Alkaloid Lindoldhamine Inhibits Acid-Sensing Ion Channel 1a and Reveals Anti-Inflammatory Properties

 ,
,
Abstract
1. Introduction
2. Results
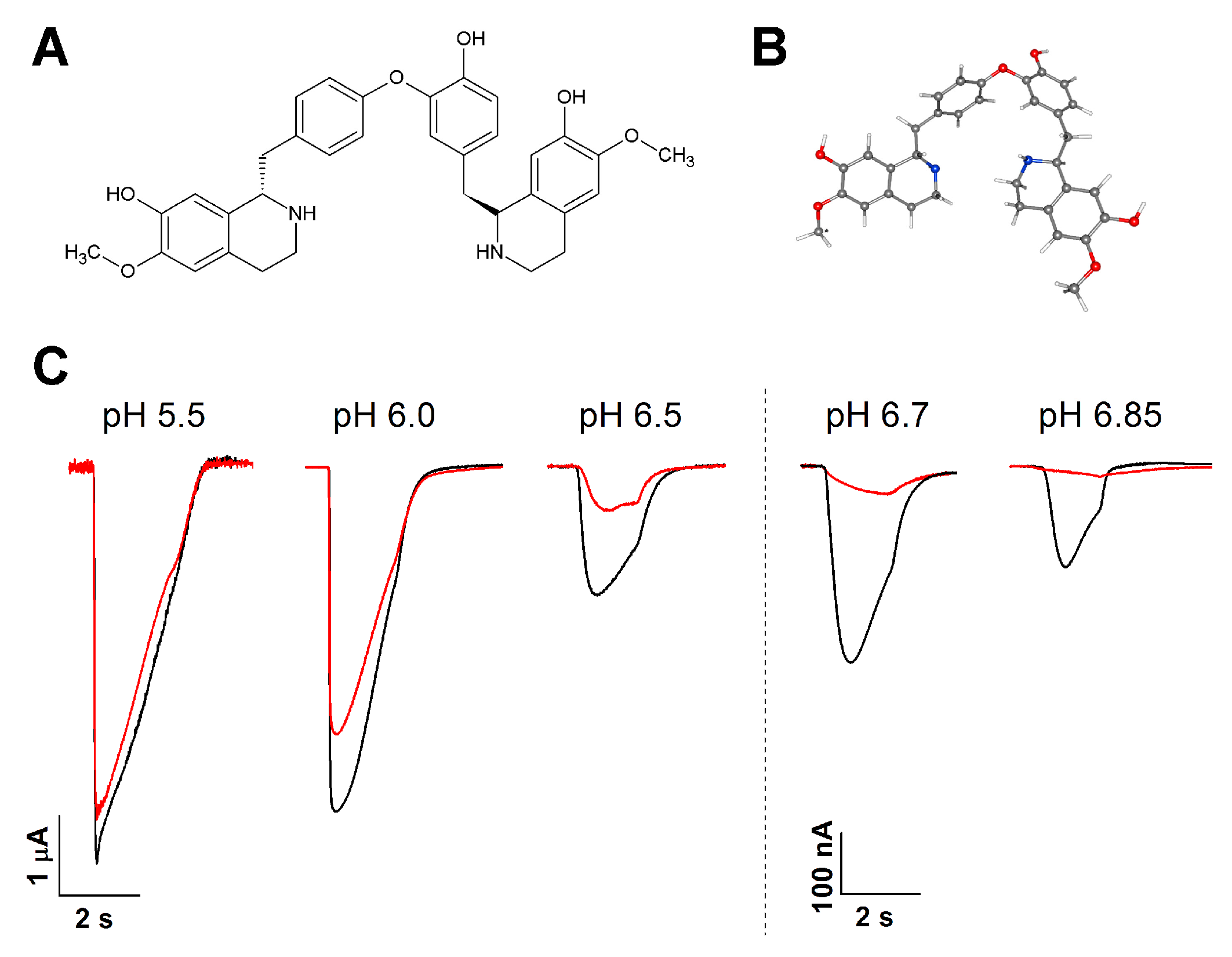
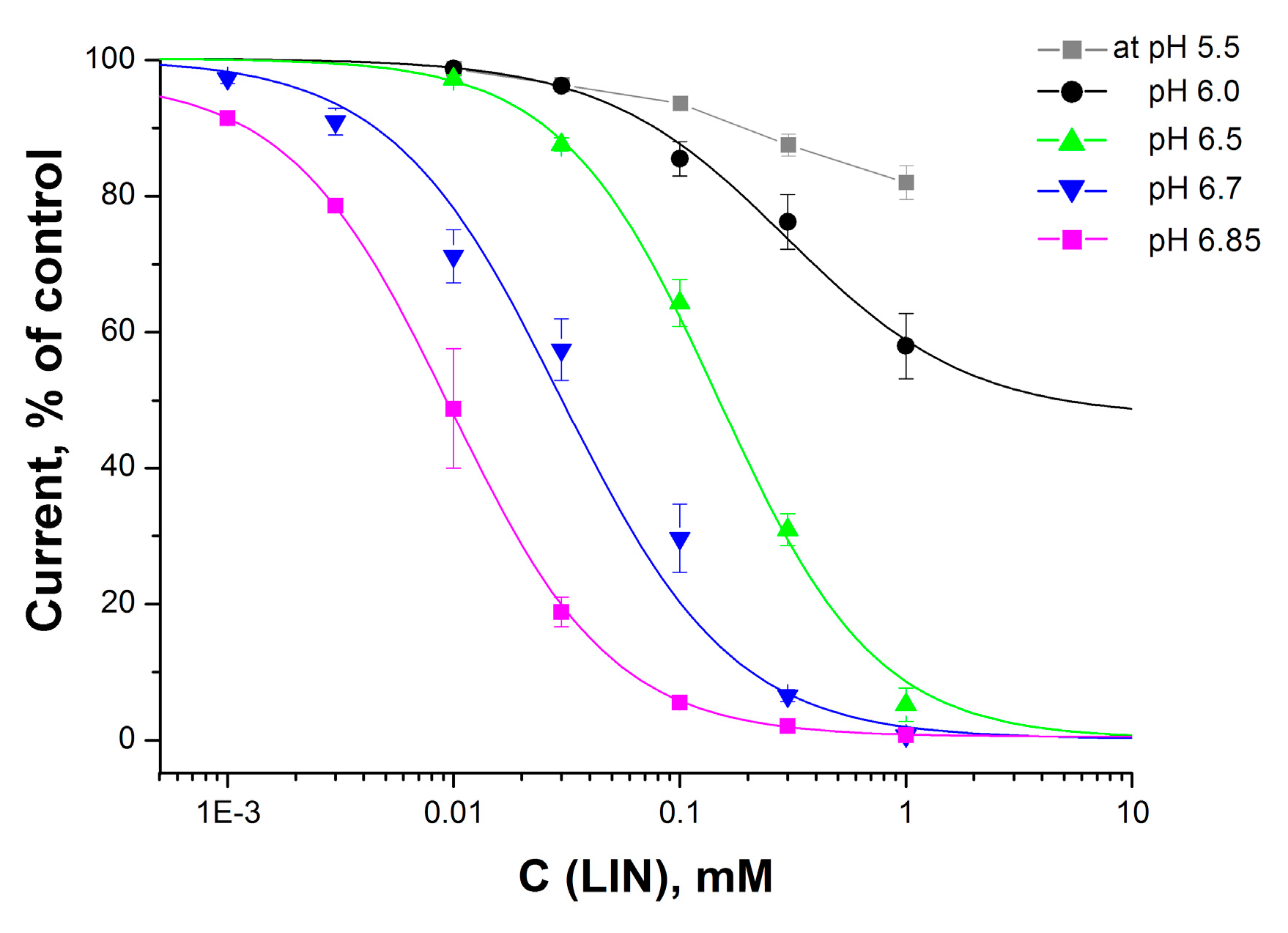
2.1. LIN Proton-Dependently Inhibits the ASIC1a Current
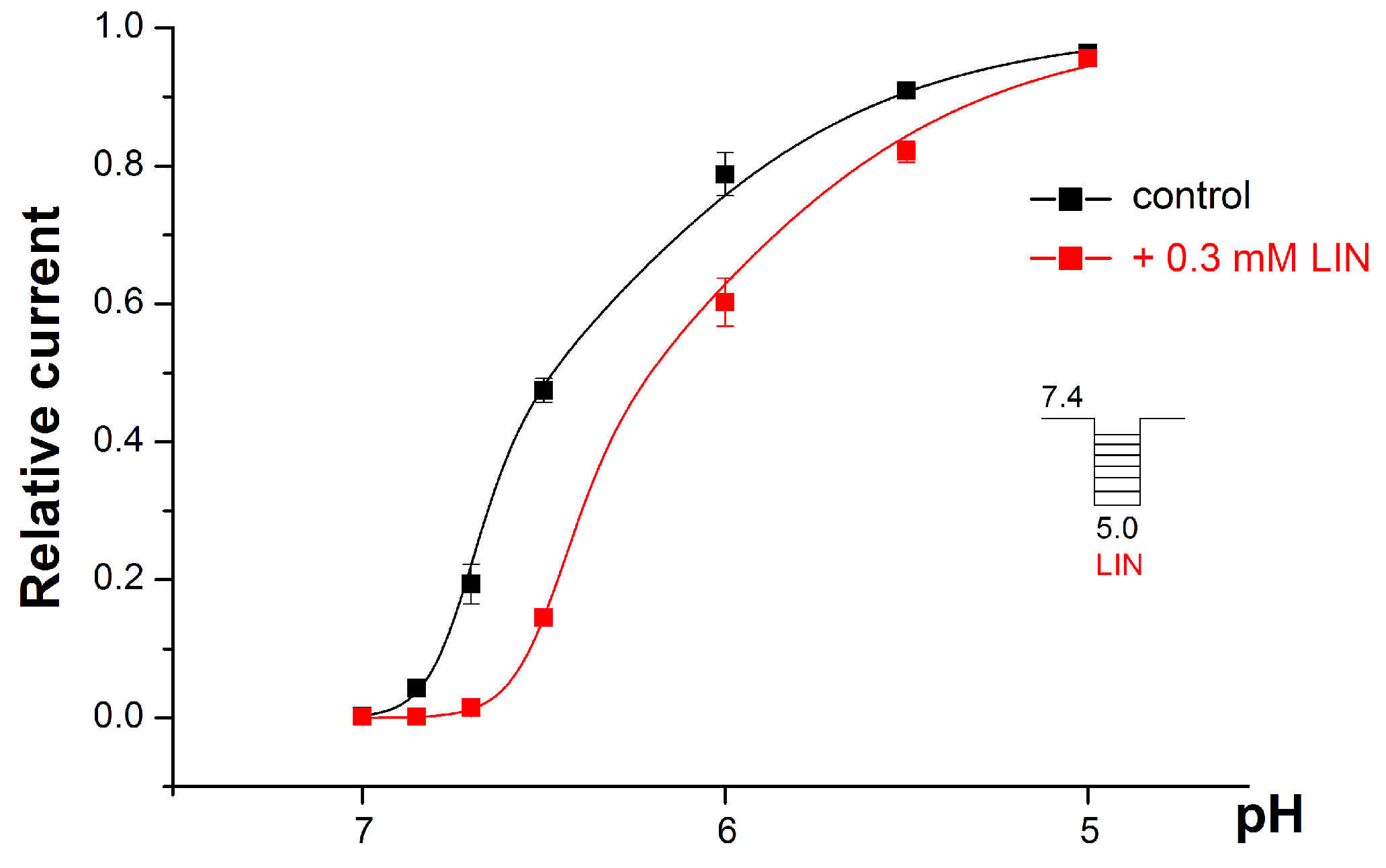
2.2. LIN’s Effect on the pH Dependence of the ASIC1a Activation
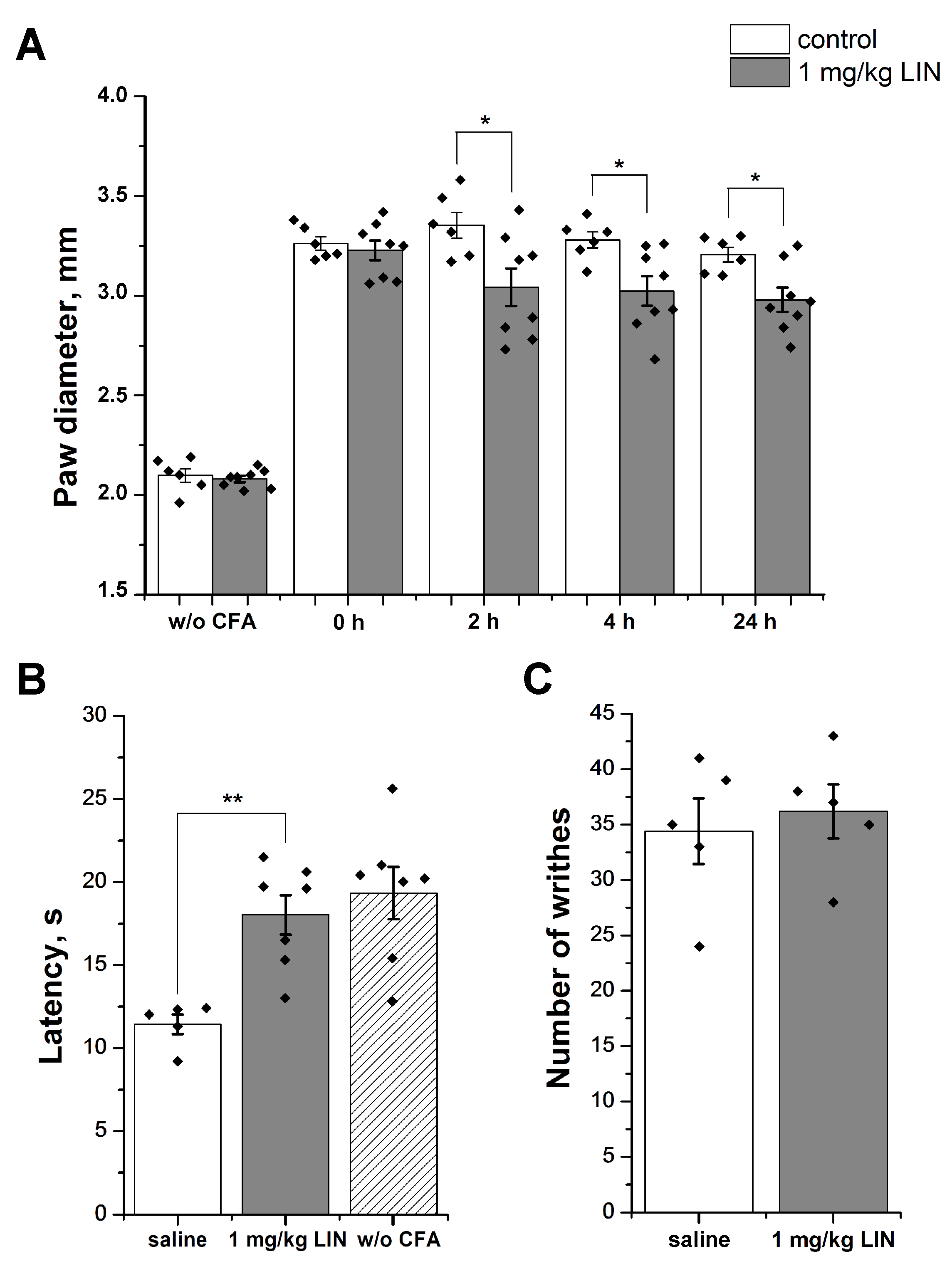
2.3. LIN Produced an Anti-Inflammatory Effect, but Failed to Decrease the Response to Acid
3. Discussion
4. Materials and Methods
4.1. Chemical Reagents and Compounds
4.2. Ethics Statement
4.3. Electrophysiological Experiments
4.4. In Vivo Assay
4.4.1. Animals
4.4.2. Complete Freund’s Adjuvant-Induced Inflammation and Thermal Hyperalgesia
4.4.3. Acetic Acid-Induced Writhing (Abdominal Constriction Test of Visceral Pain)
4.5. Data and Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Benson, C.J.; Xie, J.; Wemmie, J.A.; Price, M.P.; Henss, J.M.; Welsh, M.J.; Snyder, P.M. Heteromultimers of DEG/ENaC subunits form H+-gated channels in mouse sensory neurons. Proc. Natl. Acad. Sci. USA 2002, 99, 2338–2343. [Google Scholar] [CrossRef] [PubMed]
- Friese, M.A.; Craner, M.J.; Etzensperger, R.; Vergo, S.; Wemmie, J.A.; Welsh, M.J.; Vincent, A.; Fugger, L. Acid-sensing ion channel-1 contributes to axonal degeneration in autoimmune inflammation of the central nervous system. Nat. Med. 2007, 13, 1483–1489. [Google Scholar] [CrossRef] [PubMed]
- Osmakov, D.I.; Andreev, Y.A.; Kozlov, S.A. Acid-Sensing Ion Channels and Their Modulators. Biochemistry 2014, 79, 1528–1545. [Google Scholar] [CrossRef] [PubMed]
- Schuhmacher, L.-N.; Smith, E.S.J. Expression of acid-sensing ion channels and selection of reference genes in mouse and naked mole rat. Mol. Brain 2016, 9, 97. [Google Scholar] [CrossRef] [PubMed]
- Rash, L.D. Acid-Sensing Ion Channel Pharmacology, Past, Present, and Future. In Advances in Pharmacology; Geraghty, D.P., Rash, L.D., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; Volume 79, pp. 35–66. [Google Scholar]
- Gründer, S.; Pusch, M. Biophysical properties of acid-sensing ion channels (ASICs). Neuropharmacology 2015, 94, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Osmakov, D.I.; Koshelev, S.G.; Lyukmanova, E.N.; Shulepko, M.A.; Andreev, Y.A.; Illes, P.; Kozlov, S.A. Multiple Modulation of Acid-Sensing Ion Channel 1a by the Alkaloid Daurisoline. Biomolecules 2019, 9, 336. [Google Scholar] [CrossRef] [PubMed]
- Wemmie, J.A.; Taugher, R.J.; Kreple, C.J. Acid-sensing ion channels in pain and disease. Nat. Rev. Neurosci. 2013, 14, 461–471. [Google Scholar] [CrossRef]
- Coryell, M.W.; Wunsch, A.M.; Haenfler, J.M.; Allen, J.E.; McBride, J.L.; Davidson, B.L.; Wemmie, J.A. Restoring Acid-Sensing Ion Channel-1a in the Amygdala of Knock-Out Mice Rescues Fear Memory but Not Unconditioned Fear Responses. J. Neurosci. 2008, 28, 13738–13741. [Google Scholar] [CrossRef]
- Leng, T.; Shi, Y.; Xiong, Z.-G.; Sun, D. Proton-sensitive cation channels and ion exchangers in ischemic brain injury: New therapeutic targets for stroke? Prog. Neurobiol. 2014, 115, 189–209. [Google Scholar] [CrossRef]
- Duan, B.; Wang, Y.-Z.; Yang, T.; Chu, X.-P.; Yu, Y.; Huang, Y.; Cao, H.; Hansen, J.; Simon, R.P.; Zhu, M.X.; et al. Extracellular Spermine Exacerbates Ischemic Neuronal Injury through Sensitization of ASIC1a Channels to Extracellular Acidosis. J. Neurosci. 2011, 31, 2101–2112. [Google Scholar] [CrossRef]
- Vick, J.S.; Askwith, C.C. ASICs and neuropeptides. Neuropharmacology 2015, 94, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Osmakov, D.I.; Koshelev, S.G.; Ivanov, I.A.; Andreev, Y.A.; Kozlov, S.A. Endogenous Neuropeptide Nocistatin Is a Direct Agonist of Acid-Sensing Ion Channels (ASIC1, ASIC2 and ASIC3). Biomolecules 2019, 9, 401. [Google Scholar] [CrossRef]
- Bohlen, C.J.; Chesler, A.T.; Sharif-Naeini, R.; Medzihradszky, K.F.; Zhou, S.; King, D.; Sánchez, E.E.; Burlingame, A.L.; Basbaum, A.I.; Julius, D. A heteromeric Texas coral snake toxin targets acid-sensing ion channels to produce pain. Nature 2011, 479, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Mazzuca, M.; Heurteaux, C.; Alloui, A.; Diochot, S.; Baron, A.; Voilley, N.; Blondeau, N.; Escoubas, P.; Gélot, A.; Cupo, A.; et al. A tarantula peptide against pain via ASIC1a channels and opioid mechanisms. Nat. Neurosci. 2007, 10, 943–945. [Google Scholar] [CrossRef] [PubMed]
- Diochot, S.; Baron, A.; Salinas, M.; Douguet, D.; Scarzello, S.; Dabert-Gay, A.-S.; Debayle, D.; Friend, V.; Alloui, A.; Lazdunski, M.; et al. Black mamba venom peptides target acid-sensing ion channels to abolish pain. Nature 2012, 490, 552–555. [Google Scholar] [CrossRef] [PubMed]
- Baron, A.; Lingueglia, E. Pharmacology of acid-sensing ion channels—Physiological and therapeutical perspectives. Neuropharmacology 2015, 94, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Waltenberger, B.; Pferschy-Wenzig, E.-M.; Linder, T.; Wawrosch, C.; Uhrin, P.; Temml, V.; Wang, L.; Schwaiger, S.; Heiss, E.H.; et al. Discovery and resupply of pharmacologically active plant-derived natural products: A review. Biotechnol. Adv. 2015, 33, 1582–1614. [Google Scholar] [CrossRef]
- Khalifa, S.A.M.; De Medina, P.; Erlandsson, A.; El-Seedi, H.R.; Silvente-Poirot, S.; Poirot, M. The novel steroidal alkaloids dendrogenin A and B promote proliferation of adult neural stem cells. Biochem. Biophys. Res. Commun. 2014, 446, 681–686. [Google Scholar] [CrossRef]
- Wu, W.-N.; Wu, P.-F.; Chen, X.-L.; Zhang, Z.; Gu, J.; Yang, Y.-J.; Xiong, Q.-J.; Ni, L.; Wang, F.; Chen, J.-G. Sinomenine protects against ischaemic brain injury: Involvement of co-inhibition of acid-sensing ion channel 1a and L-type calcium channels. Br. J. Pharmacol. 2011, 164, 1445–1459. [Google Scholar] [CrossRef]
- Silva Teles, M.M.R.; Vieira Pinheiro, A.A.; Da Silva Dias, C.; Fechine Tavares, J.; Barbosa Filho, J.M.; Leitão Da Cunha, E.V. Alkaloids of the Lauraceae. In The Alkaloids: Chemistry and Biology; Knölker, H.-J., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; Volume 82, pp. 147–304. [Google Scholar]
- Murebwayire, S.; Ingkaninan, K.; Changwijit, K.; Frédérich, M.; Duez, P. Triclisia sacleuxii (Pierre) Diels (Menispermaceae), a potential source of acetylcholinesterase inhibitors. J. Pharm. Pharmacol. 2009, 61, 103–107. [Google Scholar] [CrossRef]
- Fournet, A.; Inchausti, A.; Yaluff, G.; De Arias, A.R.; Guinaudeau, H.; Bruneton, J.; Breidenbach, M.A.; Karplus, P.A.; Faerman, C.H. Trypanocidal Bisbenzylisoquinoline Alkaloids are Inhibitors of Trypanothione Reductase. J. Enzyme Inhib. 1998, 13, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chia, Y.-C.; Chang, F.-R.; Wu, C.-C.; Teng, C.-M.; Chen, K.-S.; Wu, Y.-C. Effect of Isoquinoline Alkaloids of Different Structural Types on Antiplatelet Aggregation in Vitro. Planta Med. 2006, 72, 1238–1241. [Google Scholar] [CrossRef] [PubMed]
- Osmakov, D.I.; Koshelev, S.G.; Andreev, Y.A.; Dubinnyi, M.A.; Kublitski, V.S.; Efremov, R.G.; Sobolevsky, A.I.; Kozlov, S.A. Proton-independent activation of acid-sensing ion channel 3 by an alkaloid, lindoldhamine, from Laurus nobilis. Br. J. Pharmacol. 2018, 175, 924–937. [Google Scholar] [CrossRef] [PubMed]
- Morton, D.B.; Abbot, D.; Barclay, R.; Close, B.S.; Ewbank, R.; Gask, D.; Heath, M.; Mattic, S.; Poole, T.; Seamer, J.; et al. Removal of blood from laboratory mammals and birds: First Report of the BVA/FRAME/RSPCA/UFAW Joint Working Group on Refinement. Lab. Anim. 1993, 27, 1–22. [Google Scholar] [CrossRef]
- Weber, C.; Opatz, T. Bisbenzylisoquinoline Alkaloids. In The Alkaloids: Chemistry and Biology; Knölker, H.-J., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; Volume 81, pp. 1–114. [Google Scholar]
- Bhagya, N.; Chandrashekar, K.R. Tetrandrine—A molecule of wide bioactivity. Phytochemistry 2016, 125, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Liu, X.; Li, W. Tetrandrine, a Chinese plant-derived alkaloid, is a potential candidate for cancer chemotherapy. Oncotarget 2016, 7, 40800–40815. [Google Scholar] [CrossRef] [PubMed]
- Ishikita, H. Proton-Binding Sites of Acid-Sensing Ion Channel 1. PLoS ONE 2011, 6, e16920. [Google Scholar] [CrossRef]
- Vullo, S.; Bonifacio, G.; Roy, S.; Johner, N.; Bernèche, S.; Kellenberger, S. Conformational dynamics and role of the acidic pocket in ASIC pH-dependent gating. Proc. Natl. Acad. Sci. USA 2017, 114, 3768–3773. [Google Scholar] [CrossRef] [PubMed]
- Laurent, B.; Murail, S.; Shahsavar, A.; Sauguet, L.; Delarue, M.; Baaden, M. Sites of Anesthetic Inhibitory Action on a Cationic Ligand-Gated Ion Channel. Structure 2016, 24, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Alijevic, O.; Kellenberger, S. Subtype-specific Modulation of Acid-sensing Ion Channel (ASIC) Function by 2-Guanidine-4-methylquinazoline. J. Biol. Chem. 2012, 287, 36059–36070. [Google Scholar] [CrossRef]
- Besson, T.; Lingueglia, E.; Salinas, M. Pharmacological modulation of Acid-Sensing Ion Channels 1a and 3 by amiloride and 2-guanidine-4-methylquinazoline (GMQ). Neuropharmacology 2017, 125, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, T.W.; Askwith, C.C. Endogenous arginine-phenylalanine-amide-related peptides alter steady-state desensitization of ASIC1a. J. Biol. Chem. 2008, 283, 1818–1830. [Google Scholar] [CrossRef] [PubMed]
- Stephan, G.; Huang, L.; Tang, Y.; Vilotti, S.; Fabbretti, E.; Yu, Y.; Nörenberg, W.; Franke, H.; Gölöncsér, F.; Sperlágh, B.; et al. The ASIC3/P2X3 cognate receptor is a pain-relevant and ligand-gated cationic channel. Nat. Commun. 2018, 9, 1354. [Google Scholar] [CrossRef] [PubMed]
- Voilley, N.; de Weille, J.; Mamet, J.; Lazdunski, M. Nonsteroid Anti-Inflammatory Drugs Inhibit Both the Activity and the Inflammation-Induced Expression of Acid-Sensing Ion Channels in Nociceptors. J. Neurosci. 2001, 21, 8026–8033. [Google Scholar] [CrossRef] [PubMed]
- Mamet, J.; Baron, A.; Lazdunski, M.; Voilley, N. ProInflammatory Mediators, Stimulators of Sensory Neuron Excitability via the Expression of Acid-Sensing Ion Channels. J. Neurosci. 2002, 22, 10662–10670. [Google Scholar] [CrossRef] [PubMed]
- Duan, B.; Wu, L.-J.; Yu, Y.-Q.; Ding, Y.; Jing, L.; Xu, L.; Chen, J.; Xu, T.-L. Upregulation of Acid-Sensing Ion Channel ASIC1a in Spinal Dorsal Horn Neurons Contributes to Inflammatory Pain Hypersensitivity. J. Neurosci. 2007, 27, 11139–11148. [Google Scholar] [CrossRef]
- Steen, K.; Steen, A.; Reeh, P. A dominant role of acid pH in inflammatory excitation and sensitization of nociceptors in rat skin, in vitro. J. Neurosci. 1995, 15, 3982–3989. [Google Scholar] [CrossRef]
- Lardner, A. The effects of extracellular pH on immune function. J. Leukoc. Biol. 2001, 69, 522–530. [Google Scholar] [CrossRef]
- Sluka, K.A.; Price, M.P.; Breese, N.M.; Stucky, C.L.; Wemmie, J.A.; Welsh, M.J. Chronic hyperalgesia induced by repeated acid injections in muscle is abolished by the loss of ASIC3, but not ASIC1. Pain 2003, 106, 229–239. [Google Scholar] [CrossRef]
- Osmakov, D.I.; Koshelev, S.G.; Andreev, Y.A.; Kozlov, S.A. Endogenous isoquinoline alkaloids agonists of acid-sensing ion channel type 3. Front. Mol. Neurosci. 2017, 10, 282. [Google Scholar] [CrossRef]
- Karczewski, J.; Spencer, R.H.; Garsky, V.M.; Liang, A.; Leitl, M.D.; Cato, M.J.; Cook, S.P.; Kane, S.; Urban, M.O. Reversal of acid-induced and inflammatory pain by the selective ASIC3 inhibitor, APETx2. Br. J. Pharmacol. 2010, 161, 950–960. [Google Scholar] [CrossRef] [PubMed]
- Andreev, Y.; Osmakov, D.; Koshelev, S.; Maleeva, E.; Logashina, Y.; Palikov, V.; Palikova, Y.; Dyachenko, I.; Kozlov, S. Analgesic Activity of Acid-Sensing Ion Channel 3 (ASIС3) Inhibitors: Sea Anemones Peptides Ugr9-1 and APETx2 versus Low Molecular Weight Compounds. Mar. Drugs 2018, 16, 500. [Google Scholar] [CrossRef] [PubMed]
- Frias, B.; Merighi, A. Capsaicin, Nociception and Pain. Molecules 2016, 21, 797. [Google Scholar] [CrossRef] [PubMed]
- Materazzi, S.; Benemei, S.; Fusi, C.; Gualdani, R.; De Siena, G.; Vastani, N.; Andersson, D.A.; Trevisan, G.; Moncelli, M.R.; Wei, X.; et al. Parthenolide inhibits nociception and neurogenic vasodilatation in the trigeminovascular system by targeting the TRPA1 channel. Pain 2013, 154, 2750–2758. [Google Scholar] [CrossRef] [PubMed]
- Logashina, Y.A.; Mosharova, I.V.; Korolkova, Y.V.; Shelukhina, I.V.; Dyachenko, I.A.; Palikov, V.A.; Palikova, Y.A.; Murashev, A.N.; Kozlov, S.A.; Stensvåg, K.; et al. Peptide from Sea Anemone Metridium senile Affects Transient Receptor Potential Ankyrin-repeat 1 (TRPA1) Function and Produces Analgesic Effect. J. Biol. Chem. 2017, 292, 2992–3004. [Google Scholar] [CrossRef] [PubMed]
- Logashina, Y.A.; Solstad, R.G.; Mineev, K.S.; Korolkova, Y.V.; Mosharova, I.V.; Dyachenko, I.A.; Palikov, V.A.; Palikova, Y.A.; Murashev, A.N.; Arseniev, A.S.; et al. New Disulfide-Stabilized Fold Provides Sea Anemone Peptide to Exhibit Both Antimicrobial and TRPA1 Potentiating Properties. Toxins (Basel) 2017, 9, 154. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pH 6.85 | pH 6.7 | pH 6.5 | pH 6.0 | |
|---|---|---|---|---|
| IC50, µM | 9 ± 0.3 | 25 ± 6 | 150 ± 10 | 296 ± 146 |
| nH | 1.22 ± 0.03 | 1.29 ± 0.12 | 1.25 ± 0.05 | 1.07 ± 0.15 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osmakov, D.I.; Koshelev, S.G.; Palikov, V.A.; Palikova, Y.A.; Shaykhutdinova, E.R.; Dyachenko, I.A.; Andreev, Y.A.; Kozlov, S.A. Alkaloid Lindoldhamine Inhibits Acid-Sensing Ion Channel 1a and Reveals Anti-Inflammatory Properties. Toxins 2019, 11, 542. https://doi.org/10.3390/toxins11090542
Osmakov DI, Koshelev SG, Palikov VA, Palikova YA, Shaykhutdinova ER, Dyachenko IA, Andreev YA, Kozlov SA. Alkaloid Lindoldhamine Inhibits Acid-Sensing Ion Channel 1a and Reveals Anti-Inflammatory Properties. Toxins. 2019; 11(9):542. https://doi.org/10.3390/toxins11090542
Chicago/Turabian StyleOsmakov, Dmitry I., Sergey G. Koshelev, Victor A. Palikov, Yulia A. Palikova, Elvira R. Shaykhutdinova, Igor A. Dyachenko, Yaroslav A. Andreev, and Sergey A. Kozlov. 2019. "Alkaloid Lindoldhamine Inhibits Acid-Sensing Ion Channel 1a and Reveals Anti-Inflammatory Properties" Toxins 11, no. 9: 542. https://doi.org/10.3390/toxins11090542
APA StyleOsmakov, D. I., Koshelev, S. G., Palikov, V. A., Palikova, Y. A., Shaykhutdinova, E. R., Dyachenko, I. A., Andreev, Y. A., & Kozlov, S. A. (2019). Alkaloid Lindoldhamine Inhibits Acid-Sensing Ion Channel 1a and Reveals Anti-Inflammatory Properties. Toxins, 11(9), 542. https://doi.org/10.3390/toxins11090542