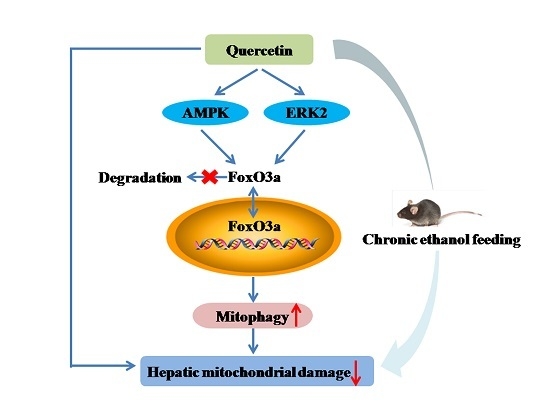
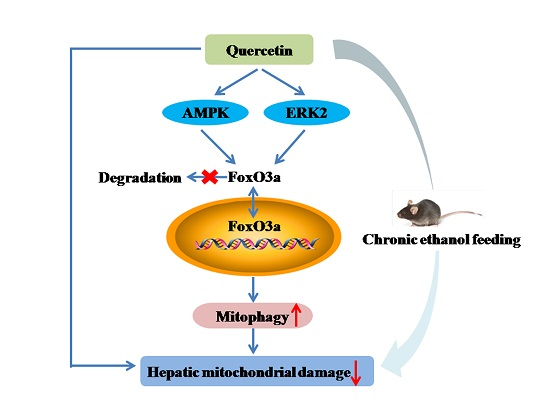
Quercetin Attenuates Chronic Ethanol-Induced Hepatic Mitochondrial Damage through Enhanced Mitophagy

 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Animal Treatment
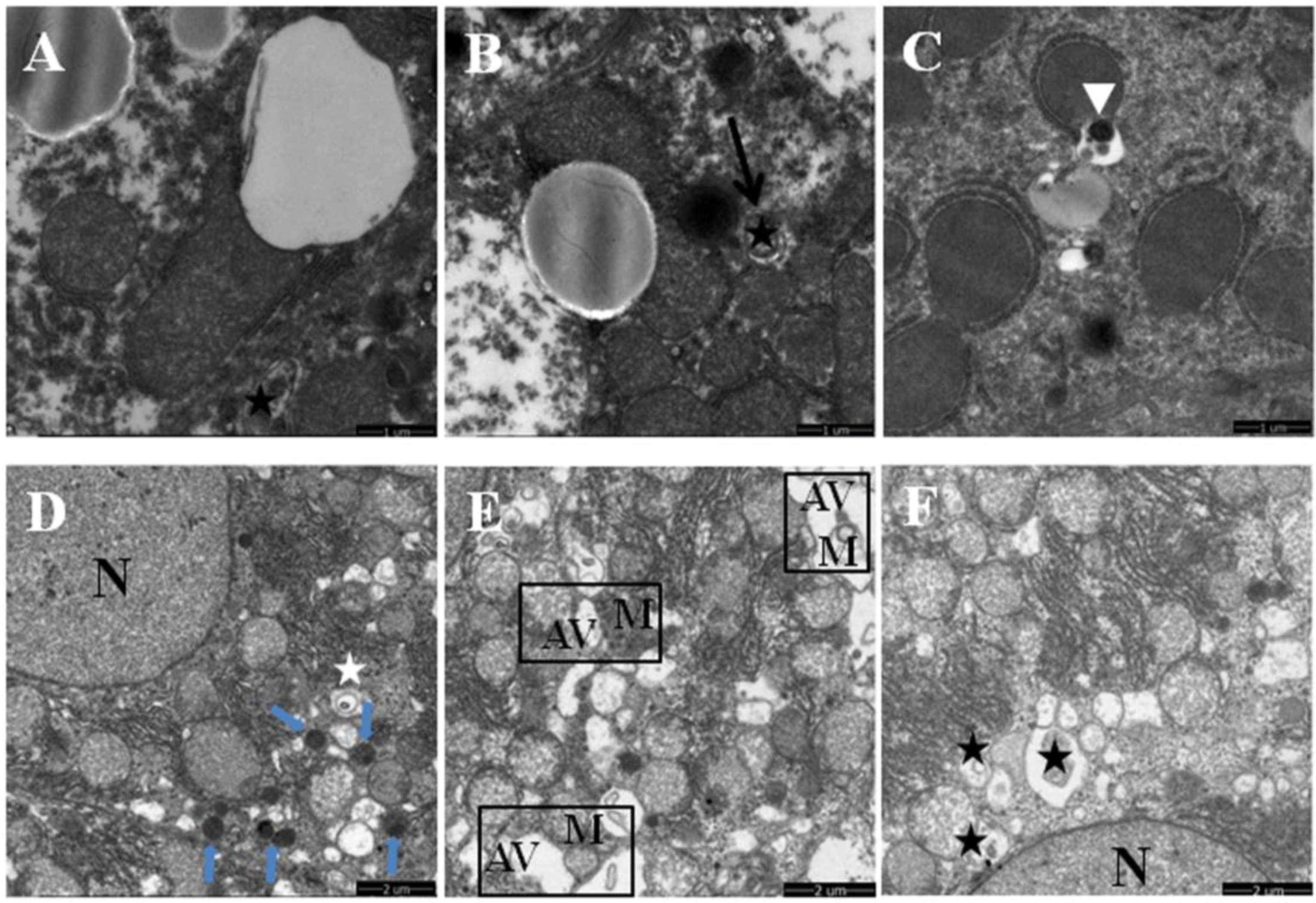
2.3. Transmission Electron Microscopy
2.4. Mitochondrial Membrane Potential
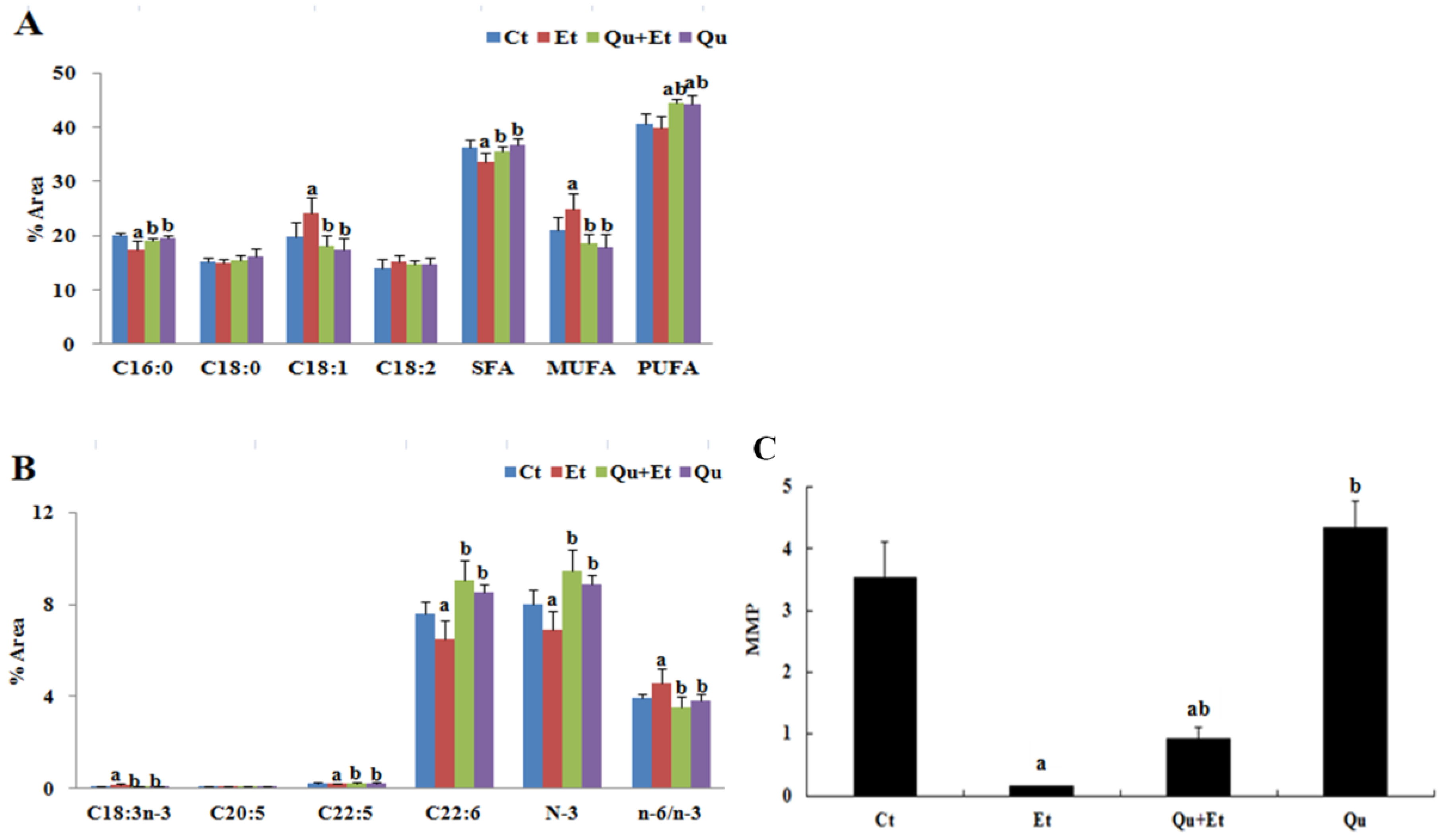
2.5. Mitochondrial Membrane Lipid Composition
2.6. Western Blot Analysis
2.7. Real-Time Quantitative Polymerase Chain Reaction Analysis
2.8. Statistical Analysis
3. Results
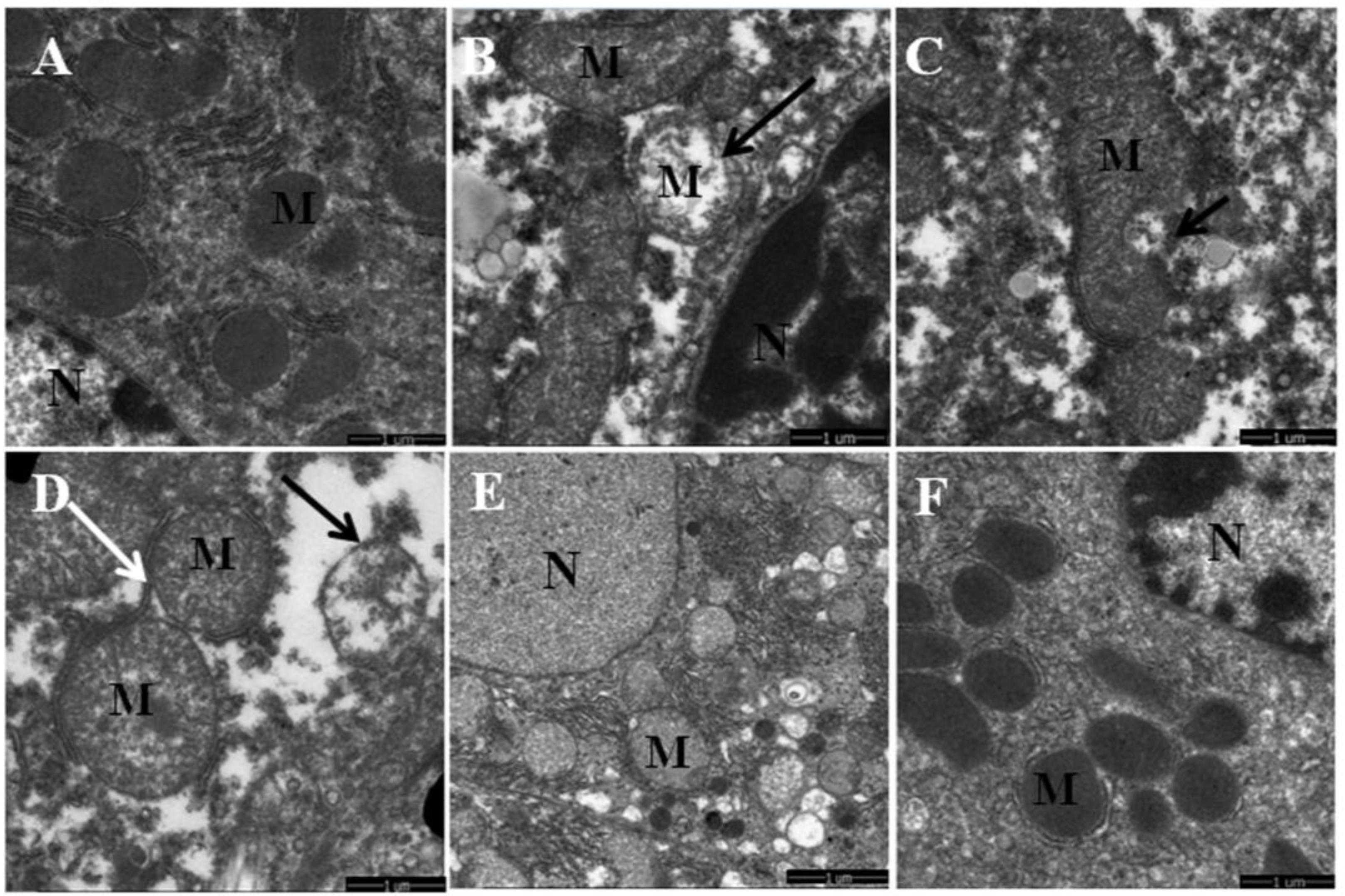
3.1. Quercetin Alleviated Chronic Ethanol-Induced Mitochondrial Damage in Mice Liver
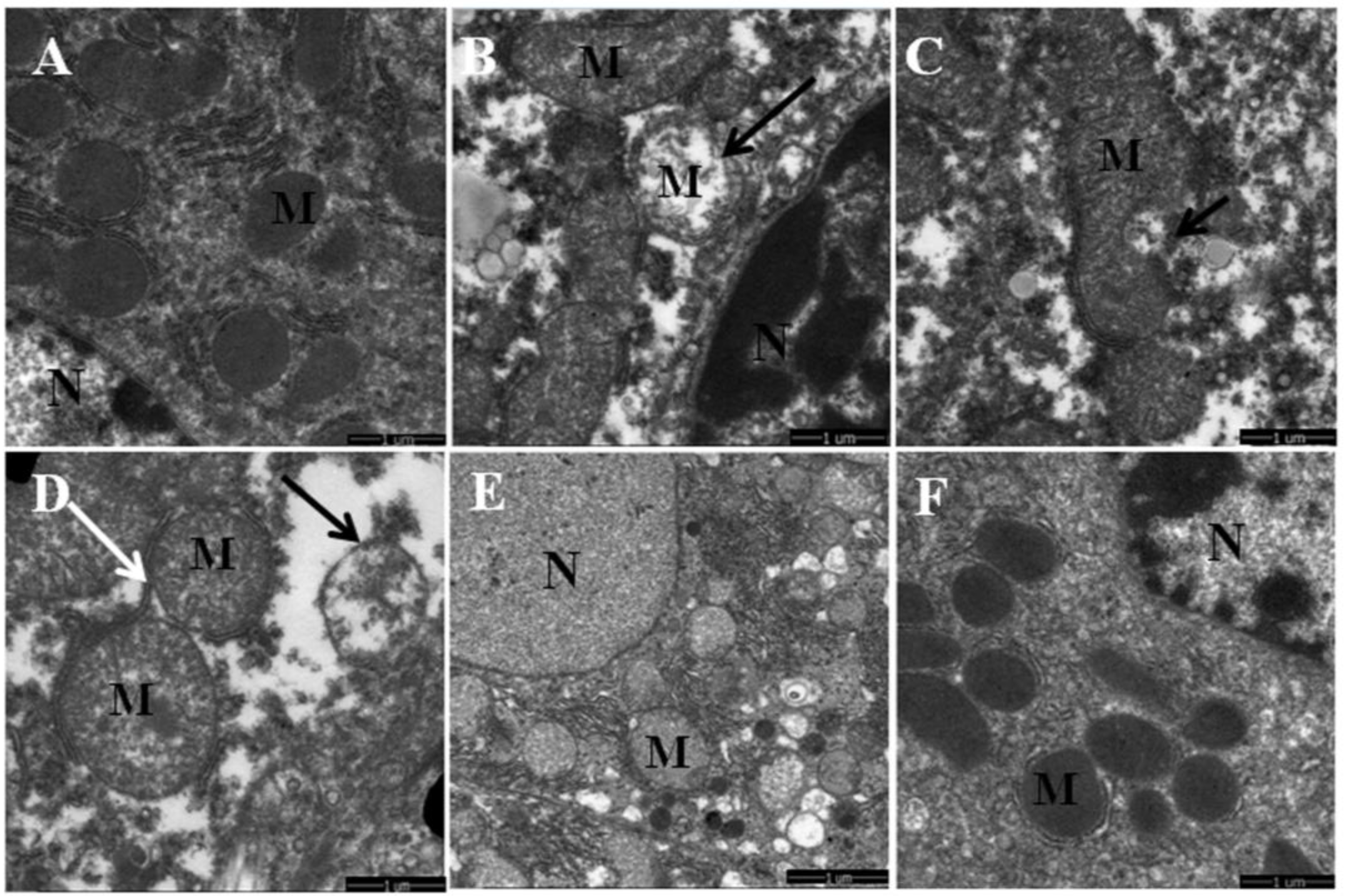
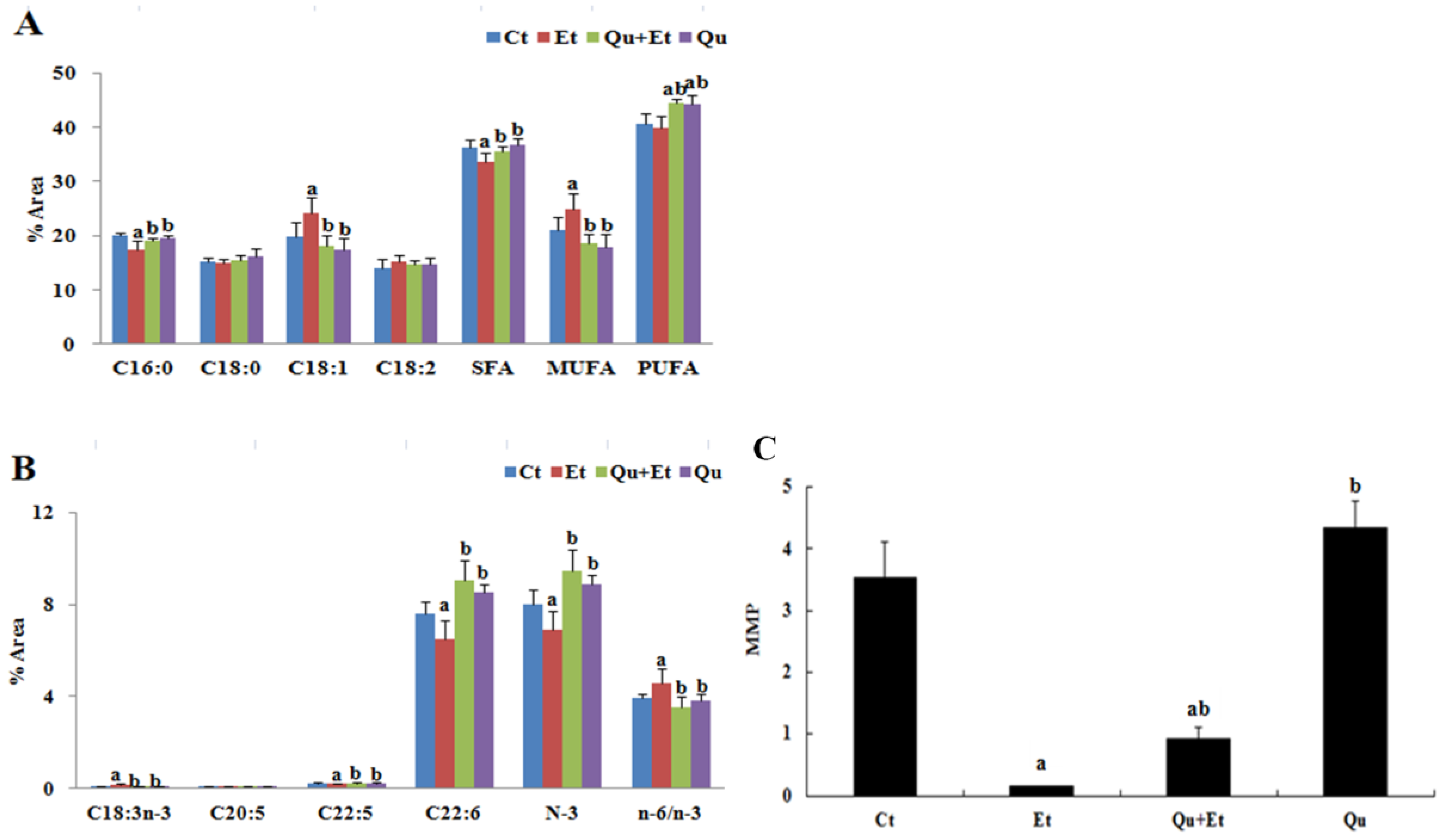
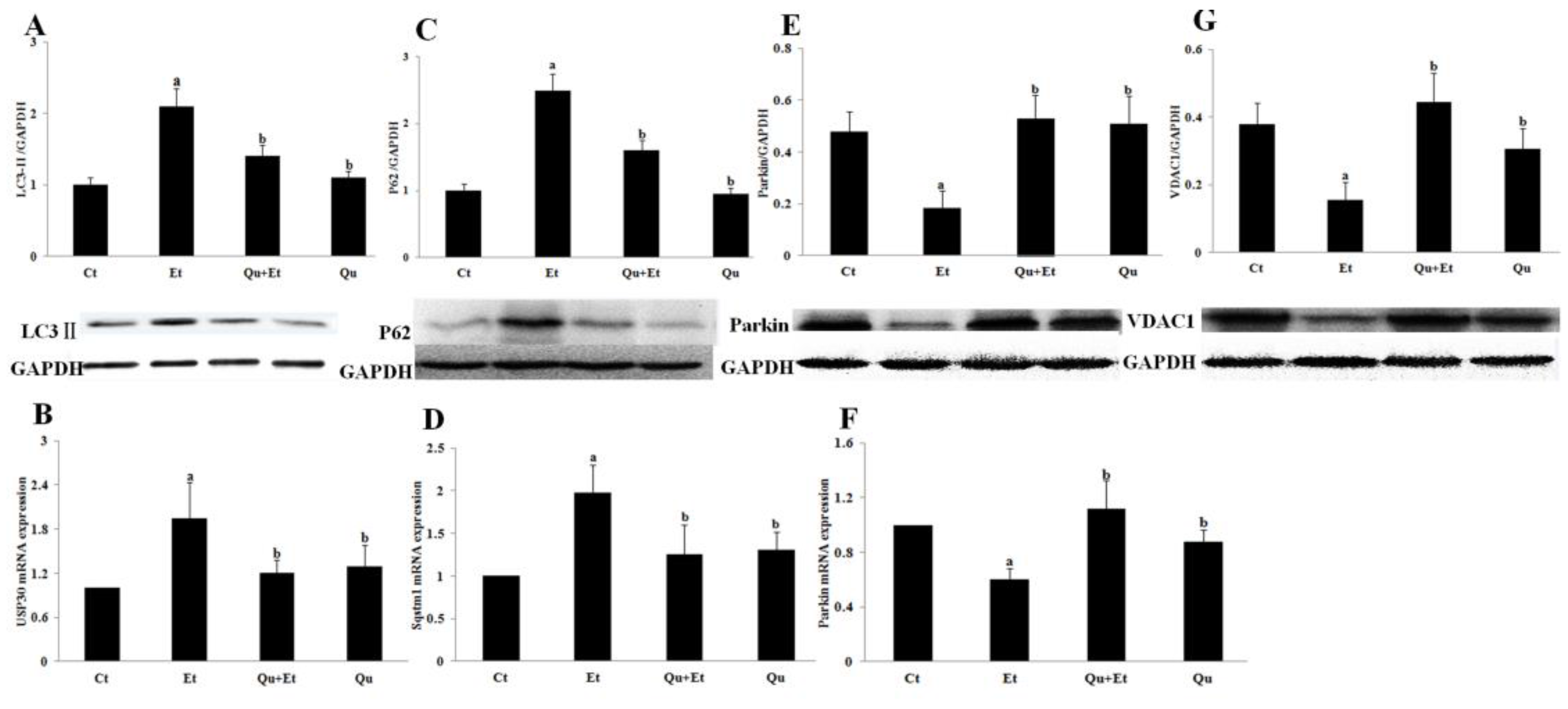
3.2. Quercetin Enhanced Chronic Ethanol-Mediated Mitophagy Suppression in Mice Liver
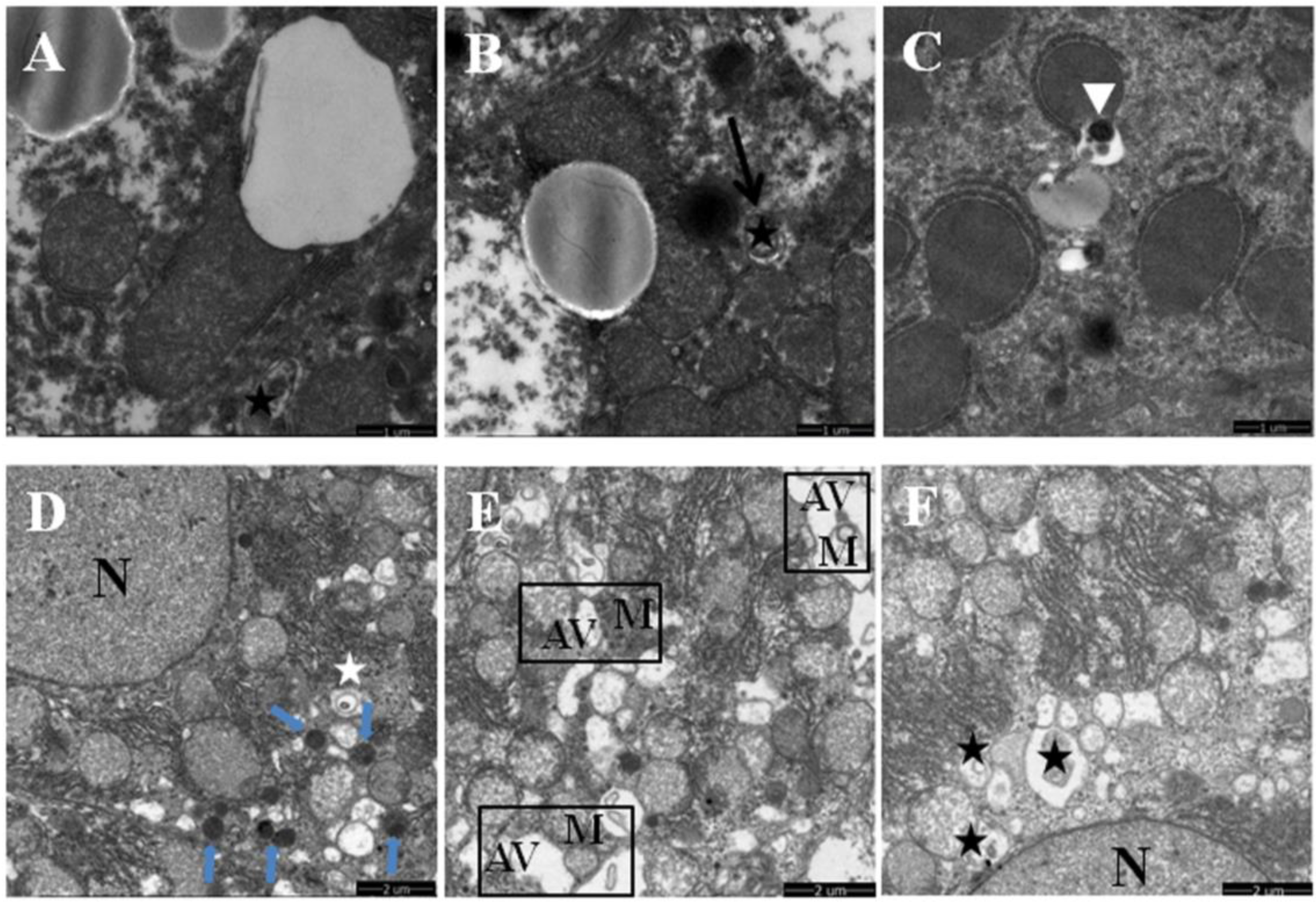
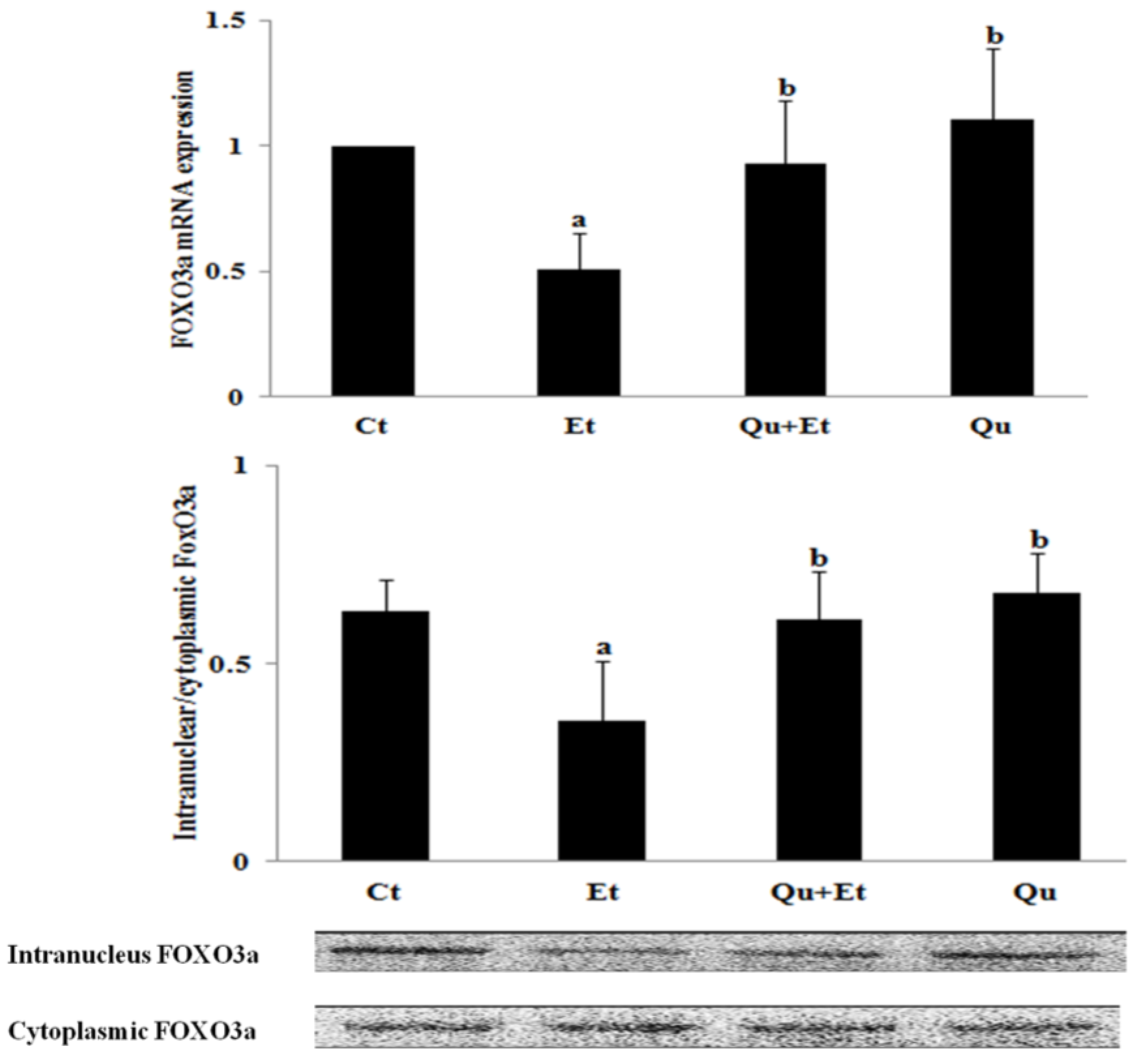
3.3. Increased Nuclear Translocation of FoxO3a Potentially Mediated the Protective Effect of Quercetin against Mitophagy Suppression
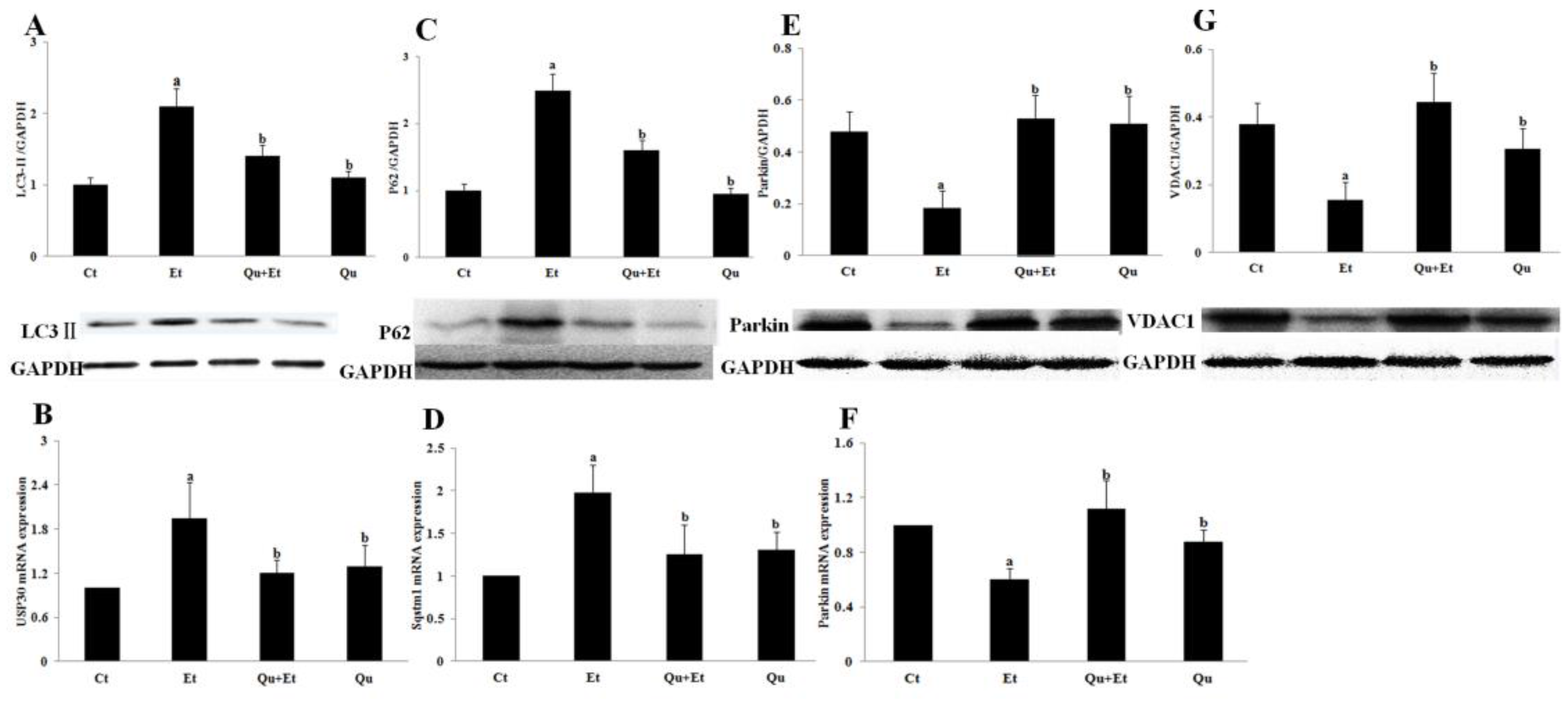

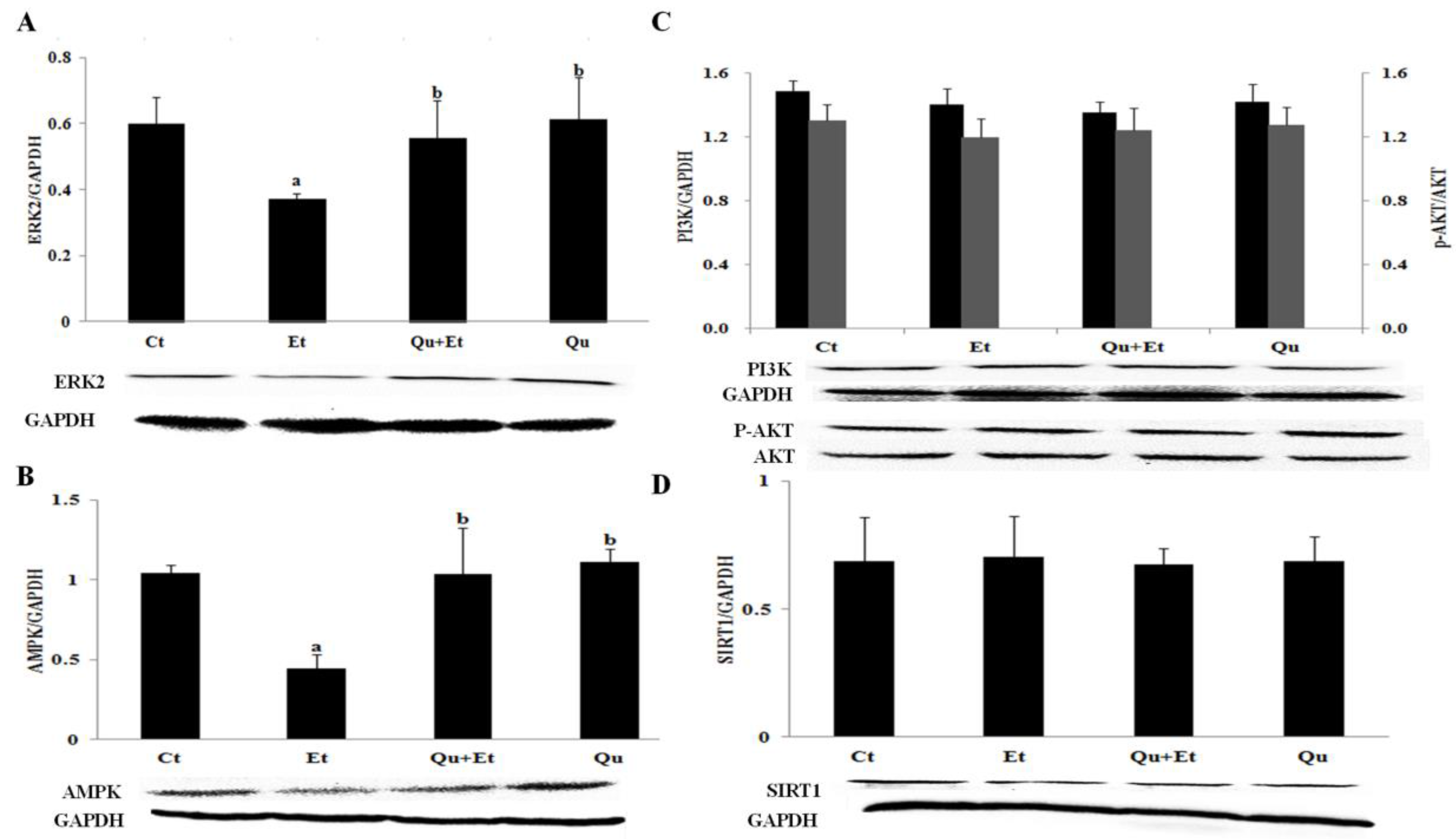
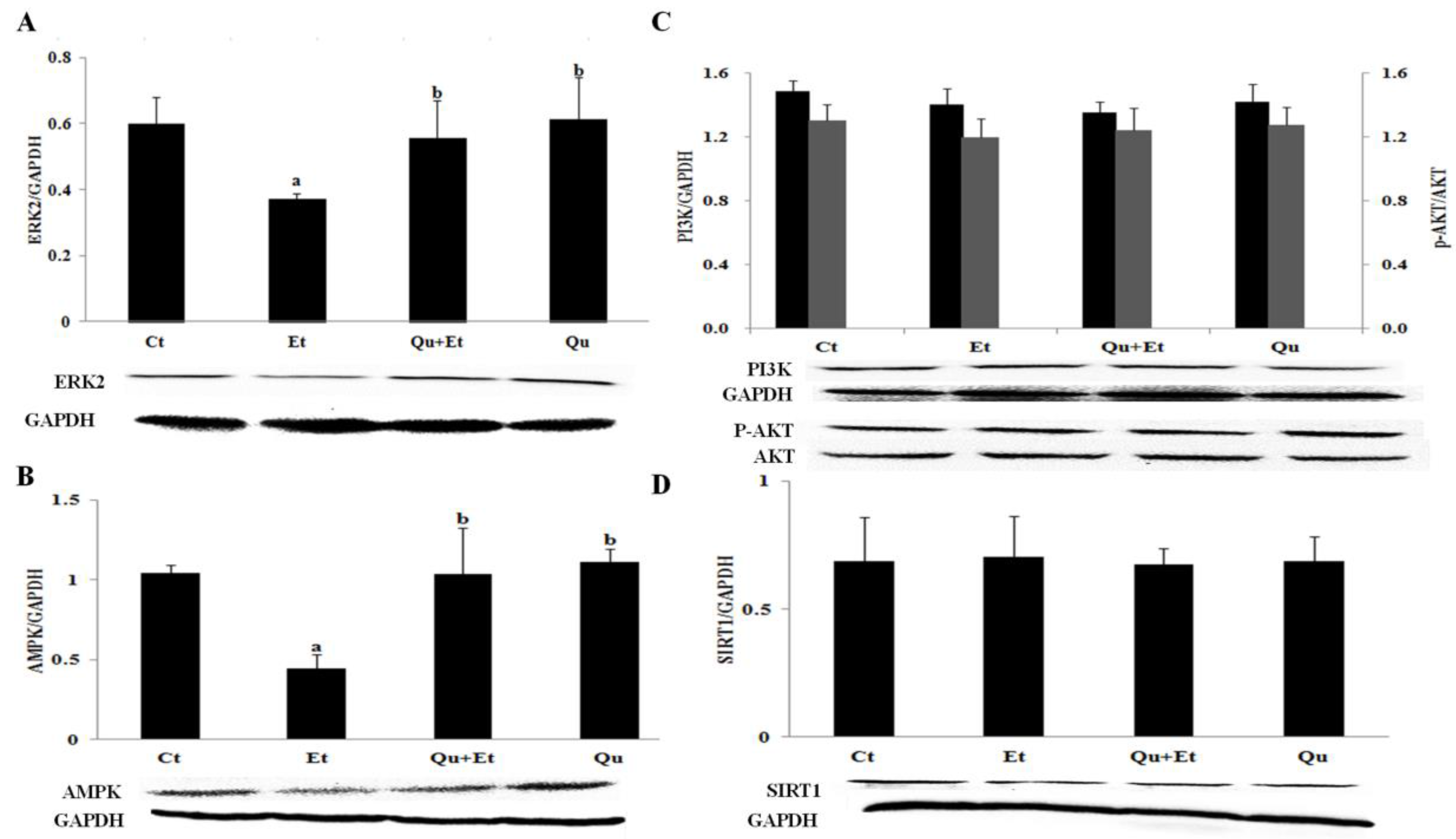
3.4. AMPK and ERK2, Instead of PI3K/AKT and SIRT1, Were Involved in Quercetin-Mediated Mitophagy Activation
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization. Global Status Report on Alcohol and Health 2014; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef] [PubMed]
- Mathurin, P.; Bataller, R. Trends in the management and burden of alcoholic liver disease. J. Hepatol. 2015, 62, S38–S46. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Ramshesh, V.K.; Rehman, H.; Liu, Q.; Theruvath, T.P.; Krishnasamy, Y.; Lemasters, J.J. Acute ethanol causes hepatic mitochondrial depolarization in mice: Role of ethanol metabolism. PLoS ONE 2014, 9, e91308. [Google Scholar] [CrossRef] [PubMed]
- Eid, N.; Ito, Y.; Maemura, K.; Otsuki, Y. Elevated autophagic sequestration of mitochondria and lipid droplets in steatotic hepatocytes of chronic ethanol-treated rats: An immunohistochemical and electron microscopic study. J. Mol. Histol. 2013, 44, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Keegan, A.; Martini, R.; Batey, R. Ethanol-related liver injury in the rat: A model of steatosis, inflammation and pericentral fibrosis. J. Hepatol. 1995, 23, 591–600. [Google Scholar] [CrossRef]
- Lakshmi Devi, S.; Anuradha, C.V. Mitochondrial damage, cytotoxicity and apoptosis in iron-potentiated alcoholic liver fibrosis: Amelioration by taurine. Amino Acids 2010, 38, 869–879. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, T.; Longato, L.; Dostalek, M.; Tong, M.; Wands, J.R.; de la Monte, S.M. Insulin resistance, ceramide accumulation and endoplasmic reticulum stress in experimental chronic alcohol-induced steatohepatitis. Alcohol Alcohol. 2013, 48, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Ni, H.M.; Ding, Y.; Ding, W.X. Parkin regulates mitophagy and mitochondrial function to protect against alcohol-induced liver injury and steatosis in mice. Am. J. Physiol. Gastr. L. 2015, 309, G324–G340. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.D.; Liu, Y.H.; Zhang, Q.L.; Zhang, B.G.; Zhao, N.; Wang, Q.L.; Wang, X.D. Pre-endurance training prevents acute alcoholic liver injury in rats through the regulation of damaged mitochondria accumulation and mitophagy balance. Hepatol. Int. 2014, 8, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Bansal, S.; Biswas, G.; Avadhani, N.G. Mitochondria-targeted heme oxygenase-1 induces oxidative stress and mitochondrial dysfunction in macrophages, kidney fibroblasts and in chronic alcohol hepatotoxicity. Redox Biol. 2014, 2, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Boesch-Saadatmandi, C.; Loboda, A.; Wagner, A.E.; Stachurska, A.; Jozkowicz, A.; Dulak, J.; Doring, F.; Wolffram, S.; Rimbach, G. Effect of quercetin and its metabolites isorhamnetin and quercetin-3-glucuronide on inflammatory gene expression: Role of miR-155. J. Nutr. Biochem. 2011, 22, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Boots, A.W.; Haenen, G.R.; Bast, A. Health effects of quercetin: From antioxidant to nutraceutical. Eur. J. Pharmacol. 2008, 585, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Kelly, G.S. Quercetin. Altern. Med. Rev. 2011, 16, 172–194. [Google Scholar] [PubMed]
- Tang, Y.; Gao, C.; Xing, M.; Li, Y.; Zhu, L.; Wang, D.; Yang, X.; Liu, L.; Yao, P. Quercetin prevents ethanol-induced dyslipidemia and mitochondrial oxidative damage. Food Chem. Toxicol. 2012, 50, 1194–1200. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.L.; Wang, L.T.; Huang, K.H.; Wang, C.C.; Chiang, C.K.; Liu, S.H. Quercetin attenuates renal ischemia/reperfusion injury via an activation of AMP-activated protein kinase-regulated autophagy pathway. J. Nutr. Biochem. 2014, 25, 1226–1234. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Moon, J.Y.; Ahn, K.S.; Cho, S.K. Quercetin induces mitochondrial mediated apoptosis and protective autophagy in human glioblastoma U373MG cells. Oxid. Med. Cell. Longev. 2013, 2013, 596496. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liu, R.; Li, J.; Mao, J.; Lei, Y.; Wu, J.; Zeng, J.; Zhang, T.; Wu, H.; Chen, L.; Huang, C.; Wei, Y. Quercetin induces protective autophagy in gastric cancer cells: Involvement of Akt-mTOR- and hypoxia-induced factor 1α-mediated signaling. Autophagy 2011, 7, 966–978. [Google Scholar]
- Das, S.; Mitrovsky, G.; Vasanthi, H.R.; Das, D.K. Antiaging properties of a grape-derived antioxidant are regulated by mitochondrial balance of fusion and fission leading to mitophagy triggered by a signaling network of Sirt1-Sirt3-Foxo3-PINK1-PARKIN. Oxid. Med. Cell. Longev. 2014, 2014, 345105. [Google Scholar] [CrossRef] [PubMed]
- Aoki, H.; Takada, Y.; Kondo, S.; Sawaya, R.; Aggarwal, B.B.; Kondo, Y. Evidence that curcumin suppresses the growth of malignant gliomas in vitro and in vivo through induction of autophagy: Role of Akt and extracellular signal-regulated kinase signaling pathways. Mol. Pharmacol. 2007, 72, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Figliomeni, M.L.; Abdel-Rahman, M.S. Ethanol does not increase the hepatotoxicity of cocaine in primary rat hepatocyte culture. Toxicology 1998, 129, 125–135. [Google Scholar] [CrossRef]
- Wieckowski, M.R.; Giorgi, C.; Lebiedzinska, M.; Duszynski, J.; Pinton, P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protoc. 2009, 4, 1582–1590. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [PubMed]
- Bicalho, B.; David, F.; Rumplel, K.; Kindt, E.; Sandra, P. Creating a fatty acid methyl ester database for lipid profiling in a single drop of human blood using high resolution capillary gas chromatography and mass spectrometry. J. Chromatogr. A 2008, 1211, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Nassir, F.; Ibdah, J.A. Role of mitochondria in alcoholic liver disease. World J. Gastroenterol. 2014, 20, 2136–2142. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.S.; Prathibha, P.; Rejitha, S.; Indira, M. Ethanol induced hepatic mitochondrial dysfunction is attenuated by all trans retinoic acid supplementation. Life Sci. 2015, 135, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Waring, A.J.; Rottenberg, H.; Ohnishi, T.; Rubin, E. Membranes and phospholipids of liver mitochondria from chronic alcoholic rats are resistant to membrane disordering by alcohol. Proc. Natl. Acad. Sci. USA 1981, 78, 2582–2586. [Google Scholar] [CrossRef] [PubMed]
- Nervi, A.M.; Peluffo, R.O.; Brenner, R.R.; Leikin, A.I. Effect of ethanol administration on fatty acid desaturation. Lipids 1980, 15, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, J.P.; Oliveira, P.J.; Jurado, A.S. Mitochondrial membrane lipid remodeling in pathophysiology: A new target for diet and therapeutic interventions. Prog. Lipid Res. 2013, 52, 513–528. [Google Scholar] [CrossRef] [PubMed]
- Kobori, M.; Masumoto, S.; Akimoto, Y.; Oike, H. Chronic dietary intake of quercetin alleviates hepatic fat accumulation associated with consumption of a Western-style diet in C57/BL6J mice. Mol. Nutr. Food Res. 2011, 55, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Stanley, W.C.; Khairallah, R.J.; Dabkowski, E.R. Update on lipids and mitochondrial function: Impact of dietary n-3 polyunsaturated fatty acids. Curr. Opin. Clin. Nutr. 2012, 15, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Toufektsian, M.C.; Salen, P.; Laporte, F.; Tonelli, C.; de Lorgeril, M. Dietary flavonoids increase plasma very long-chain (n-3) fatty acids in rats. J. Nutr. 2011, 141, 37–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Infante, J.P.; Huszagh, V.A. Secondary carnitine deficiency and impaired docosahexaenoic (22:6n-3) acid synthesis: A common denominator in the pathophysiology of diseases of oxidative phosphorylation and β-oxidation. FEBS Lett. 2000, 68, 1–5. [Google Scholar] [CrossRef]
- Herbst, E.A.F.; Paglialunga, S.; Gerling, C.; Whitfield, J.; Mukai, K.; Chabowski, A.; Heigenhauser, G.J.F.; Spriet, L.L.; Holloway, G.P. Omega-3 supplementation alters mitochondrial membrane composition and respiration kinetics in human skeletal muscle. J. Physiol. 2014, 592, 1341–1352. [Google Scholar] [CrossRef] [PubMed]
- Heijnen, C.G.; Haenen, G.R.; Minou Oostveen, R.; Stalpers, E.M.; Bast, A. Protection of flavonoids against lipid peroxidation: The structure activity relationship revisited. Free Rad. Res. 2002, 36, 575–581. [Google Scholar] [CrossRef]
- Liu, L.; Tang, Y.; Gao, C.; Li, Y.; Chen, S.; Xiong, T.; Li, J.; Du, M.; Gong, Z.Y.; Chen, H.; Liu, L.; Yao, P. Characterization and biodistribution in vivo of quercetin-loaded cationic nanostructured lipid carriers. Colloids Surf. B Biointerfaces 2014, 115, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Fiorani, M.; Guidarelli, A.; Blasa, M.; Azzolini, C.; Candiracci, M.; Piatti, E.; Cantoni, O. Mitochondria accumulate large amounts of quercetin: Prevention of mitochondrial damage and release upon oxidation of the extramitochondrial fraction of the flavonoid. J. Nutr. Biochem. 2010, 21, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.P.; Youle, R.J. Targeting mitochondrial dysfunction: Role for PINK1 and Parkin in mitochondrial quality control. Antioxid. Redox Sign. 2011, 14, 1929–1938. [Google Scholar] [CrossRef] [PubMed]
- Bingol, B.; Tea, J.S.; Phu, L.; Reichelt, M.; Bakalarski, C.E.; Song, Q.; Foreman, O.; Kirkpatrick, D.S.; Sheng, M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 2014, 510, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell. Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lu, J.F.; Mi, Y.J.; Shi, Z.; Chen, C.H.; Riley, J.; Zhou, C.M. Voltage-dependent anion channels (VDACs) promote mitophagy to protect neuron from death in an early brain injury following a subarachnoid hemorrhage in rats. Brain Res. 2014, 1573, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Rostovtseva, T.K.; Kazemi, N.; Weinrich, M.; Bezrukov, S.M. Voltage gating of VDAC is regulated by nonlamellar lipids of mitochondrial membranes. J. Biol. Chem. 2006, 281, 37496–37506. [Google Scholar] [CrossRef] [PubMed]
- Bonet-Ponce, L.; Saez-Atienzar, S.; da Casa, C.; Flores-Bellver, M.; Barcia, J.M.; Sancho-Pelluz, J.; Romero, F.J.; Jordan, J.; Galindo, M.F. On the mechanism underlying ethanol-induced mitochondrial dynamic disruption and autophagy response. BBA-Mol. Basis Dis. 2015, 1852, 1400–1409. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Liu, L.; Chen, Q. Selective removal of mitochondria via mitophagy: Distinct pathways for different mitochondrial stresses. BBA-Mol. Cell Res. 2015, 1853, 2784–2790. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.M.; Du, K.; You, M.; Ding, W.X. Critical role of FoxO3a in alcohol-induced autophagy and hepatotoxicity. Am. J. Pathol. 2013, 183, 1815–1825. [Google Scholar] [CrossRef] [PubMed]
- Manley, S.; Ni, H.M.; Williams, J.A.; Kong, B.; DiTacchio, L.; Guo, G.; Ding, W.X. Farnesoid X receptor regulates forkhead Box O3a activation in ethanol-induced autophagy and hepatotoxicity. Redox Biol. 2014, 2, 991–1002. [Google Scholar] [CrossRef] [PubMed]
- Nepal, S.; Park, P.H. Activation of autophagy by globular adiponectin attenuates ethanol-induced apoptosis in HepG2 cells: Involvement of AMPK/FoxO3A axis. BBA-Mol. Cell Res. 2013, 1833, 2111–2125. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.Y.; Zong, C.S.; Xia, W.; Yamaguchi, H.; Ding, Q.; Xie, X.; Lang, J.Y.; Lai, C.C.; Chang, C.J.; Huang, W.C.; et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat. Cell Biol. 2008, 10, 138–148. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, X.; Xu, Y.; Zhang, S.; Sun, J.; Liu, P.; Xiao, L.; Tang, Y.; Liu, L.; Yao, P. Quercetin Attenuates Chronic Ethanol-Induced Hepatic Mitochondrial Damage through Enhanced Mitophagy. Nutrients 2016, 8, 27. https://doi.org/10.3390/nu8010027
Yu X, Xu Y, Zhang S, Sun J, Liu P, Xiao L, Tang Y, Liu L, Yao P. Quercetin Attenuates Chronic Ethanol-Induced Hepatic Mitochondrial Damage through Enhanced Mitophagy. Nutrients. 2016; 8(1):27. https://doi.org/10.3390/nu8010027
Chicago/Turabian StyleYu, Xiao, Yanyan Xu, Shanshan Zhang, Jian Sun, Peiyi Liu, Lin Xiao, Yuhan Tang, Liegang Liu, and Ping Yao. 2016. "Quercetin Attenuates Chronic Ethanol-Induced Hepatic Mitochondrial Damage through Enhanced Mitophagy" Nutrients 8, no. 1: 27. https://doi.org/10.3390/nu8010027