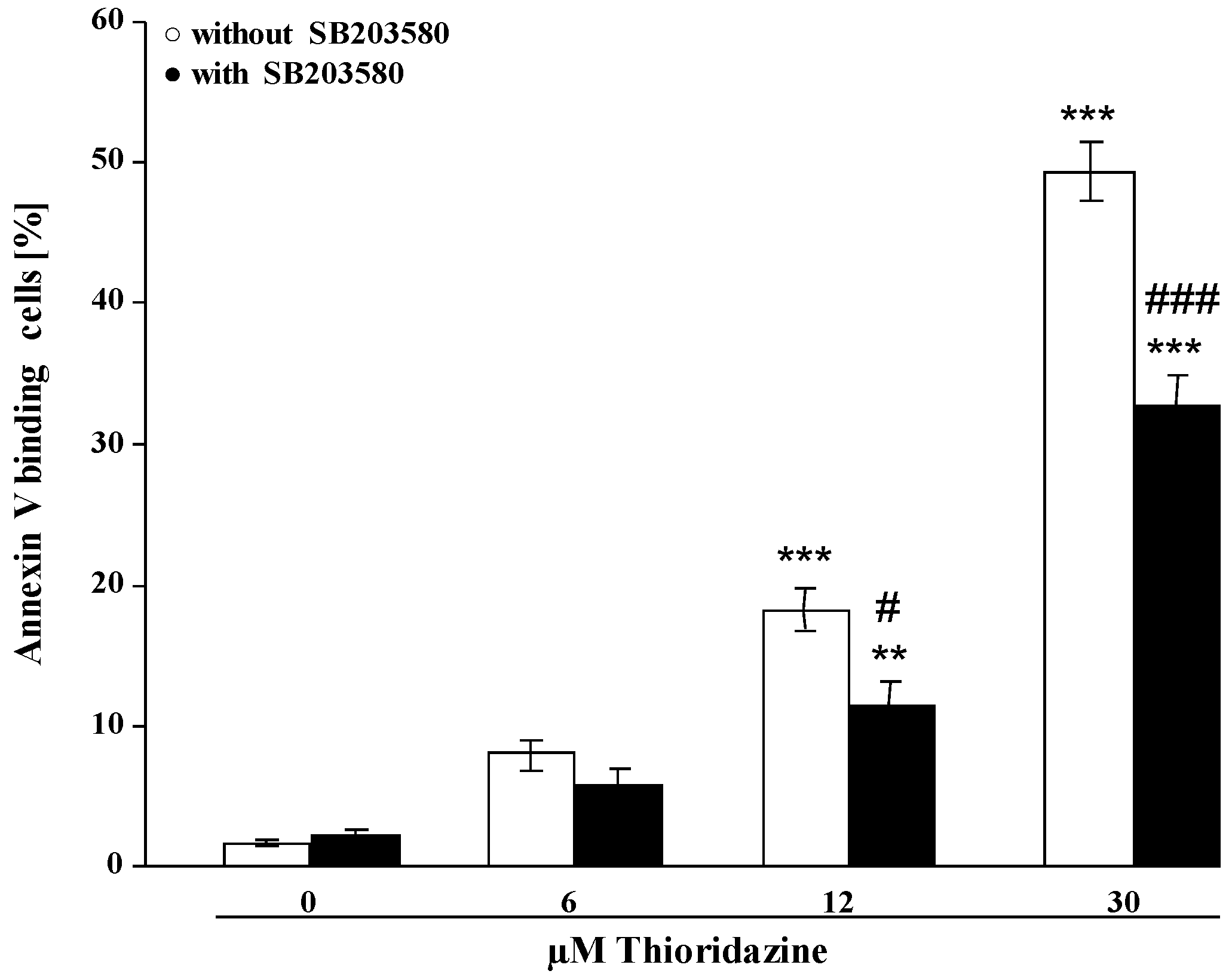
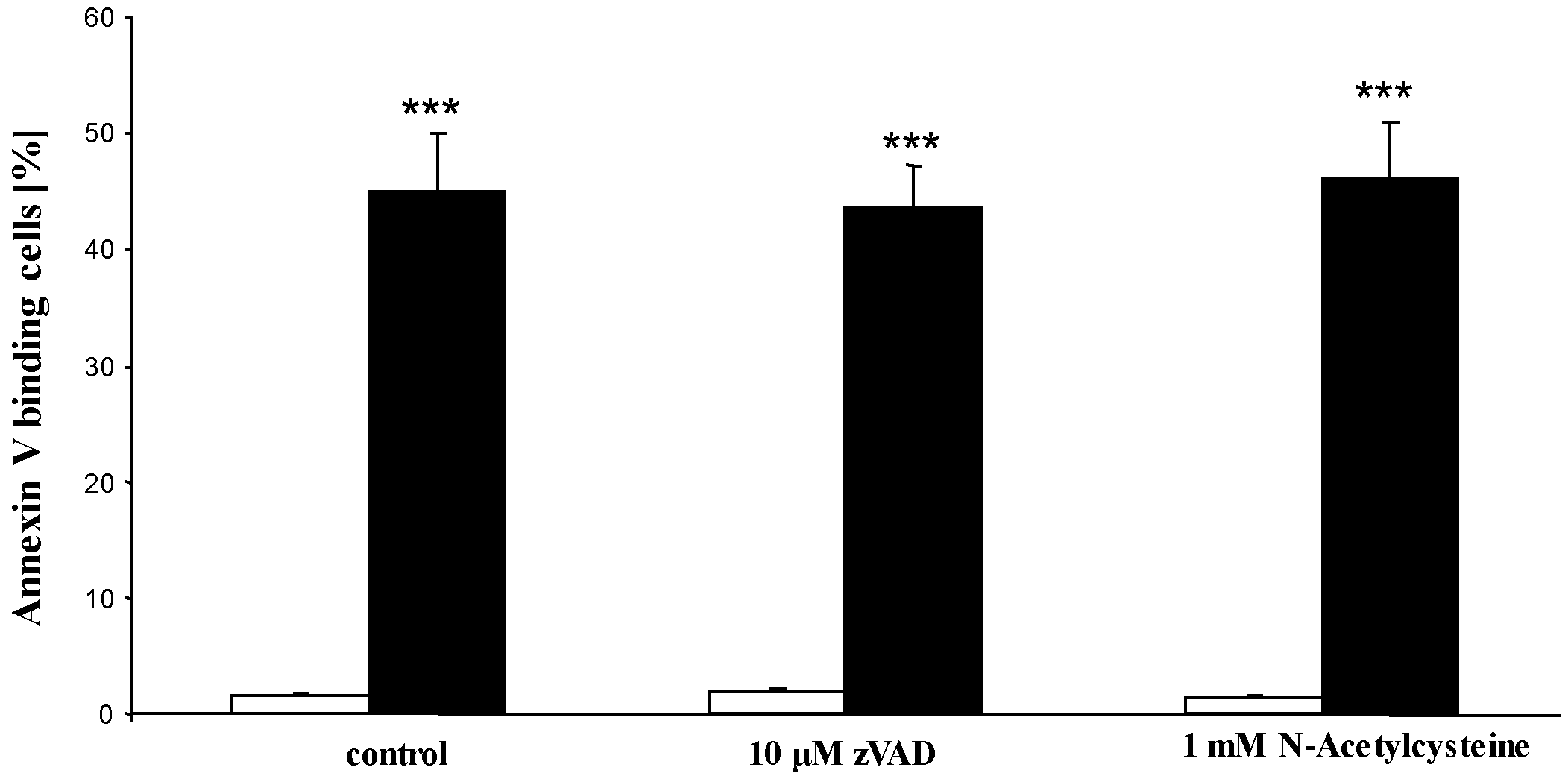
2. Results and Discussion

3. Experimental Section
3.1. Erythrocytes, Solutions and Chemicals
3.2. FACS Analysis of Annexin-V-Binding and Forward Scatter
3.3. Measurement of Intracellular Ca2+
3.4. Measurement of Hemolysis
3.5. Statistics
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Purhonen, M.; Koponen, H.; Tiihonen, J.; Tanskanen, A. Outcome of patients after market withdrawal of thioridazine: A retrospective analysis in a nationwide cohort. Pharmacoepidemiol. Drug Saf. 2012, 21, 1227–1231. [Google Scholar] [CrossRef]
- Thanacoody, R.H. Thioridazine: The good and the bad. Recent Pat. Antiinfect. Drug Discov. 2011, 6, 92–98. [Google Scholar] [CrossRef]
- Amaral, L. Thioridazine: An old neuroleptic effective against totally drug resistant tuberculosis. Acta Med. Port. 2012, 25, 118–121. [Google Scholar]
- Amaral, L.; Molnar, J. Potential therapy of multidrug-resistant and extremely drug-resistant tuberculosis with thioridazine. In Vivo 2012, 26, 231–236. [Google Scholar]
- Amaral, L.; Viveiros, M. Why thioridazine in combination with antibiotics cures extensively drug-resistant mycobacterium tuberculosis infections. Int. J. Antimicrob. Agents. 2012, 39, 376–380. [Google Scholar] [CrossRef]
- Sohaskey, C. Latent tuberculosis: Is there a role for thioridazine? Recent Pat. Antiinfect. Drug Discov. 2011, 6, 139–146. [Google Scholar] [CrossRef]
- Cooper, J.W., Jr.; Pesnell, L.H. Thioridazine-associated immune hemolytic anemia. South Med. J. 1978, 71, 1443–1444. [Google Scholar] [CrossRef]
- King, D.J.; Wager, E. Haematological safety of antipsychotic drugs. J. Psychopharmacol. 1998, 12, 283–288. [Google Scholar] [CrossRef]
- Lang, F.; Gulbins, E.; Lerche, H.; Huber, S.M.; Kempe, D.S.; Föller, M. Eryptosis, a window to systemic disease. Cell. Physiol. Biochem. 2008, 22, 373–380. [Google Scholar] [CrossRef]
- Lang, P.A.; Kaiser, S.; Myssina, S.; Wieder, T.; Lang, F.; Huber, S.M. Role of Ca2+-activated K+ channels in human erythrocyte apoptosis. Am. J. Physiol. Cell. Physiol. 2003, 285, C1553–C1560. [Google Scholar] [CrossRef]
- Föller, M.; Kasinathan, R.S.; Koka, S.; Lang, C.; Shumilina, E.; Birnbaumer, L.; Lang, F.; Huber, S.M. Trpc6 contributes to the Ca2+ leak of human erythrocytes. Cell. Physiol. Biochem. 2008, 21, 183–192. [Google Scholar] [CrossRef]
- Föller, M.; Sopjani, M.; Koka, S.; Gu, S.; Mahmud, H.; Wang, K.; Floride, E.; Schleicher, E.; Schulz, E.; Munzel, T.; et al. Regulation of erythrocyte survival by amp-activated protein kinase. FASEB J. 2009, 23, 1072–1080. [Google Scholar] [CrossRef]
- Lang, P.A.; Kaiser, S.; Myssina, S.; Birka, C.; Weinstock, C.; Northoff, H.; Wieder, T.; Lang, F.; Huber, S.M. Effect of vibrio parahaemolyticus haemolysin on human erythrocytes. Cell. Microbiol. 2004, 6, 391–400. [Google Scholar] [CrossRef]
- Brugnara, C.; de Franceschi, L.; Alper, S.L. Inhibition of Ca2+-dependent K+ transport and cell dehydration in sickle erythrocytes by clotrimazole and other imidazole derivatives. J. Clin. Invest. 1993, 92, 520–526. [Google Scholar] [CrossRef]
- Berg, C.P.; Engels, I.H.; Rothbart, A.; Lauber, K.; Renz, A.; Schlosser, S.F.; Schulze-Osthoff, K.; Wesselborg, S. Human mature red blood cells express caspase-3 and caspase-8, but are devoid of mitochondrial regulators of apoptosis. Cell. Death. Differ. 2001, 8, 1197–1206. [Google Scholar] [CrossRef]
- Lang, F.; Gulbins, E.; Lang, P.A.; Zappulla, D.; Föller, M. Ceramide in suicidal death of erythrocytes. Cell. Physiol. Biochem. 2010, 26, 21–28. [Google Scholar] [CrossRef]
- Klarl, B.A.; Lang, P.A.; Kempe, D.S.; Niemoeller, O.M.; Akel, A.; Sobiesiak, M.; Eisele, K.; Podolski, M.; Huber, S.M.; Wieder, T.; et al. Protein kinase C mediates erythrocyte “programmed cell death” following glucose depletion. Am. J. Physiol. Cell. Physiol. 2006, 290, C244–C253. [Google Scholar]
- Bhavsar, S.K.; Bobbala, D.; Xuan, N.T.; Föller, M.; Lang, F. Stimulation of suicidal erythrocyte death by alpha-lipoic acid. Cell. Physiol. Biochem. 2010, 26, 859–868. [Google Scholar] [CrossRef]
- Föller, M.; Huber, S.M.; Lang, F. Erythrocyte programmed cell death. IUBMB Life 2008, 60, 661–668. [Google Scholar] [CrossRef]
- Föller, M.; Mahmud, H.; Gu, S.; Wang, K.; Floride, E.; Kucherenko, Y.; Luik, S.; Laufer, S.; Lang, F. Participation of leukotriene C4 in the regulation of suicidal erythrocyte death. J. Physiol. Pharmacol. 2009, 60, 135–143. [Google Scholar]
- Lau, I.P.; Chen, H.; Wang, J.; Ong, H.C.; Leung, K.C.; Ho, H.P.; Kong, S.K. In vitro effect of ctab- and peg-coated gold nanorods on the induction of eryptosis/erythroptosis in human erythrocytes. Nanotoxicology 2012, 6, 847–856. [Google Scholar] [CrossRef]
- Maellaro, E.; Leoncini, S.; Moretti, D.; Del Bello, B.; Tanganelli, I.; De Felice, C.; Ciccoli, L. Erythrocyte caspase-3 activation and oxidative imbalance in erythrocytes and in plasma of type 2 diabetic patients. Acta Diabetol. 2013, 50, 489–495. [Google Scholar] [CrossRef]
- Föller, M.; Feil, S.; Ghoreschi, K.; Koka, S.; Gerling, A.; Thunemann, M.; Hofmann, F.; Schuler, B.; Vogel, J.; Pichler, B.; et al. Anemia and splenomegaly in cgki-deficient mice. Proc. Natl. Acad. Sci. USA 2008, 105, 6771–6776. [Google Scholar] [CrossRef] [Green Version]
- Bhavsar, S.K.; Gu, S.; Bobbala, D.; Lang, F. Janus kinase 3 is expressed in erythrocytes, phosphorylated upon energy depletion and involved in the regulation of suicidal erythrocyte death. Cell. Physiol. Biochem. 2011, 27, 547–556. [Google Scholar] [CrossRef]
- Kucherenko, Y.; Zelenak, C.; Eberhard, M.; Qadri, S.M.; Lang, F. Effect of casein kinase 1 α activator pyrvinium pamoate on erythrocyte ion channels. Cell. Physiol. Biochem. 2012, 30, 407–417. [Google Scholar] [CrossRef]
- Zelenak, C.; Eberhard, M.; Jilani, K.; Qadri, S.M.; Macek, B.; Lang, F. Protein kinase CK1α regulates erythrocyte survival. Cell. Physiol. Biochem. 2012, 29, 171–180. [Google Scholar] [CrossRef]
- Gatidis, S.; Zelenak, C.; Fajol, A.; Lang, E.; Jilani, K.; Michael, D.; Qadri, S.M.; Lang, F. P38 mapk activation and function following osmotic shock of erythrocytes. Cell. Physiol. Biochem. 2011, 28, 1279–1286. [Google Scholar] [CrossRef]
- Zelenak, C.; Föller, M.; Velic, A.; Krug, K.; Qadri, S.M.; Viollet, B.; Lang, F.; Macek, B. Proteome analysis of erythrocytes lacking amp-activated protein kinase reveals a role of PAK2 kinase in eryptosis. J. Proteome Res. 2011, 10, 1690–1697. [Google Scholar] [CrossRef]
- Lupescu, A.; Shaik, N.; Jilani, K.; Zelenak, C.; Lang, E.; Pasham, V.; Zbidah, M.; Plate, A.; Bitzer, M.; Föller, M.; et al. Enhanced erythrocyte membrane exposure of phosphatidylserine following sorafenib treatment: An in vivo and in vitro study. Cell. Physiol. Biochem. 2012, 30, 876–888. [Google Scholar] [CrossRef]
- Shaik, N.; Lupescu, A.; Lang, F. Sunitinib-sensitive suicidal erythrocyte death. Cell. Physiol. Biochem. 2012, 30, 512–522. [Google Scholar] [CrossRef]
- Abed, M.; Towhid, S.T.; Mia, S.; Pakladok, T.; Alesutan, I.; Borst, O.; Gawaz, M.; Gulbins, E.; Lang, F. Sphingomyelinase-induced adhesion of eryptotic erythrocytes to endothelial cells. Am. J. Physiol. Cell. Physiol. 2012, 303, C991–C999. [Google Scholar] [CrossRef]
- Abed, M.; Towhid, S.T.; Shaik, N.; Lang, F. Stimulation of suicidal death of erythrocytes by rifampicin. Toxicology 2012, 302, 123–128. [Google Scholar] [CrossRef]
- Bottger, E.; Multhoff, G.; Kun, J.F.; Esen, M. Plasmodium falciparum-infected erythrocytes induce granzyme B by NK cells through expression of host-hsp70. PLoS One 2012, 7, e33774. [Google Scholar] [CrossRef]
- Felder, K.M.; Hoelzle, K.; Ritzmann, M.; Kilchling, T.; Schiele, D.; Heinritzi, K.; Groebel, K.; Hoelzle, L.E. Hemotrophic mycoplasmas induce programmed cell death in red blood cells. Cell. Physiol. Biochem. 2011, 27, 557–564. [Google Scholar] [CrossRef] [Green Version]
- Firat, U.; Kaya, S.; Cim, A.; Buyukbayram, H.; Gokalp, O.; Dal, M.S.; Tamer, M.N. Increased caspase-3 immunoreactivity of erythrocytes in stz diabetic rats. Exp. Diabetes. Res. 2012, 2012, 316–384. [Google Scholar]
- Ganesan, S.; Chaurasiya, N.D.; Sahu, R.; Walker, L.A.; Tekwani, B.L. Understanding the mechanisms for metabolism-linked hemolytic toxicity of primaquine against glucose 6-phosphate dehydrogenase deficient human erythrocytes: Evaluation of eryptotic pathway. Toxicology 2012, 294, 54–60. [Google Scholar] [CrossRef]
- Gao, M.; Cheung, K.L.; Lau, I.P.; Yu, W.S.; Fung, K.P.; Yu, B.; Loo, J.F.; Kong, S.K. Polyphyllin D induces apoptosis in human erythrocytes through Ca2+ rise and membrane permeabilization. Arch. Toxicol. 2012, 86, 741–752. [Google Scholar] [CrossRef]
- Ghashghaeinia, M.; Cluitmans, J.C.; Akel, A.; Dreischer, P.; Toulany, M.; Koberle, M.; Skabytska, Y.; Saki, M.; Biedermann, T.; Duszenko, M.; et al. The impact of erythrocyte age on eryptosis. Br. J. Haematol. 2012, 157, 606–614. [Google Scholar] [CrossRef]
- Ghashghaeinia, M.; Toulany, M.; Saki, M.; Bobbala, D.; Fehrenbacher, B.; Rupec, R.; Rodemann, H.P.; Ghoreschi, K.; Rocken, M.; Schaller, M.; et al. The nfkb pathway inhibitors bay 11-7082 and parthenolide induce programmed cell death in anucleated erythrocytes. Cell. Physiol. Biochem. 2011, 27, 45–54. [Google Scholar] [CrossRef]
- Jilani, K.; Lupescu, A.; Zbidah, M.; Abed, M.; Shaik, N.; Lang, F. Enhanced apoptotic death of erythrocytes induced by the mycotoxin ochratoxin A. Kidney Blood Press. Res. 2012, 36, 107–118. [Google Scholar] [CrossRef]
- Jilani, K.; Lupescu, A.; Zbidah, M.; Shaik, N.; Lang, F. Withaferin a-stimulated Ca2+ entry, ceramide formation and suicidal death of erythrocytes. Toxicol. in Vitro 2012, 27, 52–58. [Google Scholar]
- Kucherenko, Y.V.; Lang, F. Inhibitory effect of furosemide on non-selective voltage-independent cation channels in human erythrocytes. Cell. Physiol. Biochem. 2012, 30, 863–875. [Google Scholar] [CrossRef]
- Lang, E.; Jilani, K.; Zelenak, C.; Pasham, V.; Bobbala, D.; Qadri, S.M.; Lang, F. Stimulation of suicidal erythrocyte death by benzethonium. Cell. Physiol. Biochem. 2011, 28, 347–354. [Google Scholar] [CrossRef]
- Lang, E.; Qadri, S.M.; Jilani, K.; Zelenak, C.; Lupescu, A.; Schleicher, E.; Lang, F. Carbon monoxide-sensitive apoptotic death of erythrocytes. Basic Clin. Pharmacol. Toxicol. 2012, 111, 348–355. [Google Scholar]
- Lang, F.; Qadri, S.M. Mechanisms and significance of eryptosis, the suicidal death of erythrocytes. Blood Purif. 2012, 33, 125–130. [Google Scholar] [CrossRef]
- Lupescu, A.; Jilani, K.; Zbidah, M.; Lang, E.; Lang, F. Enhanced Ca2+ entry, ceramide formation, and apoptotic death of erythrocytes triggered by plumbagin. J. Nat. Prod. 2012, 75, 1956–1961. [Google Scholar] [CrossRef]
- Lupescu, A.; Jilani, K.; Zbidah, M.; Lang, F. Induction of apoptotic erythrocyte death by rotenone. Toxicology 2012, 300, 132–137. [Google Scholar] [CrossRef]
- Lupescu, A.; Jilani, K.; Zelenak, C.; Zbidah, M.; Qadri, S.M.; Lang, F. Hexavalent chromium-induced erythrocyte membrane phospholipid asymmetry. Biometals 2012, 25, 309–318. [Google Scholar] [CrossRef]
- Polak-Jonkisz, D.; Purzyc, L. Ca influx versus efflux during eryptosis in uremic erythrocytes. Blood Purif. 2012, 34, 209–210. [Google Scholar] [CrossRef]
- Qadri, S.M.; Bauer, J.; Zelenak, C.; Mahmud, H.; Kucherenko, Y.; Lee, S.H.; Ferlinz, K.; Lang, F. Sphingosine but not sphingosine-1-phosphate stimulates suicidal erythrocyte death. Cell. Physiol. Biochem. 2011, 28, 339–346. [Google Scholar] [CrossRef]
- Qadri, S.M.; Kucherenko, Y.; Lang, F. Beauvericin induced erythrocyte cell membrane scrambling. Toxicology 2011, 283, 24–31. [Google Scholar] [CrossRef]
- Qadri, S.M.; Kucherenko, Y.; Zelenak, C.; Jilani, K.; Lang, E.; Lang, F. Dicoumarol activates Ca2+-permeable cation channels triggering erythrocyte cell membrane scrambling. Cell. Physiol. Biochem. 2011, 28, 857–864. [Google Scholar] [CrossRef]
- Qian, E.W.; Ge, D.T.; Kong, S.K. Salidroside protects human erythrocytes against hydrogen peroxide-induced apoptosis. J. Nat. Prod. 2012, 75, 531–537. [Google Scholar] [CrossRef]
- Shaik, N.; Zbidah, M.; Lang, F. Inhibition of Ca2+ entry and suicidal erythrocyte death by naringin. Cell. Physiol. Biochem. 2012, 30, 678–686. [Google Scholar] [CrossRef]
- Vota, D.M.; Maltaneri, R.E.; Wenker, S.D.; Nesse, A.B.; Vittori, D.C. Differential erythropoietin action upon cells induced to eryptosis by different agents. Cell. Biochem. Biophys. 2012, 65, 145–157. [Google Scholar]
- Weiss, E.; Cytlak, U.M.; Rees, D.C.; Osei, A.; Gibson, J.S. Deoxygenation-induced and Ca2+ dependent phosphatidylserine externalisation in red blood cells from normal individuals and sickle cell patients. Cell. Calcium. 2012, 51, 51–56. [Google Scholar] [CrossRef]
- Zappulla, D. Environmental stress, erythrocyte dysfunctions, inflammation, and the metabolic syndrome: Adaptations to CO2 increases? Cardiometab. Syndr. 2008, 3, 30–34. [Google Scholar] [CrossRef]
- Zbidah, M.; Lupescu, A.; Jilani, K.; Lang, F. Stimulation of suicidal erythrocyte death by fumagillin. Basic Clin. Pharmacol. Toxicol. 2012, 112, 346–351. [Google Scholar]
- Zbidah, M.; Lupescu, A.; Shaik, N.; Lang, F. Gossypol-induced suicidal erythrocyte death. Toxicology 2012, 302, 101–105. [Google Scholar] [CrossRef]
- Zelenak, C.; Pasham, V.; Jilani, K.; Tripodi, P.M.; Rosaclerio, L.; Pathare, G.; Lupescu, A.; Faggio, C.; Qadri, S.M.; Lang, F. Tanshinone IIA stimulates erythrocyte phosphatidylserine exposure. Cell. Physiol. Biochem. 2012, 30, 282–294. [Google Scholar] [CrossRef]
- Lang, E.; Qadri, S.M.; Lang, F. Killing me softly—Suicidal erythrocyte death. Int. J. Biochem. Cell. Biol. 2012, 44, 1236–1243. [Google Scholar] [CrossRef]
- Calderon-Salinas, J.V.; Munoz-Reyes, E.G.; Guerrero-Romero, J.F.; Rodriguez-Moran, M.; Bracho-Riquelme, R.L.; Carrera-Gracia, M.A.; Quintanar-Escorza, M.A. Eryptosis and oxidative damage in type 2 diabetic mellitus patients with chronic kidney disease. Mol. Cell. Biochem. 2011, 357, 171–179. [Google Scholar] [CrossRef]
- Nicolay, J.P.; Schneider, J.; Niemoeller, O.M.; Artunc, F.; Portero-Otin, M.; Haik, G., Jr.; Thornalley, P.J.; Schleicher, E.; Wieder, T.; Lang, F. Stimulation of suicidal erythrocyte death by methylglyoxal. Cell. Physiol. Biochem. 2006, 18, 223–232. [Google Scholar] [CrossRef]
- Myssina, S.; Huber, S.M.; Birka, C.; Lang, P.A.; Lang, K.S.; Friedrich, B.; Risler, T.; Wieder, T.; Lang, F. Inhibition of erythrocyte cation channels by erythropoietin. J. Am. Soc. Nephrol. 2003, 14, 2750–2757. [Google Scholar] [CrossRef]
- Lang, P.A.; Beringer, O.; Nicolay, J.P.; Amon, O.; Kempe, D.S.; Hermle, T.; Attanasio, P.; Akel, A.; Schafer, R.; Friedrich, B.; et al. Suicidal death of erythrocytes in recurrent hemolytic uremic syndrome. J. Mol. Med. 2006, 84, 378–388. [Google Scholar] [CrossRef]
- Kempe, D.S.; Akel, A.; Lang, P.A.; Hermle, T.; Biswas, R.; Muresanu, J.; Friedrich, B.; Dreischer, P.; Wolz, C.; Schumacher, U.; et al. Suicidal erythrocyte death in sepsis. J. Mol. Med. 2007, 85, 269–277. [Google Scholar]
- Bobbala, D.; Alesutan, I.; Föller, M.; Huber, S.M.; Lang, F. Effect of anandamide in plasmodium berghei-infected mice. Cell. Physiol. Biochem. 2010, 26, 355–362. [Google Scholar] [CrossRef]
- Föller, M.; Bobbala, D.; Koka, S.; Huber, S.M.; Gulbins, E.; Lang, F. Suicide for survival—Death of infected erythrocytes as a host mechanism to survive malaria. Cell. Physiol. Biochem. 2009, 24, 133–140. [Google Scholar] [CrossRef]
- Koka, S.; Bobbala, D.; Lang, C.; Boini, K.M.; Huber, S.M.; Lang, F. Influence of paclitaxel on parasitemia and survival of plasmodium berghei infected mice. Cell. Physiol. Biochem. 2009, 23, 191–198. [Google Scholar] [CrossRef]
- Lang, P.A.; Schenck, M.; Nicolay, J.P.; Becker, J.U.; Kempe, D.S.; Lupescu, A.; Koka, S.; Eisele, K.; Klarl, B.A.; Rubben, H.; et al. Liver cell death and anemia in wilson disease involve acid sphingomyelinase and ceramide. Nat. Med. 2007, 13, 164–170. [Google Scholar] [CrossRef]
- Siraskar, B.; Ballal, A.; Bobbala, D.; Föller, M.; Lang, F. Effect of amphotericin B on parasitemia and survival of plasmodium berghei-infected mice. Cell. Physiol. Biochem. 2010, 26, 347–354. [Google Scholar] [CrossRef]
- Lang, P.A.; Kasinathan, R.S.; Brand, V.B.; Duranton, C.; Lang, C.; Koka, S.; Shumilina, E.; Kempe, D.S.; Tanneur, V.; Akel, A.; et al. Accelerated clearance of plasmodium-infected erythrocytes in sickle cell trait and annexin-a7 deficiency. Cell. Physiol. Biochem. 2009, 24, 415–428. [Google Scholar] [CrossRef]
- Kempe, D.S.; Lang, P.A.; Duranton, C.; Akel, A.; Lang, K.S.; Huber, S.M.; Wieder, T.; Lang, F. Enhanced programmed cell death of iron-deficient erythrocytes. FASEB J. 2006, 20, 368–370. [Google Scholar]
- Qadri, S.M.; Mahmud, H.; Lang, E.; Gu, S.; Bobbala, D.; Zelenak, C.; Jilani, K.; Siegfried, A.; Föller, M.; Lang, F. Enhanced suicidal erythrocyte death in mice carrying a loss-of-function mutation of the adenomatous polyposis coli gene. J. Cell. Mol. Med. 2012, 16, 1085–1093. [Google Scholar] [CrossRef]
- Birka, C.; Lang, P.A.; Kempe, D.S.; Hoefling, L.; Tanneur, V.; Duranton, C.; Nammi, S.; Henke, G.; Myssina, S.; Krikov, M.; et al. Enhanced susceptibility to erythrocyte “apoptosis” following phosphate depletion. Pflüg. Arch. 2004, 448, 471–477. [Google Scholar]
- Gottschalk, L.A.; Biener, R.; Noble, E.P.; Birch, H.; Wilbert, D.E.; Heiser, J.F. Thioridazine plasma levels and clinical response. Compr. Psychiatry 1975, 16, 323–337. [Google Scholar] [CrossRef]
- Bookchin, R.M.; Ortiz, O.E.; Lew, V.L. Activation of calcium-dependent potassium channels in deoxygenated sickled red cells. Prog. Clin. Biol. Res. 1987, 240, 193–200. [Google Scholar]
- Brand, V.B.; Sandu, C.D.; Duranton, C.; Tanneur, V.; Lang, K.S.; Huber, S.M.; Lang, F. Dependence of plasmodium falciparum in vitro growth on the cation permeability of the human host erythrocyte. Cell. Physiol. Biochem. 2003, 13, 347–356. [Google Scholar] [CrossRef]
- Palacios, J.; Sepulveda, M.R.; Lee, A.G.; Mata, A.M. Ca2+ transport by the synaptosomal plasma membrane Ca2+-atpase and the effect of thioridazine. Biochemistry 2004, 43, 2353–2358. [Google Scholar] [CrossRef]
- Borst, O.; Abed, M.; Alesutan, I.; Towhid, S.T.; Qadri, S.M.; Föller, M.; Gawaz, M.; Lang, F. Dynamic adhesion of eryptotic erythrocytes to endothelial cells via cxcl16/sr-psox. Am. J. Physiol. Cell. Physiol. 2012, 302, C644–C651. [Google Scholar] [CrossRef]
- Andrews, D.A.; Low, P.S. Role of red blood cells in thrombosis. Curr. Opin. Hematol. 1999, 6, 76–82. [Google Scholar] [CrossRef]
- Closse, C.; Dachary-Prigent, J.; Boisseau, M.R. Phosphatidylserine-related adhesion of human erythrocytes to vascular endothelium. Br. J. Haematol. 1999, 107, 300–302. [Google Scholar] [CrossRef]
- Gallagher, P.G.; Chang, S.H.; Rettig, M.P.; Neely, J.E.; Hillery, C.A.; Smith, B.D.; Low, P.S. Altered erythrocyte endothelial adherence and membrane phospholipid asymmetry in hereditary hydrocytosis. Blood 2003, 101, 4625–4627. [Google Scholar] [CrossRef]
- Pandolfi, A.; Di Pietro, N.; Sirolli, V.; Giardinelli, A.; Di Silvestre, S.; Amoroso, L.; Di Tomo, P.; Capani, F.; Consoli, A.; Bonomini, M. Mechanisms of uremic erythrocyte-induced adhesion of human monocytes to cultured endothelial cells. J. Cell. Physiol. 2007, 213, 699–709. [Google Scholar]
- Wood, B.L.; Gibson, D.F.; Tait, J.F. Increased erythrocyte phosphatidylserine exposure in sickle cell disease: Flow-cytometric measurement and clinical associations. Blood 1996, 88, 1873–1880. [Google Scholar]
- Chung, S.M.; Bae, O.N.; Lim, K.M.; Noh, J.Y.; Lee, M.Y.; Jung, Y.S.; Chung, J.H. Lysophosphatidic acid induces thrombogenic activity through phosphatidylserine exposure and procoagulant microvesicle generation in human erythrocytes. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 414–421. [Google Scholar]
- Zwaal, R.F.; Comfurius, P.; Bevers, E.M. Surface exposure of phosphatidylserine in pathological cells. Cell. Mol. Life Sci. 2005, 62, 971–988. [Google Scholar] [CrossRef]
- Le Blaye, I.; Donatini, B.; Hall, M.; Krupp, P. Acute overdosage with thioridazine: A review of the available clinical exposure. Vet. Hum. Toxicol. 1993, 35, 147–150. [Google Scholar]
- Isbister, G.K.; Balit, C.R.; Kilham, H.A. Antipsychotic poisoning in young children: A systematic review. Drug Saf. 2005, 28, 1029–1044. [Google Scholar] [CrossRef]
- Schmidt, W.; Lang, K. Life-threatening dysrhythmias in severe thioridazine poisoning treated with physostigmine and transient atrial pacing. Crit. Care Med. 1997, 25, 1925–1930. [Google Scholar] [CrossRef]
- Lee, Y.M.; Cheng, P.Y.; Chen, S.Y.; Chung, M.T.; Sheu, J.R. Wogonin suppresses arrhythmias, inflammatory responses, and apoptosis induced by myocardial ischemia/reperfusion in rats. J. Cardiovasc. Pharmacol. 2011, 58, 133–142. [Google Scholar] [CrossRef]
- Surinkaew, S.; Kumphune, S.; Chattipakorn, S.; Chattipakorn, N. Inhibition of p38 mapk during ischemia, but not reperfusion, effectively attenuates fatal arrhythmia in ischemia/reperfusion heart. J. Cardiovasc. Pharmacol. 2013, 61, 133–141. [Google Scholar] [CrossRef]
- Harrison, H.E.; Bunting, H.; Ordway, N.K.; Albrink, W.S. The pathogenesis of the renal injury produced in the dog by hemoglobin or methemoglobin. J. Exp. Med. 1947, 86, 339–356. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lang, E.; Modicano, P.; Arnold, M.; Bissinger, R.; Faggio, C.; Abed, M.; Lang, F. Effect of Thioridazine on Erythrocytes. Toxins 2013, 5, 1918-1931. https://doi.org/10.3390/toxins5101918
Lang E, Modicano P, Arnold M, Bissinger R, Faggio C, Abed M, Lang F. Effect of Thioridazine on Erythrocytes. Toxins. 2013; 5(10):1918-1931. https://doi.org/10.3390/toxins5101918
Chicago/Turabian StyleLang, Elisabeth, Paola Modicano, Markus Arnold, Rosi Bissinger, Caterina Faggio, Majed Abed, and Florian Lang. 2013. "Effect of Thioridazine on Erythrocytes" Toxins 5, no. 10: 1918-1931. https://doi.org/10.3390/toxins5101918