A Novel Magnetic Molecular Imprinted Polymer for Selective Extraction of Zearalenone from Cereal Flours before Liquid Chromatography-Tandem Mass Spectrometry Determination

 ,
,  ,
,  ,
, 
Abstract
:1. Introduction
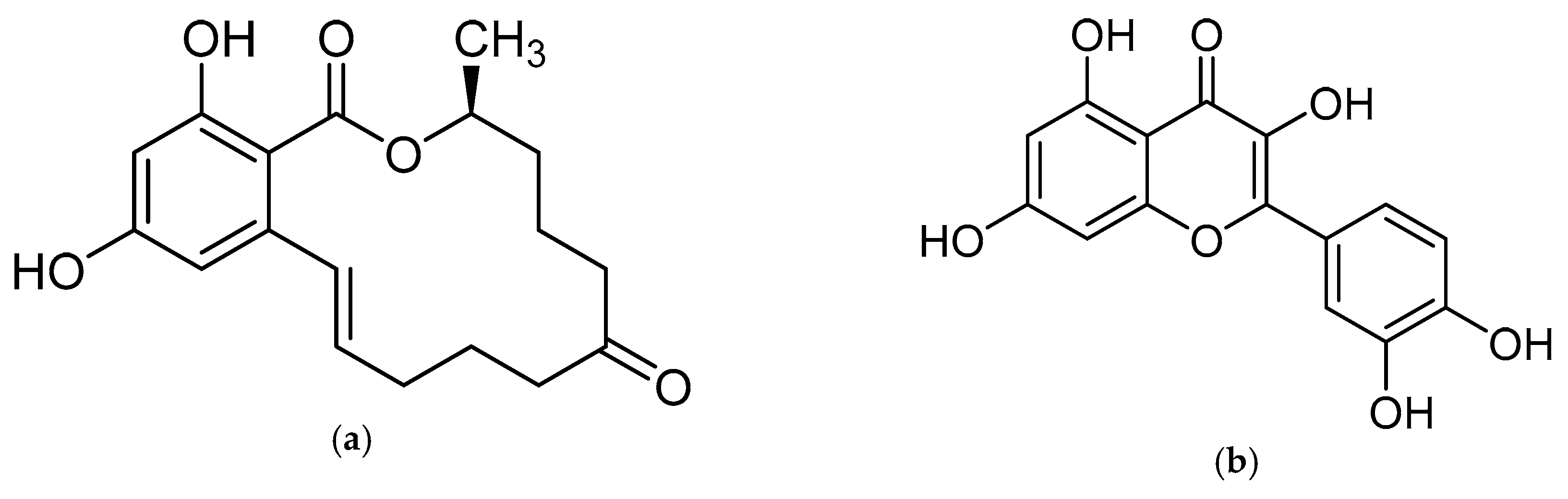
2. Results and Discussion
2.1. Preparation of mMIP
2.2. The Selectivity of the mMIP
2.3. Optimization of Sample Clean-Up Using the mMIP
2.4. Method Performance and Single-Lab Validation
2.4.1. Carry-Over
2.4.2. Extraction Recovery and Matrix Effect
2.4.3. Trueness and Precision
2.4.4. Linearity, Limit of Detection, and Limit of Quantification
2.5. Comparison with Two Literature Procedures
2.6. Cereal Sample Analysis
3. Conclusions
4. Materials and Methods
4.1. Chemicals and Materials
4.2. Preparation of the mMIP and Assessment of Its Binding Properties
4.3. Sample Preparation
4.4. UHPLC-MS/MS Analysis
4.5. The Selectivity of MIP Towards ZEN
4.6. Method Validation
4.6.1. Extraction Recovery and Matrix Effect
4.6.2. Linear Range and Calibration Curve
4.6.3. Trueness and Precision
4.6.4. LOD and LOQ
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Metzler, M.; Pfeiffer, E.; Hildebrand, A. Zearalenone and its metabolites as endocrine disrupting chemicals. World Mycotoxin J. 2010, 3, 385–401. [Google Scholar] [CrossRef]
- Laganà, A.; Bacaloni, A.; De Leva, I.; Faberi, A.; Fago, G.; Marino, A. Analytical methodologies for determining the occurrence of endocrine disrupting chemicals in sewage treatment plants and natural waters. Anal. Chim. Acta 2004, 501, 79–88. [Google Scholar] [CrossRef]
- Marin, S.; Ramos, A.J.; Cano-Sancho, G.; Sanchis, V. Mycotoxins: Occurrence, toxicology, and exposure assessment. Food Chem. Toxicol. 2013, 60, 218–237. [Google Scholar] [CrossRef] [PubMed]
- Bertero, A.; Moretti, A.; Spicer, L.J.; Caloni, F. Fusarium molds and mycotoxins: Potential species-specific effects. Toxins (Basel) 2018, 10, 244. [Google Scholar] [CrossRef] [PubMed]
- Rychlik, M.; Humpf, H.-U.; Marko, D.; Dänicke, S.; Mally, A.; Berthiller, F.; Klaffke, H.; Lorenz, N. Proposal of a comprehensive definition of modified and other forms of mycotoxins including “masked” mycotoxins. Mycotoxin Res. 2014, 30, 197–205. [Google Scholar] [CrossRef] [PubMed]
- EFSA Panel on Contaminants in the Food Chain (EFSA CONTAM Panel) Scientific opinion on the appropriateness to set a group health-based guidance value for zearalenone and its modified forms. EFSA J. 2016, 14, 4425.
- Capriotti, A.L.; Cavaliere, C.; Colapicchioni, V.; Piovesana, S.; Samperi, R.; Laganà, A. Analytical strategies based on chromatography–mass spectrometry for the determination of estrogen-mimicking compounds in food. J. Chromatogr. A 2013, 1313, 62–77. [Google Scholar] [CrossRef] [PubMed]
- Regulation (EC) No 1881/2006 of 19 December 2006 setting maximum levels for certain contaminants in foodstuffs. Off. J. Eur. Union 2006, L364, 5–24.
- EFSA Panel on Contaminants in the Food Chain (EFSA CONTAM Panel) Scientific Opinion on the risks for public health related to the presence of zearalenone in food. EFSA J. 2011, 9, 2197. [CrossRef]
- Cavaliere, C.; Samperi, R.; Foglia, P.; Caruso, G.; Laganà, A.; Capriotti, A.L. Multiclass mycotoxin analysis in food, environmental and biological matrices with chromatography/mass spectrometry. Mass Spectrom. Rev. 2011, 31, 466–503. [Google Scholar]
- Tittlemier, S.A.; Cramer, B.; Dall’Asta, C.; Iha, M.H.; Lattanzio, V.M.T.; Malone, R.J.; Maragos, C.; Solfrizzo, M.; Stranska-Zachariasova, M.; Stroka, J. Developments in mycotoxin analysis: An update for 2017–2018. World Mycotoxin J. 2019, 12, 3–29. [Google Scholar] [CrossRef]
- Cavaliere, C.; Foglia, P.; Guarino, C.; Motto, M.; Nazzari, M.; Samperi, R.; Laganà, A.; Berardo, N. Mycotoxins produced by Fusarium genus in maize: Determination by screening and confirmatory methods based on liquid chromatography tandem mass spectrometry. Food Chem. 2007, 105, 700–710. [Google Scholar] [CrossRef]
- Cavaliere, C.; D’Ascenzo, G.; Foglia, P.; Pastorini, E.; Samperi, R.; Laganà, A. Determination of type B trichothecenes and macrocyclic lactone mycotoxins in field contaminated maize. Food Chem. 2005, 92, 559–568. [Google Scholar] [CrossRef]
- De Boevre, M.; Di Mavungu, J.D.; Maene, P.; Audenaert, K.; Deforce, D.; Haesaert, G.; Eeckhout, M.; Callebaut, A.; Berthiller, F.; Van Peteghem, C.; et al. Development and validation of an LC-MS/MS method for the simultaneous determination of deoxynivalenol, zearalenone, T-2-toxin and some masked metabolites in different cereals and cereal-derived food. Food Addit. Contam. Part A 2012, 29, 819–835. [Google Scholar] [CrossRef] [PubMed]
- Sulyok, M.; Berthiller, F.; Krska, R.; Schuhmacher, R. Development and validation of a liquid chromatography/tandem mass spectrometric method for the determination of 39 mycotoxins in wheat and maize. Rapid Commun. Mass Spectrom. 2006, 20, 2649–2659. [Google Scholar] [CrossRef] [PubMed]
- Vendl, O.; Berthiller, F.; Crews, C.; Krska, R. Simultaneous determination of deoxynivalenol, zearalenone, and their major masked metabolites in cereal-based food by LC–MS–MS. Anal. Bioanal. Chem. 2009, 395, 1347–1354. [Google Scholar] [CrossRef] [PubMed]
- Llorens, A.; Mateo, R.; Mateo, J.J.; Jiménez, M. Comparison of extraction and clean-up procedures for analysis of zearalenone in corn, rice and wheat grains by high-performance liquid chromatography with photodiode array and fluorescence detection. Food Addit. Contam. 2002, 19, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Lucci, P.; Derrien, D.; Alix, F.; Pérollier, C.; Bayoudh, S. Molecularly imprinted polymer solid-phase extraction for detection of zearalenone in cereal sample extracts. Anal. Chim. Acta 2010, 672, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.; Freudenschuss, M.; Krska, R.; Mizaikoff, B. Improving methods of analysis for mycotoxins: Molecularly imprinted polymers for deoxynivalenol and zearalenone. Food Addit. Contam. 2003, 20, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Urraca, J.L.; Marazuela, M.D.; Merino, E.R.; Orellana, G.; Moreno-Bondi, M.C. Molecularly imprinted polymers with a streamlined mimic for zearalenone analysis. J. Chromatogr. A 2006, 1116, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Urraca, J.L.; Marazuela, M.D.; Moreno-Bondi, M.C. Molecularly imprinted polymers applied to the clean-up of zearalenone and α-zearalenol from cereal and swine feed sample extracts. Anal. Bioanal. Chem. 2006, 385, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Speltini, A.; Scalabrini, A.; Maraschi, F.; Sturini, M.; Profumo, A. Newest applications of molecularly imprinted polymers for extraction of contaminants from environmental and food matrices: A review. Anal. Chim. Acta 2017, 974, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Matsui, J.; Fujiwara, K.; Takeuchi, T. Atrazine-Selective Polymers Prepared by Molecular Imprinting of Trialkylmelamines as Dummy Template Species of Atrazine. Anal. Chem. 2000, 72, 1810–1813. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Xu, J.; Zheng, J.; Zhu, F.; Xie, L.; Ouyang, G. Synthesis and application of magnetic molecularly imprinted polymers in sample preparation. Anal. Bioanal. Chem. 2018, 410, 3991–4014. [Google Scholar] [CrossRef] [PubMed]
- Piovesana, S.; Benedetti, B.; Sparnacci, K.; Gianotti, V.; Laus, M.; Antonioli, D.; Laganà, A. Magnetic Molecularly Imprinted Multishell Particles for Zearalenone Pre-Concentration and Clean-Up. Polymers. (under review).
- Soleimany, F.; Jinap, S.; Faridah, A.; Khatib, A. A UPLC–MS/MS for simultaneous determination of aflatoxins, ochratoxin A, zearalenone, DON, fumonisins, T-2 toxin and HT-2 toxin, in cereals. Food Control 2012, 25, 647–653. [Google Scholar] [CrossRef]
- Barbera, G.; Capriotti, A.; Cavaliere, C.; Foglia, P.; Montone, C.; Chiozzi, R.; Laganà, A. A Rapid Magnetic Solid Phase Extraction Method Followed by Liquid Chromatography-Tandem Mass Spectrometry Analysis for the Determination of Mycotoxins in Cereals. Toxins (Basel) 2017, 9, 147. [Google Scholar] [CrossRef] [PubMed]
- Kruve, A.; Rebane, R.; Kipper, K.; Oldekop, M.-L.; Evard, H.; Herodes, K.; Ravio, P.; Leito, I. Tutorial review on validation of liquid chromatography–mass spectrometry methods: Part I. Anal. Chim. Acta 2015, 870, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Kruve, A.; Rebane, R.; Kipper, K.; Oldekop, M.-L.; Evard, H.; Herodes, K.; Ravio, P.; Leito, I. Tutorial review on validation of liquid chromatography–mass spectrometry methods: Part II. Anal. Chim. Acta 2015, 870, 8–28. [Google Scholar] [CrossRef]
- Samperi, R.; Piovesana, S.; Laganà, A.; Zenezini Chiozzi, R.; La Barbera, G.; Capriotti, A.L.; Cavaliere, C. Polydopamine-coated magnetic nanoparticles for isolation and enrichment of estrogenic compounds from surface water samples followed by liquid chromatography-tandem mass spectrometry determination. Anal. Bioanal. Chem. 2016, 408, 4011–4020. [Google Scholar] [Green Version]


{kind=link}
| Compound | Structure | Not Retained Amount (RSD, %) 1 | Compound | Structure | Not Retained Amount (RSD, %) 1 |
|---|---|---|---|---|---|
| DON |  | 112% (14) | β-ZEL |  | 3% (2) |
| DAD |  | 5% (3) | α-ZEL |  | <LOD |
| GEN |  | <LOD | ZEN |  | 4% (2) |
| H-T2 |  | 81% (9) | ZAN |  | <LOD |
| Compound | Acetonitrile/Water 50:50 (v/v) RE; ME (%) 1 | Acetonitrile/Water 80:20 (v/v) RE; ME (%) 1 | Methanol/Water 80:20 (v/v) RE; ME (%) 1 |
|---|---|---|---|
| ZEN | 80; 86 | 92; 85 | 95; 99 |
| Compound | RE% 1 (RSD) 0.5 × ML | ME% 1 (RSD) 0.5 × ML | RE% 1 (RSD) 1.0 × ML | ME% 1 (RSD) 1.0 × ML | RE% 1 (RSD) 4.0 × ML | ME% 1 (RSD) 4.0 × ML |
|---|---|---|---|---|---|---|
| ZEN | 98 (7) | 99 (3) | 94 (3) | 102 (6) | 76 (9) | 103 (4) |
| Compound | REapp, % (RSDr; RSDR) | ||
|---|---|---|---|
| Spiking level | 0.5 × ML | 1.0 × ML | 4.0 × ML |
| ZEN | 98 (10; 7) | 97 (8; 11) | 81 (9; 13) |
| Compound | LOD (ng g−1) | LOQ (ng g−1) |
|---|---|---|
| ZEN | 0.044 | 0.14 |
| Flour Sample Contamination (ng g−1) | |||||||
|---|---|---|---|---|---|---|---|
| Compound | Tapioca and Maize | Maize | Hulled Wheat | Whole Wheat | Rice | Buck Wheat | Durum Wheat |
| ZEN | 7.5 | 8.5 | 8.9 | 9.7 | 15.0 | <LOQ | <LOQ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cavaliere, C.; Antonelli, M.; Cerrato, A.; La Barbera, G.; Laganà, A.; Laus, M.; Piovesana, S.; Capriotti, A.L. A Novel Magnetic Molecular Imprinted Polymer for Selective Extraction of Zearalenone from Cereal Flours before Liquid Chromatography-Tandem Mass Spectrometry Determination. Toxins 2019, 11, 493. https://doi.org/10.3390/toxins11090493
Cavaliere C, Antonelli M, Cerrato A, La Barbera G, Laganà A, Laus M, Piovesana S, Capriotti AL. A Novel Magnetic Molecular Imprinted Polymer for Selective Extraction of Zearalenone from Cereal Flours before Liquid Chromatography-Tandem Mass Spectrometry Determination. Toxins. 2019; 11(9):493. https://doi.org/10.3390/toxins11090493
Chicago/Turabian StyleCavaliere, Chiara, Michela Antonelli, Andrea Cerrato, Giorgia La Barbera, Aldo Laganà, Michele Laus, Susy Piovesana, and Anna Laura Capriotti. 2019. "A Novel Magnetic Molecular Imprinted Polymer for Selective Extraction of Zearalenone from Cereal Flours before Liquid Chromatography-Tandem Mass Spectrometry Determination" Toxins 11, no. 9: 493. https://doi.org/10.3390/toxins11090493
APA StyleCavaliere, C., Antonelli, M., Cerrato, A., La Barbera, G., Laganà, A., Laus, M., Piovesana, S., & Capriotti, A. L. (2019). A Novel Magnetic Molecular Imprinted Polymer for Selective Extraction of Zearalenone from Cereal Flours before Liquid Chromatography-Tandem Mass Spectrometry Determination. Toxins, 11(9), 493. https://doi.org/10.3390/toxins11090493