Glycyrrhizic Acid and Compound Probiotics Supplementation Alters the Intestinal Transcriptome and Microbiome of Weaned Piglets Exposed to Deoxynivalenol

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Transcriptional Changes of Piglets with GAP Supplementation
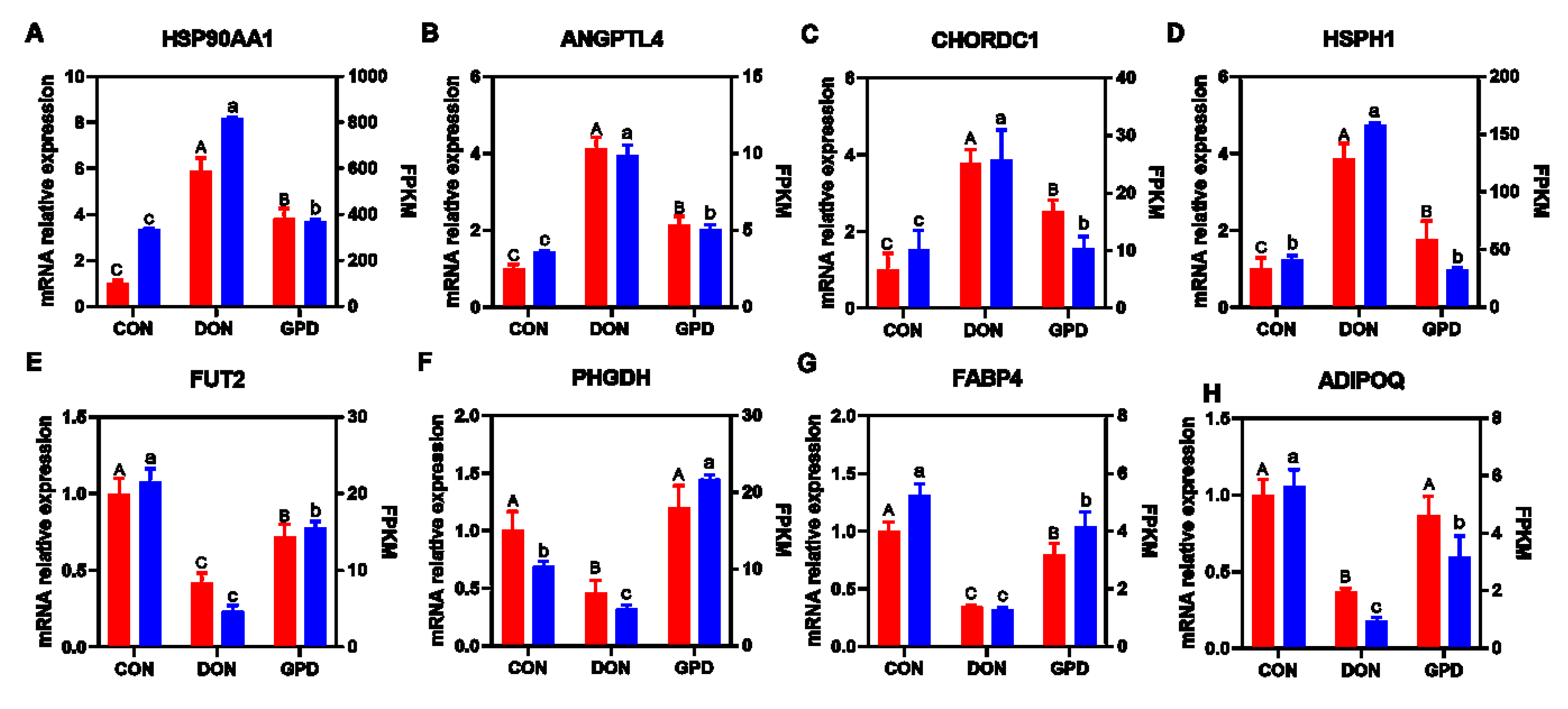
2.2. QRT-PCR Validation
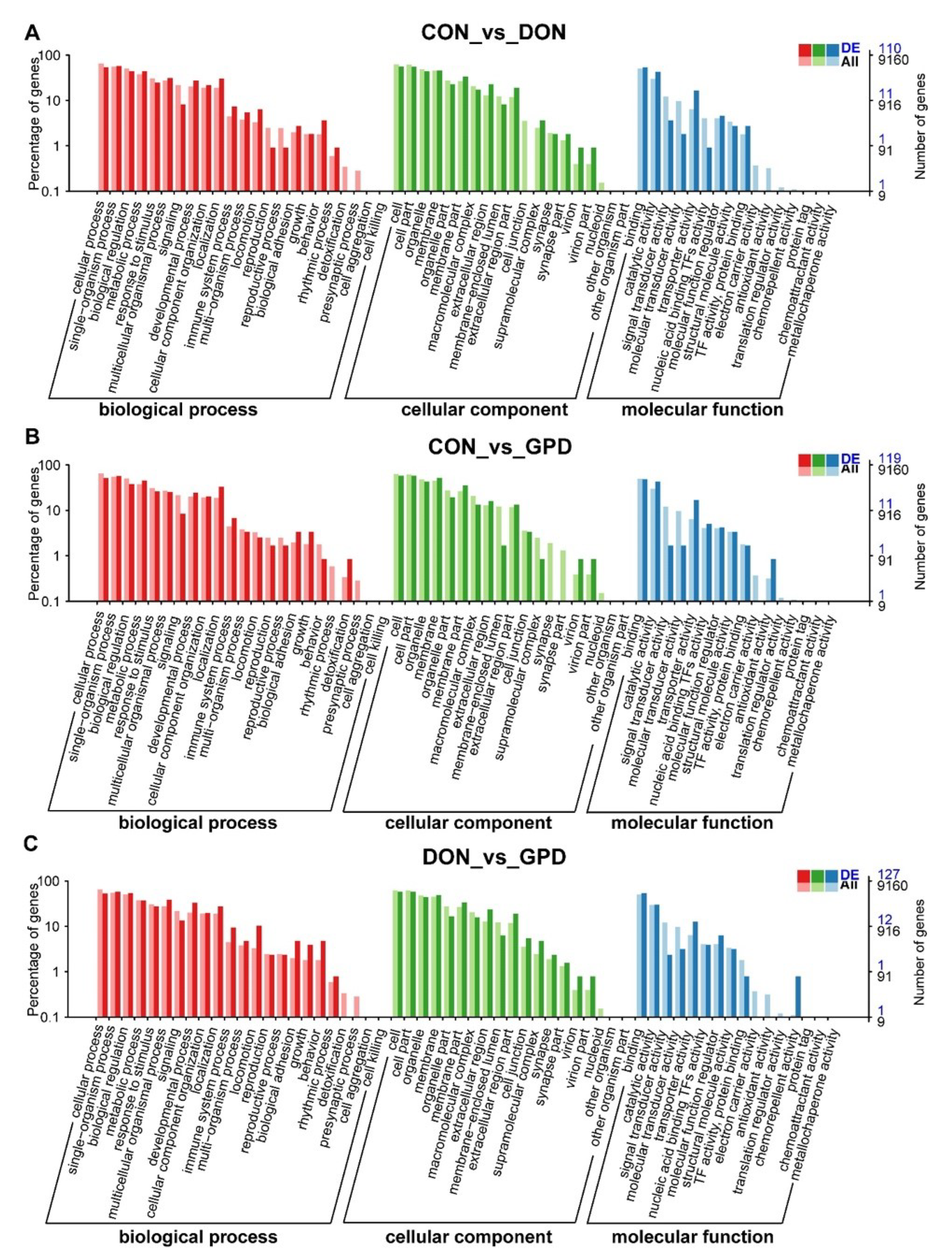
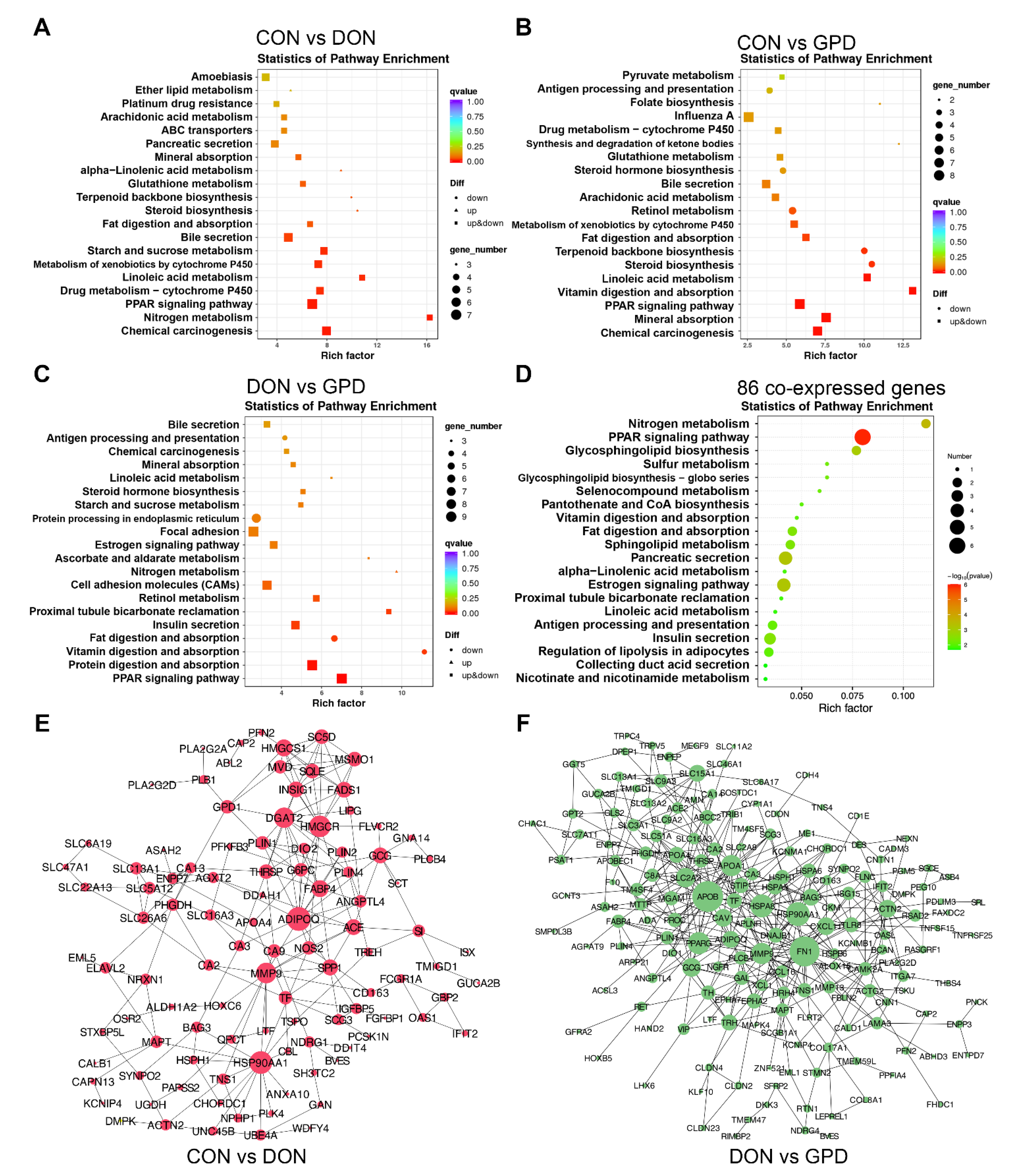
2.3. Functional Annotation Analyses
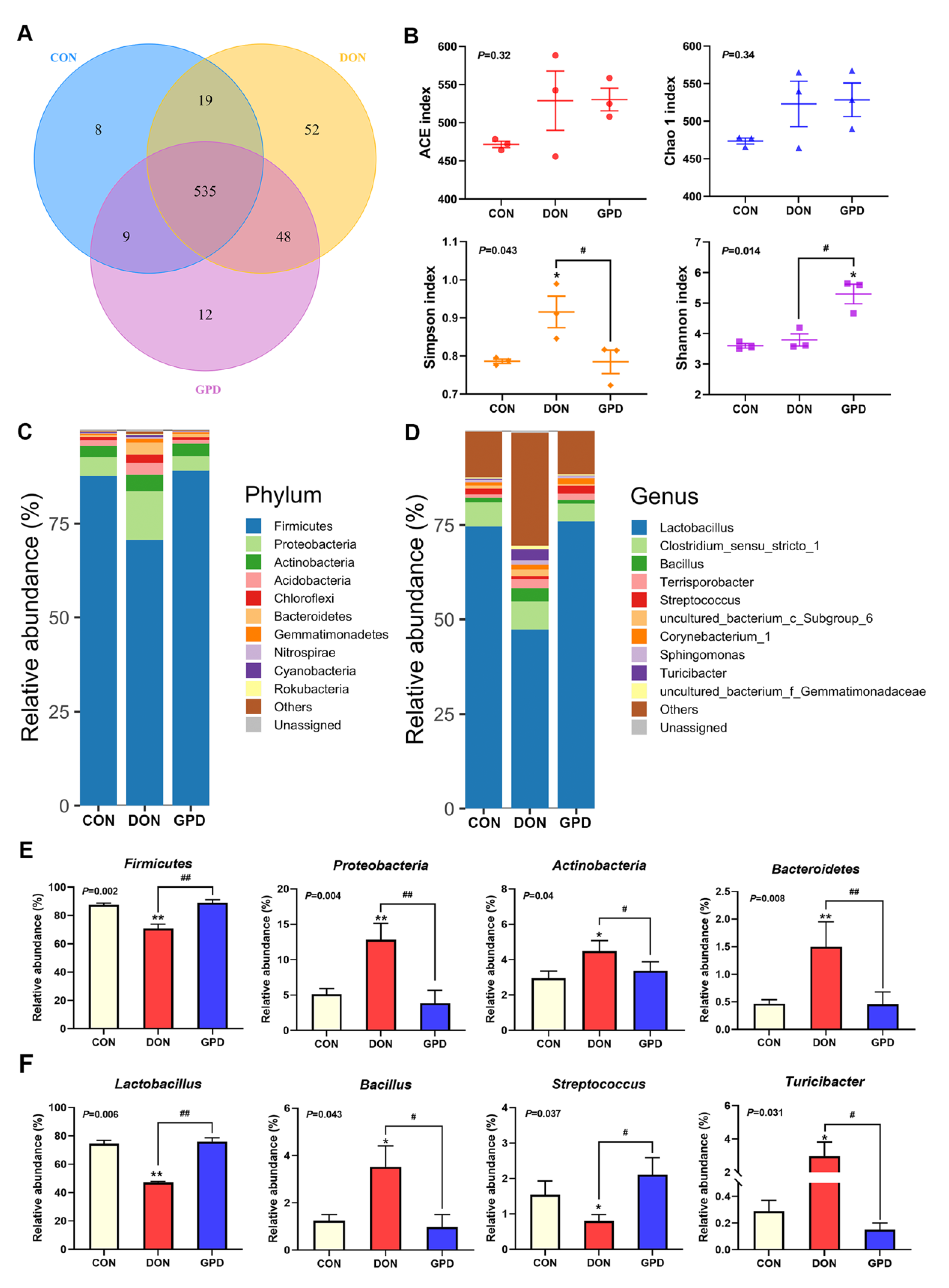
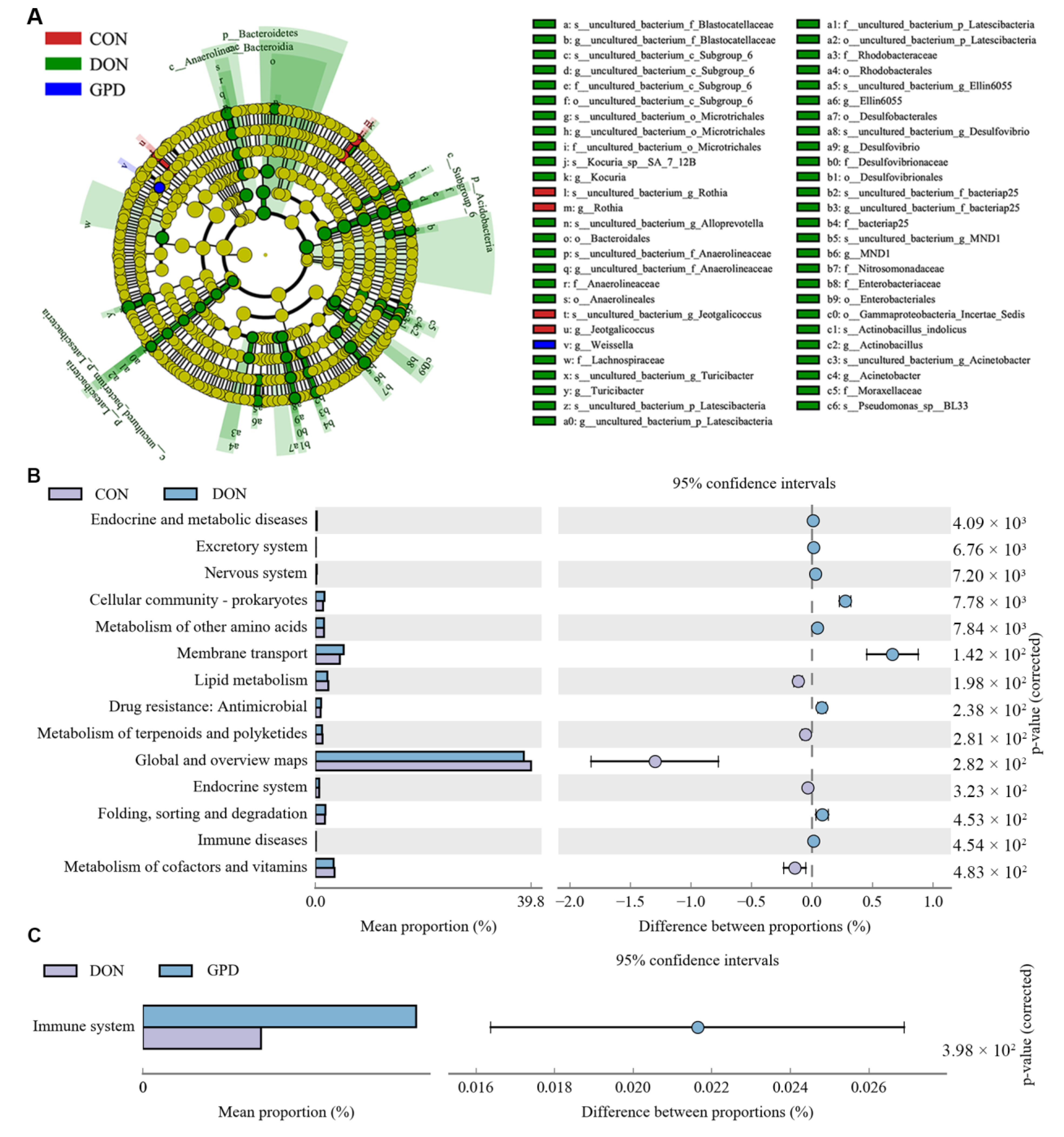
2.4. Differences of Microbiota Composition in Jejunum Contents
2.5. LEfSe and Predictive Function Analysis
2.6. Correlation Analysis between the DEGs and Gut Microbiota
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Glycyrrhizic Acid and Compound Probiotics Preparation
5.2. Animals and Managements
5.3. RNA-Seq Analysis
5.4. QRT-PCR Validation
5.5. 16S rRNA Gene Sequencing Analysis
5.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mishra, S.; Srivastava, S.; Dewangan, J.; Divakar, A.; Kumar Rath, S. Global occurrence of deoxynivalenol in food commodities and exposure risk assessment in humans in the last decade: A survey. Crit. Rev. Food Sci. Nutr. 2020, 60, 1346–1374. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.; Liu, Z.; Liu, S.; Yao, L.; Liu, Y.; Wu, Y.; Gong, Z. Natural occurrence of deoxynivalenol and its acetylated derivatives in Chinese maize and wheat collected in 2017. Toxins 2020, 12, 200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pestka, J.J. Deoxynivalenol: Mechanisms of action, human exposure, and toxicological relevance. Arch. Toxicol. 2010, 84, 663–679. [Google Scholar] [CrossRef] [PubMed]
- Pestka, J.J.; Smolinski, A.T. Deoxynivalenol: Toxicology and potential effects on humans. J. Toxicol. Environ. Health B Crit. Rev. 2005, 8, 39–69. [Google Scholar] [CrossRef]
- Sugita-Konishi, Y.; Park, B.J.; Kobayashi-Hattori, K.; Tanaka, T.; Chonan, T.; Yoshikawa, K.; Kumagai, S. Effect of cooking process on the deoxynivalenol content and its subsequent cytotoxicity in wheat products. Biosci. Biotechnol. Biochem. 2006, 70, 1764–1768. [Google Scholar] [CrossRef] [Green Version]
- Feizollahi, E.; Roopesh, M.S. Mechanisms of deoxynivalenol (DON) degradation during different treatments: A review. Crit. Rev. Food Sci. Nutr. 2022, 62, 5903–5924. [Google Scholar] [CrossRef]
- Yao, Y.; Long, M. The biological detoxification of deoxynivalenol: A review. Food Chem. Toxicol. 2020, 145, 111649. [Google Scholar] [CrossRef]
- Franco, T.S.; Garcia, S.; Hirooka, E.Y.; Ono, Y.S.; dos Santos, J.S. Lactic acid bacteria in the inhibition of fusarium graminearum and deoxynivalenol detoxification. J. Appl. Microbiol. 2011, 111, 739–748. [Google Scholar] [CrossRef]
- Massoud, R.; Zoghi, A. Potential probiotic strains with heavy metals and mycotoxins bioremoval capacity for application in foodstuffs. J. Appl. Microbiol. 2022, 133, 1288–1307. [Google Scholar] [CrossRef]
- Chlebicz, A.; Slizewska, K. In vitro detoxification of aflatoxin B1, deoxynivalenol, fumonisins, T-2 toxin and zearalenone by probiotic bacteria from genus Lactobacillus and Saccharomyces cerevisiae yeast. Probiotics Antimicrob. Proteins 2020, 12, 289–301. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, W.; Zhang, Z.; Liu, F.; Wang, J.; Yin, Y.; Wang, Z. Effects of Enterococcus faecalis on egg production, egg quality and caecal microbiota of hens during the late laying period. Arch. Anim. Nutr. 2019, 73, 208–221. [Google Scholar] [CrossRef] [PubMed]
- Alfajaro, M.M.; Kim, H.J.; Park, J.G.; Ryu, E.H.; Kim, J.Y.; Jeong, Y.J.; Kim, D.S.; Hosmillo, M.; Son, K.Y.; Lee, J.H.; et al. Anti-rotaviral effects of Glycyrrhiza uralensis extract in piglets with rotavirus diarrhea. Virol. J. 2012, 9, 310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Yang, C.; Chang, J.; Wang, P.; Yin, Q.; Liu, C.; Gao, T.; Dang, X.; Lu, F. Dietary Supplementation with Compound Probiotics and Berberine Alters Piglet Production Performance and Fecal Microbiota. Animals 2020, 10, 511. [Google Scholar] [CrossRef] [Green Version]
- Xu, X. The Molecular Mechanism of Combination of Glycyrrhizic Acid and Probiotics for Alleviating the Hazards of Weaned Piglet Growth and Gut Health Induced by Deoxynivalenol. Ph.D. Thesis, Henan Agricultural University, Zhengzhou, China, 2021. [Google Scholar] [CrossRef]
- Xu, C.; Wang, X.; Zhou, S.; Wu, J.; Geng, Q.; Ruan, D.; Qiu, Y.; Quan, J.; Ding, R.; Cai, G.; et al. Brain transcriptome analysis reveals potential transcription factors and biological pathways associated with feed efficiency in commercial DLY pigs. DNA Cell Biol. 2021, 40, 272–282. [Google Scholar] [CrossRef]
- Guo, M.; Huang, K.; Chen, S.; Qi, X.; He, X.; Cheng, W.H.; Luo, Y.; Xia, K.; Xu, W. Combination of metagenomics and culture-based methods to study the interaction between ochratoxin a and gut microbiota. Toxicol. Sci. 2014, 141, 314–323. [Google Scholar] [CrossRef]
- Pasternak, J.A.; Aiyer, V.I.A.; Hamonic, G.; Beaulieu, A.D.; Columbus, D.A.; Wilson, H.L. Molecular and physiological effects on the small intestine of weaner pigs following feeding with Deoxynivalenol-contaminated feed. Toxins 2018, 10, 40. [Google Scholar] [CrossRef] [Green Version]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef]
- Seibert, J.T.; Adur, M.K.; Schultz, R.B.; Thomas, P.Q.; Kiefer, Z.E.; Keating, A.F.; Baumgard, L.H.; Ross, J.W. Differentiating between the effects of heat stress and lipopolysaccharide on the porcine ovarian heat shock protein response1. J. Anim. Sci. 2019, 97, 4965–4973. [Google Scholar] [CrossRef]
- Zlotnik, A.; Yoshie, O. The chemokine superfamily revisited. Immunity 2012, 36, 705–716. [Google Scholar] [CrossRef]
- Zhao, Y.; Albrecht, E.; Li, Z.; Schregel, J.; Sciascia, Q.L.; Metges, C.C.; Maak, S. Distinct roles of perilipins in the intramuscular deposition of lipids in glutamine-supplemented, low-, and normal-birth-weight piglets. Front. Vet. Sci. 2021, 8, 633898. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Raza, S.H.A.; Zhao, C.; Cheng, G.; Zan, L. Overexpression of PLIN1 promotes lipid metabolism in bovine adipocytes. Animals 2020, 10, 1944. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, Y.; Zhu, Q. Cigarette smoke extract-mediated FABP4 upregulation suppresses viability and induces apoptosis, inflammation and oxidative stress of bronchial epithelial cells by activating p38 MAPK/MK2 signaling pathway. J. Inflamm. 2022, 19, 7. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Li, D.G.; Li, Q.; Huang, L.; He, Z.; Zhang, F.; Wang, C.B. Relationship between adipoq gene polymorphism and lipid levels and diabetes. J. Biol. Regul. Homeost. Agents 2015, 29, 221–227. [Google Scholar]
- Spillier, Q.; Frederick, R. Phosphoglycerate dehydrogenase (PHGDH) inhibitors: A comprehensive review 2015–2020. Expert Opin. Ther. Pat. 2021, 31, 597–608. [Google Scholar] [CrossRef]
- Kurz, S.; King, J.G.; Dinglasan, R.R.; Paschinger, K.; Wilson, I.B. The fucomic potential of mosquitoes: Fucosylated N-glycan epitopes and their cognate fucosyltransferases. Insect Biochem. Mol. Biol. 2016, 68, 52–63. [Google Scholar] [CrossRef] [Green Version]
- Pham, T.A.; Clare, S.; Goulding, D.; Arasteh, J.M.; Stares, M.D.; Browne, H.P.; Keane, J.A.; Page, A.J.; Kumasaka, N.; Kane, L.; et al. Epithelial IL-22RA1-mediated fucosylation promotes intestinal colonization resistance to an opportunistic pathogen. Cell Host Microbe 2014, 16, 504–516. [Google Scholar] [CrossRef] [Green Version]
- Robert, H.; Payros, D.; Pinton, P.; Theodorou, V.; Mercier-Bonin, M.; Oswald, I.P. Impact of mycotoxins on the intestine: Are mucus and microbiota new targets? J. Toxicol. Environ. Health B Crit. Rev. 2017, 20, 249–275. [Google Scholar] [CrossRef]
- Perez-Burillo, S.; Hinojosa-Nogueira, D.; Pastoriza, S.; Rufian-Henares, J.A. Plant extracts as natural modulators of gut microbiota community structure and functionality. Heliyon 2020, 6, e05474. [Google Scholar] [CrossRef]
- Wang, M.; Wu, H.; Lu, L.; Jiang, L.; Yu, Q. Lactobacillus reuteri promotes intestinal development and regulates mucosal immune function in newborn piglets. Front. Vet. Sci. 2020, 7, 42. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Cai, R.; Huang, A.; Wang, X.; Qu, W.; Shi, L.; Li, C.; Yan, H. Comparison of oropharyngeal microbiota in healthy piglets and piglets With respiratory disease. Front. Microbiol. 2018, 9, 3218. [Google Scholar] [CrossRef]
- Downs, I.A.; Aroniadis, O.C.; Kelly, L.; Brandt, L.J. Postinfection irritable bowel syndrome: The links between gastroenteritis, inflammation, the microbiome, and functional disease. J. Clin. Gastroenterol. 2017, 51, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Litvak, Y.; Byndloss, M.X.; Tsolis, R.M.; Baumler, A.J. Dysbiotic proteobacteria expansion: A microbial signature of epithelial dysfunction. Curr. Opin. Microbiol. 2017, 39, 1–6. [Google Scholar] [CrossRef]
- Bruns, A.N.; Lo, S.H. Tensin regulates pharyngeal pumping in Caenorhabditis elegans. Biochem. Biophys. Res. Commun. 2020, 522, 599–603. [Google Scholar] [CrossRef]
- Verardo, L.L.; Sevon-Aimonen, M.L.; Serenius, T.; Hietakangas, V.; Uimari, P. Whole-genome association analysis of pork meat pH revealed three significant regions and several potential genes in Finnish Yorkshire pigs. BMC Genet. 2017, 18, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, J.; Yan, G.; Chen, D.; Xiao, S.; Gao, J.; Zhang, Z. An association study using imputed whole-genome sequence data identifies novel significant loci for growth-related traits in a Duroc x Erhualian F2 population. J. Anim. Breed. Genet. 2019, 136, 217–228. [Google Scholar] [CrossRef]
- Reis, L.M.; Basel, D.; McCarrier, J.; Weinberg, D.V.; Semina, E.V. Compound heterozygous splicing CDON variants result in isolated ocular coloboma. Clin. Genet. 2020, 98, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Jones, F.S.; Kioussi, C.; Copertino, D.W.; Kallunki, P.; Holst, B.D.; Edelman, G.M. Barx2, a new homeobox gene of the Bar class, is expressed in neural and craniofacial structures during development. Proc. Natl. Acad. Sci. USA 1997, 94, 2632–2637. [Google Scholar] [CrossRef] [Green Version]
- Meech, R.; Edelman, D.B.; Jones, F.S.; Makarenkova, H.P. The homeobox transcription factor Barx2 regulates chondrogenesis during limb development. Development 2005, 132, 2135–2146. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Chang, J.; Wang, P.; Yin, Q.Q.; Huang, W.W.; Liu, C.Q.; Bai, X.X.; Zhu, Q.; Gao, T.Z.; Zhou, P. Effects of Saccharomyces cerevisiae on alleviating cytotoxicity of porcine jejunal epithelia cells induced by deoxynivalenol. AMB Expr. 2019, 9, 137. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Yan, G.; Chang, J.; Wang, P.; Yin, Q.; Liu, C.; Zhu, Q.; Lu, F. Comparative transcriptome analysis reveals the protective mechanism of glycyrrhinic acid for deoxynivalenol-induced inflammation and apoptosis in IPEC-J2 cells. Oxid. Med. Cell. Longev. 2020, 2020, 5974157. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Meth. 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Meth. 2010, 7, 335–336. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Accession Number | Primer Sequence (5′-3′) |
|---|---|---|
| HSP90AA1 | NM_213973.2 | F: ATCGCCCAGTTGATGTCGTT |
| R: TATCGTGAGGGTCCGGTCTT | ||
| ANGPTL4 | NM_001038644.1 | F: TGTTTGAAGAGGGAGAGCGG |
| R: TGGTTAAAGTCCACCGAGCC | ||
| CHORDC1 | NM_001113446.1 | F: GCCACCAGAACCAGTCAAAC |
| R: TGTCATTGGTTCATCTGGGCT | ||
| HSPH1 | NM_001097504.1 | F: TGAAAGTCAAGGTGCGAGTCA |
| R: GCTTCACTGTTGTCTTGCTGG | ||
| FUT2 | NM_214069.1 | F: TAAGCACTGATGTCGGCTGG |
| R: CTCCCTGTGCCTTGGAAGTG | ||
| PHDGDH | NM_001123162.1 | F: GTCCTACCAGACCTCAGTGG |
| R: ATGGAACTGGAAAGCCTCAGT | ||
| FABP4 | NM_001002817.1 | F: ACGGCTTCTTTCTCACCTTGA |
| R: AGCCCACTCCCACTTCTTTC | ||
| ADIPOQ | NM_214370.1 | F: GTTGAAGGTCCCCGAGGTTT |
| R: CCACACTGAATGCTGAACGG | ||
| GAPDH | NM_001206359.1 | F: ATGACCACAGTCCATGCCATC |
| R: CCTGCTTCACCACCTTCTTG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, X.; Chang, J.; Wang, P.; Liu, C.; Liu, M.; Zhou, T.; Yin, Q.; Yan, G. Glycyrrhizic Acid and Compound Probiotics Supplementation Alters the Intestinal Transcriptome and Microbiome of Weaned Piglets Exposed to Deoxynivalenol. Toxins 2022, 14, 856. https://doi.org/10.3390/toxins14120856
Xu X, Chang J, Wang P, Liu C, Liu M, Zhou T, Yin Q, Yan G. Glycyrrhizic Acid and Compound Probiotics Supplementation Alters the Intestinal Transcriptome and Microbiome of Weaned Piglets Exposed to Deoxynivalenol. Toxins. 2022; 14(12):856. https://doi.org/10.3390/toxins14120856
Chicago/Turabian StyleXu, Xiaoxiang, Juan Chang, Ping Wang, Chaoqi Liu, Mengjie Liu, Ting Zhou, Qingqiang Yin, and Guorong Yan. 2022. "Glycyrrhizic Acid and Compound Probiotics Supplementation Alters the Intestinal Transcriptome and Microbiome of Weaned Piglets Exposed to Deoxynivalenol" Toxins 14, no. 12: 856. https://doi.org/10.3390/toxins14120856