Target Analysis and Retrospective Screening of Multiple Mycotoxins in Pet Food Using UHPLC-Q-Orbitrap HRMS

 ,
,  , ,
, ,  ,
,  and
and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Optimization of the Ultrahigh-Performance Liquid Chromatography Coupled to High-Resolution Mass-Spectrometry (UHPLC-Q-Orbitrap HRMS) Analysis
2.2. Optimization of Sample Preparation Procedure
2.2.1. Stirring Time
2.2.2. Sonication Treatment
2.2.3. Clean-Up Step
2.2.4. Sample Amount
2.3. Method Validation
2.4. Occurrence of Mycotoxins in Pet Food Samples
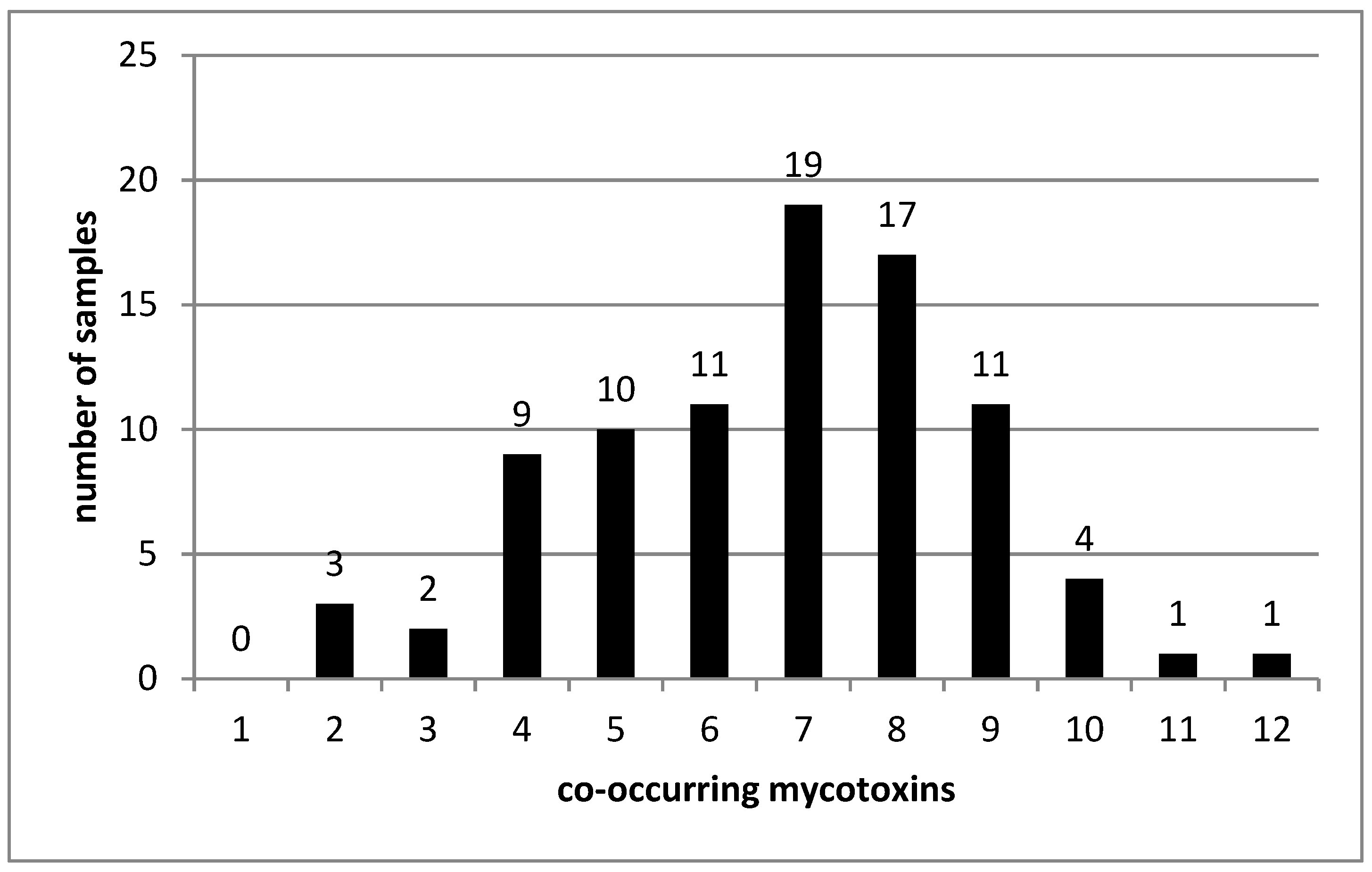
2.5. Co-occurrence of Mycotoxins in Analyzed Samples
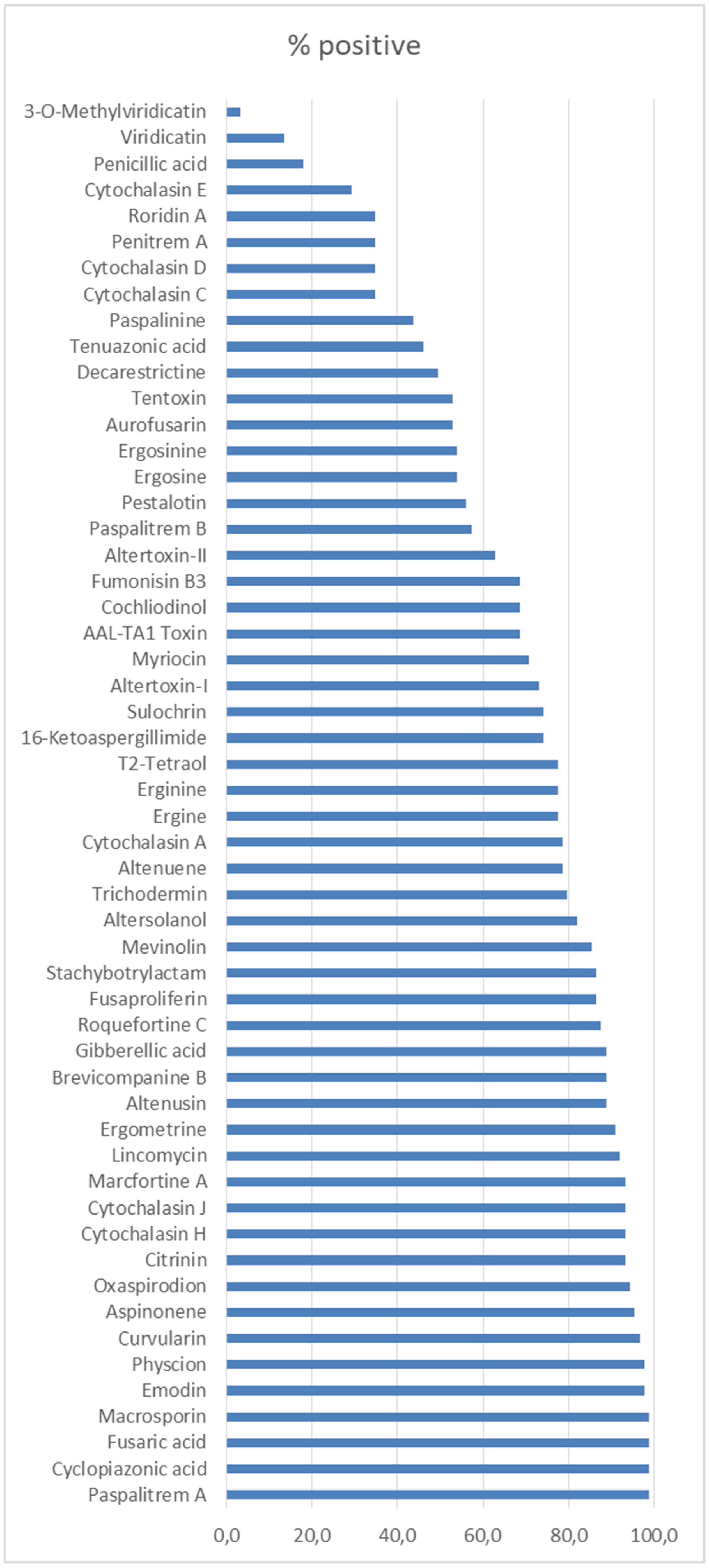
2.6. Identification of Non Target Coumpounds Based on A Retrospective Screening Analysis
3. Conclusions
4. Materials and Methods
4.1. Chemical and Reagents
4.2. Sampling
4.3. Sample Preparation
4.4. Method Validation
4.5. Quality Assurance/Quality Control
4.6. Ultrahigh-Performance Liquid Chromatography Coupled to High-Resolution Mass-Spectrometry (UHPLC-Q-Orbitrap HRMS) Analysis
4.7. Statistics and Data Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jedidi, I.; Soldevilla, C.; Lahouar, A.; Marin, P.; Gonzalez-Jaen, M.T.; Said, S. Mycoflora isolation and molecular characterization of Aspergillus and Fusarium species in Tunisian cereals. Saudi J. Biol. Sci. 2018, 25, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, S.Z.; Selamat, J.; Ariño, A. Mycotoxins in food and food products: Current status. In Food Safety; Springer: Berlin, Germany, 2016; pp. 113–123. [Google Scholar]
- Chhonker, S.; Rawat, D.; Naik, R.; Koiri, R. An Overview of Mycotoxins in Human Health with Emphasis on Development and Progression of Liver Cancer. Clin. Oncol. 2018, 3, 1408. [Google Scholar]
- Meeting, J.F.W.E.C.o.F.A. Safety Evaluation of Certain Mycotoxins in Food; Food & Agriculture Org.: Rome, Italy, 2001. [Google Scholar]
- Pascari, X.; Ramos, A.J.; Marin, S.; Sanchis, V. Mycotoxins and beer. Impact of beer production process on mycotoxin contamination. A review. Food Res. Int. 2018, 103, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Gruber-Dorninger, C.; Novak, B.; Nagl, V.; Berthiller, F. Emerging mycotoxins: Beyond traditionally determined food contaminants. J. Agric. Food Chem. 2016, 65, 7052–7070. [Google Scholar] [CrossRef] [PubMed]
- Suleiman, R.; Rosentrater, K.; Chove, B. Understanding postharvest practices, knowledge, and actual mycotoxin levels in maize in three agro-ecological zones in Tanzania. J. Stored Prod. Postharvest Res. 2017, 8, 73. [Google Scholar]
- Unusan, N. Systematic review of mycotoxins in food and feeds in Turkey. Food Control 2018, 97, 1–14. [Google Scholar] [CrossRef]
- Streit, E.; Schatzmayr, G.; Tassis, P.; Tzika, E.; Marin, D.; Taranu, I.; Tabuc, C.; Nicolau, A.; Aprodu, I.; Puel, O. Current situation of mycotoxin contamination and co-occurrence in animal feed—Focus on Europe. Toxins 2012, 4, 788–809. [Google Scholar] [CrossRef]
- Soler, L.; Oswald, I. The importance of accounting for sex in the search of proteomic signatures of mycotoxin exposure. J. Proteom. 2018, 178, 114–122. [Google Scholar] [CrossRef]
- Escrivá, L.; Font, G.; Berrada, H.; Manyes, L. Mycotoxin contamination in laboratory rat feeds and their implications in animal research. Toxicol. Mech. Methods 2016, 26, 529–537. [Google Scholar] [CrossRef]
- Vila-Donat, P.; Marín, S.; Sanchis, V.; Ramos, A. A review of the mycotoxin adsorbing agents, with an emphasis on their multi-binding capacity, for animal feed decontamination. Food Chem. Toxicol. 2018, 114, 246–259. [Google Scholar] [CrossRef]
- Rodríguez-Carrasco, Y.; Moltó, J.C.; Berrada, H.; Mañes, J. A survey of trichothecenes, zearalenone and patulin in milled grain-based products using GC–MS/MS. Food Chem. 2014, 146, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Adeyeye, S.A. Fungal mycotoxins in foods: A review. Cogent Food Agric. 2016, 2, 1213127. [Google Scholar] [CrossRef]
- Mahmoud, A.F.; Escrivá, L.; Rodríguez-Carrasco, Y.; Moltó, J.C.; Berrada, H. Determination of trichothecenes in chicken liver using gas chromatography coupled with triple-quadrupole mass spectrometry. LWT 2018, 93, 237–242. [Google Scholar] [CrossRef]
- Tolosa, J.; Rodríguez-Carrasco, Y.; Ferrer, E.; Mañes, J. Identification and Quantification of Enniatins and Beauvericin in Animal Feeds and Their Ingredients by LC-QTRAP/MS/MS. Metabolites 2019, 9, 33. [Google Scholar] [CrossRef] [PubMed]
- Rico-Yuste, A.; Walravens, J.; Urraca, J.; Abou-Hany, R.; Descalzo, A.; Orellana, G.; Rychlik, M.; De Saeger, S.; Moreno-Bondi, M. Analysis of alternariol and alternariol monomethyl ether in foodstuffs by molecularly imprinted solid-phase extraction and ultra-high-performance liquid chromatography tandem mass spectrometry. Food Chem. 2018, 243, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Kaczynski, P.; Hrynko, I.; Lozowicka, B. Evolution of novel sorbents for effective clean-up of honeybee matrix in highly toxic insecticide LC/MS/MS analysis. Ecotoxicol. Environ. Saf. 2017, 139, 124–131. [Google Scholar] [CrossRef]
- Rodríguez-Carrasco, Y.; Gaspari, A.; Graziani, G.; Sandini, A.; Ritieni, A. Fast analysis of polyphenols and alkaloids in cocoa-based products by ultra-high performance liquid chromatography and Orbitrap high resolution mass spectrometry (UHPLC-Q-Orbitrap-MS/MS). Food Res. Int. 2018, 111, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Van Wijk, X.M.; Goodnough, R.; Colby, J.M. Mass spectrometry in emergency toxicology: Current state and future applications. Crit. Rev. Clin. Lab. Sci. 2019, 56, 225–238. [Google Scholar] [CrossRef]
- Abd-Elhakim, Y.M.; El Sharkawy, N.I.; Moustafa, G.G. An investigation of selected chemical contaminants in commercial pet foods in Egypt. J. Vet. Diagn. Investig. 2016, 28, 70–75. [Google Scholar] [CrossRef]
- Błajet-Kosicka, A.; Kosicki, R.; Twarużek, M.; Grajewski, J. Determination of moulds and mycotoxins in dry dog and cat food using liquid chromatography with mass spectrometry and fluorescence detection. Food Addit. Contam. Part B 2014, 7, 302–308. [Google Scholar] [CrossRef]
- Bissoqui, L.Y.; Frehse, M.S.; Freire, R.L.; Ono, M.A.; Bordini, J.G.; Hirozawa, M.T.; de Oliveira, A.J.; Ono, E.Y. Exposure assessment of dogs to mycotoxins through consumption of dry feed. J. Sci. Food Agric. 2016, 96, 4135–4142. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.D.; Baijnath, S.; Chuturgoon, A.A. A comparison of mycotoxin contamination of premium and grocery brands of pelleted cat food in South Africa. J. S. Afr. Vet. Assoc. 2017, 88. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Teixeira, E.; Frehse, M.; Freire, R.; Ono, M.; Bordini, J.; Hirozawa, M.; Ono, E. Safety of low and high cost dry feed intended for dogs in Brazil concerning fumonisins, zearalenone and aflatoxins. World Mycotoxin J. 2017, 10, 273–283. [Google Scholar] [CrossRef]
- Mao, J.; Zheng, N.; Wen, F.; Guo, L.; Fu, C.; Ouyang, H.; Zhong, L.; Wang, J.; Lei, S. Multi-mycotoxins analysis in raw milk by ultra high performance liquid chromatography coupled to quadrupole orbitrap mass spectrometry. Food Control 2018, 84, 305–311. [Google Scholar] [CrossRef]
- Kabir, A.; Locatelli, M.; Ulusoy, H. Recent trends in microextraction techniques employed in analytical and bioanalytical sample preparation. Separations 2017, 4, 36. [Google Scholar] [CrossRef]
- Singh, S.D.; Chuturgoon, A.A. A comparative analysis of mycotoxin contamination of supermarket and premium brand pelleted dog food in Durban, South Africa. J. S. Afr. Vet. Assoc. 2017, 88, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.; Li, L.; Gu, Z.; Yao, M.; Xu, D.; Fan, W.; Yan, L.; Song, S. Mycotoxins in commercial dry pet food in China. Food Addit. Contam. Part B 2018, 11, 237–245. [Google Scholar] [CrossRef]
- Böhm, J.; Koinig, L.; Razzazi-Fazeli, E.; Blajet-Kosicka, A.; Twaruzek, M.; Grajewski, J.; Lang, C. Survey and risk assessment of the mycotoxins deoxynivalenol, zearalenone, fumonisins, ochratoxin A, and aflatoxins in commercial dry dog food. Mycotoxin Res. 2010, 26, 147–153. [Google Scholar] [CrossRef]
- Campos, S.; Keller, L.; Cavaglieri, L.; Krüger, C.; Fernández Juri, M.; Dalcero, A.; Magnoli, C.; Rosa, C. Aflatoxigenic fungi and aflatoxin B1 in commercial pet food in Brazil. World Mycotoxin J. 2009, 2, 85–90. [Google Scholar] [CrossRef]
- Tolosa, J.; Font, G.; Mañes, J.; Ferrer, E. Natural occurrence of emerging Fusarium mycotoxins in feed and fish from aquaculture. J. Agric. Food Chem. 2014, 62, 12462–12470. [Google Scholar] [CrossRef]
- Warth, B.; Parich, A.; Atehnkeng, J.; Bandyopadhyay, R.; Schuhmacher, R.; Sulyok, M.; Krska, R. Quantitation of mycotoxins in food and feed from Burkina Faso and Mozambique using a modern LC-MS/MS multitoxin method. J. Agric. Food Chem. 2012, 60, 9352–9363. [Google Scholar] [CrossRef] [PubMed]
- Streit, E.; Schwab, C.; Sulyok, M.; Naehrer, K.; Krska, R.; Schatzmayr, G. Multi-mycotoxin screening reveals the occurrence of 139 different secondary metabolites in feed and feed ingredients. Toxins 2013, 5, 504–523. [Google Scholar] [CrossRef] [PubMed]
- Alassane-Kpembi, I.; Schatzmayr, G.; Taranu, I.; Marin, D.; Puel, O.; Oswald, I.P. Mycotoxins co-contamination: Methodological aspects and biological relevance of combined toxicity studies. Crit. Rev. Food Sci. Nutr. 2017, 57, 3489–3507. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.-C.; Madec, S.; Coton, E.; Hymery, N. Natural co-occurrence of mycotoxins in foods and feeds and their in vitro combined toxicological effects. Toxins 2016, 8, 94. [Google Scholar] [CrossRef] [PubMed]
- Di Marino, D.; D‘Annessa, I.; Coletta, A.; Via, A.; Tramontano, A. Characterization of the differences in the cyclopiazonic acid binding mode to mammalian and P. Falciparum Ca2+ pumps: A computational study. Proteins Struct. Funct. Bioinform. 2015, 83, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Anjorin, S.T.; Fapohunda, S.; Sulyok, M.; Krska, R. Natural Co-occurrence of Emerging and Minor Mycotoxins on Maize Grains from Abuja, Nigeria. Ann. Agric. Environ. Sci. 2016, 1, 1. [Google Scholar]
- Ogara, I.M.; Zarafi, A.B.; Alabi, O.; Banwo, O.; Ezekiel, C.N.; Warth, B.; Sulyok, M.; Krska, R. Mycotoxin patterns in ear rot infected maize: A comprehensive case study in Nigeria. Food Control 2017, 73, 1159–1168. [Google Scholar] [CrossRef]
- Romera, D.; Mateo, E.M.; Mateo-Castro, R.; Gomez, J.V.; Gimeno-Adelantado, J.V.; Jimenez, M. Determination of multiple mycotoxins in feedstuffs by combined use of UPLC–MS/MS and UPLC–QTOF–MS. Food Chem. 2018, 267, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Antignac, J.-P.; Le Bizec, B.; Monteau, F.; Andre, F. Validation of analytical methods based on mass spectrometric detection according to the “2002/657/EC” European decision: Guideline and application. Anal. Chim. Acta 2003, 483, 325–334. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Mycotoxin | Products | a Regulated Maximum Level (mg/kg) Relative to a Feedingstuff | b Guidance Value (mg/kg) Relative to a Feedingstuff | c Guidance Value in mg/kg (ppm) Relative to a Feedingstuff with a Moisture Content of 12 % |
|---|---|---|---|---|
| AFB1 | All feed materials; complete feedingstuffs for pigs and poultry (except young animals); complementary feedingstuffs for cattle, sheep and goats (except complementary feedingstuffs for dairy animals, calves and lambs); complementary feedingstuffs for pigs and poultry (except young animals); complete feedingstuffs for cattle, sheep and goats with the exception of: | 0.02 | ||
| complete feedingstuffs for dairy animals | 0.005 | |||
| complete feedingstuffs for calves and lambs | 0.01 | |||
| DON | Maize by-products | 12 | ||
| Other cereals and cereal products | 8 | |||
| Complementary and complete feedingstuffs with the exception of: | 5 | |||
| complementary and complete feedingstuffs for pigs | 0.9 | |||
| complementary and complete feedingstuffs for calves (<4 months), lambs and kids | 2 | |||
| ZEN | Maize by-products | 2 | ||
| Other cereals and cereal products | 3 | |||
| Complementary and complete feedingstuffs for piglets and young sows | 0.1 | |||
| Complementary and complete feedingstuffs for sows and fattening pigs | 0.25 | |||
| Complementary and complete feedingstuffs for calves, dairy cattle, sheep and goats | 0.50 | |||
| OTA | Cereals and cereal products | 0.25 | ||
| Complementary and complete feedingstuffs for pigs | 0.05 | |||
| Complementary and complete feedingstuffs for poultry | 0.1 | |||
| FBs | Maize and maize products | 60 | ||
| Complementary and complete feedingstuffs for pigs, horses, rabbits and pet animals | 5 | |||
| Complementary and complete feedingstuffs for fish | 10 | |||
| Complementary and complete feedingstuffs for poultry, calves (<4 months), lambs and kids | 20 | |||
| Complementary and complete feedingstuffs for adult ruminants (>4 months) and mink | 50 | |||
| T-2 + HT-2 toxin | Compound feed for cats | 0.05 | ||
| Mycotoxins | Retention Time (min) | Elemental Composition | Adduct Ion | Theoretical Mass (m/z) | Product Ion | Collision Energy (eV) |
|---|---|---|---|---|---|---|
| AFB1 | 4.64 | C17H12O6 | [M + H]+ | 313.07066 | 285.07489; 269.04373 | 36 |
| AFB2 | 4.98 | C17H14O6 | [M + H]+ | 315.08631 | 287.09064; 259.05945 | 36 |
| AFG1 | 4.79 | C17H12O7 | [M + H]+ | 329.06558 | 243.06467; 200.04640 | 40 |
| AFG2 | 4.61 | C17H14O7 | [M + H]+ | 331.08123 | 313.07010; 245.08032 | 37 |
| OTA | 6.50 | C20H18NO6Cl | [M + H]+ | 404.08954 | 358.08304; 341.05658 | 16 |
| FB1 | 6.03 | C34H59NO15 | [M + H]+ | 722.39575 | 352.32010, 334.30963 | 48 |
| FB2 | 6.78 | C34H59NO14 | [M + H]+ | 706.40083 | 336.32547; 318.31488 | 58 |
| DON | 4.18 | C15H20O6 | [M + HCOOH]− | 341.12451 | 295.1189; 265.10822 | −12 |
| 3-ADON | 3.83 | C17H22O7 | [M + H]+ | 339.14383 | 231.10118; 203.10638 | 20 |
| 15-ADON | 4.02 | C17H22O7 | [M + H]+ | 339.14383 | 261.11154; 137.05957 | 20 |
| HT-2 | 5.63 | C22H32O8 | [M + NH4]+ | 442.24354 | 263.12744; 215.10641 | 27 |
| T-2 | 6.13 | C24H34O9 | [M + NH4]+ | 484.25411 | 215.10603; 185.09561 | 23 |
| NEO | 4.32 | C19H26O8 | [M + NH4]+ | 400.19659 | 305.13803; 141.0053 | 10 |
| DAS | 5.11 | C19H26O7 | [M + NH4]+ | 384.20168 | 307.15329; 105.06977 | 15 |
| FUS-X | 4.28 | C17H22O8 | [M + Na]+ | 377.12073 | 228.16002; 175.07550 | 20 |
| ZEN | 6.55 | C18H22O5 | [M − H]− | 317.13945 | 175.03989; 131.05008 | −32 |
| α-ZEL | 4.87 | C18H24O5 | [M − H]− | 319.15510 | 174.95630; 129.01947 | 36 |
| β-ZEL | 4.98 | C18H24O5 | [M − H]− | 319.15510 | 174.95604; 160.97665 | 36 |
| α-ZAL | 4.81 | C18H26O5 | [M − H]− | 321.17044 | 259.09497; 91.00272 | 29 |
| β-ZAL | 4.94 | C18H26O5 | [M − H]− | 321.17044 | 259.09497; 91.00272 | 40 |
| ZAN | 5.00 | C18H24O5 | [M − H]− | 319.15510 | 273.01187; 131.05020 | 35 |
| BEA | 5.77 | C45H57N3O9 | [M + NH4]+ | 801.44331 | 262.76715; 244.18239 | 70 |
| ENN A | 8.17 | C36H63N3O9 | [M + NH4]+ | 699.49026 | 228.15900; 210.14847 | 43 |
| ENN A1 | 8.16 | C35H61N3O9 | [M + NH4]+ | 685.47461 | 228.15900; 210.14847 | 48 |
| ENN B | 7.87 | C33H57N3O9 | [M + NH4]+ | 657.44331 | 214.14320; 196.13280 | 50 |
| ENN B1 | 8.06 | C34H59N3O9 | [M + NH4]+ | 671.45986 | 214.14343; 196.13295 | 48 |
| AOH | 5.88 | C14H10O5 | [M − H]− | 257.04555 | 215.03490; 213.05569 | −32 |
| AME | 6.82 | C15H12O5 | [M − H]− | 271.06120 | 256.03751; 228.04276 | −36 |
| Analyzed Samples (n) | Analytes Investigated | Mycotoxins | Positive Samples (%) | a Range or Average (μg/kg) | Detection Methods | LOQ (μg/kg) | Reference |
|---|---|---|---|---|---|---|---|
| 89 | 28 | AFB1 | 25.8 | 3.3–7.9 | UHPLC-Q-Orbitrap | 0.013 | This work |
| AFB2 | 5.6 | 1.8–16.6 | 0.013 | ||||
| AFG1 | 1.1 | 11.1 | |||||
| AFG2 | 5.6 | 1.7–31.6 | 0.125 | ||||
| OTA | 2.2 | 1.4–1.5 | 1.25 | ||||
| ZEN | 91.0 | 0.9–60.6 | 0.013 | ||||
| Σ α + β-ZEL | 87.6 | 0.9–58.9 | α-ZEL = 1.25; β-ZEL = 0.125 | ||||
| Σ α + β -ZAL | 79.8 | <LOQ | α-ZAL = 1.25; β-ZAL = 0.125 | ||||
| ZAN | n.f. | n.f. | 0.125 | ||||
| DON | 30.3 | 7.6–297.3 | 1.25 | ||||
| Σ 3 + 15 AcDON | 5.6 | 10.9–63.2 | 3-AcDON = 1.25; 15-AcDON = 1.25 | ||||
| NEO | n.f. | n.f. | 0.188 | ||||
| HT2 | 32.6 | 3.3–110.1 | 1.25 | ||||
| T2 | 47.2 | 0.7–9.0 | 0.125 | ||||
| BEA | 86.5 | 0.8–176.1 | 0.013 | ||||
| ENNA | 10.1 | 0.3–9.6 | 0.125 | ||||
| ENNA1 | 22.5 | 0.4–28.1 | 0.125 | ||||
| ENNB | 93.3 | 0.4–212.4 | 0.125 | ||||
| ENNB1 | 58.4 | 0.3–71.8 | 0.013 | ||||
| AOH | 82.0 | 0.2–12.8 | 0.125 | ||||
| AME | 84.3 | 0.1–15.6 | 0.125 | ||||
| FB1 | 66.3 | 11.8–990.1 | 0.125 | ||||
| FB2 | 52.8 | 10.5–556.3 | 0.250 | ||||
| DAS | n.f. | n.f. | |||||
| FUS-X | n.f. | n.f. | |||||
| 48 | 5 | ENNA | n.a. | n.a. | LC-MS/MS | 5 | Tolosa et al., 2019 [16] |
| ENNA1 | 41.5 | 8.1–11.9 | 1 | ||||
| ENNB | 89 | 2.0–89.5 | 1 | ||||
| ENNB1 | 64 | 7.4–28.8 | 1 | ||||
| BEA | 62 | 4.6–129.6 | 5 | ||||
| 32 | 8 | AFB1 | 47.7 | 30.3–242.7 | LC-MS/MS | 1.7 | Shao et al., 2018 [29] |
| AFG1 | 13.9 | 13.9 | 0.7 | ||||
| OTA | 16.2 | 15.1–17.3 | 10.7 | ||||
| ZEN | 54.5 | 14.5–389.2 | 2.5 | ||||
| DON | 66.3 | 22.8–421.3 | 16.5 | ||||
| T-2 | 15.4 | 15.4 | 3.3 | ||||
| BEA | 19.1 | 0.2–153.4 | 2.5 | ||||
| FB1 | 87.2 | 6.6–191.9 | 10.0 | ||||
| 12 | 6 | AFB1 | n.a. | 83.3 | HPLC-FLD | 41.57 | Singh et al., 2017a [24] |
| AFB2 | n.a. | 9.0 | 11.77 | ||||
| OTA | n.a. | 1.0 | - | ||||
| ZEN | n.a. | 5.7 | - | ||||
| FB1 | n.a. | 106.3 | 202.53 | ||||
| FB2 | n.a. | 61.9 | 118.37 | ||||
| 49 | 3 | AFs | 100 | 0.16–5.39 | HPLC-FLD | AFB1 = 0.13; AFB2 = 0.59; AFG1 = 0.03; AFG2 = 0.22 | Teixeira et al., 2017 [25] |
| ZEN | 95.9 | 4.07–98.3 | 3.95 | ||||
| FBs | 77.6 | 37.4–1015 | FB1 = 27.5; FB2 = 35.3 | ||||
| 100 | 4 | AFLs | 68 | 0.34–3.88 | HPLC-FLD | B1=0.13; G1 = 0.03; B2 = 0.59; G2 = 0.22 | Bissoqui et al., 2016 [23] |
| ZEN | 95 | 5.45–442.2 | 3.95 | ||||
| FB1 | 68 | 20.0–220 | 27.5 | ||||
| FB2 | 35 | 40.0–160 | 35.3 | ||||
| 20 | 3 | AFs | n.a. | n.a. | ELISA-UV | 5 | Yasmina et al., 2016 [21] |
| AFB1 | 15 | 2.6–18.4 | 1 | ||||
| OTA | 70 | 2.62–6.65 | 2.5 | ||||
| ZEN | 20 | 148–1170 | 1.75 | ||||
| 49 | 2 | AFB1 | 8.2 | <0.05–0.21 | HPLC-FLD | 0.15 | Błajet-Kosicka et al., 2014 [22] |
| OTA | 46.9 | <0.13–3 | 0.40 | ||||
| 5 | DON | 100 | 22.7–436 | LC-MS/MS | 20.0 | ||
| T-2 | 87.7 | <0.5–13.3 | 1.50 | ||||
| HT-2 | 83.7 | <1.60–19.6 | 5.00 | ||||
| ZEN | 100 | 1.81–123 | 0.30 | ||||
| 7 | FBs | 28.6 | <5–108 | FB1 = 1.60; FB2 = 1.60; FB3 = 1.60 | |||
| 76 | 4 | DON | 97 | >250 | ELISA-UV | - | Böhm et al., 2010 [30] |
| OTA | 5 | 3.5 | - | ||||
| ZEN | 47 | 80 | - | ||||
| FBs | 42 | 178 | - | ||||
| 29 | 3 | DON | 83 | 409 | HPLC-FLD | 25 | |
| 22 | ZEN | 68 | 185 | 20 | |||
| 3 | FBs | 67 | 69 | 15 | |||
| 180 | 1 | AFB1 | 70.5 | 0.3–9.43 | HPLC-FLD | 0.1 | Campos et al., 2008 [31] |
| Composition (%) | Dog (n = 55) | Cat (n = 34) |
|---|---|---|
| Proteins | 25.8 ± 5.2 | 33.8 ± 3.8 |
| Fats | 14.9 ± 3.4 | 15.2 ± 4.4 |
| Fibers | 6.8 ± 1.2 | 7.5 ± 1.1 |
| Total minerals | 3.0 ± 2.1 | 3.9 ± 2.9 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castaldo, L.; Graziani, G.; Gaspari, A.; Izzo, L.; Tolosa, J.; Rodríguez-Carrasco, Y.; Ritieni, A. Target Analysis and Retrospective Screening of Multiple Mycotoxins in Pet Food Using UHPLC-Q-Orbitrap HRMS. Toxins 2019, 11, 434. https://doi.org/10.3390/toxins11080434
Castaldo L, Graziani G, Gaspari A, Izzo L, Tolosa J, Rodríguez-Carrasco Y, Ritieni A. Target Analysis and Retrospective Screening of Multiple Mycotoxins in Pet Food Using UHPLC-Q-Orbitrap HRMS. Toxins. 2019; 11(8):434. https://doi.org/10.3390/toxins11080434
Chicago/Turabian StyleCastaldo, Luigi, Giulia Graziani, Anna Gaspari, Luana Izzo, Josefa Tolosa, Yelko Rodríguez-Carrasco, and Alberto Ritieni. 2019. "Target Analysis and Retrospective Screening of Multiple Mycotoxins in Pet Food Using UHPLC-Q-Orbitrap HRMS" Toxins 11, no. 8: 434. https://doi.org/10.3390/toxins11080434
APA StyleCastaldo, L., Graziani, G., Gaspari, A., Izzo, L., Tolosa, J., Rodríguez-Carrasco, Y., & Ritieni, A. (2019). Target Analysis and Retrospective Screening of Multiple Mycotoxins in Pet Food Using UHPLC-Q-Orbitrap HRMS. Toxins, 11(8), 434. https://doi.org/10.3390/toxins11080434