Methoxistasis: Integrating the Roles of Homocysteine and Folic Acid in Cardiovascular Pathobiology


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methionine-Homocysteine Cycle Regulates Redox and Methylation Reactions
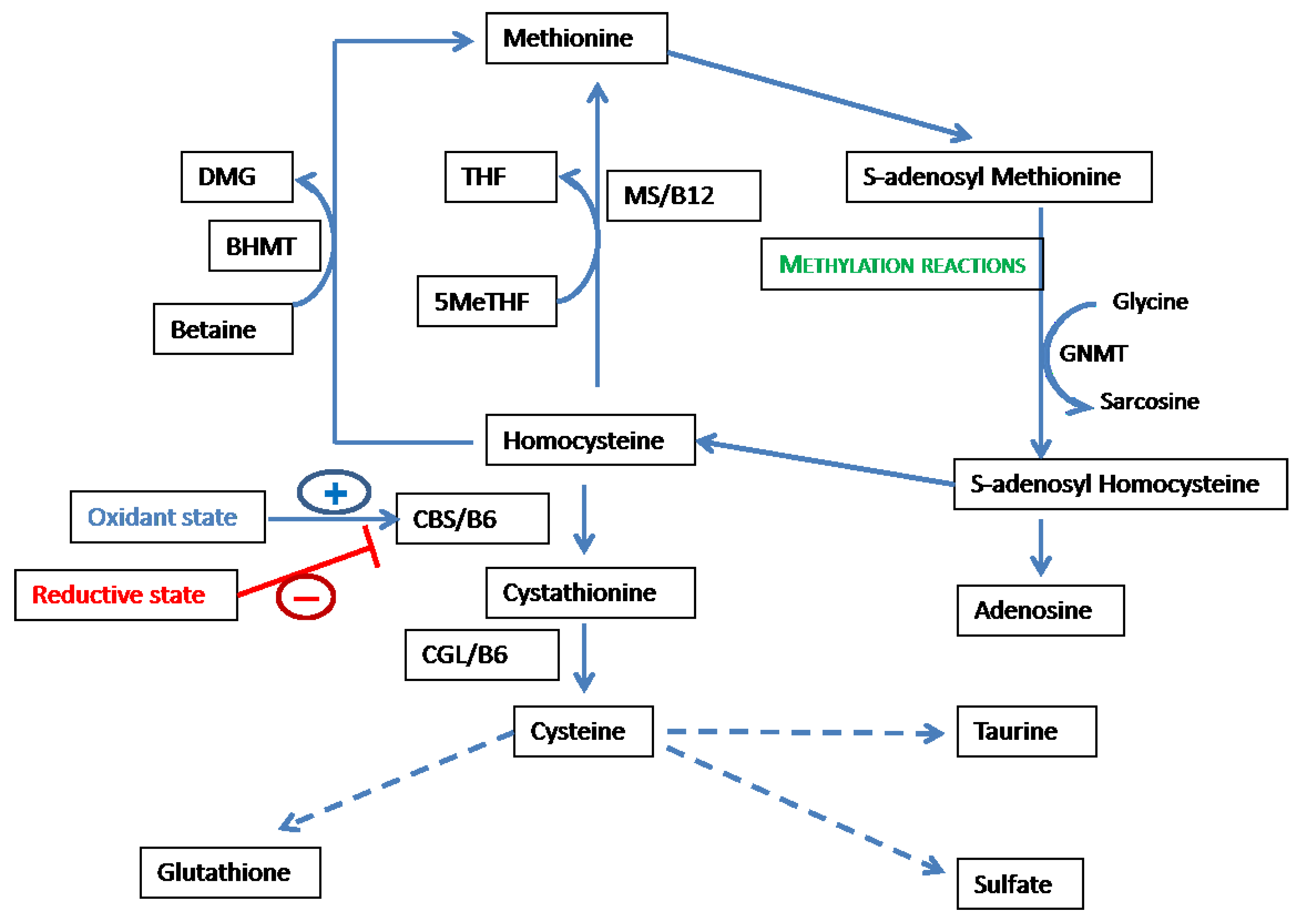
2.1. Overview of the Methionine-Homocysteine Cycle and Sulfur-Containing Amino Acid Metabolism
2.2. Methionine-Homocysteine Cycle and Redox Potential
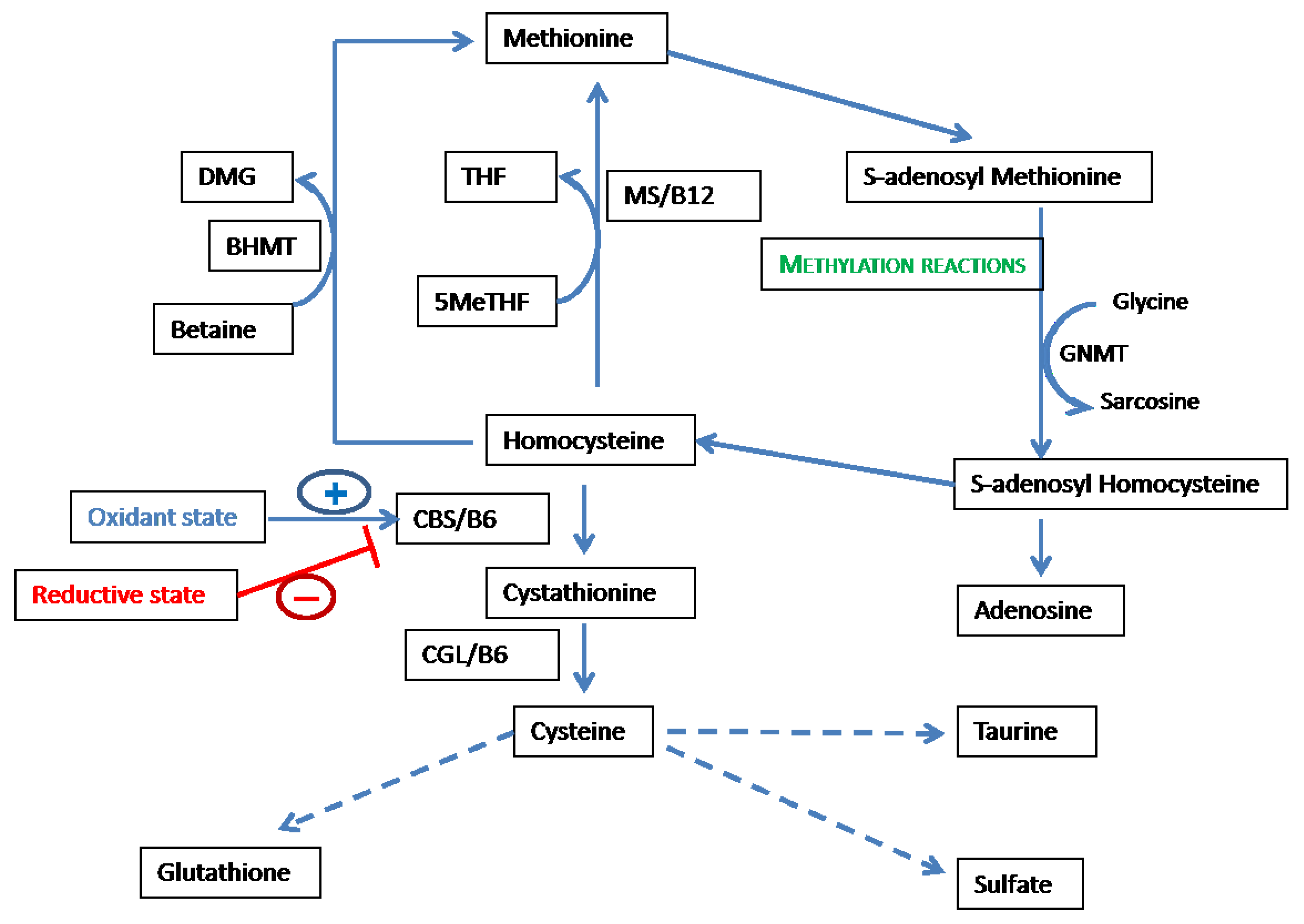
2.3. Feedback of Redox Status on the Methionine-Homocysteine Cycle
2.4. Methionine-Homocysteine Cycle and Methylation Reactions
3. Folate, Methionine-Homocysteine Cycle, and Redox-Methylation Balance
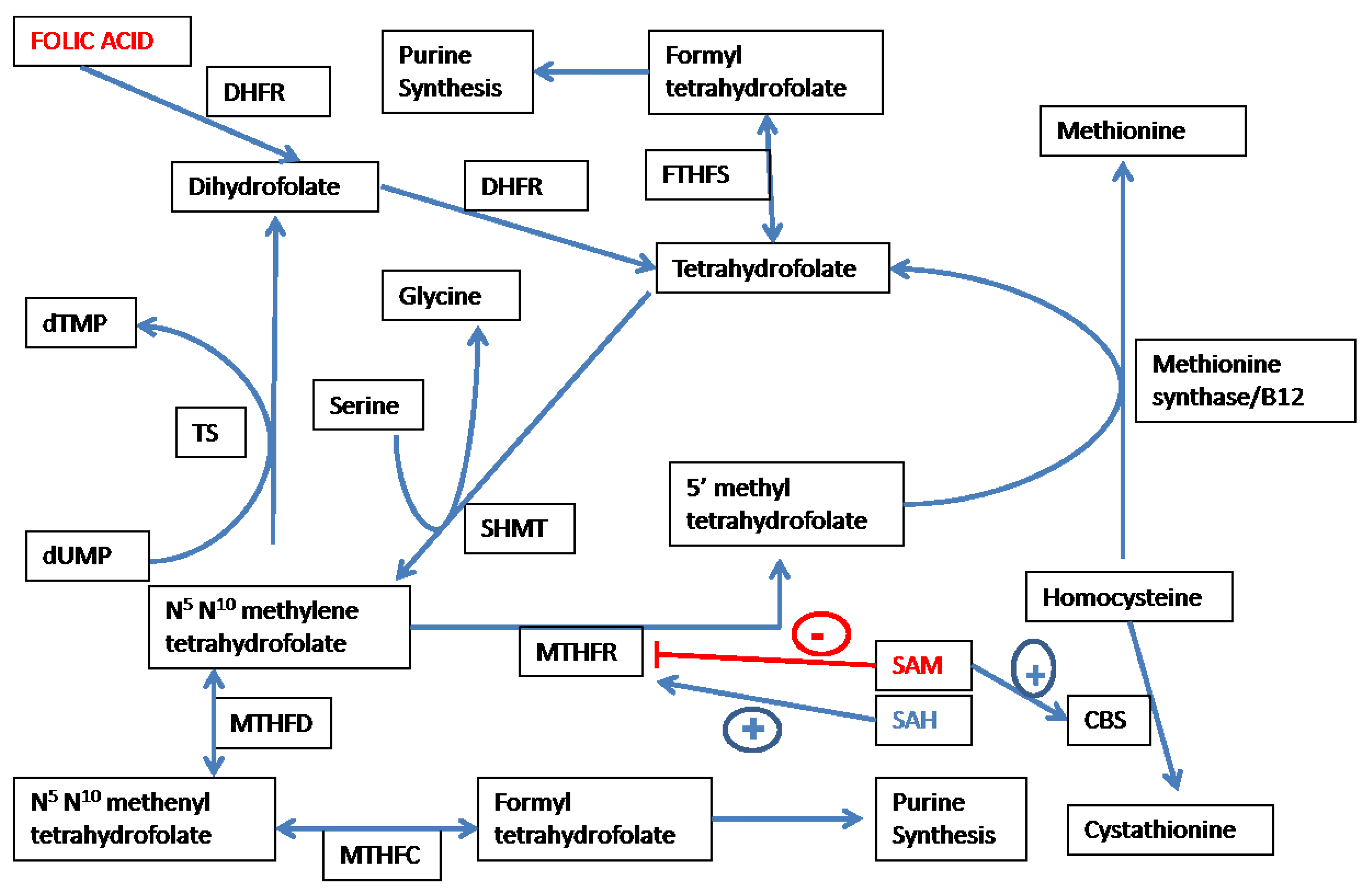
3.1. Overview of Folate-Dependent One Carbon Metabolism
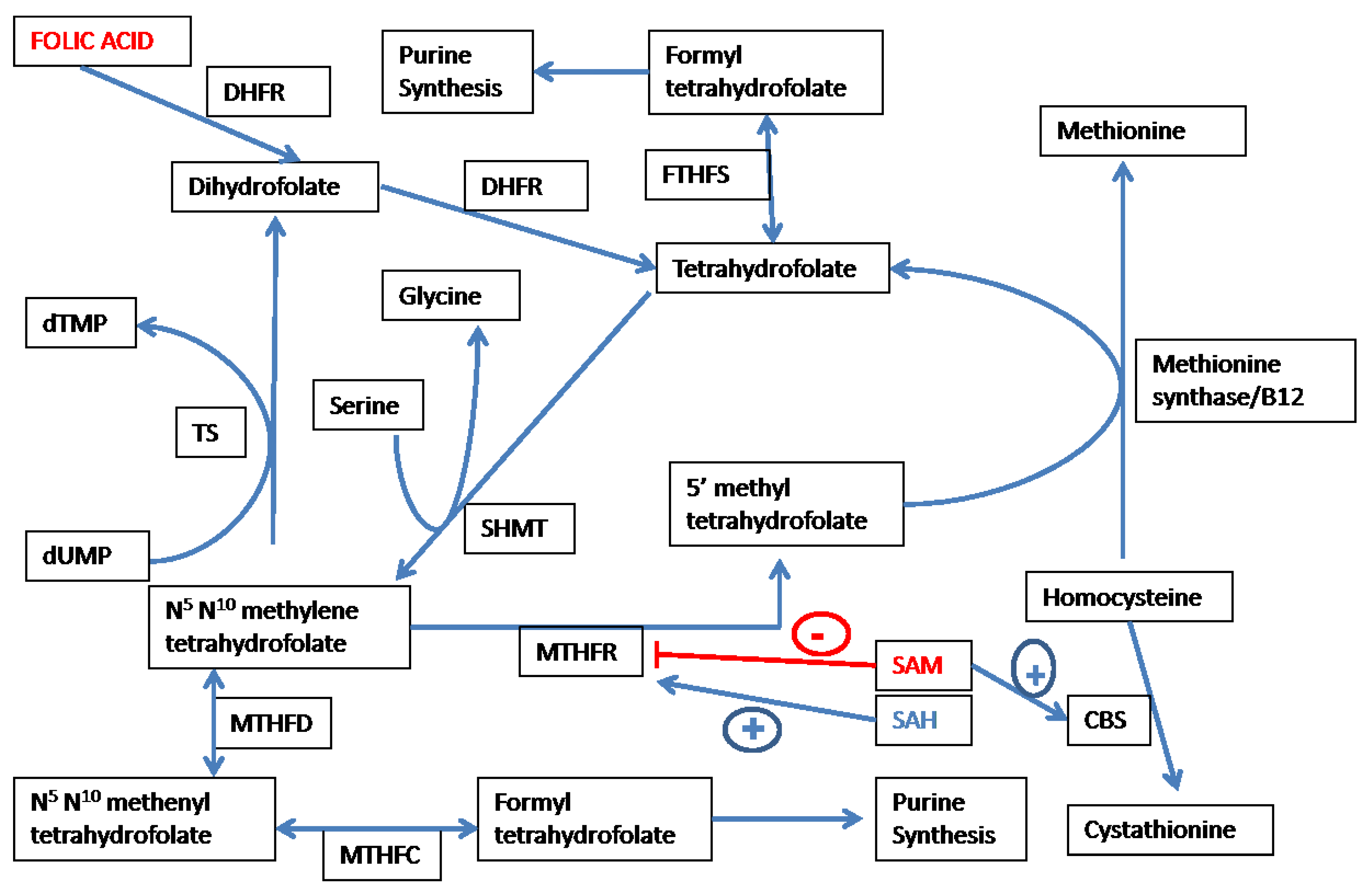
3.2. Folate Affects Redox-Methylation Balance via the Methionine-Homocysteine Cycle
4. Clinical Evidence Connecting Methionine-Homocysteine Cycle, Folate Metabolism, and Methylation
5. Clinical Evidence Connecting the Methionine-Homocysteine Cycle, Folate Metabolism and Redox Balance
6. Clinical Trials of B-Vitamin Supplementation in Cardiovascular Disease
7. Methionine-Homocysteine Cycle, Folate Metabolism, and Heart Failure: Novel Cardiovascular Targets for Nutritional Manipulation
7.1. Clinical Heart Failure and B-Vitamin Deficiency
7.2. Clinical and Epidemiologic Evidence Linking Hyperhomocysteinemia to Heart Failure
7.3. Preclinical Evidence Linking the Methionine-Homocysteine Cycle and Heart Failure
7.4. Effect of Homocysteine-Lowering Therapy on Myocardial Structure and Heart Failure
8. Methoxistasis—A Novel Paradigm
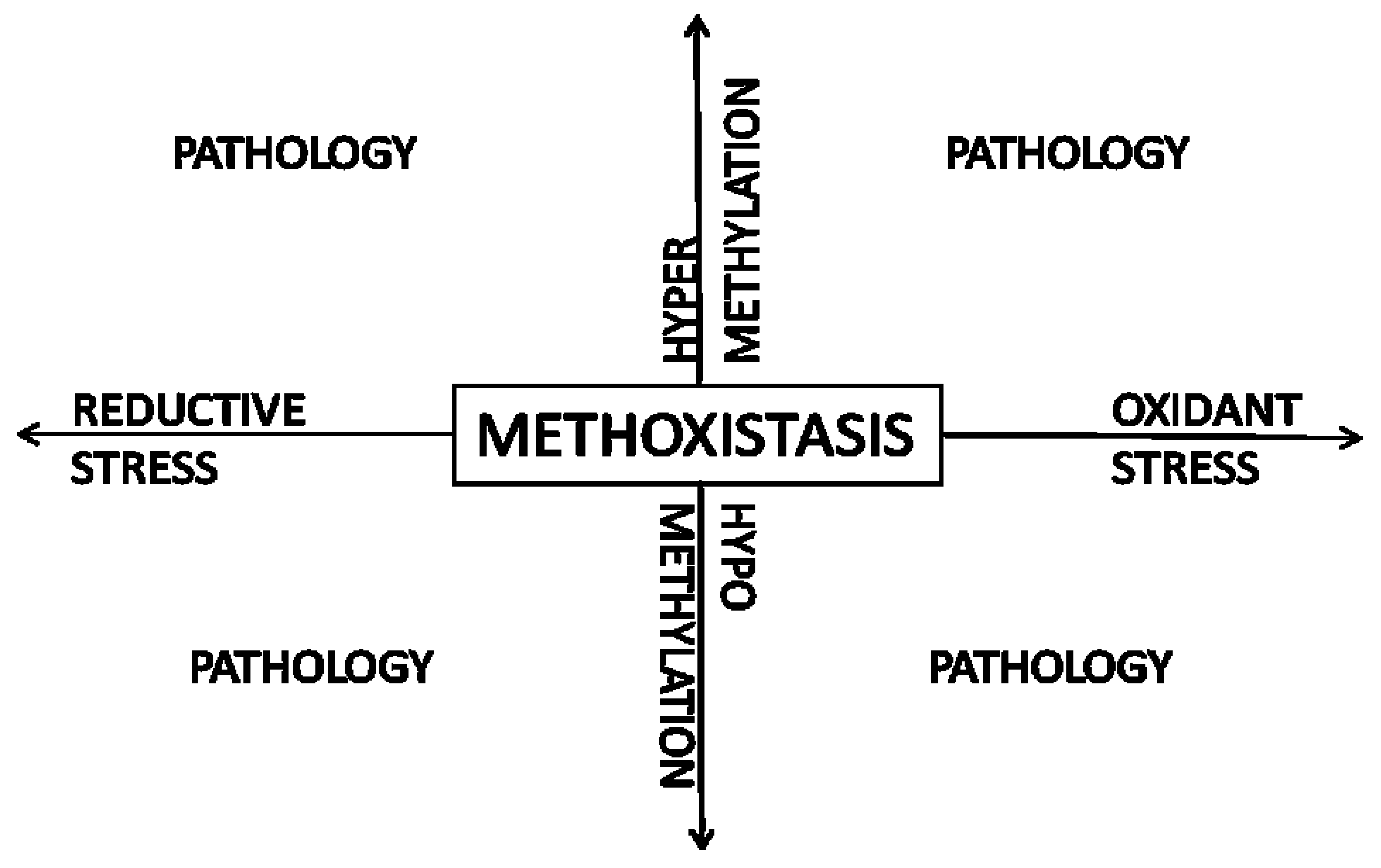
9. Conclusions
Acknowledgments
Conflict of Interest
References
- McCully, K.S. Vascular pathology of homocysteinemia: Implications for the pathogenesis of arteriosclerosis. Am. J. Pathol. 1969, 56, 111–128. [Google Scholar]
- Marti-Carvajal, A.J.; Sola, I.; Lathyris, D.; Karakitsiou, D.E.; Simancas-Racines, D. Homocysteine-lowering interventions for preventing cardiovascular events. Cochrane Database Syst. Rev. 2013, 1, CD006612. [Google Scholar]
- Yang, Q.; Botto, L.D.; Erickson, J.D.; Berry, R.J.; Sambell, C.; Johansen, H.; Friedman, J.M. Improvement in stroke mortality in canada and the united states, 1990 to 2002. Circulation 2006, 113, 1335–1343. [Google Scholar] [CrossRef]
- Bonaa, K.H.; Njolstad, I.; Ueland, P.M.; Schirmer, H.; Tverdal, A.; Steigen, T.; Wang, H.; Nordrehaug, J.E.; Arnesen, E.; Rasmussen, K. Homocysteine lowering and cardiovascular events after acute myocardial infarction. N. Engl. J. Med. 2006, 354, 1578–1588. [Google Scholar] [CrossRef]
- Lange, H.; Suryapranata, H.; de Luca, G.; Borner, C.; Dille, J.; Kallmayer, K.; Pasalary, M.N.; Scherer, E.; Dambrink, J.H. Folate therapy and in-stent restenosis after coronary stenting. N. Engl. J. Med. 2004, 350, 2673–2681. [Google Scholar] [CrossRef]
- Rafeq, Z.; Roh, J.D.; Guarino, P.; Kaufman, J.; Joseph, J. Adverse myocardial effects of B-vitamin therapy in subjects with chronic kidney disease and hyperhomocysteinaemia. Nutr. Metab. Cardiovasc. Dis. 2012. [Google Scholar] [CrossRef]
- Ebbing, M.; Bonaa, K.H.; Arnesen, E.; Ueland, P.M.; Nordrehaug, J.E.; Rasmussen, K.; Njolstad, I.; Nilsen, D.W.; Refsum, H.; Tverdal, A.; et al. Combined analyses and extended follow-up of two randomized controlled homocysteine-lowering B-vitamin trials. J. Intern. Med. 2010, 268, 367–382. [Google Scholar] [CrossRef]
- Loland, K.H.; Bleie, O.; Blix, A.J.; Strand, E.; Ueland, P.M.; Refsum, H.; Ebbing, M.; Nordrehaug, J.E.; Nygard, O. Effect of homocysteine-lowering B vitamin treatment on angiographic progression of coronary artery disease: A western norway B vitamin intervention trial (wenbit) substudy. Am. J. Cardiol. 2010, 105, 1577–1584. [Google Scholar] [CrossRef]
- Joseph, J.; Handy, D.E.; Loscalzo, J. Quo vadis: Whither homocysteine research? Cardiovasc. Toxicol. 2009, 9, 53–63. [Google Scholar] [CrossRef]
- Joseph, J. Fattening by deprivation: Methyl balance and perinatal cardiomyopathy. J. Pathol. 2011, 225, 315–317. [Google Scholar] [CrossRef]
- Stipanuk, M.H. Sulfur amino acid metabolism: Pathways for production and removal of homocysteine and cysteine. Annu. Rev. Nutr. 2004, 24, 539–577. [Google Scholar] [CrossRef]
- Gueant, J.L.; Namour, F.; Gueant-Rodriguez, R.M.; Daval, J.L. Folate and fetal programming: A play in epigenomics? Trends Endocrinol. Metab. 2013, 24, 279–289. [Google Scholar] [CrossRef]
- Azizi-Namini, P.; Ahmed, M.; Yan, A.T.; Keith, M. The role of b vitamins in the management of heart failure. Nutr. Clin. Pract. 2012, 27, 363–374. [Google Scholar] [CrossRef]
- Mudd, S.H.; Poole, J.R. Labile methyl balances for normal humans on various dietary regimens. Metabolism 1975, 24, 721–735. [Google Scholar] [CrossRef]
- Luka, Z.; Mudd, S.H.; Wagner, C. Glycine N-methyltransferase and regulation of S-adenosylmethionine levels. J. Biol. Chem. 2009, 284, 22507–22511. [Google Scholar] [CrossRef]
- Eloranta, T.O. Tissue distribution of S-adenosylmethionine and S-adenosylhomocysteine in the rat. Effect of age, sex and methionine administration on the metabolism of S-adenosylmethionine, S-adenosylhomocysteine and polyamines. Biochem. J. 1977, 166, 521–529. [Google Scholar]
- Elrod, P.; Zhang, J.; Yang, X.; Yin, D.; Hu, Y.; Borchardt, R.T.; Schowen, R.L. Contributions of active site residues to the partial and overall catalytic activities of human S-adenosylhomocysteine hydrolase. Biochemistry 2002, 41, 8134–8142. [Google Scholar] [CrossRef]
- Finkelstein, J.D.; Harris, B.J.; Kyle, W.E. Methionine metabolism in mammals: Kinetic study of betaine-homocysteine methyltransferase. Arch. Biochem. Biophys. 1972, 153, 320–324. [Google Scholar] [CrossRef]
- Mudd, S.H.; Finkelstein, J.D.; Irreverre, F.; Laster, L. Transsulfuration in mammals. Microassays and tissue distributions of three enzymes of the pathway. J. Biol. Chem. 1965, 240, 4382–4392. [Google Scholar]
- Garcia, R.A.; Stipanuk, M.H. The splanchnic organs, liver and kidney have unique roles in the metabolism of sulfur amino acids and their metabolites in rats. J. Nutr. 1992, 122, 1693–1701. [Google Scholar]
- Stipanuk, M.H.; Coloso, R.M.; Garcia, R.A.; Banks, M.F. Cysteine concentration regulates cysteine metabolism to glutathione, sulfate and taurine in rat hepatocytes. J. Nutr. 1992, 122, 420–427. [Google Scholar]
- Ottaviano, F.G.; Handy, D.E.; Loscalzo, J. Redox regulation in the extracellular environment. Circ. J. 2008, 72, 1–16. [Google Scholar] [CrossRef]
- Moriarty-Craige, S.E.; Jones, D.P. Extracellular thiols and thiol/disulfide redox in metabolism. Annu. Rev. Nutr. 2004, 24, 481–509. [Google Scholar] [CrossRef]
- Chen, P.; Poddar, R.; Tipa, E.V.; Dibello, P.M.; Moravec, C.D.; Robinson, K.; Green, R.; Kruger, W.D.; Garrow, T.A.; Jacobsen, D.W. Homocysteine metabolism in cardiovascular cells and tissues: Implications for hyperhomocysteinemia and cardiovascular disease. Adv. Enzyme Regul. 1999, 39, 93–109. [Google Scholar] [CrossRef]
- Fukagawa, N.K.; Ajami, A.M.; Young, V.R. Plasma methionine and cysteine kinetics in response to an intravenous glutathione infusion in adult humans. Am. J. Physiol. 1996, 270, E209–E214. [Google Scholar]
- Wang, X.; Cui, L.; Joseph, J.; Jiang, B.; Pimental, D.; Handy, D.E.; Liao, R.; Loscalzo, J. Homocysteine induces cardiomyocyte dysfunction and apoptosis through p38 MAPK-mediated increase in oxidant stress. J. Mol. Cell. Cardiol. 2012, 52, 753–760. [Google Scholar] [CrossRef]
- Forgione, M.A.; Cap, A.; Liao, R.; Moldovan, N.I.; Eberhardt, R.T.; Lim, C.C.; Jones, J.; Goldschmidt-Clermont, P.J.; Loscalzo, J. Heterozygous cellular glutathione peroxidase deficiency in the mouse: Abnormalities in vascular and cardiac function and structure. Circulation 2002, 106, 1154–1158. [Google Scholar] [CrossRef]
- Joseph, J.; Joseph, L.; Devi, S.; Kennedy, R.H. Effect of anti-oxidant treatment on hyperhomocysteinemia-induced myocardial fibrosis and diastolic dysfunction. J. Heart Lung Transplant. 2008, 27, 1237–1241. [Google Scholar] [CrossRef]
- Loscalzo, J. The oxidant stress of hyperhomocyst(e)inemia. J. Clin. Investig. 1996, 98, 5–7. [Google Scholar] [CrossRef]
- Weiss, N.; Zhang, Y.Y.; Heydrick, S.; Bierl, C.; Loscalzo, J. Overexpression of cellular glutathione peroxidase rescues homocyst(e)ine-induced endothelial dysfunction. Proc. Natl. Acad. Sci. USA 2001, 98, 12503–12508. [Google Scholar] [CrossRef]
- Taoka, S.; Ohja, S.; Shan, X.; Kruger, W.D.; Banerjee, R. Evidence for heme-mediated redox regulation of human cystathionine β-synthase activity. J. Biol. Chem. 1998, 273, 25179–25184. [Google Scholar]
- Meier, M.; Janosik, M.; Kery, V.; Kraus, J.P.; Burkhard, P. Structure of human cystathionine beta-synthase: A unique pyridoxal 5′-phosphate-dependent heme protein. EMBO J. 2001, 20, 3910–3916. [Google Scholar] [CrossRef]
- Kabil, O.; Toaka, S.; LoBrutto, R.; Shoemaker, R.; Banerjee, R. Pyridoxal phosphate binding sites are similar in human heme-dependent and yeast heme-independent cystathionine β-synthases. Evidence from 31p NMR and pulsed epr spectroscopy that heme and plp cofactors are not proximal in the human enzyme. J. Biol. Chem. 2001, 276, 19350–19355. [Google Scholar] [CrossRef]
- Mosharov, E.; Cranford, M.R.; Banerjee, R. The quantitatively important relationship between homocysteine metabolism and glutathione synthesis by the transsulfuration pathway and its regulation by redox changes. Biochemistry 2000, 39, 13005–13011. [Google Scholar] [CrossRef]
- Zou, C.G.; Banerjee, R. Tumor necrosis factor-α-induced targeted proteolysis of cystathionine β-synthase modulates redox homeostasis. J. Biol. Chem. 2003, 278, 16802–16808. [Google Scholar] [CrossRef]
- Olteanu, H.; Banerjee, R. Redundancy in the pathway for redox regulation of mammalian methionine synthase: Reductive activation by the dual flavoprotein, novel reductase 1. J. Biol. Chem. 2003, 278, 38310–38314. [Google Scholar] [CrossRef]
- Castro, C.; Millian, N.S.; Garrow, T.A. Liver betaine-homocysteine S-methyltransferase activity undergoes a redox switch at the active site zinc. Arch. Biochem. Biophys. 2008, 472, 26–33. [Google Scholar] [CrossRef]
- Corrales, F.J.; Perez-Mato, I.; Sanchez Del Pino, M.M.; Ruiz, F.; Castro, C.; Garcia-Trevijano, E.R.; Latasa, U.; Martinez-Chantar, M.L.; Martinez-Cruz, A.; Avila, M.A.; et al. Regulation of mammalian liver methionine adenosyltransferase. J. Nutr. 2002, 132, 2377S–2381S. [Google Scholar]
- Sullivan, D.M.; Hoffman, J.L. Fractionation and kinetic properties of rat liver and kidney methionine adenosyltransferase isozymes. Biochemistry 1983, 22, 1636–1641. [Google Scholar] [CrossRef]
- Kotb, M.; Geller, A.M. Methionine adenosyltransferase: Structure and function. Pharmacol. Ther. 1993, 59, 125–143. [Google Scholar] [CrossRef]
- Mudd, S.H.; Ebert, M.H.; Scriver, C.R. Labile methyl group balances in the human: The role of sarcosine. Metabolism 1980, 29, 707–720. [Google Scholar] [CrossRef]
- Hoffman, D.R.; Cornatzer, W.E.; Duerre, J.A. Relationship between tissue levels of S-adenosylmethionine, S-adenylhomocysteine, and transmethylation reactions. Can. J. Biochem. 1979, 57, 56–65. [Google Scholar] [CrossRef]
- Kerr, S.J. Competing methyltransferase systems. J. Biol. Chem. 1972, 247, 4248–4252. [Google Scholar]
- Takata, Y.; Huang, Y.; Komoto, J.; Yamada, T.; Konishi, K.; Ogawa, H.; Gomi, T.; Fujioka, M.; Takusagawa, F. Catalytic mechanism of glycine N-methyltransferase. Biochemistry 2003, 42, 8394–8402. [Google Scholar] [CrossRef]
- Yi, P.; Melnyk, S.; Pogribna, M.; Pogribny, I.P.; Hine, R.J.; James, S.J. Increase in plasma homocysteine associated with parallel increases in plasma S-adenosylhomocysteine and lymphocyte DNA hypomethylation. J. Biol. Chem. 2000, 275, 29318–29323. [Google Scholar]
- Billings, R.E.; Noker, P.E.; Tephly, T.R. The role of methionine in regulating folate-dependent reactions in isolated rat hepatocytes. Arch. Biochem. Biophys. 1981, 208, 108–120. [Google Scholar] [CrossRef]
- Kutzbach, C.; Stokstad, E.L. Mammalian methylenetetrahydrofolate reductase. Partial purification, properties, and inhibition by S-adenosylmethionine. Biochim. Biophys. Acta 1971, 250, 459–477. [Google Scholar]
- Yeo, E.J.; Wagner, C. Purification and properties of pancreatic glycine N-methyltransferase. J. Biol. Chem. 1992, 267, 24669–24674. [Google Scholar]
- Yeo, E.J.; Briggs, W.T.; Wagner, C. Inhibition of glycine N-methyltransferase by 5-methyltetrahydrofolate pentaglutamate. J. Biol. Chem. 1999, 274, 37559–37564. [Google Scholar] [CrossRef]
- Janosik, M.; Kery, V.; Gaustadnes, M.; Maclean, K.N.; Kraus, J.P. Regulation of human cystathionine β-synthase by S-adenosyl-l-methionine: Evidence for two catalytically active conformations involving an autoinhibitory domain in the C-terminal region. Biochemistry 2001, 40, 10625–10633. [Google Scholar] [CrossRef]
- Kang, S.S.; Wong, P.W.; Bock, H.G.; Horwitz, A.; Grix, A. Intermediate hyperhomocysteinemia resulting from compound heterozygosity of methylenetetrahydrofolate reductase mutations. Am. J. Hum. Genet. 1991, 48, 546–551. [Google Scholar]
- Kluijtmans, L.A.; van den Heuvel, L.P.; Boers, G.H.; Frosst, P.; Stevens, E.M.; van Oost, B.A.; den Heijer, M.; Trijbels, F.J.; Rozen, R.; Blom, H.J. Molecular genetic analysis in mild hyperhomocysteinemia: A common mutation in the methylenetetrahydrofolate reductase gene is a genetic risk factor for cardiovascular disease. Am. J. Hum. Genet. 1996, 58, 35–41. [Google Scholar]
- Jung, A.Y.; Smulders, Y.; Verhoef, P.; Kok, F.J.; Blom, H.; Kok, R.M.; Kampman, E.; Durga, J. No effect of folic acid supplementation on global DNA methylation in men and women with moderately elevated homocysteine. PLoS One 2011, 6, e24976. [Google Scholar]
- Pizzolo, F.; Blom, H.J.; Choi, S.W.; Girelli, D.; Guarini, P.; Martinelli, N.; Stanzial, A.M.; Corrocher, R.; Olivieri, O.; Friso, S. Folic acid effects on S-adenosylmethionine, S-adenosylhomocysteine, and DNA methylation in patients with intermediate hyperhomocysteinemia. J. Am. Coll. Nutr. 2011, 30, 11–18. [Google Scholar] [CrossRef]
- Figueiredo, J.C.; Grau, M.V.; Wallace, K.; Levine, A.J.; Shen, L.; Hamdan, R.; Chen, X.; Bresalier, R.S.; McKeown-Eyssen, G.; Haile, R.W.; et al. Global DNA hypomethylation (line-1) in the normal colon and lifestyle characteristics and dietary and genetic factors. Cancer Epidemiol. Biomarkers Prev. 2009, 18, 1041–1049. [Google Scholar] [CrossRef]
- Pufulete, M.; Al-Ghnaniem, R.; Khushal, A.; Appleby, P.; Harris, N.; Gout, S.; Emery, P.W.; Sanders, T.A. Effect of folic acid supplementation on genomic DNA methylation in patients with colorectal adenoma. Gut 2005, 54, 648–653. [Google Scholar] [CrossRef]
- Rampersaud, G.C.; Kauwell, G.P.; Hutson, A.D.; Cerda, J.J.; Bailey, L.B. Genomic DNA methylation decreases in response to moderate folate depletion in elderly women. Am. J. Clin. Nutr. 2000, 72, 998–1003. [Google Scholar]
- Svingen, G.F.; Ueland, P.M.; Pedersen, E.K.; Schartum-Hansen, H.; Seifert, R.; Ebbing, M.; Loland, K.H.; Tell, G.S.; Nygard, O. Plasma dimethylglycine and risk of incident acute myocardial infarction in patients with stable angina pectoris. Arterioscler. Thromb. Vasc. Biol. 2013. [Google Scholar] [CrossRef]
- Wotherspoon, F.; Laight, D.W.; Turner, C.; Meeking, D.R.; Allard, S.E.; Munday, L.J.; Shaw, K.M.; Cummings, M.H. The effect of oral folic acid upon plasma homocysteine, endothelial function and oxidative stress in patients with type 1 diabetes and microalbuminuria. Int. J. Clin. Pract. 2008, 62, 569–574. [Google Scholar] [CrossRef]
- Alvares Delfino, V.D.; de Andrade Vianna, A.C.; Mocelin, A.J.; Barbosa, D.S.; Mise, R.A.; Matsuo, T. Folic acid therapy reduces plasma homocysteine levels and improves plasma antioxidant capacity in hemodialysis patients. Nutrition 2007, 23, 242–247. [Google Scholar] [CrossRef]
- Antoniades, C.; Shirodaria, C.; Warrick, N.; Cai, S.; de Bono, J.; Lee, J.; Leeson, P.; Neubauer, S.; Ratnatunga, C.; Pillai, R.; et al. 5-Methyltetrahydrofolate rapidly improves endothelial function and decreases superoxide production in human vessels: Effects on vascular tetrahydrobiopterin availability and endothelial nitric oxide synthase coupling. Circulation 2006, 114, 1193–1201. [Google Scholar] [CrossRef]
- Ullegaddi, R.; Powers, H.J.; Gariballa, S.E. B-group vitamin supplementation mitigates oxidative damage after acute ischaemic stroke. Clin. Sci. (Lond.) 2004, 107, 477–484. [Google Scholar]
- Ullegaddi, R.; Powers, H.J.; Gariballa, S.E. Antioxidant supplementation with or without B-group vitamins after acute ischemic stroke: A randomized controlled trial. J. Parenter. Enter. Nutr. 2006, 30, 108–114. [Google Scholar] [CrossRef]
- Moat, S.J.; Hill, M.H.; McDowell, I.F.; Pullin, C.H.; Ashfield-Watt, P.A.; Clark, Z.E.; Whiting, J.M.; Newcombe, R.G.; Lewis, M.J.; Powers, H.J. Reduction in plasma total homocysteine through increasing folate intake in healthy individuals is not associated with changes in measures of antioxidant activity or oxidant damage. Eur. J. Clin. Nutr. 2003, 57, 483–489. [Google Scholar] [CrossRef]
- Andersson, A.; Jonasson, T.; Ohlin, H.; Lindgren, A.; Hultberg, B. Vitamin supplementation normalizes total plasma homocysteine concentration but not plasma homocysteine redox status in patients with acute coronary syndromes and hyperhomocysteinemia. Clin. Chem. Lab. Med. 2002, 40, 554–558. [Google Scholar]
- Toole, J.F.; Malinow, M.R.; Chambless, L.E.; Spence, J.D.; Pettigrew, L.C.; Howard, V.J.; Sides, E.G.; Wang, C.H.; Stampfer, M. Lowering homocysteine in patients with ischemic stroke to prevent recurrent stroke, myocardial infarction, and death: The vitamin intervention for stroke prevention (visp) randomized controlled trial. JAMA 2004, 291, 565–575. [Google Scholar] [CrossRef]
- Ebbing, M.; Bleie, O.; Ueland, P.M.; Nordrehaug, J.E.; Nilsen, D.W.; Vollset, S.E.; Refsum, H.; Pedersen, E.K.; Nygard, O. Mortality and cardiovascular events in patients treated with homocysteine-lowering B vitamins after coronary angiography: A randomized controlled trial. JAMA 2008, 300, 795–804. [Google Scholar] [CrossRef]
- Lonn, E.; Yusuf, S.; Arnold, M.J.; Sheridan, P.; Pogue, J.; Micks, M.; McQueen, M.J.; Probstfield, J.; Fodor, G.; Held, C.; et al. Homocysteine lowering with folic acid and B vitamins in vascular disease. N. Engl. J. Med. 2006, 354, 1567–1577. [Google Scholar] [CrossRef]
- Albert, C.M.; Cook, N.R.; Gaziano, J.M.; Zaharris, E.; MacFadyen, J.; Danielson, E.; Buring, J.E.; Manson, J.E. Effect of folic acid and b vitamins on risk of cardiovascular events and total mortality among women at high risk for cardiovascular disease: A randomized trial. JAMA 2008, 299, 2027–2036. [Google Scholar] [CrossRef]
- Jamison, R.L.; Hartigan, P.; Kaufman, J.S.; Goldfarb, D.S.; Warren, S.R.; Guarino, P.D.; Gaziano, J.M. Effect of homocysteine lowering on mortality and vascular disease in advanced chronic kidney disease and end-stage renal disease: A randomized controlled trial. JAMA 2007, 298, 1163–1170. [Google Scholar] [CrossRef]
- Zoungas, S.; McGrath, B.P.; Branley, P.; Kerr, P.G.; Muske, C.; Wolfe, R.; Atkins, R.C.; Nicholls, K.; Fraenkel, M.; Hutchison, B.G.; et al. Cardiovascular morbidity and mortality in the atherosclerosis and folic acid supplementation trial (asfast) in chronic renal failure: A multicenter, randomized, controlled trial. J. Am. Coll. Cardiol. 2006, 47, 1108–1116. [Google Scholar] [CrossRef]
- Heinz, J.; Kropf, S.; Domrose, U.; Westphal, S.; Borucki, K.; Luley, C.; Neumann, K.H.; Dierkes, J. B vitamins and the risk of total mortality and cardiovascular disease in end-stage renal disease: Results of a randomized controlled trial. Circulation 2010, 121, 1432–1438. [Google Scholar] [CrossRef]
- Roger, V.L.; Go, A.S.; Lloyd-Jones, D.M.; Benjamin, E.J.; Berry, J.D.; Borden, W.B.; Bravata, D.M.; Dai, S.; Ford, E.S.; Fox, C.S.; et al. Heart disease and stroke statistics—2012 update: A report from the american heart association. Circulation 2012, 125, e2–e220. [Google Scholar] [CrossRef]
- Keith, M.E.; Walsh, N.A.; Darling, P.B.; Hanninen, S.A.; Thirugnanam, S.; Leong-Poi, H.; Barr, A.; Sole, M.J. B-vitamin deficiency in hospitalized patients with heart failure. J. Am. Diet. Assoc. 2009, 109, 1406–1410. [Google Scholar] [CrossRef]
- Seligmann, H.; Halkin, H.; Rauchfleisch, S.; Kaufmann, N.; Motro, M.; Vered, Z.; Ezra, D. Thiamine deficiency in patients with congestive heart failure receiving long-term furosemide therapy: A pilot study. Am. J. Med. 1991, 91, 151–155. [Google Scholar] [CrossRef]
- Morrow, L.E.; Grimsley, E.W. Long-term diuretic therapy in hypertensive patients: Effects on serum homocysteine, vitamin B6, vitamin B12, and red blood cell folate concentrations. South. Med. J. 1999, 92, 866–870. [Google Scholar] [CrossRef]
- Brady, J.A.; Rock, C.L.; Horneffer, M.R. Thiamin status, diuretic medications, and the management of congestive heart failure. J. Am. Diet. Assoc. 1995, 95, 541–544. [Google Scholar] [CrossRef]
- Gorelik, O.; Almoznino-Sarafian, D.; Feder, I.; Wachsman, O.; Alon, I.; Litvinjuk, V.; Roshovsky, M.; Modai, D.; Cohen, N. Dietary intake of various nutrients in older patients with congestive heart failure. Cardiology 2003, 99, 177–181. [Google Scholar] [CrossRef]
- Arcand, J.; Floras, V.; Ahmed, M.; Al-Hesayen, A.; Ivanov, J.; Allard, J.P.; Newton, G.E. Nutritional inadequacies in patients with stable heart failure. J. Am. Diet. Assoc. 2009, 109, 1909–1913. [Google Scholar] [CrossRef]
- Catapano, G.; Pedone, C.; Nunziata, E.; Zizzo, A.; Passantino, A.; Incalzi, R.A. Nutrient intake and serum cytokine pattern in elderly people with heart failure. Eur. J. Heart Fail. 2008, 10, 428–434. [Google Scholar] [CrossRef]
- Gao, C.; Zhong, L.; Gao, Y.; Li, X.; Zhang, M.; Wei, S. Cystatin C levels are associated with the prognosis of systolic heart failure patients. Arch. Cardiovasc. Dis. 2011, 104, 565–571. [Google Scholar] [CrossRef]
- Agoston-Coldea, L.; Mocan, T.; Gatfosse, M.; Lupu, S.; Dumitrascu, D.L. Plasma homocysteine and the severity of heart failure in patients with previous myocardial infarction. Cardiol. J. 2011, 18, 55–62. [Google Scholar]
- Okuyan, E.; Uslu, A.; Cakar, M.A.; Sahin, I.; Onur, I.; Enhos, A.; Biter, H.I.; Cetin, S.; Dinckal, M.H. Homocysteine levels in patients with heart failure with preserved ejection fraction. Cardiology 2010, 117, 21–27. [Google Scholar] [CrossRef]
- Washio, T.; Nomoto, K.; Watanabe, I.; Tani, S.; Nagao, K.; Hirayama, A. Relationship between plasma homocysteine levels and congestive heart failure in patients with acute myocardial infarction. Homocysteine and congestive heart failure. Int. Heart J. 2011, 52, 224–228. [Google Scholar] [CrossRef]
- Herrmann, M.; Muller, S.; Kindermann, I.; Gunther, L.; Konig, J.; Bohm, M.; Herrmann, W. Plasma B vitamins and their relation to the severity of chronic heart failure. Am. J. Clin. Nutr. 2007, 85, 117–123. [Google Scholar]
- Maurer, M.; Burri, S.; de Marchi, S.; Hullin, R.; Martinelli, M.; Mohacsi, P.; Hess, O.M. Plasma homocysteine and cardiovascular risk in heart failure with and without cardiorenal syndrome. Int. J. Cardiol. 2010, 141, 32–38. [Google Scholar] [CrossRef]
- Naruszewicz, M.; Jankowska, E.A.; Zymlinski, R.; Bukowska, H.; Millo, B.; Banasiak, W.; Ponikowski, P. Hyperhomocysteinemia in patients with symptomatic chronic heart failure: Prevalence and prognostic importance—Pilot study. Atherosclerosis 2007, 194, 408–414. [Google Scholar] [CrossRef]
- Gueant-Rodriguez, R.M.; Juilliere, Y.; Nippert, M.; Abdelmouttaleb, I.; Herbeth, B.; Aliot, E.; Danchin, N.; Gueant, J.L. Left ventricular systolic dysfunction is an independent predictor of homocysteine in angiographically documented patients with or without coronary artery lesions. J. Thromb. Haemost. 2007, 5, 1209–1216. [Google Scholar] [CrossRef]
- Alter, P.; Rupp, H.; Rominger, M.B.; Figiel, J.H.; Renz, H.; Klose, K.J.; Maisch, B. Association of hyperhomocysteinemia with left ventricular dilatation and mass in human heart. Clin. Chem. Lab. Med. 2010, 48, 555–560. [Google Scholar]
- Nasir, K.; Tsai, M.; Rosen, B.D.; Fernandes, V.; Bluemke, D.A.; Folsom, A.R.; Lima, J.A. Elevated homocysteine is associated with reduced regional left ventricular function: The multi-ethnic study of atherosclerosis. Circulation 2007, 115, 180–187. [Google Scholar]
- Sundstrom, J.; Sullivan, L.; Selhub, J.; Benjamin, E.J.; D’Agostino, R.B.; Jacques, P.F.; Rosenberg, I.H.; Levy, D.; Wilson, P.W.; Vasan, R.S. Relations of plasma homocysteine to left ventricular structure and function: The framingham heart study. Eur. Heart J. 2004, 25, 523–530. [Google Scholar] [CrossRef]
- Vasan, R.S.; Beiser, A.; D’Agostino, R.B.; Levy, D.; Selhub, J.; Jacques, P.F.; Rosenberg, I.H.; Wilson, P.W. Plasma homocysteine and risk for congestive heart failure in adults without prior myocardial infarction. JAMA 2003, 289, 1251–1257. [Google Scholar] [CrossRef]
- Joseph, J.; Washington, A.; Joseph, L.; Koehler, L.; Fink, L.M.; Hauer-Jensen, M.; Kennedy, R.H. Hyperhomocysteinemia leads to adverse cardiac remodeling in hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H2567–H2574. [Google Scholar]
- Joseph, J.; Joseph, L.; Shekhawat, N.S.; Devi, S.; Wang, J.; Melchert, R.B.; Hauer-Jensen, M.; Kennedy, R.H. Hyperhomocysteinemia leads to pathological ventricular hypertrophy in normotensive rats. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H679–H686. [Google Scholar]
- Kennedy, R.H.; Melchert, R.B.; Joseph, J. Cardiovascular effects of hyperhomocysteinemia in conscious unrestrained rats. Am. J. Hypertens. 2006, 19, 94–97. [Google Scholar] [CrossRef]
- Devi, S.; Kennedy, R.H.; Joseph, L.; Shekhawat, N.S.; Melchert, R.B.; Joseph, J. Effect of long-term hyperhomocysteinemia on myocardial structure and function in hypertensive rats. Cardiovasc. Pathol. 2006, 15, 75–82. [Google Scholar]
- Miller, A.; Mujumdar, V.; Palmer, L.; Bower, J.D.; Tyagi, S.C. Reversal of endocardial endothelial dysfunction by folic acid in homocysteinemic hypertensive rats. Am. J. Hypertens. 2002, 15, 157–163. [Google Scholar] [CrossRef]
- Walker, E.; Black, J.; Parris, C.; Bryda, E.C.; Cansino, S.; Hunt, L.; Chappell, J.; Wehner, P.; Studeny, M.; Wright, G.L. Effect of experimental hyperhomocysteinemia on cardiac structure and function in the rat. Ann. Clin. Lab. Sci. 2004, 34, 175–180. [Google Scholar]
- Joseph, J.; Pencina, M.J.; Wang, T.J.; Hayes, L.; Tofler, G.H.; Jacques, P.; Selhub, J.; Levy, D.; D’Agostino, R.B., Sr.; Benjamin, E.J.; et al. Cross-sectional relations of multiple biomarkers representing distinct biological pathways to plasma markers of collagen metabolism in the community. J. Hypertens. 2009, 27, 1317–1324. [Google Scholar] [CrossRef]
- Herrmann, M.; Stanger, O.; Paulweber, B.; Hufnagl, C.; Herrmann, W. Effect of folate supplementation on N-terminal pro-brain natriuretic peptide. Int. J. Cardiol. 2007, 118, 267–269. [Google Scholar] [CrossRef]
- Witte, K.K.; Nikitin, N.P.; Parker, A.C.; von Haehling, S.; Volk, H.D.; Anker, S.D.; Clark, A.L.; Cleland, J.G. The effect of micronutrient supplementation on quality-of-life and left ventricular function in elderly patients with chronic heart failure. Eur. Heart J. 2005, 26, 2238–2244. [Google Scholar] [CrossRef]
- Loscalzo, J. Oxidant stress: A key determinant of atherothrombosis. Biochem. Soc. Trans. 2003, 31, 1059–1061. [Google Scholar] [CrossRef]
- Rajasekaran, N.S.; Connell, P.; Christians, E.S.; Yan, L.J.; Taylor, R.P.; Orosz, A.; Zhang, X.Q.; Stevenson, T.J.; Peshock, R.M.; Leopold, J.A.; et al. Human αB-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell 2007, 130, 427–439. [Google Scholar] [CrossRef]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef]
- Metes-Kosik, N.; Luptak, I.; Dibello, P.M.; Handy, D.E.; Tang, S.S.; Zhi, H.; Qin, F.; Jacobsen, D.W.; Loscalzo, J.; Joseph, J. Both selenium deficiency and modest selenium supplementation lead to myocardial fibrosis in mice via effects on redox-methylation balance. Mol. Nutr. Food Res. 2012, 56, 1812–1824. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Joseph, J.; Loscalzo, J. Methoxistasis: Integrating the Roles of Homocysteine and Folic Acid in Cardiovascular Pathobiology. Nutrients 2013, 5, 3235-3256. https://doi.org/10.3390/nu5083235
Joseph J, Loscalzo J. Methoxistasis: Integrating the Roles of Homocysteine and Folic Acid in Cardiovascular Pathobiology. Nutrients. 2013; 5(8):3235-3256. https://doi.org/10.3390/nu5083235
Chicago/Turabian StyleJoseph, Jacob, and Joseph Loscalzo. 2013. "Methoxistasis: Integrating the Roles of Homocysteine and Folic Acid in Cardiovascular Pathobiology" Nutrients 5, no. 8: 3235-3256. https://doi.org/10.3390/nu5083235