Metabolic Inflammation-Differential Modulation by Dietary Constituents

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Mechanisms of Metabolic-Inflammation in Adipose Tissue
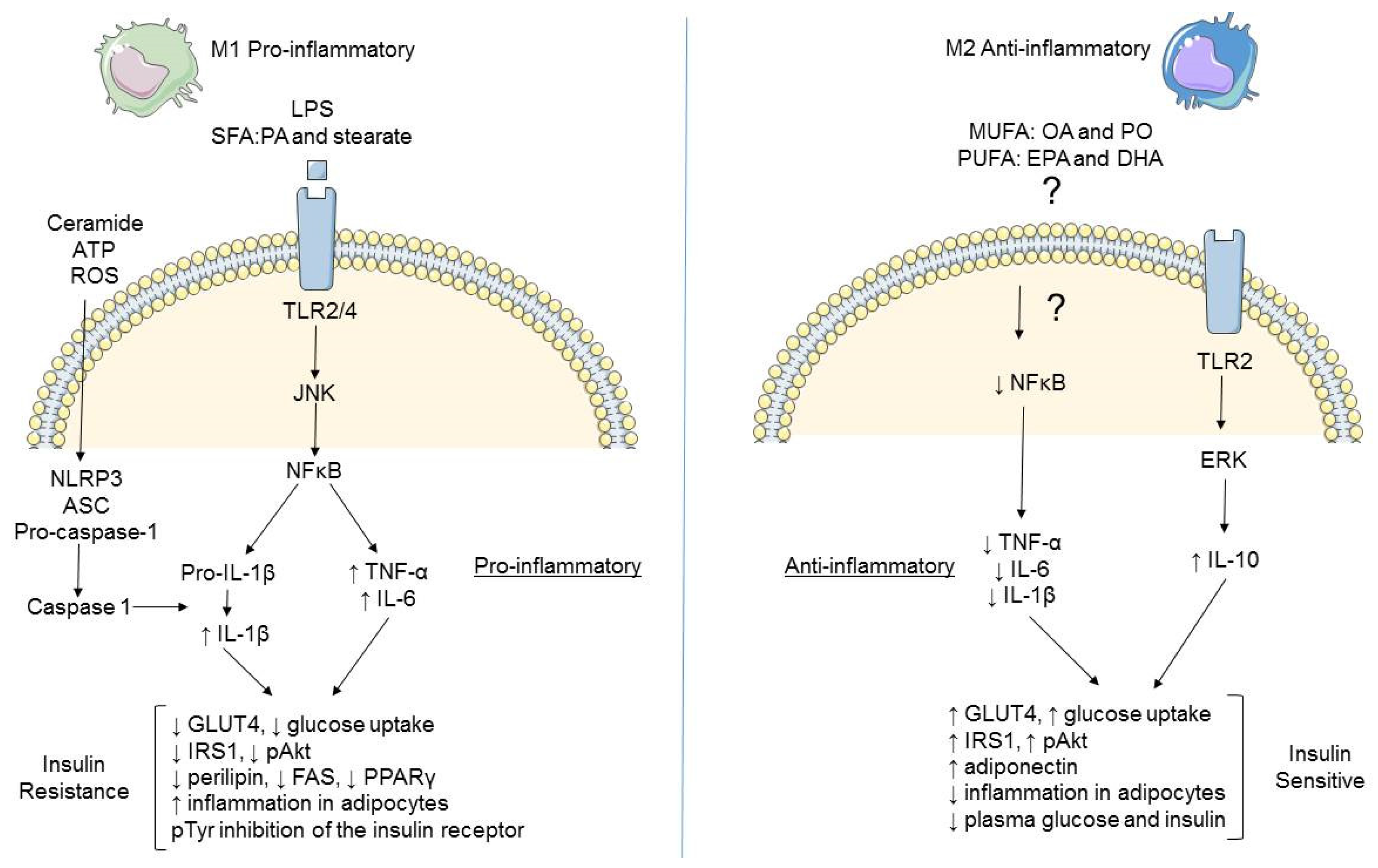
2.1. Immune Cell Infiltration
2.2. Adipose Tissue Macrophages (ATM)
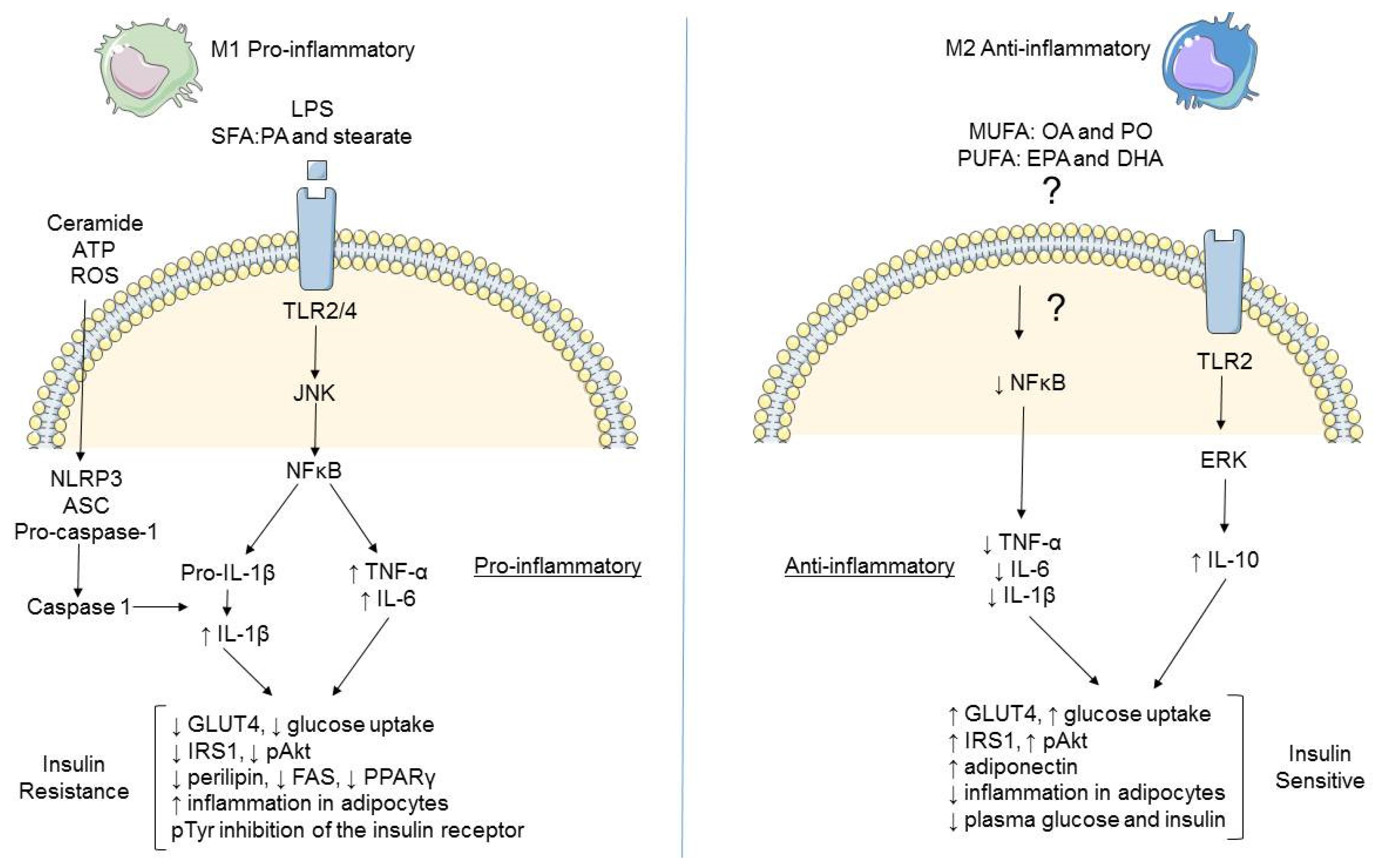
2.3. Differential Modulation of Inflammatory Mediators in Obesity
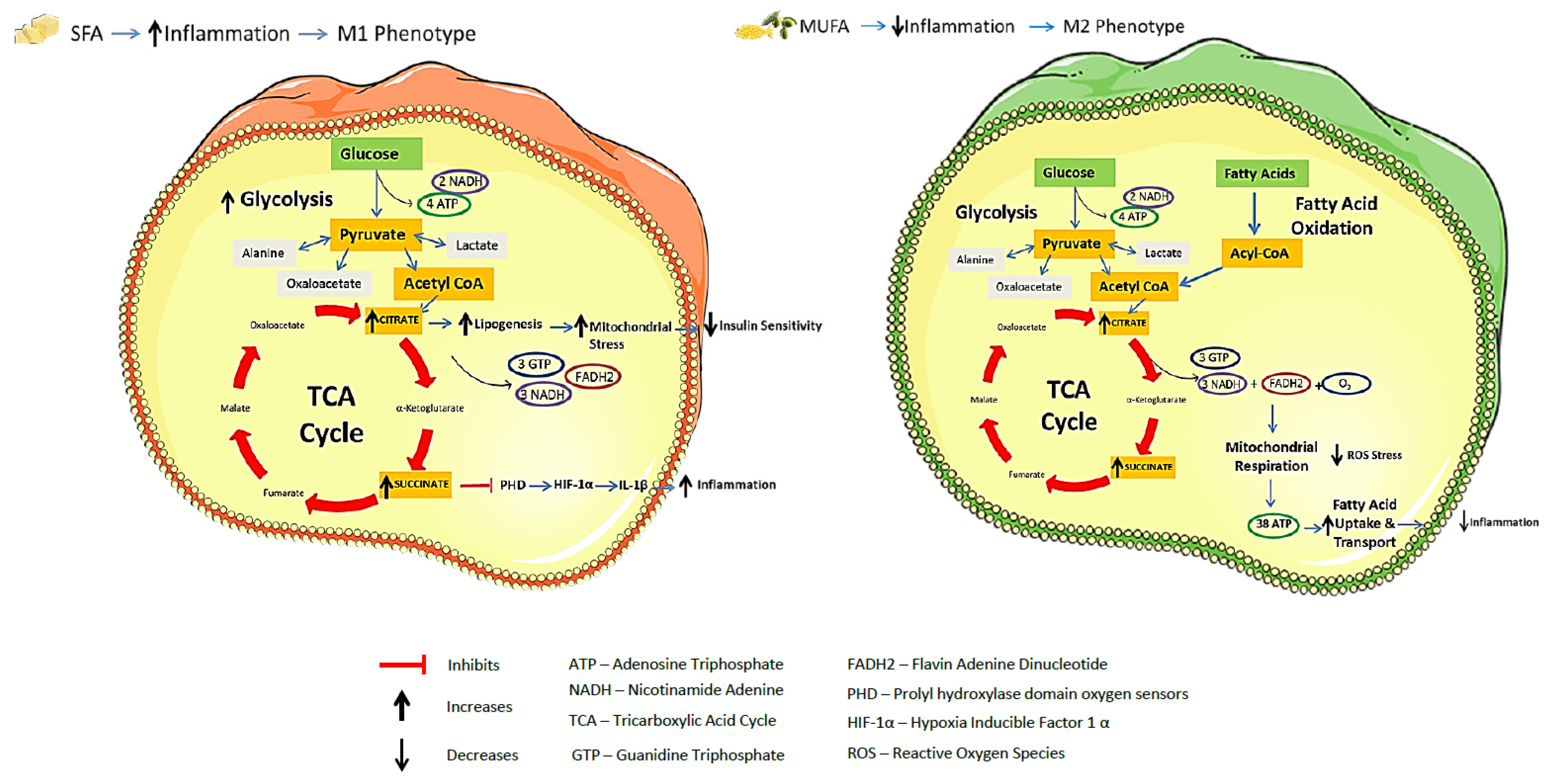
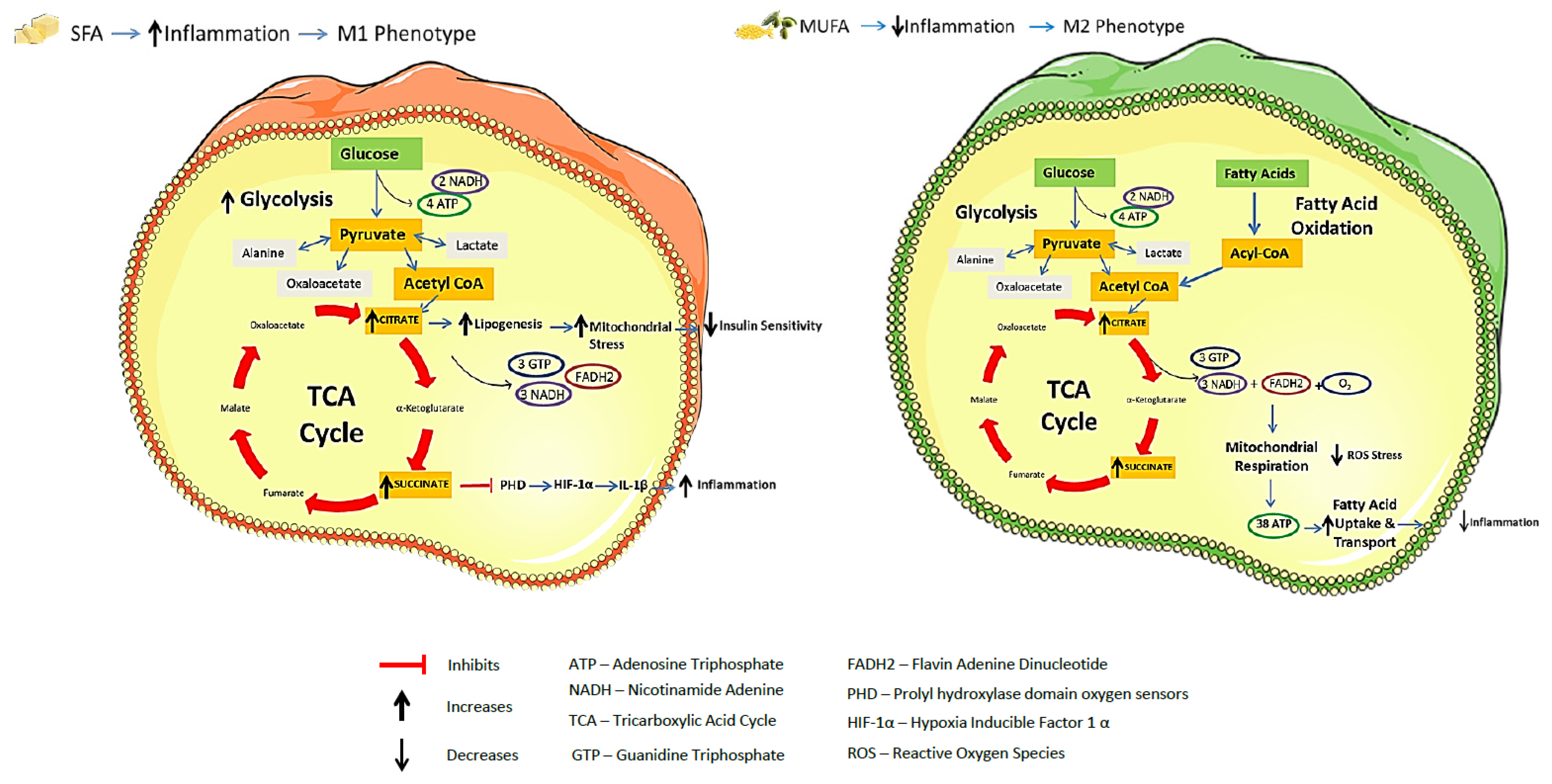
2.4. Integration of Metabolism and Immune Responses
2.5. Role of AMPK in Metabolic Inflammation
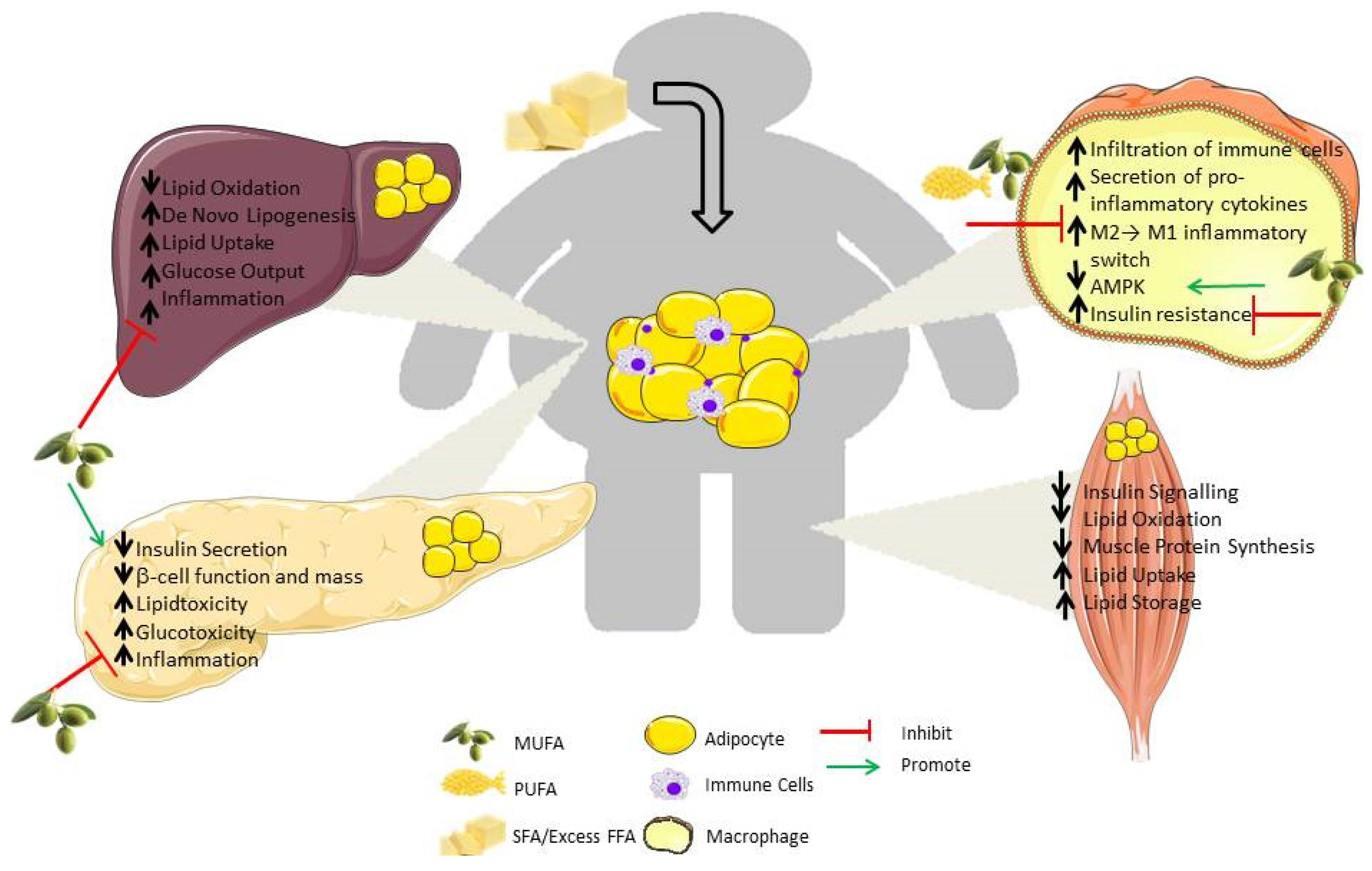
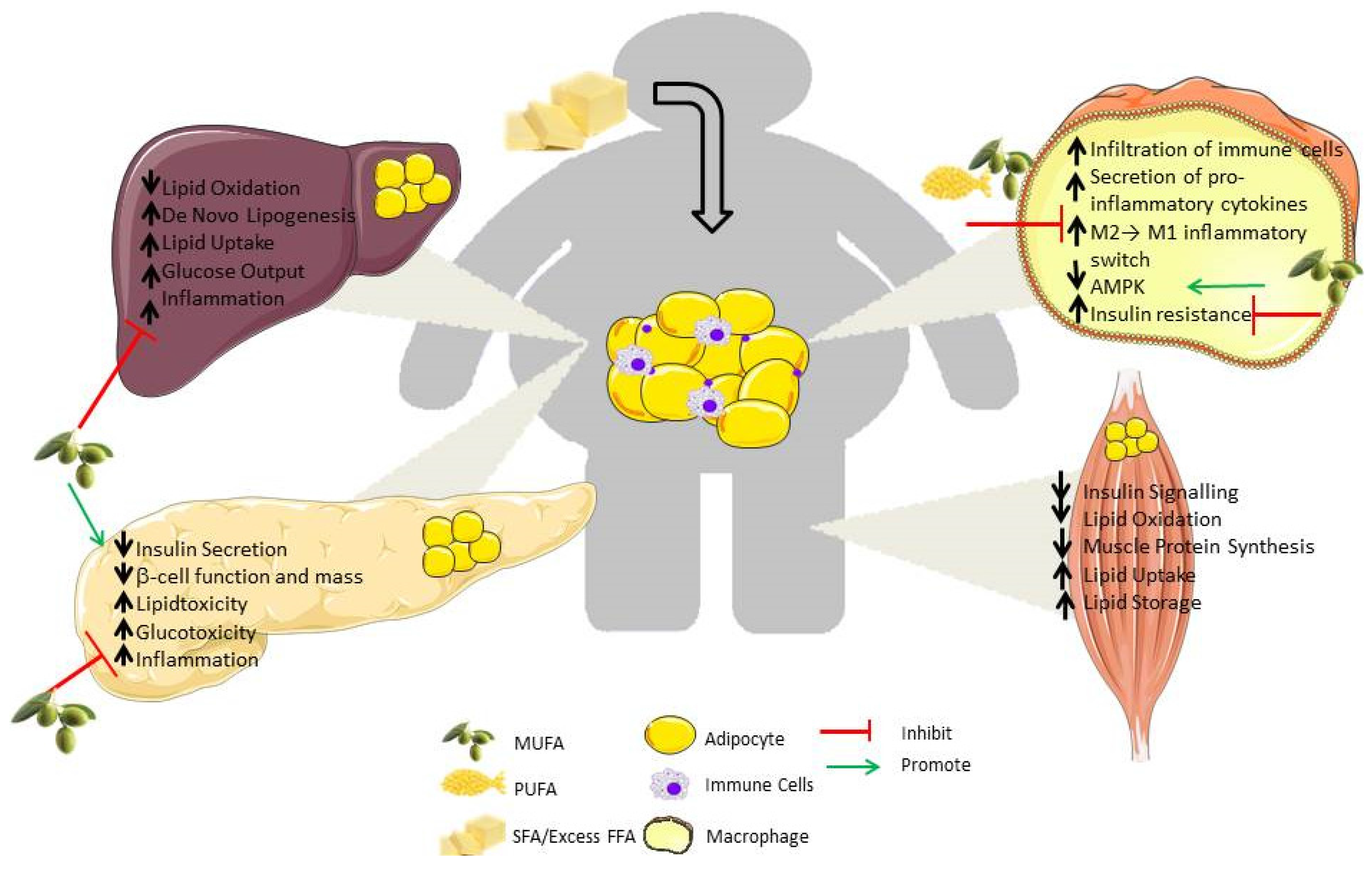
3. Consequence of “Meta-Inflammation” in Metabolic Organs
3.1. Liver
3.2. Muscle
3.3. Pancreas
4. Therapies to Improve Inflammation and Metabolic Health
4.1. Pharmaceuticals That Target Metabolic Inflammation
4.2. Weight Loss and Improved Insulin Sensitivity
4.3. Manipulation of Dietary Fat
4.4. Supplements/Functional Foods
5. Future Perspectives and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef] [PubMed]
- Van Herpen, N.A.; Schrauwen-Hinderling, V.B. Lipid accumulation in non-adipose tissue and lipotoxicity. Physiol. Behav. 2008, 94, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Krahmer, N.; Farese, R.V.; Walther, T.C. Balancing the fat: Lipid droplets and human disease. EMBO Mol. Med. 2013, 5, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Servier Medical Art. Available online: http://www.servier.fr/servier-medical-art (accessed on 15 January 2016).
- Stienstra, R.; Joosten, L.A.B.; Koenen, T.; van Tits, B.; van Diepen, J.A.; van den Berg, S.A.A.; Rensen, P.C.N.; Voshol, P.J.; Fantuzzi, G.; Hijmans, A.; et al. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab. 2010, 12, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Finucane, O.M.; Lyons, C.L.; Murphy, A.M.; Reynolds, C.M.; Klinger, R.; Healy, N.P.; Cooke, A.A.; Coll, R.C.; McAllan, L.; Nilaweera, K.N.; et al. Monounsaturated fatty acid-enriched high-fat diets impede adipose NLRP3 inflammasome-mediated IL-1β secretion and insulin resistance despite obesity. Diabetes 2015, 64, 2116–2128. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Divoux, A.; Sun, J.; Zhang, J.; Clément, K.; Glickman, J.N.; Sukhova, G.K.; Wolters, P.J.; Du, J.; Gorgun, C.Z.; et al. Genetic deficiency and pharmacological stabilization of mast cells reduce diet-induced obesity and diabetes in mice. Nat. Med. 2009, 15, 940–945. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, S.; Manabe, I.; Nagasaki, M.; Eto, K.; Yamashita, H.; Ohsugi, M.; Otsu, M.; Hara, K.; Ueki, K.; Sugiura, S.; et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat. Med. 2009, 15, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.S.; Prakken, B.; Kalkhoven, E.; Boes, M. Adipose tissue-resident immune cells: Key players in immunometabolism. Trends Endocrinol. Metab. 2012, 23, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Cinti, S. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J. Lipid Res. 2005, 46, 2347–2355. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Kamei, N.; Tobe, K.; Suzuki, R.; Ohsugi, M.; Watanabe, T.; Kubota, N.; Ohtsuka-Kowatari, N.; Kumagai, K.; Sakamoto, K.; Kobayashi, M.; et al. Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J. Biol. Chem. 2006, 281, 26602–26614. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.M.; Perrard, X.-Y.; Brunner, G.; Lui, H.; Sparks, L.M.; Smith, S.R.; Wang, X.; Shi, Z.-Z.; Lewis, D.E.; Wu, H.; et al. Intermuscular and perimuscular fat expansion in obesity correlates with skeletal muscle T cell and macrophage infiltration and insulin resistance. Int. J. Obes. 2015, 39, 1607–1618. [Google Scholar] [CrossRef] [PubMed]
- Morinaga, H.; Mayoral, R.; Heinrichsdorff, J.; Osborn, O.; Franck, N.; Hah, N.; Walenta, E.; Bandyopadhyay, G.; Pessentheiner, A.R.; Chi, T.J.; et al. Characterization of distinct subpopulations of hepatic macrophages in HFD/obese mice. Diabetes 2015, 64, 1120–1130. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, C.M.; McGillicuddy, F.C.; Harford, K.A.; Finucane, O.M.; Mills, K.H.G.; Roche, H.M. Dietary saturated fatty acids prime the NLRP3 inflammasome via TLR4 in dendritic cells-implications for diet-induced insulin resistance. Mol. Nutr. Food Res. 2012, 56, 1212–1222. [Google Scholar] [CrossRef] [PubMed]
- Feuerer, M.; Herrero, L.; Cipolletta, D.; Naaz, A.; Wong, J.; Nayer, A.; Lee, J.; Goldfine, A.B.; Benoist, C.; Shoelson, S.; et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 2009, 15, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Cancello, R.; Henegar, C.; Viguerie, N.; Taleb, S.; Poitou, C.; Rouault, C.; Coupaye, M.; Pelloux, V.; Hugol, D.; Bouillot, J.-L.; et al. Reduction of Macrophage Infiltration and Chemoattractant Gene Expression Changes in White Adipose Tissue of Morbidly Obese Subjects after Surgery-Induced Weight Loss. Diabetes 2005, 54, 2277–2286. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.L.; Pillon, N.J.; Sivaloganathan, D.M.; Costford, S.R.; Liu, Z.; Théret, M.; Chazaud, B.; Klip, A. Palmitoleate Reverses High Fat-induced Proinflammatory Macrophage Polarization via AMP-activated Protein Kinase (AMPK). J. Biol. Chem. 2015, 290, 16979–16988. [Google Scholar] [CrossRef] [PubMed]
- Kratz, M.; Coats, B.R.; Hisert, K.B.; Hagman, D.; Mutskov, V.; Peris, E.; Schoenfelt, K.Q.; Kuzma, J.N.; Larson, I.; Billing, P.S.; et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. 2014, 20, 614–625. [Google Scholar] [CrossRef] [PubMed]
- McGillicuddy, F.C.; Harford, K.A.; Reynolds, C.M.; Oliver, E.; Claessens, M.; Mills, K.H.G.; Roche, H.M. Lack of interleukin-1 receptor I (IL-1RI) protects mice from high-fat diet-induced adipose tissue inflammation coincident with improved glucose homeostasis. Diabetes 2011, 60, 1688–1698. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.; Shargill, N.; Spiegelman, B. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Suganami, T.; Tanimoto-Koyama, K.; Nishida, J.; Itoh, M.; Yuan, X.; Mizuarai, S.; Kotani, H.; Yamaoka, S.; Miyake, K.; Aoe, S.; et al. Role of the Toll-like receptor 4/NF-kappaB pathway in saturated fatty acid-induced inflammatory changes in the interaction between adipocytes and macrophages. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Budavari, A.; Murray, D.; Spiegelman, B.M. Reduced tyrosine kinase activity of the insulin receptor in obesity-diabetes. Central role of tumor necrosis factor-alpha. J. Clin. Investig. 1994, 94, 1543–1549. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.-H.; Brickey, W.J.; Ting, J.P.-Y. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Koenen, T.B.; Stienstra, R.; van Tits, L.J.; Joosten, L.A.B.; van Velzen, J.F.; Hijmans, A.; Pol, J.A.; van der Vliet, J.A.; Netea, M.G.; Tack, C.J.; et al. The inflammasome and caspase-1 activation: A new mechanism underlying increased inflammatory activity in human visceral adipose tissue. Endocrinology 2011, 152, 3769–3778. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.W.; Hsu, K.C.; Lee, J.-W.; Ham, M.; Huh, J.Y.; Shin, H.J.; Kim, W.S.; Kim, J.B. Berberine suppresses proinflammatory responses through AMPK activation in macrophages. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E955–E964. [Google Scholar] [CrossRef] [PubMed]
- Weldon, S.M.; Mullen, A.C.; Loscher, C.E.; Hurley, L.A.; Roche, H.M. Docosahexaenoic acid induces an anti-inflammatory profile in lipopolysaccharide-stimulated human THP-1 macrophages more effectively than eicosapentaenoic acid. J. Nutr. Biochem. 2007, 18, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Oliver, E.; McGillicuddy, F.C.; Harford, K.A.; Reynolds, C.M.; Phillips, C.M.; Ferguson, J.F.; Roche, H.M. Docosahexaenoic acid attenuates macrophage-induced inflammation and improves insulin sensitivity in adipocytes-specific differential effects between LC n-3 PUFA. J. Nutr. Biochem. 2012, 23, 1192–1200. [Google Scholar] [CrossRef] [PubMed]
- Anthony, J.S.; Karnovskyi, M.L. The Biochemical Basis of Phagocytosis. J. Biol. Chem. 1959, 234, 1355–1362. [Google Scholar]
- Warburg, O.; Wind, F.; Negelein, E.I. Killing-Off of Tumor Cells in vitro. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Hard, G.C. Some biochemical aspects of the immune macrophage. Br. J. Exp. Pathol. 1970, 51, 97–105. [Google Scholar] [PubMed]
- Palsson-McDermott, E.M.; Curtis, A.M.; Goel, G.; Lauterbach, M.A.R.; Sheedy, F.J.; Gleeson, L.E.; van den Bosch, M.W.M.; Quinn, S.R.; Domingo-Fernandez, R.; Johnston, D.G.W.; et al. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 2015, 21, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.K.; Huang, S.C.-C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network Integration of Parallel Metabolic and Transcriptional Data Reveals Metabolic Modules that Regulate Macrophage Polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Norata, G.D.; Caligiuri, G.; Chavakis, T.; Matarese, G.; Netea, M.G.; Nicoletti, A.; O’Neill, L.A.J.; Marelli-Berg, F.M. The Cellular and Molecular Basis of Translational Immunometabolism. Immunity 2015, 43, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Sadagopan, N.; Li, W.; Roberds, S.L.; Major, T.; Preston, G.M.; Yu, Y.; Tones, M.A. Circulating succinate is elevated in rodent models of hypertension and metabolic disease. Am. J. Hypertens. 2007, 20, 1209–1215. [Google Scholar] [PubMed]
- Vats, D.; Mukundan, L.; Odegaard, J.I.; Zhang, L.; Smith, K.L.; Morel, C.R.; Greaves, D.R.; Murray, P.J.; Chawla, A. Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation. Cell Metab. 2006, 4, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. The AMP-activated protein kinase pathway—New players upstream and downstream. J. Cell Sci. 2004, 117, 5479–5487. [Google Scholar] [CrossRef] [PubMed]
- Galic, S.; Fullerton, M.D.; Schertzer, J.D.; Sikkema, S.; Marcinko, K.; Walkley, C.R.; Izon, D.; Honeyman, J.; Chen, Z.-P.; van Denderen, B.J.; et al. Hematopoietic AMPK β1 reduces mouse adipose tissue macrophage inflammation and insulin resistance in obesity. J. Clin. Investig. 2011, 121, 4903–4915. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Wahl, D.R.; Byersdorfer, C.A.; Ferrara, J.L.M.; Opipari, A.W.; Glick, G.D. Distinct metabolic programs in activated T cells: Opportunities for selective immunomodulation. Immunol. Rev. 2012, 249, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Green, D.R. Metabolic reprogramming and metabolic dependency in T cells. Immunol. Rev. 2012, 249, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.; Denanglaire, S.; Viollet, B.; Leo, O.; Andris, F. AMP-activated protein kinase regulates lymphocyte responses to metabolic stress but is largely dispensable for immune cell development and function. Eur. J. Immunol. 2008, 38, 948–956. [Google Scholar] [CrossRef] [PubMed]
- Lord, G.M.; Matarese, G.; Howard, J.K.; Baker, R.J.; Bloom, S.R.; Lechler, R.I. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature 1998, 394, 897–901. [Google Scholar] [PubMed]
- Lee, J.; Walsh, M.C.; Hoehn, K.L.; James, D.E.; Wherry, E.J.; Choi, Y. Regulator of Fatty Acid Metabolism, Acetyl Coenzyme A Carboxylase 1, Controls T Cell Immunity. J. Immunol. 2014, 192, 3190–3199. [Google Scholar] [CrossRef] [PubMed]
- Knowler, W.C.; Barrett-Connor, E.; Fowler, S.E.; Hamman, R.F.; Lachin, J.M.; Walker, E.A.; Nathan, D.M. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N. Engl. J. Med. 2002, 346, 393–403. [Google Scholar] [PubMed]
- Gauthier, M.-S.; O’Brien, E.L.; Bigornia, S.; Mott, M.; Cacicedo, J.M.; Xu, X.J.; Gokce, N.; Apovian, C.; Ruderman, N. Decreased AMP-activated protein kinase activity is associated with increased inflammation in visceral adipose tissue and with whole-body insulin resistance in morbidly obese humans. Biochem. Biophys. Res. Commun. 2011, 404, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Cacicedo, J.M.; Yagihashi, N.; Keaney, J.F.; Ruderman, N.B.; Ido, Y. AMPK inhibits fatty acid-induced increases in NF-kappaB transactivation in cultured human umbilical vein endothelial cells. Biochem. Biophys. Res. Commun. 2004, 324, 1204–1209. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Kahn, B.B.; Shi, H.; Xue, B.Z. Macrophageα1 AMP-activated protein kinase (α1AMPK) antagonizes fatty acid-induced inflammation through SIRT1. J. Biol. Chem. 2010, 285, 19051–19059. [Google Scholar] [CrossRef] [PubMed]
- Bechmann, L.P.; Hannivoort, R.A.; Gerken, G.; Hotamisligil, G.S.; Trauner, M.; Canbay, A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J. Hepatol. 2012, 56, 952–964. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A. Insulin resistance, lipotoxicity, type 2 diabetes and atherosclerosis: The missing links. The Claude Bernard Lecture 2009. Diabetologia 2010, 53, 1270–1287. [Google Scholar] [CrossRef] [PubMed]
- Unger, R.H.; Orci, L. Lipotoxic diseases of nonadipose tissues in obesity. Int. J. Obes. Relat. Metab. Disord. 2000, 24, S28–S32. [Google Scholar] [CrossRef] [PubMed]
- Tovar, A.R.; Torres, N. The role of dietary protein on lipotoxicity. Biochim. Biophys. Acta 2010, 1801, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Trauner, M.; Arrese, M.; Wagner, M. Fatty liver and lipotoxicity. Biochim. Biophys. Acta 2010, 1801, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Petersen, K.F.; Shulman, G.I. Lipid-induced insulin resistance: Unravelling the mechanism. Lancet 2010, 375, 2267–2277. [Google Scholar] [CrossRef]
- Cai, D.; Yuan, M.; Frantz, D.F.; Melendez, P.A.; Hansen, L.; Lee, J.; Shoelson, S.E. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat. Med. 2005, 11, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Klover, P.J.; Zimmers, T.A.; Koniaris, L.G.; Mooney, R.A. Chronic exposure to interleukin-6 causes hepatic insulin resistance in mice. Diabetes 2003, 52, 2784–2789. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology 2010, 52, 774–788. [Google Scholar] [CrossRef] [PubMed]
- Bettermann, K.; Hohensee, T.; Haybaeck, J. Steatosis and steatohepatitis: Complex disorders. Int. J. Mol. Sci. 2014, 15, 9924–9944. [Google Scholar] [CrossRef] [PubMed]
- Jarrar, M.H.; Baranova, A.; Collantes, R.; Ranard, B.; Stepanova, M.; Bennett, C.; Fang, Y.; Elariny, H.; Goodman, Z.; Chandhoke, V.; et al. Adipokines and cytokines in non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2008, 27, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.F.; Dufour, S.; Befroy, D.; Lehrke, M.; Hendler, R.E.; Shulman, G.I. Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes 2005, 54, 603–608. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Tripathy, D. Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 2009, 32, S157–S163. [Google Scholar] [CrossRef] [PubMed]
- Savage, D.B.; Petersen, K.F.; Shulman, G.I. Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol. Rev. 2007, 87, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.A.; Lillioja, S.; Kriketos, A.D.; Milner, M.R.; Baur, L.A.; Bogardus, C.; Jenkins, A.B.; Storlien, L.H. Skeletal Muscle Triglyceride Levels Are Inversely Related to Insulin Action. Diabetes 1997, 46, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Roden, M.; Price, T.B.; Perseghin, G.; Petersen, K.F.; Rothman, D.L.; Cline, G.W.; Shulman, G.I. Mechanism of free fatty acid-induced insulin resistance in humans. J. Clin. Investig. 1996, 97, 2859–2865. [Google Scholar] [CrossRef] [PubMed]
- Boden, G. Role of Fatty Acids in the Pathogenesis of Insulin Resistance and NIDDM. Diabetes 1997, 46, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Schmitz-Peiffer, C.; Craig, D.L.; Biden, T.J. Ceramide Generation Is Sufficient to Account for the Inhibition of the Insulin-stimulated PKB Pathway in C2C12 Skeletal Muscle Cells Pretreated with Palmitate. J. Biol. Chem. 1999, 274, 24202–24210. [Google Scholar] [CrossRef] [PubMed]
- Dresner, A.; Laurent, D.; Marcucci, M.; Griffin, M.E.; Dufour, S.; Cline, G.W.; Slezak, L.A.; Andersen, D.K.; Hundal, R.S.; Rothman, D.L.; et al. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J. Clin. Investig. 1999, 103, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Itani, S.I.; Ruderman, N.B.; Schmieder, F.; Boden, G. Lipid-Induced Insulin Resistance in Human Muscle Is Associated With Changes in Diacylglycerol, Protein Kinase C, and IκB-α. Diabetes 2002, 51, 2005–2011. [Google Scholar] [CrossRef] [PubMed]
- Hommelberg, P.P.H.; Plat, J.; Langen, R.C.J.; Schols, A.M.W.J.; Mensink, R.P. Fatty acid-induced NF-kappaB activation and insulin resistance in skeletal muscle are chain length dependent. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E114–E120. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Pratipanawatr, T.; Berria, R.; Wang, E.; DeFronzo, R.A.; Sullards, M.C.; Mandarino, L.J. Ceramide Content Is Increased in Skeletal Muscle From Obese Insulin-Resistant Humans. Diabetes 2003, 53, 25–31. [Google Scholar] [CrossRef]
- Proud, C.G. Regulation of protein synthesis by insulin. Biochem. Soc. Trans. 2006, 34, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Stephens, F.B.; Chee, C.; Wall, B.T.; Murton, A.J.; Shannon, C.E.; van Loon, L.J.C.; Tsintzas, K. Lipid-induced insulin resistance is associated with an impaired skeletal muscle protein synthetic response to amino acid ingestion in healthy young men. Diabetes 2015, 64, 1615–1620. [Google Scholar] [CrossRef] [PubMed]
- Guillet, C.; Delcourt, I.; Rance, M.; Giraudet, C.; Walrand, S.; Bedu, M.; Duche, P.; Boirie, Y. Changes in basal and insulin and amino acid response of whole body and skeletal muscle proteins in obese men. J. Clin. Endocrinol. Metab. 2009, 94, 3044–3050. [Google Scholar] [CrossRef] [PubMed]
- Masgrau, A.; Mishellany-Dutour, A.; Murakami, H.; Beaufrère, A.-M.; Walrand, S.; Giraudet, C.; Migné, C.; Gerbaix, M.; Metz, L.; Courteix, D.; et al. Time-course changes of muscle protein synthesis associated with obesity-induced lipotoxicity. J. Physiol. 2012, 590, 5199–5210. [Google Scholar] [CrossRef] [PubMed]
- Bensellam, M.; Laybutt, D.R.; Jonas, J.-C. The molecular mechanisms of pancreatic β-cell glucotoxicity: Recent findings and future research directions. Mol. Cell. Endocrinol. 2012, 364, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Fex, M.; Nitert, M.D.; Wierup, N.; Sundler, F.; Ling, C.; Mulder, H. Enhanced mitochondrial metabolism may account for the adaptation to insulin resistance in islets from C57BL/6J mice fed a high-fat diet. Diabetologia 2007, 50, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Poitout, V.; Amyot, J.; Semache, M.; Zarrouki, B.; Hagman, D.; Fontés, G. Glucolipotoxicity of the pancreatic beta cell. Biochim. Biophys. Acta 2010, 1801, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Robertson, R.P.; Harmon, J.; Tran, P.O.; Tanaka, Y.; Takahashi, H. Glucose Toxicity in β-Cells: Type 2 Diabetes, Good Radicals Gone Bad, and the Glutathione Connection. Diabetes 2003, 52, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Robertson, R.P.; Harmon, J.; Tran, P.O.T.; Poitout, V. β-Cell Glucose Toxicity, Lipotoxicity, and Chronic Oxidative Stress in Type 2 Diabetes. Diabetes 2004, 53, S119–S124. [Google Scholar] [CrossRef] [PubMed]
- Maedler, K.; Oberholzer, J.; Bucher, P.; Spinas, G.A.; Donath, M.Y. Monounsaturated Fatty Acids Prevent the Deleterious Effects of Palmitate and High Glucose on Human Pancreatic β-Cell Turnover and Function. Diabetes 2003, 52, 726–733. [Google Scholar] [CrossRef] [PubMed]
- El-Assaad, W.; Buteau, J.; Peyot, M.-L.; Nolan, C.; Roduit, R.; Hardy, S.; Joly, E.; Dbaibo, G.; Rosenberg, L.; Prentki, M. Saturated fatty acids synergize with elevated glucose to cause pancreatic beta-cell death. Endocrinology 2003, 144, 4154–4163. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Kawamata, Y.; Harada, M.; Kobayashi, M.; Fujii, R.; Fukusumi, S.; Ogi, K.; Hosoya, M.; Tanaka, Y.; Uejima, H.; et al. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature 2003, 422, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Masters, S.L.; Dunne, A.; Subramanian, S.L.; Hull, R.L.; Tannahill, G.M.; Sharp, F.A.; Becker, C.; Franchi, L.; Yoshihara, E.; Chen, Z.; et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nat. Immunol. 2010, 11, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.-H.; Adijiang, A.; Vandanmagsar, B.; Burk, D.; Ravussin, A.; Dixit, V.D. Elimination of the NLRP3-ASC inflammasome protects against chronic obesity-induced pancreatic damage. Endocrinology 2011, 152, 4039–4045. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Obesity and Overweight Factsheet. Available online: http://www.who.int/mediacentre/factsheets/fs311/en/ (accessed on 20 January 2016).
- Lan, J.; Zhao, Y.; Dong, F.; Yan, Z.; Zheng, W.; Fan, J.; Sun, G. Meta-analysis of the effect and safety of berberine in the treatment of type 2 diabetes mellitus, hyperlipemia and hypertension. J. Ethnopharmacol. 2015, 161, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Yki-Järvinen, H. Thiazolidinediones. N. Engl. J. Med. 2004, 351, 1106–1118. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Role of endoplasmic reticulum stress and c-Jun NH2-terminal kinase pathways in inflammation and origin of obesity and diabetes. Diabetes 2005, 54, S73–S78. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Waltenberger, B.; Pferschy-Wenzig, E.-M.; Blunder, M.; Liu, X.; Malainer, C.; Blazevic, T.; Schwaiger, S.; Rollinger, J.M.; Heiss, E.H.; et al. Natural product agonists of peroxisome proliferator-activated receptor gamma (PPARγ): A review. Biochem. Pharmacol. 2014, 92, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Konstantopoulos, N.; Lee, J.; Hansen, L.; Li, Z.W.; Karin, M.; Shoelson, S.E. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science 2001, 293, 1673–1677. [Google Scholar] [CrossRef] [PubMed]
- Shoelson, S.E.; Lee, J.; Goldfine, A.B. Inflammation and insulin resistance. J. Clin. Investig. 2006, 116, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Kim, Y.; Fillmore, J.J.; Chen, Y.; Moore, I.; Lee, J.; Yuan, M.; Li, Z.W.; Karin, M.; Perret, P.; et al. Prevention of fat-induced insulin resistance by salicylate. J. Clin. Investig. 2001, 108, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Hundal, R.S.; Petersen, K.F.; Mayerson, A.B.; Randhawa, P.S.; Inzucchi, S.; Shoelson, S.E.; Shulman, G.I. Mechanism by which high-dose aspirin improves glucose metabolism in type 2 diabetes. J. Clin. Investig. 2002, 109, 1321–1326. [Google Scholar] [CrossRef] [PubMed]
- Larsen, C.M.; Faulenbach, M.; Vaag, A.; Vølund, A.; Ehses, J.A.; Seifert, B.; Mandrup-Poulsen, T.; Donath, M.Y. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N. Engl. J. Med. 2007, 356, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.E.; Haffner, S.M.; Heise, M.A.; Herman, W.H.; Holman, R.R.; Jones, N.P.; Kravitz, B.G.; Lachin, J.M.; O’Neill, M.C.; Zinman, B.; et al. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N. Engl. J. Med. 2006, 355, 2427–2443. [Google Scholar] [CrossRef] [PubMed]
- Coll, R.C.; Robertson, A.; Butler, M.; Cooper, M.; O’Neill, L.A.J. The cytokine release inhibitory drug CRID3 targets ASC oligomerisation in the NLRP3 and AIM2 inflammasomes. PLoS ONE 2011, 6, e29539. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, M. Development of anti-TNF therapy for rheumatoid arthritis. Nat. Rev. Immunol. 2002, 2, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Bongartz, T.; Sutton, A.J.; Sweeting, M.J.; Buchan, I.; Matteson, E.L.; Montori, V. Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: Systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA 2006, 295, 2275–2285. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.L.; Hollingsworth, K.G.; Aribisala, B.S.; Chen, M.J.; Mathers, J.C.; Taylor, R. Reversal of type 2 diabetes: Normalisation of beta cell function in association with decreased pancreas and liver triacylglycerol. Diabetologia 2011, 54, 2506–2514. [Google Scholar] [CrossRef] [PubMed]
- Borges, R.L.; Ribeiro-Filho, F.F.; Carvalho, K.M.B.; Zanella, M.T. Impact of weight loss on adipocytokines, C-reactive protein and insulin sensitivity in hypertensive women with central obesity. Arq. Bras. Cardiol. 2007, 89, 409–414. [Google Scholar] [PubMed]
- Tuomilehto, J.; Lindström, J.; Eriksson, J.G.; Valle, T.T.; Hämäläinen, H.; Ilanne-Parikka, P.; Keinänen-Kiukaanniemi, S.; Laakso, M.; Louheranta, A.; Rastas, M.; et al. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N. Engl. J. Med. 2001, 344, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Kanat, M.; DeFronzo, R.A.; Abdul-Ghani, M.A. Treatment of prediabetes. World J. Diabetes 2015, 6, 1207–1222. [Google Scholar] [PubMed]
- Baggio, L.L.; Drucker, D.J. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157. [Google Scholar] [CrossRef] [PubMed]
- Pi-Sunyer, X.; Astrup, A.; Fujioka, K.; Greenway, F.; Halpern, A.; Krempf, M.; Wilding, J.P.H. A Randomized, Controlled Trial of 3.0 mg of Liraglutide in Weight Management. N. Engl. J. Med. 2015, 373, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Muscelli, E.; Mingrone, G.; Camastra, S.; Manco, M.; Pereira, J.A.; Pareja, J.C.; Ferrannini, E. Differential effect of weight loss on insulin resistance in surgically treated obese patients. Am. J. Med. 2005, 118, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Chrysohoou, C.; Panagiotakos, D.B.; Pitsavos, C.; Das, U.N.; Stefanadis, C. Adherence to the Mediterranean diet attenuates inflammation and coagulation process in healthy adults: The ATTICA Study. J. Am. Coll. Cardiol. 2004, 44, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Vessby, B.; Uusitupa, M.; Hermansen, K.; Riccardi, G.; Rivellese, A.A.; Tapsell, L.C.; Nälsén, C.; Berglund, L.; Louheranta, A.; Rasmussen, B.M.; et al. Substituting dietary saturated for monounsaturated fat impairs insulin sensitivity in healthy men and women: The KANWU study. Diabetologia 2001, 44, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Tierney, A.C.; McMonagle, J.; Shaw, D.I.; Gulseth, H.L.; Helal, O.; Saris, W.H.M.; Paniagua, J.A.; Gołąbek-Leszczyñska, I.; Defoort, C.; Williams, C.M.; et al. Effects of dietary fat modification on insulin sensitivity and on other risk factors of the metabolic syndrome—LIPGENE: A European randomized dietary intervention study. Int. J. Obes. 2011, 35, 800–809. [Google Scholar] [CrossRef] [PubMed]
- Yubero-Serrano, E.M.; Delgado-Lista, J.; Tierney, A.C.; Perez-Martinez, P.; Garcia-Rios, A.; Alcala-Diaz, J.F.; Lopez-Miranda, J. Insulin resistance determines a differential response to changes in dietary fat modification on metabolic syndrome risk factors: The LIPGENE study. Am. J. Clin. Nutr. 2015, 102, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Oliver, E.; McGillicuddy, F.; Phillips, C.; Toomey, S.; Roche, H.M. The role of inflammation and macrophage accumulation in the development of obesity-induced type 2 diabetes mellitus and the possible therapeutic effects of long-chain n-3 PUFA. Proc. Nutr. Soc. 2010, 69, 232–243. [Google Scholar] [CrossRef] [PubMed]
- Pischon, T.; Hankinson, S.E.; Hotamisligil, G.S.; Rifai, N.; Willett, W.C.; Rimm, E.B. Habitual dietary intake of n-3 and n-6 fatty acids in relation to inflammatory markers among US men and women. Circulation 2003, 108, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Oz, H.S.; Barve, S.; de Villiers, W.J.S.; McClain, C.J.; Varilek, G.W. The Green Tea Polyphenol (−)-Epigallocatechin-3-Gallate Blocks Nuclear Factor-kappa B Activation by Inhibiting Ikappa B Kinase Activity in the Intestinal Epithelial Cell Line IEC-6. Mol. Pharmacol. 2001, 60, 528–533. [Google Scholar] [PubMed]
- Bose, M.; Lambert, J.D.; Ju, J.; Reuhl, K.R.; Shapses, S.A.; Yang, C.S. The Major Green Tea Polyphenol, (−)-Epigallocatechin-3-Gallate, Inhibits Obesity, Metabolic Syndrome, and Fatty Liver Disease in High-Fat-Fed Mice. J. Nutr. 2008, 138, 1677–1683. [Google Scholar] [PubMed]
- Lu, C.-C.; Yen, G.-C. Antioxidative and anti-inflammatory activity of functional foods. Curr. Opin. Food Sci. 2015, 2, 1–8. [Google Scholar] [CrossRef]
- Wang, S.; Moustaid-Moussa, N.; Chen, L.; Mo, H.; Shastri, A.; Su, R.; Bapat, P.; Kwun, I.; Shen, C.-L. Novel insights of dietary polyphenols and obesity. J. Nutr. Biochem. 2014, 25, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, M.M.; Fjeldborg, K.; Ornstrup, M.J.; Kjær, T.N.; Nøhr, M.K.; Pedersen, S.B. Resveratrol and inflammation: Challenges in translating pre-clinical findings to improved patient outcomes. Biochim. Biophys. Acta 2015, 1852, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Bakker, G.C.; van Erk, M.J.; Pellis, L.; Wopereis, S.; Rubingh, C.M.; Cnubben, N.H.; Kooistra, T.; van Ommen, B.; Hendriks, H.F. An antiinflammatory dietary mix modulates inflammation and oxidative and metabolic stress in overweight men: A nutrigenomics approach. Am. J. Clin. Nutr. 2010, 91, 1044–1059. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lyons, C.L.; Kennedy, E.B.; Roche, H.M. Metabolic Inflammation-Differential Modulation by Dietary Constituents. Nutrients 2016, 8, 247. https://doi.org/10.3390/nu8050247
Lyons CL, Kennedy EB, Roche HM. Metabolic Inflammation-Differential Modulation by Dietary Constituents. Nutrients. 2016; 8(5):247. https://doi.org/10.3390/nu8050247
Chicago/Turabian StyleLyons, Claire L., Elaine B. Kennedy, and Helen M. Roche. 2016. "Metabolic Inflammation-Differential Modulation by Dietary Constituents" Nutrients 8, no. 5: 247. https://doi.org/10.3390/nu8050247
APA StyleLyons, C. L., Kennedy, E. B., & Roche, H. M. (2016). Metabolic Inflammation-Differential Modulation by Dietary Constituents. Nutrients, 8(5), 247. https://doi.org/10.3390/nu8050247