Experimental Alcohol-Related Peripheral Neuropathy: Role of Insulin/IGF Resistance

Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Chronic Ethanol Exposure Model
2.3. Electrophysiology
2.4. Histology and Morphometric Analysis
2.5. Quantitative Reverse Transcriptase Polymerase Chain Reaction (qRT-PCR) Assays of Gene Expression
2.6. Enzyme-Linked Immunosorbant Assay (ELISA)
2.7. Statistical Analysis
3. Results
3.1. General Effects of Ethanol Feeding

{kind=link}
{kind=link}
| Variable | Control | Ethanol | p-Value |
|---|---|---|---|
| Initial body wt (g) | 361.0 ± 31.5 | 353.1 ± 24.7 | |
| Final body wt (g) | 454.1 ± 18.4 | 423.8 ± 39.6 | |
| % Body wt gain | 21.04 ± 6.2 | 19.59 ± 8.4 | |
| Blood alcohol (mg/dL) | 2.62 ± 0.9 | 129.9 ± 12.0 | p < 0.0001 |
3.2. Effects of Ethanol on Nerve Conduction
| Nerve | Control | Ethanol | Control | Ethanol |
|---|---|---|---|---|
| Latency | Latency | Amplitude | Amplitude | |
| Plantar | 0.91 ± 0.01 | 0.96 ± 0.03 | 77.29 ± 8.92 | 78.49 ± 11.18 |
| Tibialis (Ankle) | 1.24 ± 0.06 | 1.18 ± 0.08 | 4.37 ± 1.28 | 4.85 ± 0.45 |
| Tibialis (Knee) | 1.69 ± 0.05 | 2.00 ± 0.07 * | 3.97 ± 1.13 | 4.58 ± 0.50 |
| Peroneal (Ankle) | 1.23 ± 0.07 | 1.11 ± 0.08 | 5.68 ± 0.81 | 5.76 ± 0.86 |
| Peroneal (Knee) | 1.71 ± 0.09 | 1.81 ± 0.08 | 4.94 ± 0.58 | 5.42 ± 0.83 |
3.3. Histopathology of Alcohol-Related Polyneuropathy
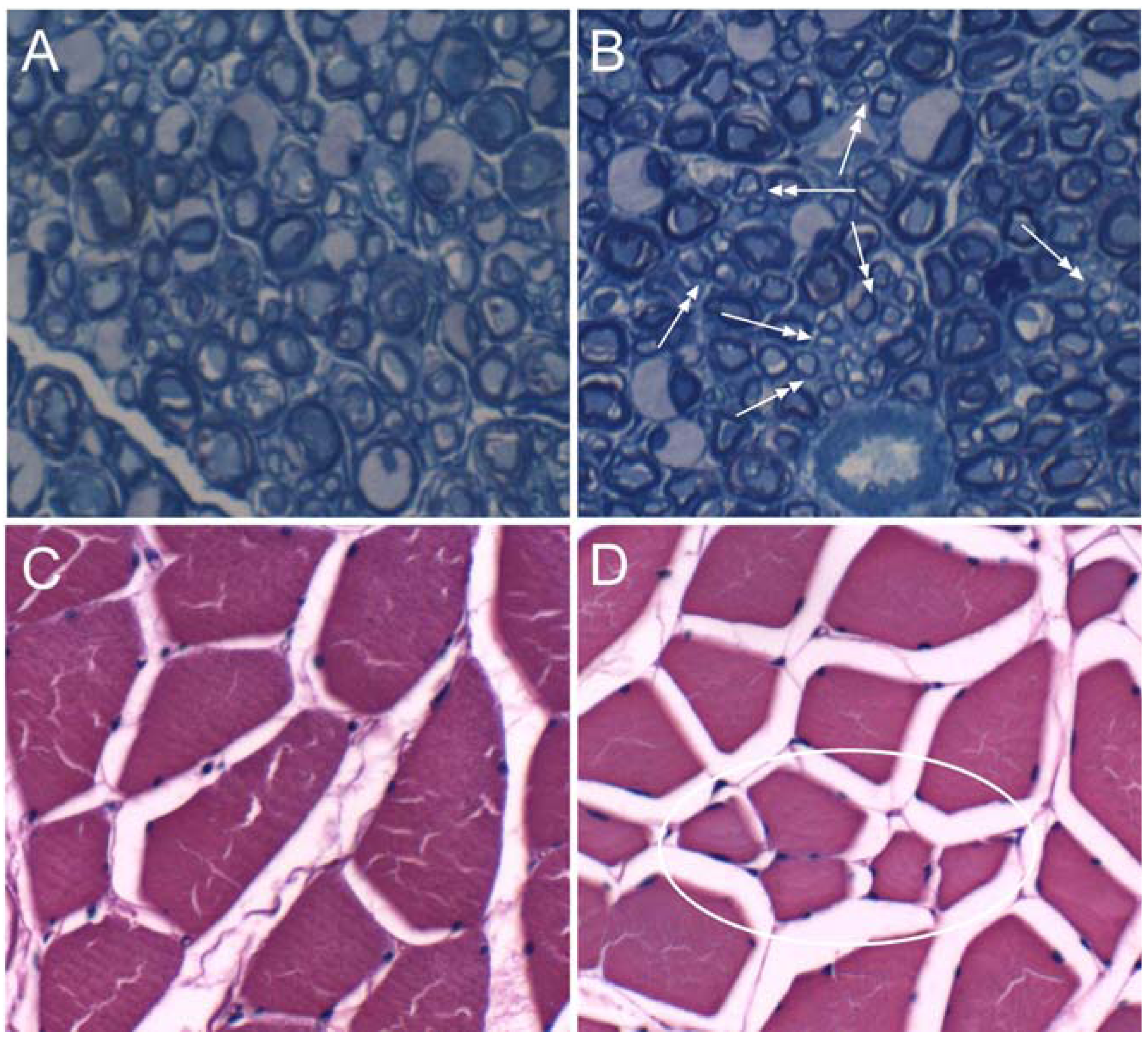
| Variable | Control | Ethanol | p-Value |
|---|---|---|---|
| Sciatic nerve diameter (µm) | 16.48 ± 1.92 | 11.48 ± 0.91 | 0.007 |
| Gastrocnemius fiber diameter (µm) | 45.83 ± 5.08 | 25.34 ± 4.38 | 0.0001 |
| Gastrocnemius fiber area (µm2) | 2338.64 ± 643.04 | 695.08 ± 163.35 | 0.0005 |
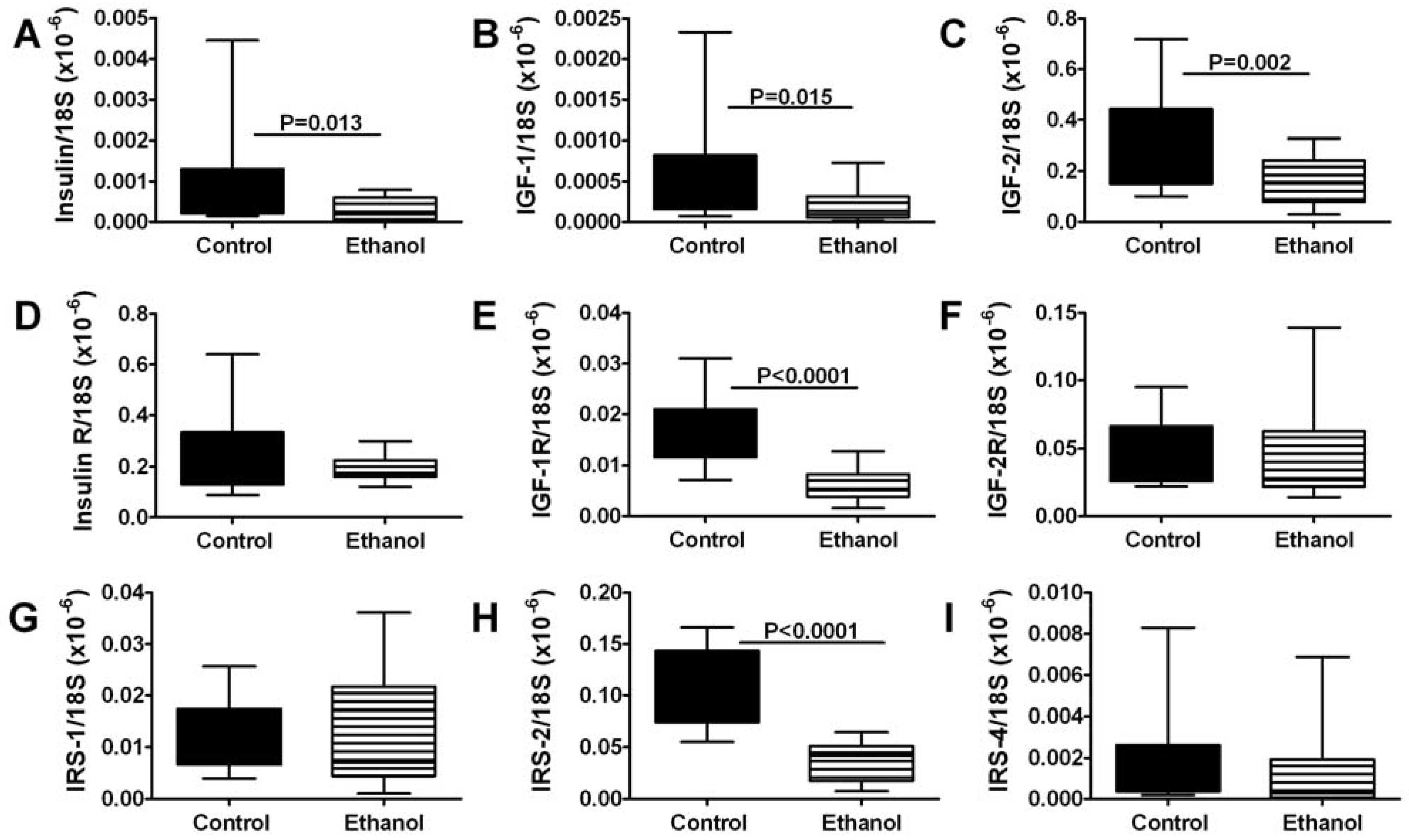
3.4. Ethanol Effects on Insulin/IGF Signaling Pathway Genes-qRT-PCR Studies
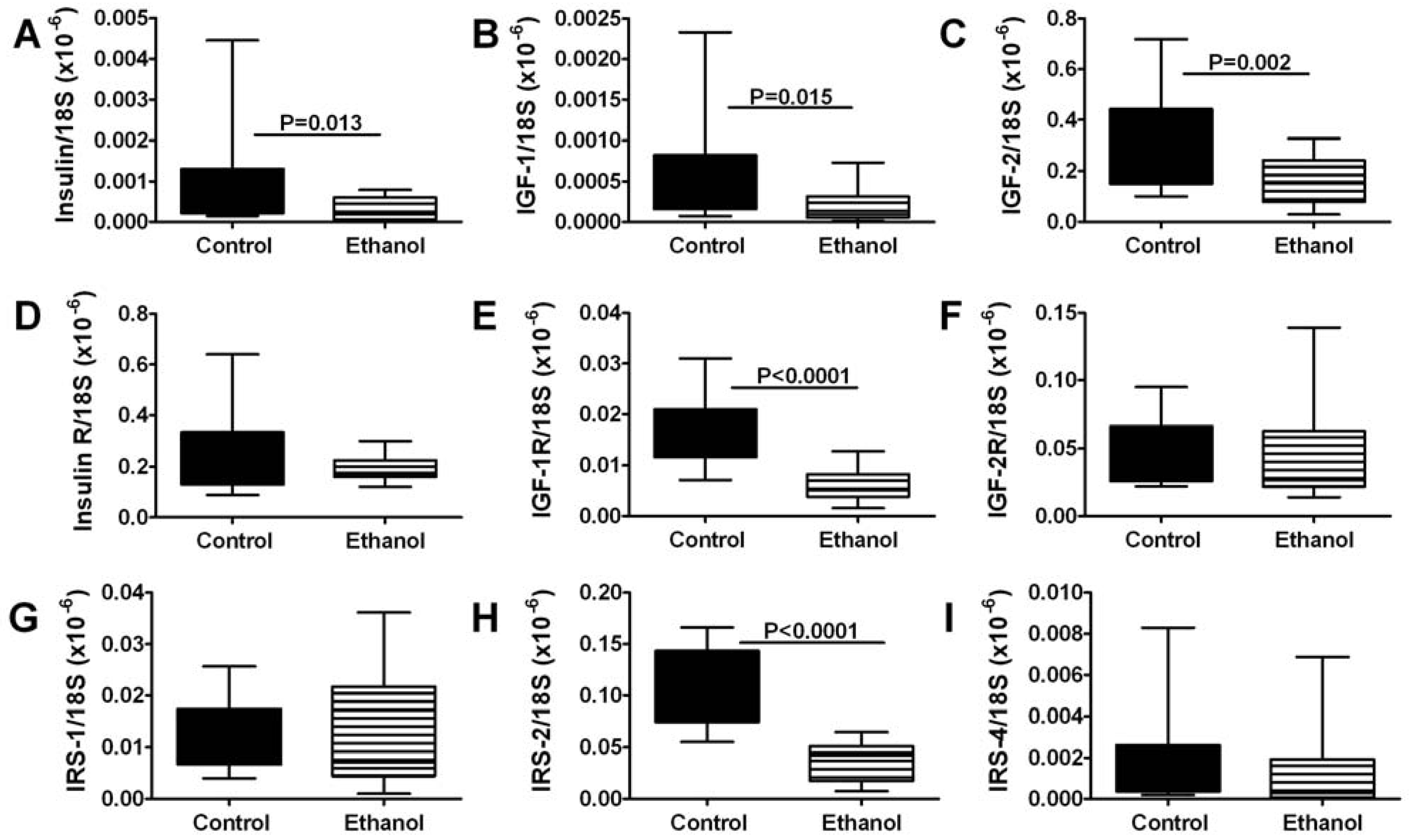
3.5. Ethanol Effects on Insulin/IGF Signaling Molecules-ELISA Studies
| Protein | Control | Ethanol | p-Value |
|---|---|---|---|
| INS-R | 37839.22 ± 2652.53 | 34612.68 ± 2319.60 | 2.41056 × 10−5 |
| IGF-1R | 39189.66 ± 2245.76 | 36973.84 ± 2254.17 | 0.000678 |
| IRS-1 | 1254.47 ± 83.49 | 1220.34 ± 38.77 | 0.0379 |
| IRS-2 | 1774.17 ± 201.19 | 1691.24 ± 148.64 | 0.0556 |
| IRS-4 | 2022.31 ± 217.02 | 1798.44 ± 273.98 | 0.0015 |
| MAG-1 | 21651.73 ± 1481.85 | 20425.26 ± 1157.55 | 0.001268 |
| Tau | 40046.09 ± 6460.38 | 35090.79 ± 5809.52 | 0.003782 |
3.6. Ethanol Effects on Peripheral Nerve Structural Proteins and Oxidative Stress
4. Discussion
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Mellion, M.; Gilchrist, J.M.; Nguyen, V.A.; de la Monte, S.M. Alcohol-related peripheral neuropathy (ALN): Nutritional, toxic or both? Muscle Nerve 2011, 43, 306–316. [Google Scholar]
- Laker, S.A.; Sullivan, W.J. Alcoholic neuropathy. Available online: http://emedicine.medscape.com/article/315159 (accessed on 19 October 2010).
- Ammendola, A.; Gemini, D.; Iannaccone, S.; Argenzio, F.; Ciccone, G.; Ammendola, E.; Serio, L.; Ugolini, G.; Bravaccio, F. Gender and peripheral neuropathy in chronic alcoholism: A clinical-electroneurographic study. Alcohol Alcohol. 2000, 35, 368–371. [Google Scholar]
- Bales, J.G.; Meals, R. Peripheral neuropathy of the upper extremity: Medical comorbidity that confounds common orthopedic pathology. Orthopedics 2009, 32. [Google Scholar] [CrossRef]
- Monforte, R.; Estruch, R.; Valls-Sole, J.; Nicolas, J.; Villalta, J.; Urbano-Marquez, A. Autonomic and peripheral neuropathies in patients with chronic alcoholism. A dose-related toxic effect of alcohol. Arch. Neurol. 1995, 52, 45–51. [Google Scholar] [CrossRef]
- Preedy, V.R.; Adachi, J.; Ueno, Y.; Ahmed, S.; Mantle, D.; Mullatti, N.; Rajendram, R.; Peters, T.J. Alcoholic skeletal muscle myopathy: Definitions, features, contribution of neuropathy, impact and diagnosis. Eur. J. Neurol. 2001, 8, 677–687. [Google Scholar] [CrossRef]
- Schuckit, M.A. Alcohol-use disorders. Lancet 2009, 373, 492–501. [Google Scholar]
- Hattori, N.; Koike, H.; Sobue, G. Metabolic and nutritional neuropathy. Rinsho Shinkeigaku 2008, 48, 1026–1027. [Google Scholar] [CrossRef]
- Koike, H.; Iijima, M.; Sugiura, M.; Mori, K.; Hattori, N.; Ito, H.; Hirayama, M.; Sobue, G. Alcoholic neuropathy is clinicopathologically distinct from thiamine-deficiency neuropathy. Ann. Neurol. 2003, 54, 19–29. [Google Scholar] [CrossRef]
- Koike, H.; Ito, S.; Morozumi, S.; Kawagashira, Y.; Iijima, M.; Hattori, N.; Tanaka, F.; Sobue, G. Rapidly developing weakness mimicking Guillain-Barre syndrome in beriberi neuropathy: Two case reports. Nutrition 2008, 24, 776–780. [Google Scholar] [CrossRef]
- Koike, H.; Iijima, M.; Mori, K.; Hattori, N.; Ito, H.; Hirayama, M.; Sobue, G. Postgastrectomy polyneuropathy with thiamine deficiency is identical to beriberi neuropathy. Nutrition 2004, 20, 961–966. [Google Scholar] [CrossRef]
- Hoyumpa, A.M., Jr.; Breen, K.J.; Schenker, S.; Wilson, F.A. Thiamine transport across the rat intestine. II. Effect of ethanol. J. Lab. Clin. Med. 1975, 86, 803–816. [Google Scholar]
- Thomson, A.D.; Baker, H.; Leevy, C.M. Patterns of 35S-thiamine hydrochloride absorption in the malnourished alcoholic patient. J. Lab. Clin. Med. 1970, 76, 34–45. [Google Scholar]
- Tomasulo, P.A.; Kater, R.M.; Iber, F.L. Impairment of thiamine absorption in alcoholism. Am. J. Clin. Nutr. 1968, 21, 1341–1344. [Google Scholar]
- Frank, O.; Luisada-Opper, A.; Sorrell, M.F.; Thomson, A.D.; Baker, H. Vitamin deficits in severe alcoholic fatty liver of man calculated from multiple reference units. Exp. Mol. Pathol. 1971, 15, 191–197. [Google Scholar] [CrossRef]
- Leevy, C.M.; Baker, H.; Tenhove, W.; Frank, O.; Cherrick, G.R. B-complex vitamins in liver disease of the alcoholic. Am. J. Clin. Nutr. 1965, 16, 339–346. [Google Scholar]
- Abe, T.; Itokawa, Y. Effect of ethanol administration on thiamine metabolism and transketolase activity in rats. Int. J. Vitam. Nutr. Res. 1977, 47, 307–314. [Google Scholar]
- Fennelly, J.; Frank, O.; Baker, H.; Leevy, C.M. Red blood cell-transketolase activity in malnourished alcoholics with cirrhosis. Am. J. Clin. Nutr. 1967, 20, 946–949. [Google Scholar]
- Rindi, G.; Comincioli, V.; Reggiani, C.; Patrini, C. Nervous tissue thiamine metabolism in vivo. III. Influence of ethanol intake on the dynamics of thiamine and its phosphoesters in different brain regions and sciatic nerve of the rat. Brain Res. 1987, 413, 23–35. [Google Scholar] [CrossRef]
- Ang, C.D.; Alviar, M.J.; Dans, A.L.; Bautista-Velez, G.G.; Villaruz-Sulit, M.V.; Tan, J.J.; Co, H.U.; Bautista, M.R.; Roxas, A.A. Vitamin B for treating peripheral neuropathy. Cochrane Database Syst. Rev. 2008. [Google Scholar] [CrossRef]
- Peters, T.J.; Kotowicz, J.; Nyka, W.; Kozubski, W.; Kuznetsov, V.; Vanderbist, F.; de Niet, S.; Marcereuil, D.; Coffiner, M. Treatment of alcoholic polyneuropathy with vitamin B complex: A randomised controlled trial. Alcohol Alcohol. 2006, 41, 636–642. [Google Scholar]
- Woelk, H.; Lehrl, S.; Bitsch, R.; Kopcke, W. Benfotiamine in treatment of alcoholic polyneuropathy: An 8-week randomized controlled study (BAP I Study). Alcohol Alcohol. 1998, 33, 631–638. [Google Scholar]
- Koike, H.; Misu, K.; Hattori, N.; Ito, S.; Ichimura, M.; Ito, H.; Hirayama, M.; Nagamatsu, M.; Sasaki, I.; Sobue, G. Postgastrectomy polyneuropathy with thiamine deficiency. J. Neurol. Neurosurg. Psychiatry 2001, 71, 357–362. [Google Scholar] [CrossRef]
- Koike, H.; Mori, K.; Misu, K.; Hattori, N.; Ito, H.; Hirayama, M.; Sobue, G. Painful alcoholic polyneuropathy with predominant small-fiber loss and normal thiamine status. Neurology 2001, 56, 1727–1732. [Google Scholar] [CrossRef]
- Koike, H.; Sobue, G. Alcoholic neuropathy. Curr. Opin. Neurol. 2006, 19, 481–486. [Google Scholar] [CrossRef]
- Cohen, A.C.; Tong, M.; Wands, J.R.; de la Monte, S.M. Insulin and insulin-like growth factor resistance with neurodegeneration in an adult chronic ethanol exposure model. Alcohol. Clin. Exp. Res. 2007, 31, 1558–1573. [Google Scholar] [CrossRef]
- De la Monte, S.M.; Longato, L.; Tong, M.; DeNucci, S.; Wands, J.R. The liver-brain axis of alcohol-mediated neurodegeneration: role of toxic lipids. Int. J. Environ. Res. Public Health 2009, 6, 2055–2075. [Google Scholar]
- De la Monte, S.M.; Tong, M.; Cohen, A.C.; Sheedy, D.; Harper, C.; Wands, J.R. Insulin and insulin-like growth factor resistance in alcoholic neurodegeneration. Alcohol. Clin. Exp. Res. 2008, 32, 1630–1644. [Google Scholar] [CrossRef]
- De la Monte, S.M.; Yeon, J.E.; Tong, M.; Longato, L.; Chaudhry, R.; Pang, M.Y.; Duan, K.; Wands, J.R. Insulin resistance in experimental alcohol-induced liver disease. J. Gastroenterol. Hepatol. 2008, 23, e477–e486. [Google Scholar] [CrossRef]
- De la Monte, S.M.; Wands, J.R. Review of insulin and insulin-like growth factor expression, signaling, and malfunction in the central nervous system: Relevance to Alzheimer’s disease. J. Alzheimers Dis. 2005, 7, 45–61. [Google Scholar]
- Chesik, D.; de Keyser, J.; Wilczak, N. Insulin-like growth factor system regulates oligodendroglial cell behavior: Therapeutic potential in CNS. J. Mol. Neurosci. 2008, 35, 81–90. [Google Scholar] [CrossRef]
- D’Ercole, A.J.; Ye, P. Expanding the mind: Insulin-like growth factor I and brain development. Endocrinology 2008, 149, 5958–5962. [Google Scholar] [CrossRef]
- Ogata, T.; Yamamoto, S.; Nakamura, K.; Tanaka, S. Signaling axis in schwann cell proliferation and differentiation. Mol. Neurobiol. 2006, 33, 51–62. [Google Scholar] [CrossRef]
- Pillion, D.J.; Kim, S.J.; Kim, H.; Meezan, E. Insulin signal transduction: The role of protein phosphorylation. Am. J. Med. Sci. 1992, 303, 40–52. [Google Scholar] [CrossRef]
- Jung-Testas, I.; Baulieu, E.E. Steroid hormone receptors and steroid action in rat glial cells of the central and peripheral nervous system. J. Steroid Biochem. Mol. Biol. 1998, 65, 243–251. [Google Scholar] [CrossRef]
- Lancaster, F.E. Alcohol and white matter development—A review. Alcohol. Clin. Exp. Res. 1994, 18, 644–647. [Google Scholar] [CrossRef]
- Lewohl, J.M.; Dodd, P.R.; Mayfield, R.D.; Harris, R.A. Application of DNA microarrays to study human alcoholism. J. Biomed. Sci. 2001, 8, 28–36. [Google Scholar]
- Lewohl, J.M.; Wang, L.; Miles, M.F.; Zhang, L.; Dodd, P.R.; Harris, R.A. Gene expression in human alcoholism: Microarray analysis of frontal cortex. Alcohol. Clin. Exp. Res. 2000, 24, 1873–1882. [Google Scholar] [CrossRef]
- Butt, A.M.; Berry, M. Oligodendrocytes and the control of myelination in vivo: New insights from the rat anterior medullary velum. J. Neurosci. Res. 2000, 59, 477–488. [Google Scholar]
- D’Ercole, A.J.; Ye, P.; Calikoglu, A.S.; Gutierrez-Ospina, G. The role of the insulin-like growth factors in the central nervous system. Mol. Neurobiol. 1996, 13, 227–255. [Google Scholar] [CrossRef]
- McMorris, F.A.; McKinnon, R.D. Regulation of oligodendrocyte development and CNS myelination by growth factors: Prospects for therapy of demyelinating disease. Brain Pathol. 1996, 6, 313–329. [Google Scholar] [CrossRef]
- Zumkeller, W. The effect of insulin-like growth factors on brain myelination and their potential therapeutic application in myelination disorders. Eur. J. Paediatr. Neurol. 1997, 1, 91–101. [Google Scholar] [CrossRef]
- Liang, G.; Cline, G.W.; Macica, C.M. IGF-1 stimulates de novo fatty acid biosynthesis by Schwann cells during myelination. Glia 2007, 55, 632–641. [Google Scholar] [CrossRef]
- De la Monte, S.M.; Pang, M.; Chaudhry, R.; Duan, K.; Longato, L.; Carter, J.J.; Ouh, J.; Wands, J.R. Peroxisome proliferator-activated receptor agonist treatment of alcohol-induced hepatic insulin resistance. Hepatol. Res. 2011, 41, 386–398. [Google Scholar] [CrossRef]
- De la Monte, S.M.; Wands, J.R. Role of central nervous system insulin resistance in fetal alcohol spectrum disorders. J. Popul. Ther. Clin. Pharmacol. 2010, 17, e390–e404. [Google Scholar]
- Pang, M.; de la Monte, S.M.; Longato, L.; Tong, M.; He, J.; Chaudhry, R.; Duan, K.; Ouh, J.; Wands, J.R. PPARdelta agonist attenuates alcohol-induced hepatic insulin resistance and improves liver injury and repair. J. Hepatol. 2009, 50, 1192–1201. [Google Scholar] [CrossRef]
- DeNucci, S.; Tong, M.; Longato, L.; Lawton, M.; Setshedi, M.; Carlson, R.; Wands, J.R.; de la Monte, S.M. Rat strain differences in susceptibility to alcohol-induced chronic liver injury and hepatic insulin resistance. Gastroenterol. Res. Pract. 2010, 2010. [Google Scholar] [CrossRef]
- Kurokawa, K.; de Almeida, D.F.; Zhang, Y.; Hebert, C.D.; Page, J.G.; Schweikart, K.M.; Oh, S.J. Sensory nerve conduction of the plantar nerve compared with other nerve conduction tests in rats. Clin. Neurophysiol. 2004, 115, 1677–1682. [Google Scholar] [CrossRef]
- Oh, S.J. Electromyographic studies in peripheral nerve injuries. South Med. J. 1976, 69, 177–182. [Google Scholar] [CrossRef]
- Gundogan, F.; Elwood, G.; Longato, L.; Tong, M.; Feijoo, A.; Carlson, R.I.; Wands, J.R.; de la Monte, S.M. Impaired placentation in fetal alcohol syndrome. Placenta 2008, 29, 148–157. [Google Scholar] [CrossRef]
- Soscia, S.J.; Tong, M.; Xu, X.J.; Cohen, A.C.; Chu, J.; Wands, J.R.; de la Monte, S.M. Chronic gestational exposure to ethanol causes insulin and IGF resistance and impairs acetylcholine homeostasis in the brain. Cell. Mol. Life Sci. 2006, 63, 2039–2056. [Google Scholar] [CrossRef]
- Zambelis, T.; Karandreas, N.; Tzavellas, E.; Kokotis, P.; Liappas, J. Large and small fiber neuropathy in chronic alcohol-dependent subjects. J. Peripher. Nerv. Syst. 2005, 10, 375–381. [Google Scholar] [CrossRef]
- Hoyumpa, A.M., Jr. Mechanisms of thiamin deficiency in chronic alcoholism. Am. J. Clin. Nutr. 1980, 33, 2750–2761. [Google Scholar]
- Ronis, M.J.; Wands, J.R.; Badger, T.M.; de la Monte, S.M.; Lang, C.H.; Calissendorff, J. Alcohol-induced disruption of endocrine signaling. Alcohol. Clin. Exp. Res. 2007, 31, 1269–1285. [Google Scholar] [CrossRef]
- Schumacher, M.; Jung-Testas, I.; Robel, P.; Baulieu, E.E. Insulin-like growth factor I: A mitogen for rat Schwann cells in the presence of elevated levels of cyclic AMP. Glia 1993, 8, 232–240. [Google Scholar] [CrossRef]
- Bambrick, L.L.; Braun, P.E. Phosphorylation of myelin-associated glycoprotein in cultured oligodendrocytes. Dev. Neurosci. 1991, 13, 412–416. [Google Scholar] [CrossRef]
- Hong, M.; Lee, V.M. Insulin and insulin-like growth factor-1 regulate tau phosphorylation in cultured human neurons. J. Biol. Chem. 1997, 272, 19547–19553. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nguyen, V.A.; Le, T.; Tong, M.; Mellion, M.; Gilchrist, J.; De la Monte, S.M. Experimental Alcohol-Related Peripheral Neuropathy: Role of Insulin/IGF Resistance. Nutrients 2012, 4, 1042-1057. https://doi.org/10.3390/nu4081042
Nguyen VA, Le T, Tong M, Mellion M, Gilchrist J, De la Monte SM. Experimental Alcohol-Related Peripheral Neuropathy: Role of Insulin/IGF Resistance. Nutrients. 2012; 4(8):1042-1057. https://doi.org/10.3390/nu4081042
Chicago/Turabian StyleNguyen, Van Anh, Tran Le, Ming Tong, Michelle Mellion, James Gilchrist, and Suzanne M. De la Monte. 2012. "Experimental Alcohol-Related Peripheral Neuropathy: Role of Insulin/IGF Resistance" Nutrients 4, no. 8: 1042-1057. https://doi.org/10.3390/nu4081042