Inflammation and Premature Ageing in Chronic Kidney Disease

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction—CKD, Inflammation, and Premature Ageing
2. Inflammation in CKD
2.1. Uremic Inflammation
2.2. Consequences of Inflammation on Premature Ageing in CKD
3. Premature Ageing in CKD
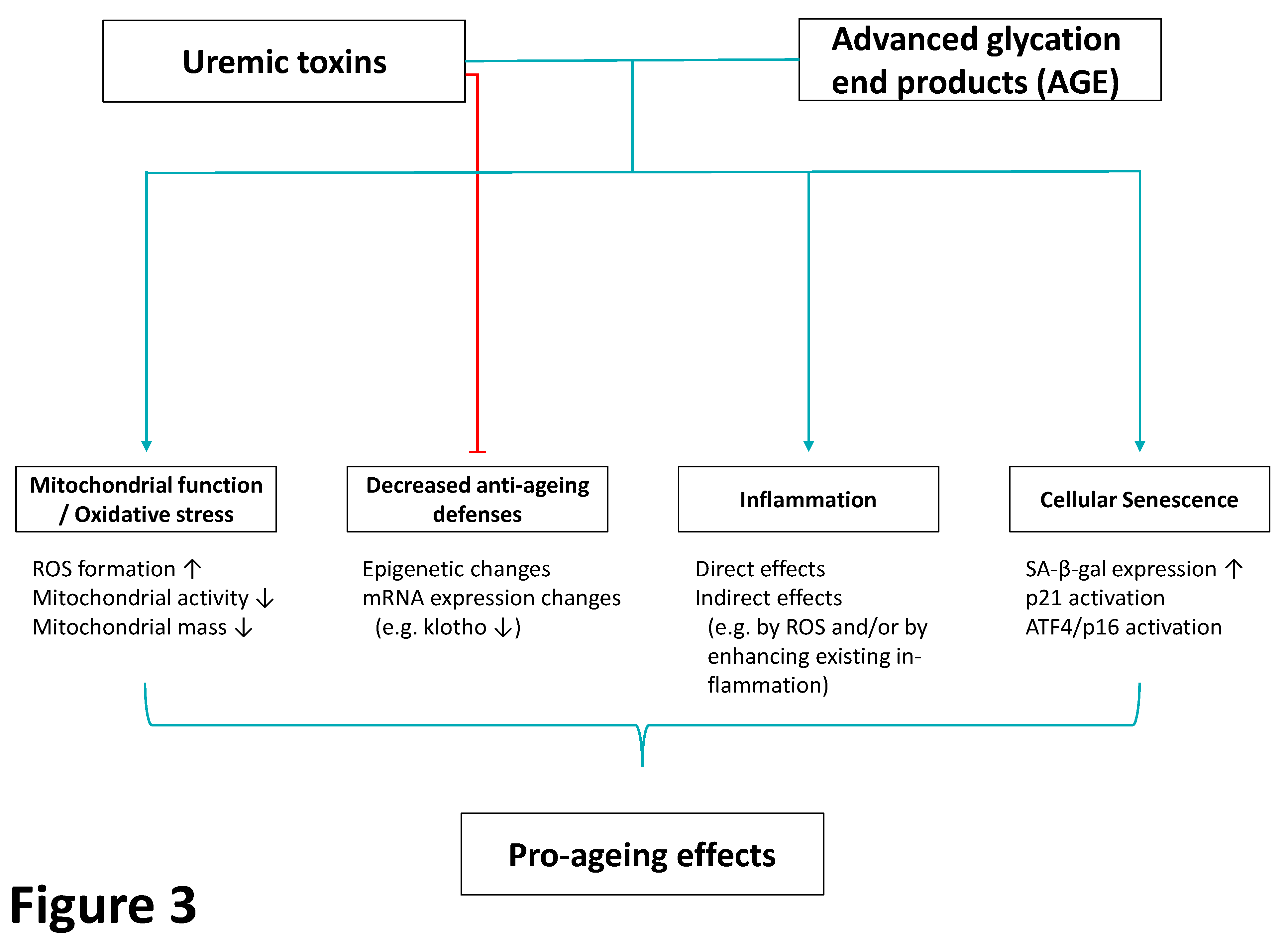
3.1. Uremic Toxins and Premature Ageing in CKD
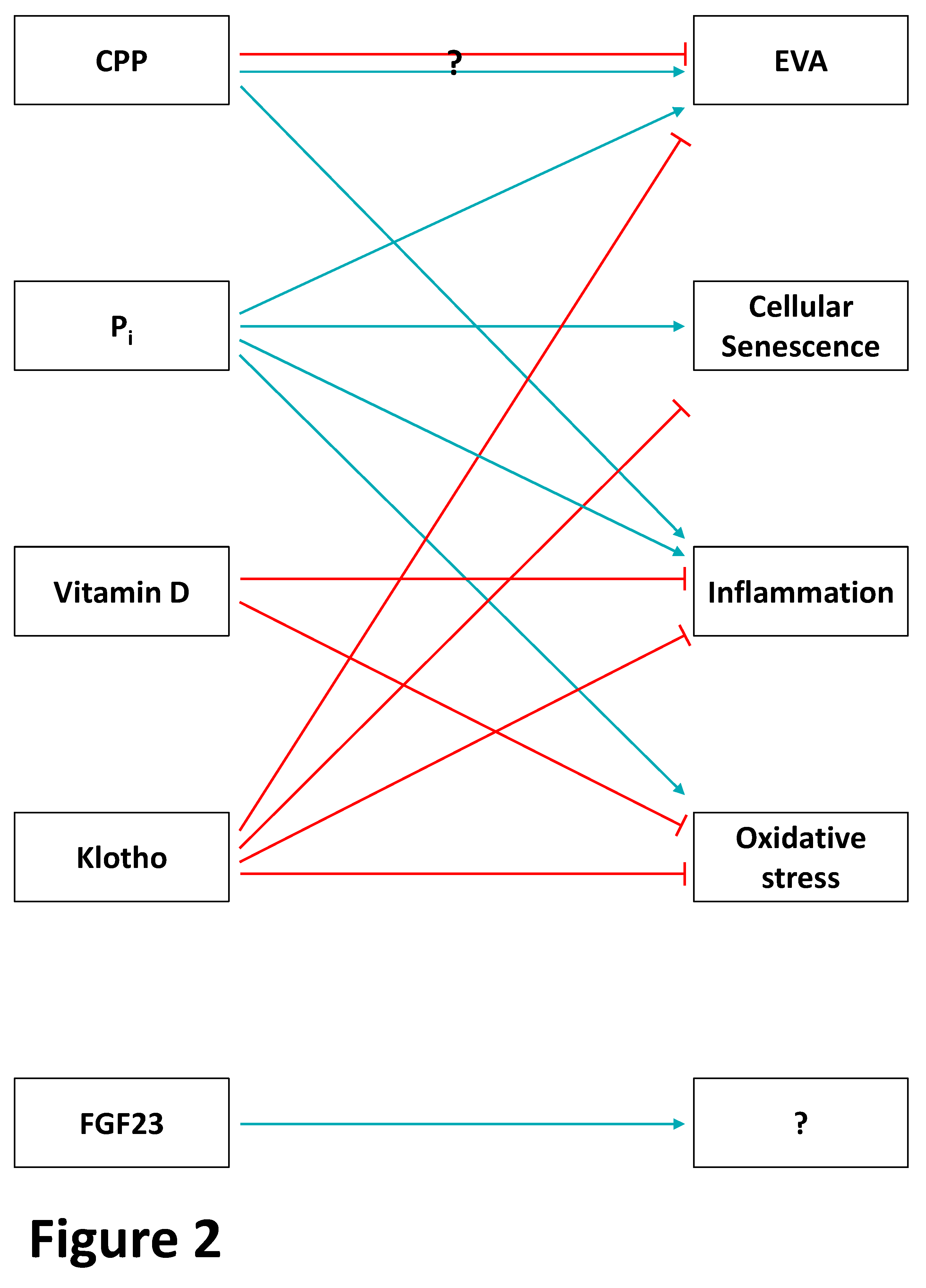
3.2. The Endocrine Phosphate-FGF-23–Klotho Axis and Premature Ageing in CKD
3.3. Mitochondrial Dysfunction/Oxidative Stress and Premature Ageing in CKD
4. Secondary Premature Ageing by CKD-Causing Diseases and CKD Treatment
4.1. Glomerular Diseases Excluding Diabetic Kidney Disease (DKD)
4.2. Polycystic Kidney Disease as a Model of Adverse Anti-Senescence
4.3. Obesity, Diabetes Mellitus, and DKD
4.4. CKD-/ESKD Treatment-Associated Ageing (Dialysis, Transplantation)
5. Approaches for Handling of Inflammation and Premature Ageing in CKD
5.1. NRF2–KEAP1 Signaling Pathway
5.2. Klotho Pathway
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bikbov, B.; Purcell, C.A.; Levey, A.S.; Smith, M.; Abdoli, A.; Abebe, M.; Adebayo, O.M.; Afarideh, M.; Agarwal, S.K.; Agudelo-Botero, M.; et al. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the global burden of disease study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Ortega, M.; Rayego-Mateos, S.; Lamas, S.; Ortiz, A.; Rodrigues-Diez, R.R. Targeting the progression of chronic kidney disease. Nat. Rev. Nephrol. 2020, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; MacFadyen, J.G.; Glynn, R.J.; Koenig, W.; Libby, P.; Everett, B.M.; Lefkowitz, M.; Thuren, T.; Cornel, J.H. Inhibition of interleukin-1β by canakinumab and cardiovascular outcomes in patients with chronic kidney disease. J. Am. Coll. Cardiol. 2018, 71, 2405–2414. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Qureshi, A.R.; Witasp, A.; Lindholm, B.; Stenvinkel, P. Early vascular ageing and cellular senescence in chronic kidney disease. Comput. Struct. Biotechnol. J. 2019, 17, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Hobson, S.; Arefin, S.; Kublickiene, K.; Shiels, P.G.; Stenvinkel, P. Senescent cells in early vascular ageing and bone disease of chronic kidney disease—A novel target for treatment. Toxins 2019, 11, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kooman, J.P.; Kotanko, P.; Schols, A.M.W.J.; Shiels, P.G.; Stenvinkel, P. Chronic kidney disease and premature ageing. Nat. Rev. Nephrol. 2014, 10, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Stenvinkel, P.; Meyer, C.J.; Block, G.A.; Chertow, G.M.; Shiels, P.G. Understanding the role of the cytoprotective transcription factor nuclear factor erythroid 2–related factor 2—Lessons from evolution, the animal kingdom and rare progeroid syndromes. Nephrol. Dial. Transplant. 2019. [Google Scholar] [CrossRef] [Green Version]
- Stenvinkel, P.; Painer, J.; Kuro-o, M.; Lanaspa, M.; Arnold, W.; Ruf, T.; Shiels, P.G.; Johnson, R.J. Novel treatment strategies for chronic kidney disease: Insights from the animal kingdom. Nat. Rev. Nephrol. 2018, 14, 265–284. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; León, R.; López, M.G.; Oliva, B.; et al. Transcription factor NRF2 as a therapeutic target for chronic diseases: A systems medicine approach. Pharmacol. Rev. 2018, 70, 348–383. [Google Scholar] [CrossRef] [Green Version]
- Kooman, J.P.; Dekker, M.J.; Usvyat, L.A.; Kotanko, P.; van der Sande, F.M.; Schalkwijk, C.G.; Shiels, P.G.; Stenvinkel, P. Inflammation and premature aging in advanced chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2017, 313, F938–F950. [Google Scholar] [CrossRef] [Green Version]
- Cobo, G.; Lindholm, B.; Stenvinkel, P. Chronic inflammation in end-stage renal disease and dialysis. Nephrol. Dial. Transplant. 2018, 33, iii35–iii40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, Y.; Yanagita, M. Immunology of the ageing kidney. Nat. Rev. Nephrol. 2019, 15, 625–640. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune—Metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Pawelzik, S.-C.; Avignon, A.; Idborg, H.; Boegner, C.; Stanke-Labesque, F.; Jakobsson, P.-J.; Sultan, A.; Bäck, M. Urinary prostaglandin D2 and E2 metabolites associate with abdominal obesity, glucose metabolism, and triglycerides in obese subjects. Prostaglandins Other Lipid Mediat. 2019, 145, 106361. [Google Scholar] [CrossRef]
- Ferenbach, D.A.; Bonventre, J.V. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol. 2015, 11, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Kirkland, J.L.; Tchkonia, T. Cellular senescence: A translational perspective. EBioMedicine 2017, 21, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Hickey, N.A.; Shalamanova, L.; Whitehead, K.A.; Dempsey-Hibbert, N.; van der Gast, C.; Taylor, R.L. Exploring the putative interactions between chronic kidney disease and chronic periodontitis. Crit. Rev. Microbiol. 2020, 12, 1–17. [Google Scholar] [CrossRef]
- Van Maldeghem, I.; Nusman, C.M.; Visser, D.H. Soluble CD14 subtype (sCD14-ST) as biomarker in neonatal early-onset sepsis and late-onset sepsis: A systematic review and meta-analysis. BMC Immunol. 2019, 20, 17. [Google Scholar] [CrossRef]
- Carracedo, M.; Artiach, G.; Witasp, A.; Clària, J.; Carlström, M.; Laguna-Fernandez, A.; Stenvinkel, P.; Bäck, M. The G-protein coupled receptor ChemR23 determines smooth muscle cell phenotypic switching to enhance high phosphate-induced vascular calcification. Cardiovasc. Res. 2019, 115, 1557–1566. [Google Scholar] [CrossRef] [Green Version]
- Abbasian, N.; Burton, J.O.; Herbert, K.E.; Tregunna, B.-E.; Brown, J.R.; Ghaderi-Najafabadi, M.; Brunskill, N.J.; Goodall, A.H.; Bevington, A. Hyperphosphatemia, phosphoprotein phosphatases, and microparticle release in vascular endothelial cells. J. Am. Soc. Nephrol. 2015, 26, 2152–2162. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.-H.; Gou, B.-D.; Zhang, T.-L.; Wang, K. Lanthanum chloride bidirectionally influences calcification in bovine vascular smooth muscle cells. J. Cell. Biochem. 2012, 113, 1776–1786. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Ji, Y.; Ju, H.; Chen, H.; Sun, M. Circulating fetuin-A and risk of all-cause mortality in patients with chronic kidney disease: A systematic review and meta-analysis. Front. Physiol. 2019, 10, 966. [Google Scholar] [CrossRef] [PubMed]
- Carrero, J.J.; Stenvinkel, P.; Fellström, B.; Qureshi, A.R.; Lamb, K.; Heimbürger, O.; Bárány, P.; Radhakrishnan, K.; Lindholm, B.; Soveri, I.; et al. Telomere attrition is associated with inflammation, low fetuin-A levels and high mortality in prevalent haemodialysis patients. J. Intern. Med. 2008, 263, 302–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voelkl, J.; Tuffaha, R.; Luong, T.T.D.; Zickler, D.; Masyout, J.; Feger, M.; Verheyen, N.; Blaschke, F.; Kuro-o, M.; Tomaschitz, A.; et al. Zinc inhibits phosphate-induced vascular calcification through TNFAIP3-Mediated suppression of NF-κB. J. Am. Soc. Nephrol. 2018, 29, 1636–1648. [Google Scholar] [CrossRef] [Green Version]
- Mikolajczyk, T.P.; Guzik, T.J. Adaptive immunity in hypertension. Curr. Hypertens. Rep. 2019, 21, 68. [Google Scholar] [CrossRef] [Green Version]
- Stinghen, A.E.M.; Massy, Z.A.; Vlassara, H.; Striker, G.E.; Boullier, A. Uremic toxicity of advanced glycation end products in CKD. J. Am. Soc. Nephrol. 2016, 27, 354–370. [Google Scholar] [CrossRef] [Green Version]
- Pedruzzi, L.M.; Cardozo, L.F.M.F.; Daleprane, J.B.; Stockler-Pinto, M.B.; Monteiro, E.B.; Leite, M.; Vaziri, N.D.; Mafra, D. Systemic inflammation and oxidative stress in hemodialysis patients are associated with down-regulation of Nrf2. J. Nephrol. 2015, 28, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Mercier, N.; Pawelzik, S.-C.; Pirault, J.; Carracedo, M.; Persson, O.; Wollensack, B.; Franco-Cereceda, A.; Bäck, M. Semicarbazide-Sensitive Amine Oxidase Increases in Calcific Aortic Valve Stenosis and Contributes to Valvular Interstitial Cell Calcification. Oxid. Med. Cell. Longev. 2020, 5197376. [Google Scholar] [CrossRef]
- Daenen, K.; Andries, A.; Mekahli, D.; Van Schepdael, A.; Jouret, F.; Bammens, B. Oxidative stress in chronic kidney disease. Pediatr. Nephrol. 2019, 34, 975–991. [Google Scholar] [CrossRef] [Green Version]
- Huang, M.; Zheng, L.; Xu, H.; Tang, D.; Lin, L.; Zhang, J.; Li, C.; Wang, W.; Yuan, Q.; Tao, L.; et al. Oxidative stress contributes to vascular calcification in patients with chronic kidney disease. J. Mol. Cell. Cardiol. 2020, 138, 256–268. [Google Scholar] [CrossRef] [Green Version]
- Rabbani, N.; Thornalley, P.J. Advanced glycation end products in the pathogenesis of chronic kidney disease. Kidney Int. 2018, 93, 803–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snelson, M.; Coughlan, M.T. Dietary advanced glycation end products: Digestion, metabolism and modulation of gut microbial ecology. Nutrients 2019, 11, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, S.; Mick, M.; Hall, P.; Mejia, C.; Sue, S.; Wase, B.A.; Nguyen, M.A.; Whisenant, E.C.; Wilcox, S.H.; Winden, D.; et al. Cigarette smoke extract induces oral squamous cell carcinoma cell invasion in a receptor for advanced glycation end-products-dependent manner. Eur. J. Oral Sci. 2018, 126, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Nowotny, K.; Jung, T.; Höhn, A.; Weber, D.; Grune, T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules 2015, 5, 194–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanajou, D.; Ghorbani Haghjo, A.; Argani, H.; Aslani, S. Age-Rage axis blockade in diabetic nephropathy: Current status and future directions. Eur. J. Pharmacol. 2018, 833, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Osorio, F.G.; Soria-Valles, C.; Santiago-Fernández, O.; Freije, J.M.P.; López-Otín, C. Chapter Four—NF-κB signaling as a driver of ageing. In International Review of Cell and Molecular Biology; Jeon, K.W., Galluzzi, L., Eds.; Academic Press: Cambridge, MA, USA, 2016; Volume 326, pp. 133–174. [Google Scholar]
- Costantino, S.; Paneni, F.; Cosentino, F. Ageing, metabolism and cardiovascular disease. J. Physiol. 2016, 594, 2061–2073. [Google Scholar] [CrossRef]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Rane, G.; Dai, X.; Shanmugam, M.K.; Arfuso, F.; Samy, R.P.; Lai, M.K.P.; Kappei, D.; Kumar, A.P.; Sethi, G. Ageing and the telomere connection: An intimate relationship with inflammation. Ageing Res. Rev. 2016, 25, 55–69. [Google Scholar] [CrossRef]
- Chen, R.; Zhang, K.; Chen, H.; Zhao, X.; Wang, J.; Li, L.; Cong, Y.; Ju, Z.; Xu, D.; Williams, B.R.G.; et al. Telomerase deficiency causes alveolar stem cell senescence-associated low-grade inflammation in lungs. J. Biol. Chem. 2015, 290, 30813–30829. [Google Scholar] [CrossRef] [Green Version]
- Romagnani, P.; Remuzzi, G.; Glassock, R.; Levin, A.; Jager, K.J.; Tonelli, M.; Massy, Z.; Wanner, C.; Anders, H.-J. Chronic kidney disease. Nat. Rev. Dis. Primer 2017, 3, 17088. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Peel, N.M.; Krosch, M.; Hubbard, R.E. Frailty and chronic kidney disease: A systematic review. Arch. Gerontol. Geriatr. 2017, 68, 135–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drew, D.A.; Weiner, D.E.; Sarnak, M.J. Cognitive impairment in CKD: Pathophysiology, management, and prevention. Am. J. Kidney Dis. 2019, 74, 782–790. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Underwood, C.F.; Hildreth, C.M.; Wyse, B.F.; Boyd, R.; Goodchild, A.K.; Phillips, J.K. Uraemia: An unrecognized driver of central neurohumoral dysfunction in chronic kidney disease? Acta Physiol. 2017, 219, 305–323. [Google Scholar] [CrossRef] [PubMed]
- Duranton, F.; Cohen, G.; Smet, R.D.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A.; European Uremic Toxin Work Group. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef] [Green Version]
- Fujii, H.; Goto, S.; Fukagawa, M. Role of uremic toxins for kidney, cardiovascular, and bone dysfunction. Toxins 2018, 10, 202. [Google Scholar] [CrossRef] [Green Version]
- Mutsaers, H.A.M.; Wilmer, M.J.G.; Reijnders, D.; Jansen, J.; van den Broek, P.H.H.; Forkink, M.; Schepers, E.; Glorieux, G.; Vanholder, R.; van den Heuvel, L.P.; et al. Uremic toxins inhibit renal metabolic capacity through interference with glucuronidation and mitochondrial respiration. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2013, 1832, 142–150. [Google Scholar] [CrossRef]
- Lee, W.-C.; Li, L.-C.; Chen, J.-B.; Chang, H.-W. Indoxyl sulfate-induced oxidative stress, mitochondrial dysfunction, and impaired biogenesis are partly protected by vitamin C and N-Acetylcysteine. Sci. World J. 2015, 2015, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Muteliefu, G.; Shimizu, H.; Enomoto, A.; Nishijima, F.; Takahashi, M.; Niwa, T. Indoxyl sulfate promotes vascular smooth muscle cell senescence with upregulation of p53, p21, and prelamin A through oxidative stress. Am. J. Physiol. Cell Physiol. 2012, 303, C126–C134. [Google Scholar]
- Sun, C.-Y.; Cheng, M.-L.; Pan, H.-C.; Lee, J.-H.; Lee, C.-C. Protein-bound uremic toxins impaired mitochondrial dynamics and functions. Oncotarget 2017, 8, 77722–77733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carracedo, M.; Persson, O.; Saliba-Gustafsson, P.; Artiach, G.; Ehrenborg, E.; Eriksson, P.; Franco-Cereceda, A.; Bäck, M. Upregulated autophagy in calcific aortic valve stenosis confers protection of valvular interstitial cells. Int. J. Mol. Sci. 2019, 20, 1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirakawa, Y.; Jao, T.-M.; Inagi, R. Pathophysiology and therapeutics of premature ageing in chronic kidney disease, with a focus on glycative stress. Clin. Exp. Pharmacol. Physiol. 2017, 44, 70–77. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zhang, X.; Zhang, H.; Liu, T.; Zhang, H.; Teng, J.; Ji, J.; Ding, X. Indoxyl sulfate enhance the hypermethylation of klotho and promote the process of vascular calcification in chronic kidney disease. Int. J. Biol. Sci. 2016, 12, 1236–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asai, M.; Kumakura, S.; Kikuchi, M. Review of the efficacy of AST-120 (KREMEZIN®) on renal function in chronic kidney disease patients. Ren. Fail. 2019, 41, 47–56. [Google Scholar] [CrossRef] [Green Version]
- Schulman, G.; Berl, T.; Beck, G.J.; Remuzzi, G.; Ritz, E.; Arita, K.; Kato, A.; Shimizu, M. Randomized placebo-controlled EPPIC trials of AST-120 in CKD. J. Am. Soc. Nephrol. 2015, 26, 1732–1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulman, G.; Berl, T.; Beck, G.J.; Remuzzi, G.; Ritz, E.; Shimizu, M.; Shobu, Y.; Kikuchi, M. The effects of AST-120 on chronic kidney disease progression in the United States of America: A post hoc subgroup analysis of randomized controlled trials. BMC Nephrol. 2016, 17, 141. [Google Scholar] [CrossRef] [Green Version]
- Koska, J.; Saremi, A.; Howell, S.; Bahn, G.; Courten, B.D.; Ginsberg, H.; Beisswenger, P.J.; Reaven, P.D. Advanced glycation end products, oxidation products, and incident cardiovascular events in patients with type 2 diabetes. Diabetes Care 2018, 41, 570–576. [Google Scholar] [CrossRef] [Green Version]
- De Vos, L.C.; Lefrandt, J.D.; Dullaart, R.P.F.; Zeebregts, C.J.; Smit, A.J. Advanced glycation end products: An emerging biomarker for adverse outcome in patients with peripheral artery disease. Atherosclerosis 2016, 254, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Chaudhuri, J.; Bains, Y.; Guha, S.; Kahn, A.; Hall, D.; Bose, N.; Gugliucci, A.; Kapahi, P. The role of advanced glycation end products in aging and metabolic diseases: Bridging association and causality. Cell Metab. 2018, 28, 337–352. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Huang, K.; Cai, G.-Y.; Chen, X.-M.; Yang, J.-R.; Lin, L.-R.; Yang, J.; Huo, B.-G.; Zhan, J.; He, Y.-N. Receptor for advanced glycation end-products promotes premature senescence of proximal tubular epithelial cells via activation of endoplasmic reticulum stress-dependent p21 signaling. Cell Signal. 2014, 26, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, J.-R.; Chen, X.-M.; Cai, G.-Y.; Lin, L.-R.; He, Y.-N. Impact of ER stress-regulated ATF4/p16 signaling on the premature senescence of renal tubular epithelial cells in diabetic nephropathy. Am. J. Physiol. Cell Physiol. 2015, 308, C621–C630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, M.; Yang, S.; Zhu, X.; Sun, D.; Sun, D.; Jiang, X.; Zhang, C.; Wang, L. The RAGE/STAT5/autophagy axis regulates senescence in mesangial cells. Cell Signal. 2019, 62, 109334. [Google Scholar] [CrossRef] [PubMed]
- Knöpfel, T.; Himmerkus, N.; Günzel, D.; Bleich, M.; Hernando, N.; Wagner, C.A. Paracellular transport of phosphate along the intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 317, G233–G241. [Google Scholar] [CrossRef] [PubMed]
- Florenzano, P.; Cipriani, C.; Roszko, K.L.; Fukumoto, S.; Collins, M.T.; Minisola, S.; Pepe, J. Approach to patients with hypophosphataemia. Lancet Diabetes Endocrinol. 2020, 8, 163–174. [Google Scholar] [CrossRef]
- Hernando, N.; Wagner, C.A. Mechanisms and regulation of intestinal phosphate absorption. Compr. Physiol. 2018, 8, 1065–1090. [Google Scholar]
- Jacquillet, G.; Unwin, R.J. Physiological regulation of phosphate by vitamin D, parathyroid hormone (PTH) and phosphate (Pi). Pflügers Arch. Eur. J. Physiol. 2019, 471, 83–98. [Google Scholar] [CrossRef] [Green Version]
- Vervloet, M. Renal and extrarenal effects of fibroblast growth factor 23. Nat. Rev. Nephrol. 2019, 15, 109–120. [Google Scholar] [CrossRef]
- Kuro-o, M. The klotho proteins in health and disease. Nat. Rev. Nephrol. 2019, 15, 27–44. [Google Scholar] [CrossRef]
- Ebert, T.; Gebhardt, C.; Scholz, M.; Wohland, T.; Schleinitz, D.; Fasshauer, M.; Blüher, M.; Stumvoll, M.; Kovacs, P.; Tönjes, A. Relationship between 12 adipocytokines and distinct components of the metabolic syndrome. J. Clin. Endocrinol. Metab. 2018, 103, 1015–1023. [Google Scholar] [CrossRef]
- Zou, D.; Wu, W.; He, Y.; Ma, S.; Gao, J. The role of klotho in chronic kidney disease. BMC Nephrol. 2018, 19, 285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bäck, M.; Aranyi, T.; Cancela, M.L.; Carracedo, M.; Conceição, N.; Leftheriotis, G.; Macrae, V.; Martin, L.; Nitschke, Y.; Pasch, A.; et al. Endogenous calcification inhibitors in the prevention of vascular calcification: A consensus statement from the COST action EuroSoftCalcNet. Front. Cardiovasc. Med. 2019, 5, 196. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, C.; Houben, A.J.H.M.; Malhotra, R.; Berendschot, T.T.J.M.; Dagnelie, P.C.; Kooman, J.P.; Webers, C.A.; Stehouwer, C.D.A.; Ix, J.H. Serum phosphate and microvascular function in a population-based cohort. Clin. J. Am. Soc. Nephrol. 2019, 14, 1626–1633. [Google Scholar] [CrossRef] [PubMed]
- Rahabi-Layachi, H.; Ourouda, R.; Boullier, A.; Massy, Z.A.; Amant, C. Distinct effects of inorganic phosphate on cell cycle and apoptosis in human vascular smooth muscle cells. J. Cell. Physiol. 2015, 230, 347–355. [Google Scholar] [CrossRef]
- Zhao, M.-M.; Xu, M.-J.; Cai, Y.; Zhao, G.; Guan, Y.; Kong, W.; Tang, C.; Wang, X. Mitochondrial reactive oxygen species promote p65 nuclear translocation mediating high-phosphate-induced vascular calcification in vitro and in vivo. Kidney Int. 2011, 79, 1071–1079. [Google Scholar] [CrossRef] [Green Version]
- Vervloet, M.G.; Sezer, S.; Massy, Z.A.; Johansson, L.; Cozzolino, M.; Fouque, D.; ERA–EDTA Working Group on Chronic Kidney Disease–Mineral and Bone Disorders and the European Renal Nutrition Working Group. The role of phosphate in kidney disease. Nat. Rev. Nephrol. 2017, 13, 27–38. [Google Scholar] [CrossRef]
- McClelland, R.; Christensen, K.; Mohammed, S.; McGuinness, D.; Cooney, J.; Bakshi, A.; Demou, E.; MacDonald, E.; Caslake, M.; Stenvinkel, P.; et al. Accelerated ageing and renal dysfunction links lower socioeconomic status and dietary phosphate intake. Aging 2017, 8, 1135–1149. [Google Scholar] [CrossRef] [Green Version]
- Yao, L.; Sun, Y.; Sun, W.; Xu, T.; Ren, C.; Fan, X.; Sun, L.; Liu, L.; Feng, J.; Ma, J.; et al. High phosphorus level leads to aortic calcification via β-catenin in chronic kidney disease. Am. J. Nephrol. 2015, 41, 28–36. [Google Scholar] [CrossRef]
- Cai, T.; Sun, D.; Duan, Y.; Wen, P.; Dai, C.; Yang, J.; He, W. WNT/β-catenin signaling promotes VSMCs to osteogenic transdifferentiation and calcification through directly modulating Runx2 gene expression. Exp. Cell Res. 2016, 345, 206–217. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-X.; Huang, C.; Duan, Z.-B.; Xu, C.-Y.; Chen, Y. Klotho/FGF23 axis mediates high phosphate-induced vascular calcification in vascular smooth muscle cells via Wnt7b/β-catenin pathway. Kaohsiung J. Med. Sci. 2019, 35, 393–400. [Google Scholar] [CrossRef]
- Chen, T.; Mao, H.; Chen, C.; Wu, L.; Wang, N.; Zhao, X.; Qian, J.; Xing, C. The Role and Mechanism of α-Klotho in the Calcification of Rat Aortic Vascular Smooth Muscle Cells. Available online: https://www.hindawi.com/journals/bmri/2015/194362/ (accessed on 28 February 2020).
- Mencke, R.; Hillebrands, J.-L. The role of the anti-ageing protein Klotho in vascular physiology and pathophysiology. Ageing Res. Rev. 2017, 35, 124–146. [Google Scholar] [CrossRef] [PubMed]
- Aghagolzadeh, P.; Bachtler, M.; Bijarnia, R.; Jackson, C.; Smith, E.R.; Odermatt, A.; Radpour, R.; Pasch, A. Calcification of vascular smooth muscle cells is induced by secondary calciprotein particles and enhanced by tumor necrosis factor-α. Atherosclerosis 2016, 251, 404–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viegas, C.S.B.; Santos, L.; Macedo, A.L.; Matos, A.A.; Silva, A.P.; Neves, P.L.; Staes, A.; Gevaert, K.; Morais, R.; Vermeer, C.; et al. Chronic kidney disease circulating calciprotein particles and extracellular vesicles promote vascular calcification. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 575–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barreto, F.C.; Barreto, D.V.; Massy, Z.A.; Drüeke, T.B. Strategies for phosphate control in patients with CKD. Kidney Int. Rep. 2019, 4, 1043–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elder, G.J.; Malik, A.; Lambert, K. Role of dietary phosphate restriction in chronic kidney disease. Nephrology 2018, 23, 1107–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, W.-C.; Wu, H.-Y.; Peng, Y.-S.; Hsu, S.-P.; Chiu, Y.-L.; Chen, H.-Y.; Yang, J.-Y.; Ko, M.-J.; Pai, M.-F.; Tu, Y.-K.; et al. Effects of lower versus higher phosphate diets on fibroblast growth factor-23 levels in patients with chronic kidney disease: A systematic review and meta-analysis. Nephrol. Dial. Transplant. 2018, 33, 1977–1983. [Google Scholar] [CrossRef] [Green Version]
- Tsai, W.-C.; Wu, H.-Y.; Peng, Y.-S.; Hsu, S.-P.; Chiu, Y.-L.; Yang, J.-Y.; Chen, H.-Y.; Pai, M.-F.; Lin, W.-Y.; Hung, K.-Y.; et al. Short-term effects of very-low-phosphate and low-phosphate diets on fibroblast growth factor 23 in hemodialysis patients: A randomized crossover trial. Clin. J. Am. Soc. Nephrol. 2019, 14, 1475–1483. [Google Scholar] [CrossRef]
- Czarnik, T.; Czarnik, A.; Gawda, R.; Gawor, M.; Piwoda, M.; Marszalski, M.; Maj, M.; Chrzan, O.; Said, R.; Rusek-Skora, M.; et al. Vitamin D kinetics in the acute phase of critical illness: A prospective observational study. J. Crit. Care 2018, 43, 294–299. [Google Scholar] [CrossRef]
- Norris, K.C.; Olabisi, O.; Barnett, M.E.; Meng, Y.-X.; Martins, D.; Obialo, C.; Lee, J.E.; Nicholas, S.B. The role of vitamin D and oxidative stress in chronic kidney disease. Int. J. Environ. Res. Public Health 2018, 15, 2701. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J. Vitamin D deficiency accelerates ageing and age-related diseases: A novel hypothesis. J. Physiol. 2017, 595, 6825–6836. [Google Scholar] [CrossRef]
- Haussler, M.R.; Whitfield, G.K.; Haussler, C.A.; Sabir, M.S.; Khan, Z.; Sandoval, R.; Jurutka, P.W. Chapter eight–1,25-Dihydroxyvitamin D and Klotho: A tale of two renal hormones coming of age. In Vitamins & Hormones; Litwack, G., Ed.; Vitamin D Hormone; Academic Press: Cambridge, MA, USA, 2016; Volume 100, pp. 165–230. [Google Scholar]
- Takenaka, T.; Inoue, T.; Ohno, Y.; Miyazaki, T.; Nishiyama, A.; Ishii, N.; Suzuki, H. Calcitriol supplementation improves endothelium-dependent vasodilation in rat hypertensive renal injury. Kidney Blood Press. Res. 2014, 39, 17–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jebreal Azimzadeh, M.; Shidfar, F.; Jazayeri, S.; Hosseini, A.F.; Ranjbaran, F. Effect of vitamin D supplementation on klotho protein, antioxidant status and nitric oxide in the elderly: A randomized, double-blinded, placebo-controlled clinical trial. Eur. J. Integr. Med. 2020, 35, 101089. [Google Scholar] [CrossRef]
- Carvalho, J.T.G.; Schneider, M.; Cuppari, L.; Grabulosa, C.C.; Aoike, D.T.; Redublo, B.M.Q.; Batista, M.C.; Cendoroglo, M.; Moyses, R.M.; Dalboni, M.A. Cholecalciferol decreases inflammation and improves vitamin D regulatory enzymes in lymphocytes in the uremic environment: A randomized controlled pilot trial. PLoS ONE 2017, 12, e0179540. [Google Scholar] [CrossRef] [PubMed]
- Mansournia, M.A.; Ostadmohammadi, V.; Doosti-Irani, A.; Ghayour-Mobarhan, M.; Ferns, G.; Akbari, H.; Ghaderi, A.; Talari, H.R.; Asemi, Z. The effects of vitamin d supplementation on biomarkers of inflammation and oxidative stress in diabetic patients: A systematic review and meta-analysis of randomized controlled trials. Horm. Metab. Res. 2018, 50, 429–440. [Google Scholar] [CrossRef] [Green Version]
- Kruit, A.; Zanen, P. The association between vitamin D and C-reactive protein levels in patients with inflammatory and non-inflammatory diseases. Clin. Biochem. 2016, 49, 534–537. [Google Scholar] [CrossRef]
- Hu, C.; Wu, X. Effect of vitamin D supplementation on vascular function and inflammation in patients with chronic kidney disease: A controversial issue. Ther. Apher. Dial. 2019. [Google Scholar] [CrossRef]
- Rodríguez, A.J.; Scott, D.; Srikanth, V.; Ebeling, P. Effect of vitamin D supplementation on measures of arterial stiffness: A systematic review and meta-analysis of randomized controlled trials. Clin. Endocrinol. (Oxf.) 2016, 84, 645–657. [Google Scholar] [CrossRef] [Green Version]
- Hussin, A.M.; Ashor, A.W.; Schoenmakers, I.; Hill, T.; Mathers, J.C.; Siervo, M. Effects of vitamin D supplementation on endothelial function: A systematic review and meta-analysis of randomised clinical trials. Eur. J. Nutr. 2017, 56, 1095–1104. [Google Scholar] [CrossRef]
- Gutiérrez, O.M.; Mannstadt, M.; Isakova, T.; Rauh-Hain, J.A.; Tamez, H.; Shah, A.; Smith, K.; Lee, H.; Thadhani, R.; Jüppner, H.; et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N. Engl. J. Med. 2008, 359, 584–592. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Grabner, A.; Yanucil, C.; Schramm, K.; Czaya, B.; Krick, S.; Czaja, M.J.; Bartz, R.; Abraham, R.; Di Marco, G.S.; et al. Fibroblast growth factor 23 directly targets hepatocytes to promote inflammation in chronic kidney disease. Kidney Int. 2016, 90, 985–996. [Google Scholar] [CrossRef] [Green Version]
- Silswal, N.; Touchberry, C.D.; Daniel, D.R.; McCarthy, D.L.; Zhang, S.; Andresen, J.; Stubbs, J.R.; Wacker, M.J. FGF23 directly impairs endothelium-dependent vasorelaxation by increasing superoxide levels and reducing nitric oxide bioavailability. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E426–E436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verkaik, M.; Juni, R.P.; van Loon, E.P.M.; van Poelgeest, E.M.; Kwekkeboom, R.F.J.; Gam, Z.; Richards, W.G.; Ter Wee, P.M.; Hoenderop, J.G.; Eringa, E.C.; et al. FGF23 impairs peripheral microvascular function in renal failure. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1414–H1424. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, K.; Olauson, H.; Amin, R.; Ponnusamy, A.; Goetz, R.; Taylor, R.F.; Mohammadi, M.; Canfield, A.; Kublickiene, K.; Larsson, T.E. Arterial klotho expression and FGF23 effects on vascular calcification and function. PLoS ONE 2013, 8, e60658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simic, P.; Kim, W.; Zhou, W.; Pierce, K.A.; Chang, W.; Sykes, D.B.; Aziz, N.B.; Elmariah, S.; Ngo, D.; Pajevic, P.D.; et al. Glycerol-3-phosphate is an FGF23 regulator derived from the injured kidney. J. Clin. Investig. 2020, 130, 1513–1526. [Google Scholar] [CrossRef] [Green Version]
- Haruna, Y.; Kashihara, N.; Satoh, M.; Tomita, N.; Namikoshi, T.; Sasaki, T.; Fujimori, T.; Xie, P.; Kanwar, Y.S. Amelioration of progressive renal injury by genetic manipulation of Klotho gene. Proc. Natl. Acad. Sci. USA 2007, 104, 2331–2336. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Ortiz, M.E.; Díaz-Tocados, J.M.; Muñoz-Castañeda, J.R.; Herencia, C.; Pineda, C.; Martínez-Moreno, J.M.; Montes de Oca, A.; López-Baltanás, R.; Alcalá-Díaz, J.; Ortiz, A.; et al. Inflammation both increases and causes resistance to FGF23 in normal and uremic rats. Clin. Sci. 2020, 134, 15–32. [Google Scholar] [CrossRef]
- Zhou, L.; Li, Y.; Zhou, D.; Tan, R.J.; Liu, Y. Loss of klotho contributes to kidney injury by derepression of Wnt/β-catenin signaling. J. Am. Soc. Nephrol. 2013, 24, 771–785. [Google Scholar] [CrossRef] [Green Version]
- Vila Cuenca, M.; Hordijk, P.L.; Vervloet, M.G. Most exposed: The endothelium in chronic kidney disease. Nephrol. Dial. Transplant. 2019. [Google Scholar] [CrossRef]
- Ter Braake, A.D.; Smit, A.E.; Bos, C.; van Herwaarden, A.E.; Alkema, W.; van Essen, H.W.; Bravenboer, N.; Vervloet, M.G.; Hoenderop, J.G.J.; de Baaij, J.H.F. Magnesium prevents vascular calcification in Klotho deficiency. Kidney Int. 2020, 97, 487–501. [Google Scholar] [CrossRef]
- Leibrock, C.B.; Alesutan, I.; Voelkl, J.; Pakladok, T.; Michael, D.; Schleicher, E.; Kamyabi-Moghaddam, Z.; Quintanilla-Martinez, L.; Kuro-o, M.; Lang, F. NH4Cl treatment prevents tissue calcification in klotho deficiency. J. Am. Soc. Nephrol. 2015, 26, 2423–2433. [Google Scholar] [CrossRef] [Green Version]
- Ravarotto, V.; Simioni, F.; Pagnin, E.; Davis, P.A.; Calò, L.A. Oxidative stress—Chronic kidney disease—Cardiovascular disease: A vicious circle. Life Sci. 2018, 210, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.-L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Gidlund, E.; Witasp, A.; Qureshi, A.R.; Söderberg, M.; Thorell, A.; Nader, G.A.; Barany, P.; Stenvinkel, P.; von Walden, F. Reduced skeletal muscle expression of mitochondrial derived peptides humanin and MOTS-C and Nrf2 in chronic kidney disease. Am. J. Physiol.-Ren. Physiol. 2019, 317, F1122–F1131. [Google Scholar]
- Kubben, N.; Zhang, W.; Wang, L.; Voss, T.C.; Yang, J.; Qu, J.; Liu, G.-H.; Misteli, T. Repression of the antioxidant NRF2 pathway in premature aging. Cell 2016, 165, 1361–1374. [Google Scholar] [CrossRef] [Green Version]
- Volonte, D.; Liu, Z.; Musille, P.M.; Stoppani, E.; Wakabayashi, N.; Di, Y.-P.; Lisanti, M.P.; Kensler, T.W.; Galbiati, F. Inhibition of nuclear factor-erythroid 2–related factor (Nrf2) by caveolin-1 promotes stress-induced premature senescence. Mol. Biol. Cell 2013, 24, 1852–1862. [Google Scholar] [CrossRef] [PubMed]
- Fulop, G.A.; Kiss, T.; Tarantini, S.; Balasubramanian, P.; Yabluchanskiy, A.; Farkas, E.; Bari, F.; Ungvari, Z.; Csiszar, A. Nrf2 deficiency in aged mice exacerbates cellular senescence promoting cerebrovascular inflammation. GeroScience 2018, 40, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.; Hipólito-Luengo, Á.S.; Villalobos, L.A.; Vallejo, S.; Valencia, I.; Michalska, P.; Pajuelo-Lozano, N.; Sánchez-Pérez, I.; León, R.; Bartha, J.L.; et al. The angiotensin-(1-7)/Mas receptor axis protects from endothelial cell senescence via klotho and Nrf2 activation. Aging Cell 2019, 18, e12913. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Kang, Y.; Zhou, C.; Cui, R.; Jia, M.; Hu, S.; Ji, X.; Yuan, J.; Cui, H.; Shi, G. Amelioratory effects of testosterone propionate on age-related renal fibrosis via suppression of TGF-β1/Smad signaling and activation of Nrf2-ARE signaling. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Stockler-Pinto, M.B.; Soulage, C.O.; Borges, N.A.; Cardozo, L.F.M.F.; Dolenga, C.J.; Nakao, L.S.; Pecoits-Filho, R.; Fouque, D.; Mafra, D. From bench to the hemodialysis clinic: Protein-bound uremic toxins modulate NF-κB/Nrf2 expression. Int. Urol. Nephrol. 2018, 50, 347–354. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, J.; Wang, Y.; Gao, C.; Gu, Y.; Huang, J.; Wang, J.; Zhang, Z. Salvianolic acid a protects the kidney against oxidative stress by activating the Akt/GSK-3 β/Nrf2 signaling pathway and inhibiting the NF- κ B signaling pathway in 5/6 nephrectomized rats. Oxid. Med. Cell. Longev. 2019, 2019, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Nagasu, H.; Sogawa, Y.; Kidokoro, K.; Itano, S.; Yamamoto, T.; Satoh, M.; Sasaki, T.; Suzuki, T.; Yamamoto, M.; Wigley, W.C.; et al. Bardoxolone methyl analog attenuates proteinuria-induced tubular damage by modulating mitochondrial function. FASEB J. 2019, 33, 12253–12263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, K.N.; Tang, S.C.W.; Schena, F.P.; Novak, J.; Tomino, Y.; Fogo, A.B.; Glassock, R.J. IgA nephropathy. Nat. Rev. Dis. Primer 2016, 2, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Sturmlechner, I.; Durik, M.; Sieben, C.J.; Baker, D.J.; van Deursen, J.M. Cellular senescence in renal ageing and disease. Nat. Rev. Nephrol. 2017, 13, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Doi, S.; Nakashima, A.; Kawaoka, K.; Ueno, T.; Doi, T.; Yokoyama, Y.; Arihiro, K.; Kohno, N.; Masaki, T. Expression of age-related factors during the development of renal damage in patients with IgA nephropathy. Clin. Exp. Nephrol. 2015, 19, 830–837. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Liang, L.; Qin, J.; Lu, Y.; Li, B.; Wang, Y.; Lin, C.; Zhou, Q.; Feng, S.; Yip, S.H.; et al. Functional networks of aging markers in the glomeruli of IgA nephropathy: A new therapeutic opportunity. Oncotarget 2016, 7, 33616–33626. [Google Scholar] [CrossRef]
- Cokan Vujkovac, A.; Novaković, S.; Vujkovac, B.; Števanec, M.; Škerl, P.; Šabovič, M. Aging in fabry disease: Role of telomere length, telomerase activity, and kidney disease. Nephron 2020, 144, 5–13. [Google Scholar] [CrossRef]
- Kooman, J.P.; Stenvinkel, P.; Shiels, P.G. Fabry disease: A new model of premature ageing? Nephron 2020, 144, 1–4. [Google Scholar] [CrossRef]
- Bergmann, C.; Guay-Woodford, L.M.; Harris, P.C.; Horie, S.; Peters, D.J.M.; Torres, V.E. Polycystic kidney disease. Nat. Rev. Dis. Primer 2018, 4, 1–24. [Google Scholar] [CrossRef]
- Lu, D.; Rauhauser, A.; Li, B.; Ren, C.; McEnery, K.; Zhu, J.; Chaki, M.; Vadnagara, K.; Elhadi, S.; Jetten, A.M.; et al. Loss of Glis2/NPHP7 causes kidney epithelial cell senescence and suppresses cyst growth in the Kif3a mouse model of cystic kidney disease. Kidney Int. 2016, 89, 1307–1323. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Zhang, Y.; Liu, D.; Wang, S.S.; Ding, Q.; Rastogi, P.; Purvis, M.; Wang, A.; Elhadi, S.; Ren, C.; et al. Innate immune signaling contributes to tubular cell senescence in the Glis2 knockout mouse model of nephronophthisis. Am. J. Pathol. 2020, 190, 176–189. [Google Scholar] [CrossRef]
- Larsson, S.C.; Bäck, M.; Rees, J.M.B.; Mason, A.M.; Burgess, S. Body mass index and body composition in relation to 14 cardiovascular conditions in UK Biobank: A Mendelian randomization study. Eur. Heart J. 2020, 41, 221–226. [Google Scholar] [CrossRef]
- Blüher, M. Obesity: Global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.J.; Miller, J.D.; LeBrasseur, N.K. Cellular senescence: Implications for metabolic disease. Mol. Cell. Endocrinol. 2017, 455, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wu, K.K.L.; Jiang, X.; Xu, A.; Cheng, K.K.Y. The role of adipose tissue senescence in obesity- and ageing-related metabolic disorders. Clin. Sci. 2020, 134, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Ebert, T.; Roth, I.; Richter, J.; Tönjes, A.; Kralisch, S.; Lossner, U.; Kratzsch, J.; Blüher, M.; Stumvoll, M.; Fasshauer, M. Different associations of adipokines in lean and healthy adults. Horm. Metab. Res. 2014, 46, 41–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, A.K.; Xu, M.; Zhu, Y.; Pirtskhalava, T.; Weivoda, M.M.; Hachfeld, C.M.; Prata, L.G.; van Dijk, T.H.; Verkade, E.; Casaclang-Verzosa, G.; et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 2019, 18, e12950. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, M.; Witasp, A.; Qureshi, A.R.; Laguna-Fernandez, A.; Brismar, T.; Stenvinkel, P.; Bäck, M. Chemerin inhibits vascular calcification through ChemR23 and is associated with lower coronary calcium in chronic kidney disease. J. Intern. Med. 2019, 286, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Melk, A.; Schmidt, B.M.W.; Vongwiwatana, A.; Rayner, D.C.; Halloran, P.F. Increased expression of senescence-associated cell cycle inhibitor p16INK4a in deteriorating renal transplants and diseased native kidney. Am. J. Transplant. 2005, 5, 1375–1382. [Google Scholar] [CrossRef]
- Baisantry, A.; Berkenkamp, B.; Rong, S.; Bhayadia, R.; Sörensen-Zender, I.; Schmitt, R.; Melk, A. Time-dependent p53 inhibition determines senescence attenuation and long-term outcome after renal ischemia-reperfusion. Am. J. Physiol. Ren. Physiol. 2019, 316, F1124–F1132. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [Green Version]
- Luttropp, K.; Nordfors, L.; McGuinness, D.; Wennberg, L.; Curley, H.; Quasim, T.; Genberg, H.; Sandberg, J.; Sönnerborg, I.; Schalling, M.; et al. Increased telomere attrition after renal transplantation—Impact of antimetabolite therapy. Transplant. Direct 2016, 2, e116. [Google Scholar] [CrossRef] [PubMed]
- Toppe, C.; Möllsten, A.; Waernbaum, I.; Schön, S.; Gudbjörnsdottir, S.; Landin-Olsson, M.; Dahlquist, G. Decreasing cumulative incidence of end-stage renal disease in young patients with type 1 diabetes in Sweden: A 38-year prospective nationwide study. Diabetes Care 2019, 42, 27–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helve, J.; Sund, R.; Arffman, M.; Harjutsalo, V.; Groop, P.-H.; Grönhagen-Riska, C.; Finne, P. Incidence of end-stage renal disease in patients with type 1 diabetes. Diabetes Care 2018, 41, 434–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, B.; Matsushita, K.; Abate, K.H.; Al-Aly, Z.; Ärnlöv, J.; Asayama, K.; Atkins, R.; Badawi, A.; Ballew, S.H.; Banerjee, A.; et al. Global cardiovascular and renal outcomes of reduced GFR. J. Am. Soc. Nephrol. 2017, 28, 2167–2179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [Green Version]
- Abreu, C.C.; Cardozo, L.F.M.F.; Stockler-Pinto, M.B.; Esgalhado, M.; Barboza, J.E.; Frauches, R.; Mafra, D. Does resistance exercise performed during dialysis modulate Nrf2 and NF-κB in patients with chronic kidney disease? Life Sci. 2017, 188, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Vega, M.R.; de la Chapman, E.; Zhang, D.D. NRF2 and the hallmarks of cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Qiao, X.; Rao, P.; Zhang, Y.; Liu, L.; Pang, M.; Wang, H.; Hu, M.; Tian, X.; Zhang, J.; Zhao, Y.; et al. Redirecting TGF-β signaling through the β-Catenin/Foxo complex prevents kidney fibrosis. J. Am. Soc. Nephrol. 2018, 29, 557–570. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, X. Klotho: A novel biomarker for cancer. J. Cancer Res. Clin. Oncol. 2015, 141, 961–969. [Google Scholar] [CrossRef]
- Dote-Montero, M.; Amaro-Gahete, F.J.; De-la-O, A.; Jurado-Fasoli, L.; Gutierrez, A.; Castillo, M.J. Study of the association of DHEAS, testosterone and cortisol with S-Klotho plasma levels in healthy sedentary middle-aged adults. Exp. Gerontol. 2019, 121, 55–61. [Google Scholar] [CrossRef]
- Pedersen, L.; Christensen, L.L.; Pedersen, S.M.; Andersen, M. Reduction of calprotectin and phosphate during testosterone therapy in aging men: A randomized controlled trial. J. Endocrinol. Investig. 2017, 40, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Neyra, J.A.; Hu, M.C. Potential application of klotho in human chronic kidney disease. Bone 2017, 100, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Gregg, L.P.; Hedayati, S.S. Management of traditional cardiovascular risk factors in CKD: What are the data? Am. J. Kidney Dis. 2018, 72, 728–744. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ebert, T.; Pawelzik, S.-C.; Witasp, A.; Arefin, S.; Hobson, S.; Kublickiene, K.; Shiels, P.G.; Bäck, M.; Stenvinkel, P. Inflammation and Premature Ageing in Chronic Kidney Disease. Toxins 2020, 12, 227. https://doi.org/10.3390/toxins12040227
Ebert T, Pawelzik S-C, Witasp A, Arefin S, Hobson S, Kublickiene K, Shiels PG, Bäck M, Stenvinkel P. Inflammation and Premature Ageing in Chronic Kidney Disease. Toxins. 2020; 12(4):227. https://doi.org/10.3390/toxins12040227
Chicago/Turabian StyleEbert, Thomas, Sven-Christian Pawelzik, Anna Witasp, Samsul Arefin, Sam Hobson, Karolina Kublickiene, Paul G. Shiels, Magnus Bäck, and Peter Stenvinkel. 2020. "Inflammation and Premature Ageing in Chronic Kidney Disease" Toxins 12, no. 4: 227. https://doi.org/10.3390/toxins12040227