Alcohol Exposure Alters Mouse Lung Inflammation in Response to Inhaled Dust

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Mice
2.2. Organic Dust Exposure
2.3. Alcohol Feeding
2.4. Bronchoalveolar Lavage (BAL)
2.5. Lung Histology
2.6. PKC Isoform Assay
2.7. Cytokine Assays
2.8. Lung Slice Protocol
2.9. Statistical Analyses
3. Results
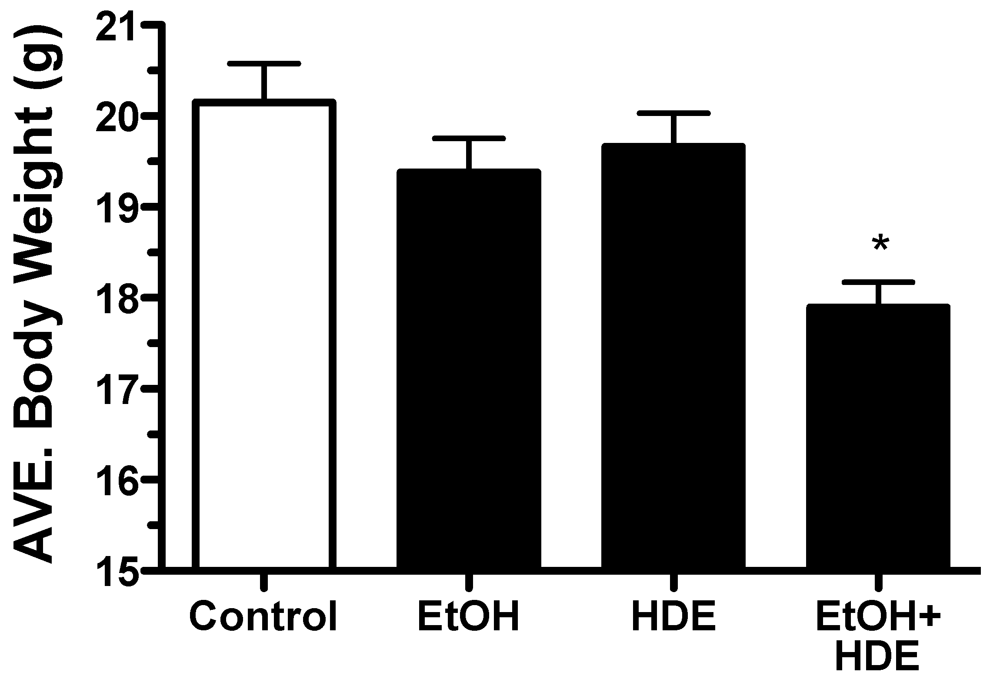
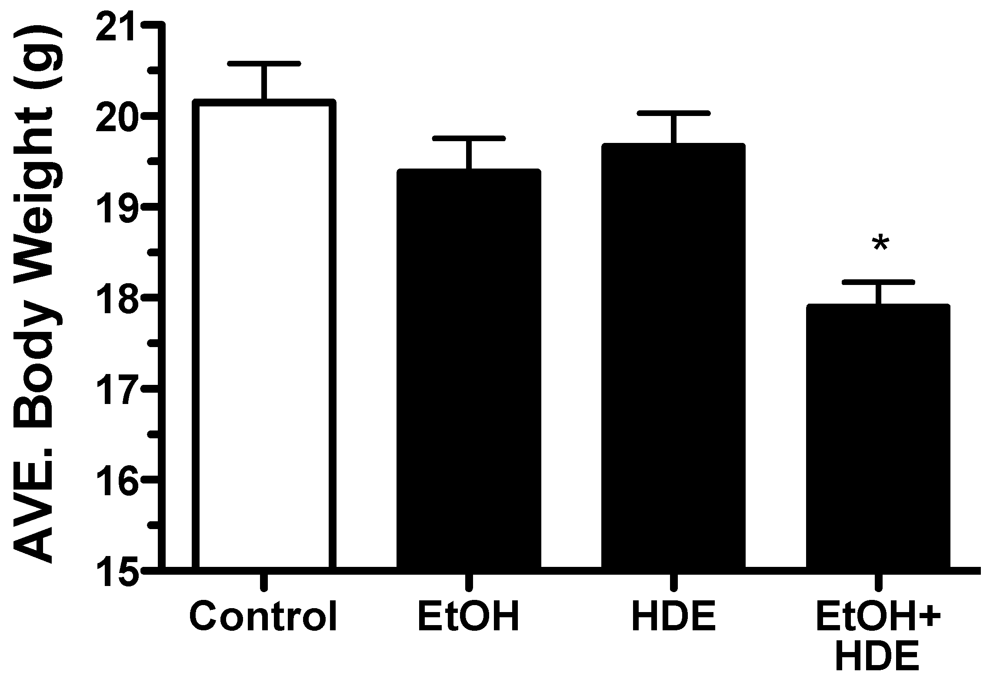
3.1. Body Weights and Mortality
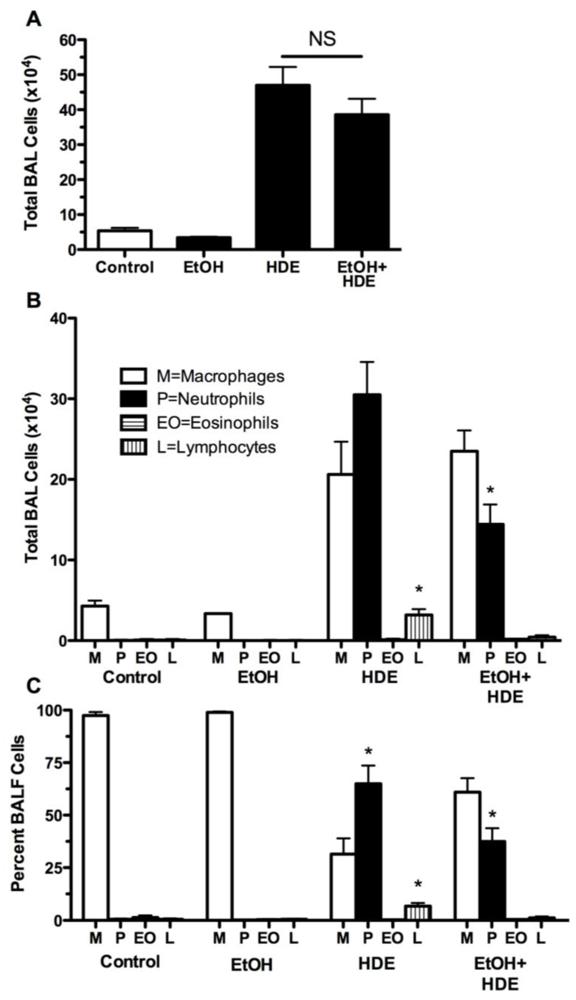
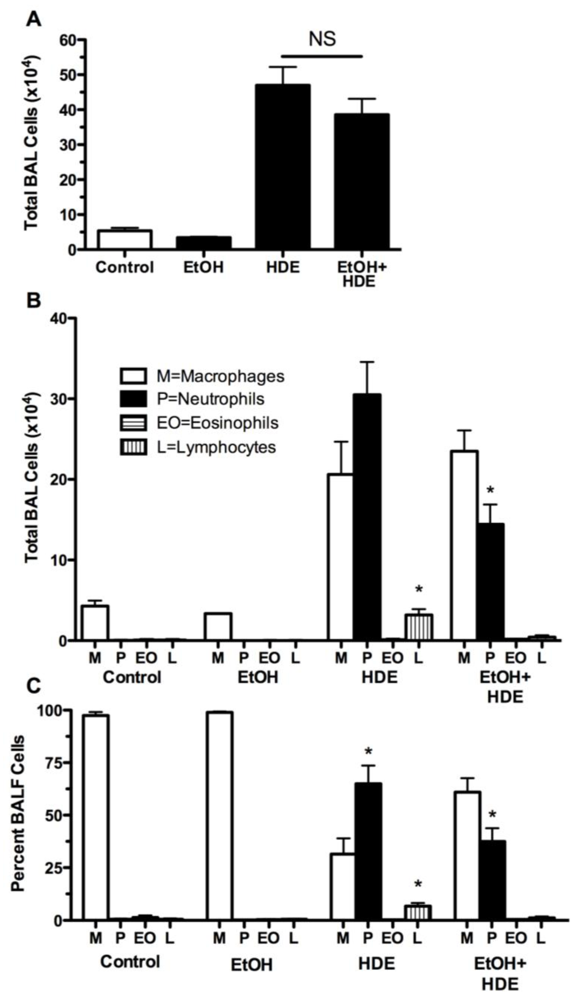
3.2. Inflammatory Cells
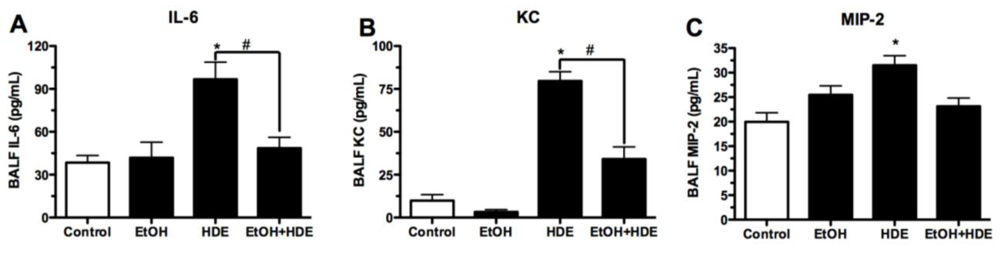
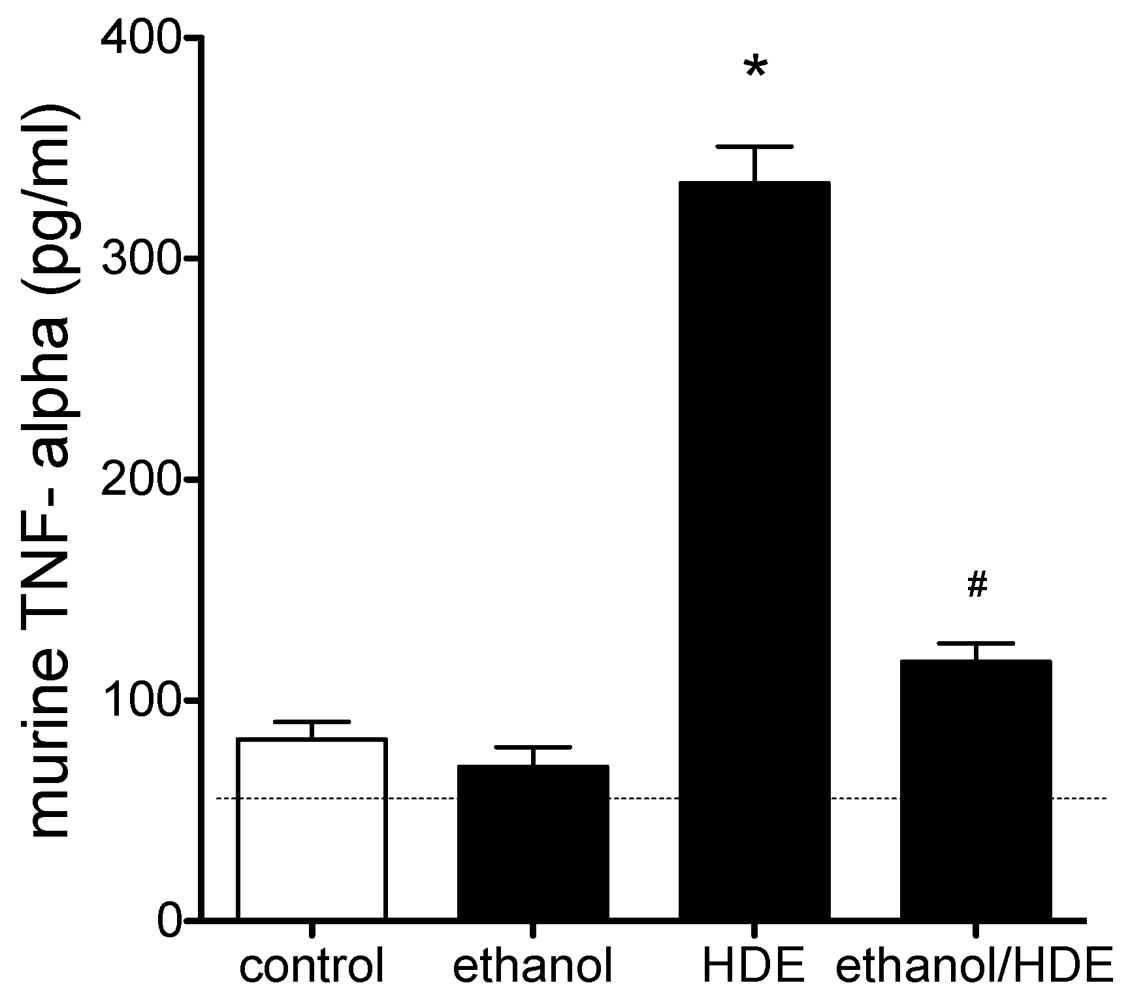
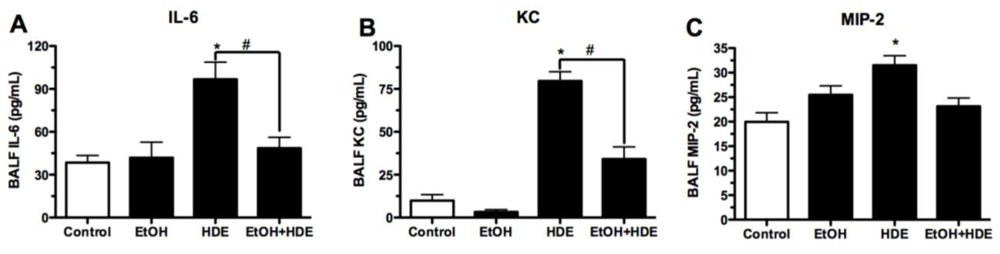
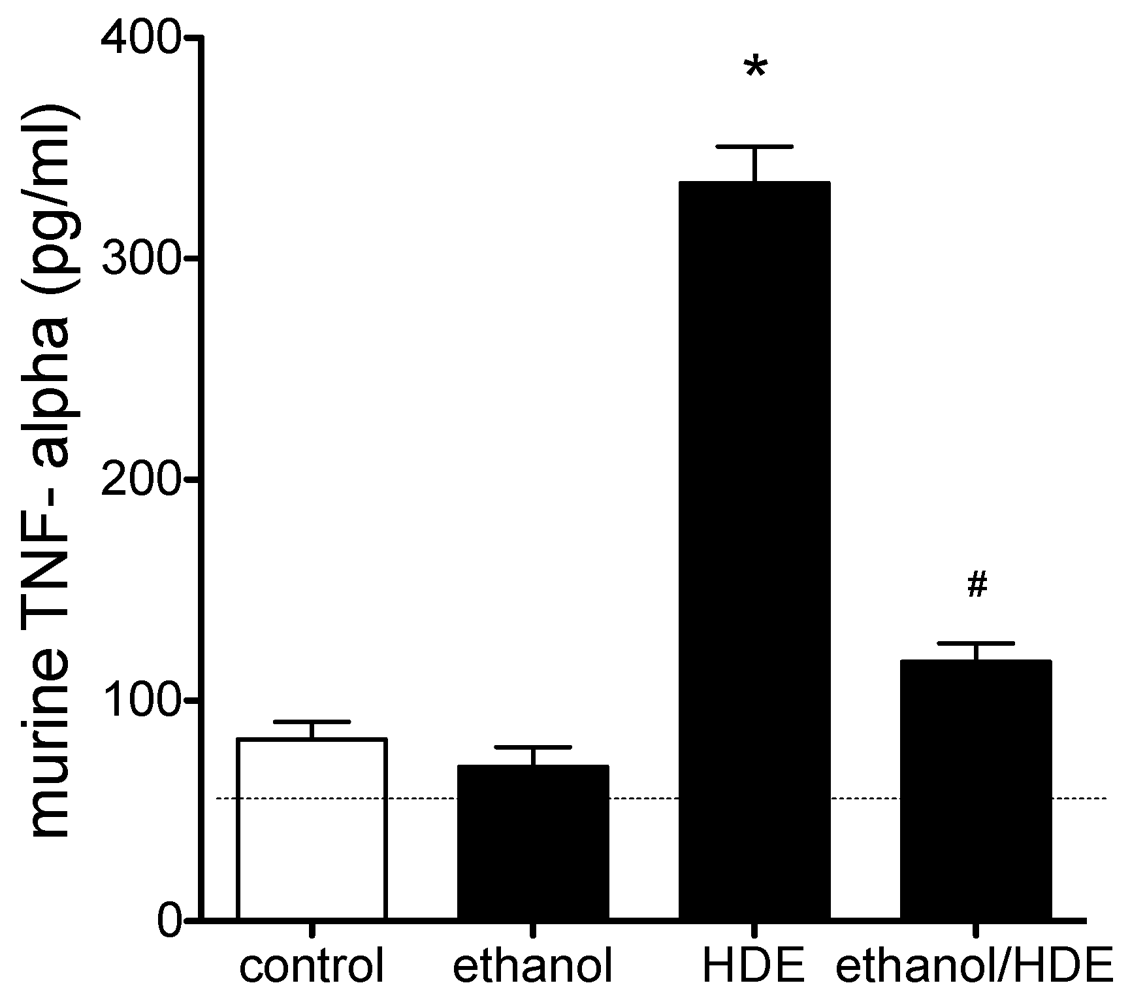
3.3. Cytokines
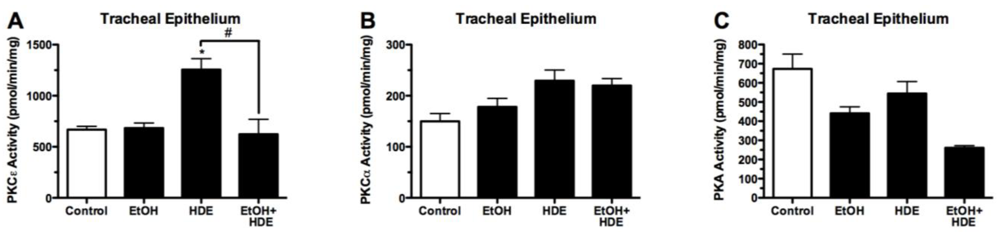
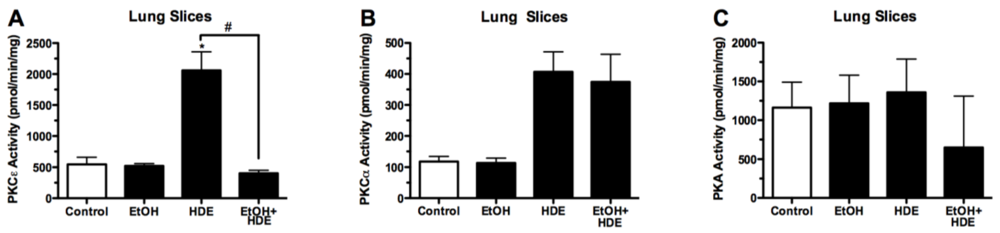
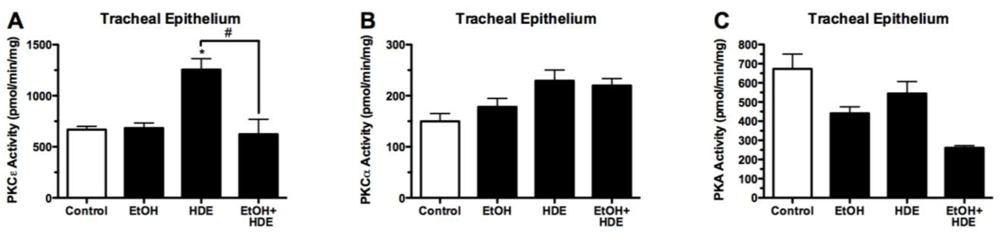
3.4. Protein Kinase Activity

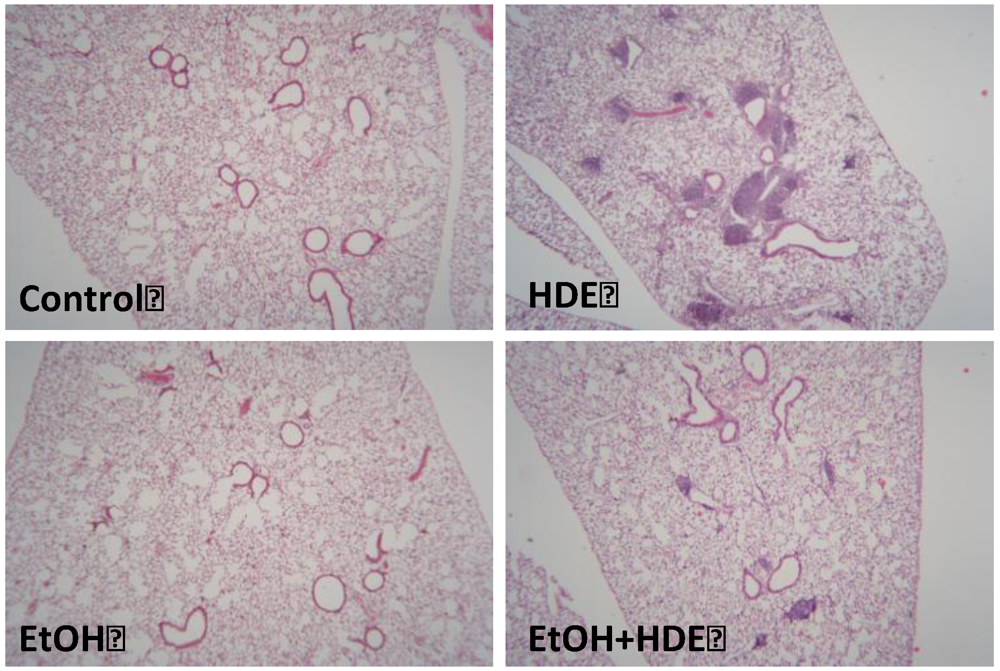
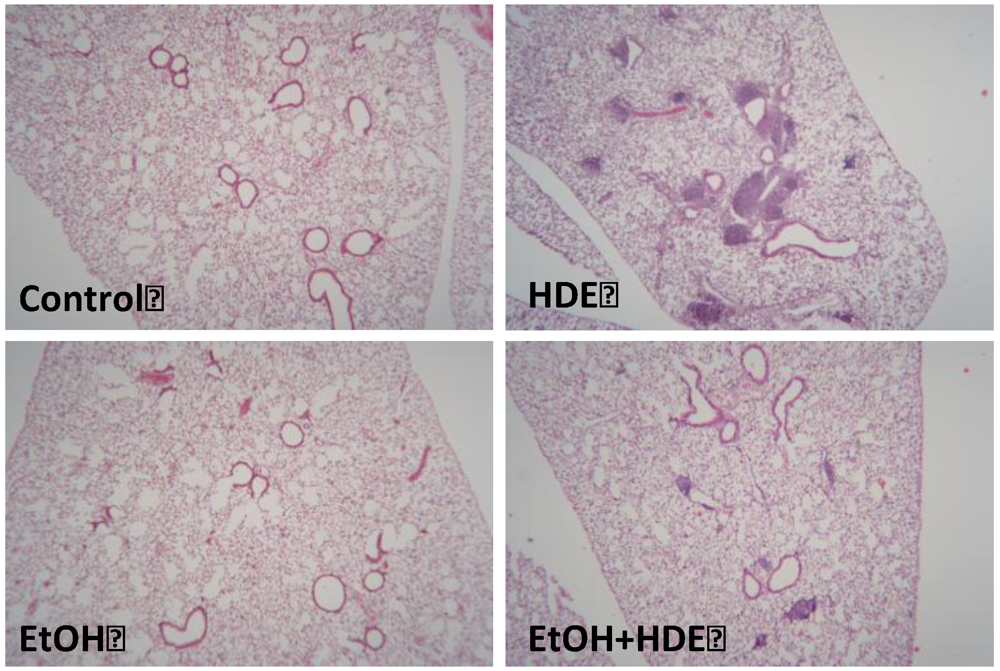
3.5. Lung Histology
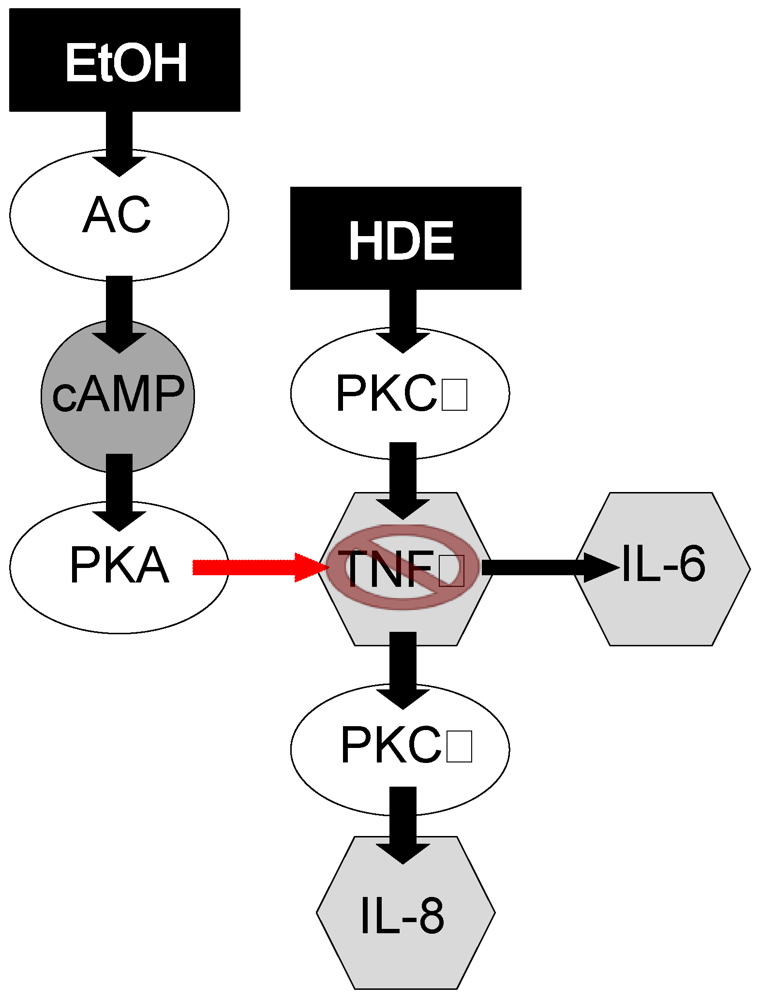
4. Discussion
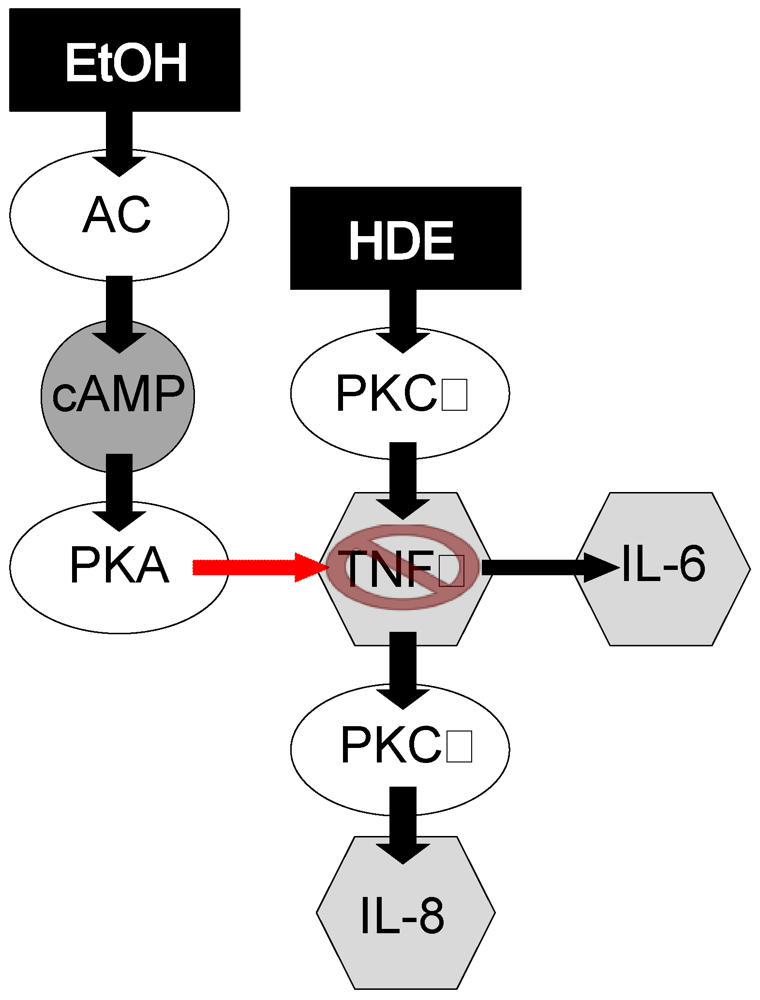
5. Conclusion
Acknowledgments
Conflict of Interest
References
- Waldschmidt, T.J.; Cook, R.T.; Kovacs, E.J. Alcohol and inflammation and immune responses: Summary of the 2006 Alcohol and Immunology Research Interest Group (AIRIG) meeting. Alcohol 2008, 42, 137–142. [Google Scholar] [CrossRef]
- Zhang, P.; Bagby, G.J.; Happel, K.I.; Raasch, C.E.; Nelson, S. Alcohol abuse, immunosuppression, and pulmonary infection. Curr. Drug Abuse Rev. 2008, 1, 56–67. [Google Scholar]
- Bailey, K.L.; Sisson, J.H.; Romberger, D.J.; Robinson, J.E.; Wyatt, T.A. Alcohol up-regulates TLR2 through a NO/cGMP dependent pathway. Alcohol. Clin. Exp. Res. 2010, 34, 51–56. [Google Scholar] [CrossRef]
- Elliott, M.K.; Sisson, J.H.; Wyatt, T.A. Effects of cigarette smoke and alcohol on ciliated tracheal epithelium and inflammatory cell recruitment. Am. J. Respir. Cell Mol. Biol. 2007, 36, 452–459. [Google Scholar]
- Jacobsen, E. The metabolism of ethyl alcohol. Pharmacol. Rev. 1952, 4, 107–135. [Google Scholar]
- Bechara, R.I.; Pelaez, A.; Palacio, A.; Joshi, P.C.; Hart, C.M.; Brown, L.A.S.; Raynor, R.; Guidot, D.M. Angiotensin II mediates glutathione depletion, transforming growth factor-β1 expression, and epithelial barrier dysfunction in the alcoholic rat lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L363–L370. [Google Scholar] [CrossRef]
- Brown, L.A.; Harris, F.L.; Guidot, D.M. Chronic alcohol ingestion potentiates TNF-alpha-mediated oxidative stress and apoptosis in rat type II cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 281, L377–L386. [Google Scholar]
- Spurzem, J.R.; Gupta, J.; Veys, T.; Kneifl, K.R.; Rennard, S.I.; Wyatt, T.A. Activation of protein kinase a accelerates bovine bronchial epithelial cell migration. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L1108–L1116. [Google Scholar]
- Goral, J.; Choudhry, M.A.; Kovacs, E.J. Acute alcohol exposure inhibits macrophage IL-6 production: Role of p38 and ERK1/2 MAPK. J. Leukoc. Biol. 2004, 75, 553–559. [Google Scholar]
- Glover, M.; Cheng, B.; Fan, R.; Pruett, S. The role of stress mediators in modulation of cytokine production by alcohol. Toxicol. Appl. Pharmacol. 2009, 239, 98–105. [Google Scholar] [CrossRef]
- Song, K.; Zhao, X.J.; Marrero, L.; Oliver, P.; Nelson, S.; Kolls, J.K. Alcohol reversibly disrupts TNF-alpha/TACE interactions in the cell membrane. Respir. Res. 2005, 24. [Google Scholar] [CrossRef]
- Zhao, X.; Marrero, L.; Song, K.; Oliver, P.; Chin, S.Y.; Simon, H.; Schurr, J.R.; Zhang, Z.; Thoppil, D.; Lee, S.; et al. Acute alcohol inhibits TNF-α processing in human monocytes by inhibiting TNF/TNF-α-converting enzyme interactions in the cell membrane. J. Immunol. 2003, 170, 2923–2931. [Google Scholar]
- Eduard, W.; Pearce, N.; Douwes, J. Chronic bronchitis, COPD, and lung function in farmers: The role of biological agents. Chest 2009, 136, 716–725. [Google Scholar] [CrossRef]
- Dosman, J.A. Respiratory response to endotoxin and dust predicts evidence of inflammatory response in volunteers in a swine barn. Am. J. Ind. Med. 2006, 49, 761–766. [Google Scholar] [CrossRef]
- Poole, J.A.; Wyatt, T.A.; Kielian, T.; Oldenburg, P.; Gleason, A.M.; Bauer, A.; Golden, G.; West, W.W.; Sisson, J.H.; Romberger, D.J. Toll-Like Receptor 2 (TLR2) regulates organic dust-induced airway inflammation. Am. J. Respir. Cell Mol. Biol. 2011, 45, 711–719. [Google Scholar] [CrossRef]
- Slager, R.E.; Allen-Gipson, D.S.; Sammut, A.; Heires, A.; de Vasure, J.; von Essen, S.G.; Romberger, D.J.; Wyatt, T.A. Hog barn dust slows airway epithelial cell migration in vitro through a PKCα-dependent mechanism. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L1469–L1474. [Google Scholar] [CrossRef]
- Wyatt, T.A.; Slager, R.E.; Heires, A.J.; de Vasure, J.M.; von Essen, S.G.; Poole, J.A.; Romberger, D.J. Sequential activation of protein kinase C isoforms by organic dust is mediated by tumor necrosis factor. Am. J. Respir. Cell Mol. Biol. 2010, 42, 706–715. [Google Scholar] [CrossRef]
- Wang, Z.; Malmberg, P.; Ek, A.; Larsson, K.; Palmberg, L. Swine dust induces cytokine secretion from human epithelial cells and alveolar macrophages. Clin. Exp. Immunol. 1999, 115, 6–12. [Google Scholar] [CrossRef]
- Palmberg, L.; Larsson, B.; Malmberg, P.; Larsson, K. Induction of IL-8 production in human alveolar macrophages and human bronchial epithelial cells in vitro by swine dust. Thorax 1998, 53, 260–264. [Google Scholar] [CrossRef]
- Stallones, L.; Xiang, H. Alcohol consumption patterns and work-related injuries among colorado farm residents. Am. J. Prev. Med. 2003, 25, 25–30. [Google Scholar] [CrossRef]
- Schoeneberger, M.L.; Leukefeld, C.G.; Hiller, M.L.; Godlaski, T. Substance abuse among rural and very rural drug users at treatment entry. Am. J. Drug Alcohol Abuse 2006, 32, 87–110. [Google Scholar] [CrossRef]
- Substance Abuse and Mental Health Services Administration, Results from the 2008 National Survey on Drug use and Health: National Findings; Substance Abuse and Mental Health Services Administration: Rockville, MD, USA, 2009; NSDUH Series H-36, HHS Publication No. SMA 09-4434.
- Glover, M.; Pruett, S.B. Role of corticosterone in immunosuppressive effects of acute alcohol exposure on toll-like receptor mediated cytokine production. J. Neuroimmune Pharmacol. 2006, 1, 435–442. [Google Scholar] [CrossRef]
- Pruett, S.B.; Fan, R.; Zheng, Q.; Myers, L.P.; Hébert, P. Modeling and predicting immunological effects of chemical stressors: Characterization of a quantitative biomarker for immunological changes caused by atrazine and alcohol. Toxicol. Sci. 2003, 75, 343–354. [Google Scholar] [CrossRef]
- Romberger, D.J.; Bodlak, V.; von Essen, S.G.; Mathisen, T.; Wyatt, T.A. Hog barn dust extract stimulates IL-8 and IL-6 release in human bronchial epithelial cells via PKC activation. J. Appl. Physiol. 2002, 93, 289–296. [Google Scholar]
- Poole, J.A.; Wyatt, T.A.; Oldenburg, P.J.; Elliott, M.K.; West, W.W.; Sisson, J.H.; von Essen, S.G.; Romberger, D.J. Intranasal organic dust exposure-induced airway adaptation response marked by persistent lung inflammation and pathology in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L1085–L1095. [Google Scholar] [CrossRef]
- Song, K.; Coleman, R.A.; Zhu, X.; Alber, C.; Ballas, Z.K.; Waldschmidt, T.J.; Cook, R.T. Chronic alcohol consumption by mice results in activated splenic T cells. J. Leukoc. Biol. 2002, 72, 1109–1116. [Google Scholar]
- Spitzer, J.H.; Meadows, G.G. Modulation of perforin, granzyme A, and granzyme B in murine Natural Killer (NK), IL2 stimulated NK, and lymphokine-activated killer cells by alcohol consumption. Cell. Immunol. 1999, 194, 205–212. [Google Scholar] [CrossRef]
- Elliott, M.K.; Sisson, J.H.; West, W.W.; Wyatt, T.A. Differential in vivo effects of whole cigarette smoke exposure versus cigarette smoke extract on mouse ciliated tracheal epithelium. Exp. Lung Res. 2006, 32, 99–118. [Google Scholar] [CrossRef]
- Wyatt, T.A.; Spurzem, J.R.; May, K.; Sisson, J.H. Regulation of ciliary beat frequency by both PKA and PKG in bovine airway epithelial cells. Am. J. Physiol. 1998, 275, L827–L835. [Google Scholar]
- Wyatt, T.A.; Schmidt, S.C.; Rennard, S.I.; Tuma, D.J.; Sisson, J.H. Acetaldehyde-stimulated PKC activity in airway epithelial cells treated with smoke extract from normal and smokeless cigarettes. Proc. Soc. Exp. Biol. Med. 2000, 225, 91–97. [Google Scholar] [CrossRef]
- Albert, C.J.; Ford, D.A. Protein kinase C translocation and PKC-dependent protein phosphorylation during myocardial ischemia. Am. J. Physiol. 1999, 276, H642–H650. [Google Scholar]
- Wyatt, T.A.; Forget, M.A.; Sisson, J.H. Alcohol stimulates ciliary beating by dual cyclic nucleotide kinase activation in bovine bronchial epithelial cells. Am. J. Pathol. 2003, 163, 1157–1166. [Google Scholar] [CrossRef]
- De Forge, L.E.; Remick, D.G. Kinetics of TNF, IL-6, and IL-8 gene expression in LPS-stimulated human whole blood. Biochem. Biophys. Res. Commun. 1991, 174, 18–24. [Google Scholar] [CrossRef]
- Bailey, K.L.; Wyatt, T.A.; Romberger, D.J.; Sisson, J.H. Alcohol functionally upregulates toll-like receptor 2 in airway epithelial cells. Alcohol. Clin. Exp. Res. 2009, 33, 499–504. [Google Scholar] [CrossRef]
- Wyatt, T.A.; Sisson, J.H.; von Essen, S.G.; Poole, J.A.; Romberger, D.J. Exposure to hog barn dust alters airway epithelial ciliary beating. Eur. Respir. J. 2008, 31, 1249–1255. [Google Scholar] [CrossRef]
- Thiele, T.E.; Willis, B.; Stadler, J.; Reynolds, J.G.; Bernstein, I.L.; McKnight, G.S. High alcohol consumption and low sensitivity to alcohol-induced sedation in protein kinase A-mutant mice. J. Neurosci. 2000, 20, RC75:1–RC75:6. [Google Scholar]
- Wyatt, T.A.; Kharbanda, K.K.; Tuma, D.J.; Sisson, J.H. Malondialdehyde-acetaldehyde-adducted bovine serum albumin activates protein kinase C and stimulates interleukin-8 release in bovine bronchial epithelial cells. Alcohol 2001, 25, 159–166. [Google Scholar] [CrossRef]
- Hammond, M.E.; Lapointe, G.R.; Feucht, P.H.; Hilt, S.; Gallegos, C.A.; Gordon, C.A.; Giedlin, M.A.; Mullenbach, G.; Tekamp-Olson, P. IL-8 induces neutrophil chemotaxis predominantly via type I IL-8 receptors. J. Immunol. 1995, 155, 1428–1433. [Google Scholar]
- De Larco, J.E.; Wuertz, B.R.; Furcht, L.T. The potential role of neutrophils in promoting the metastatic phenotype of tumors releasing interleukin-8. Clin. Cancer Res. 2004, 10, 4895–4900. [Google Scholar]
- Kunkel, S.L.; Standiford, T.; Kasahara, K.; Strieter, R.M. Interleukin-8 (IL-8): The major neutrophil chemotactic factor in the lung. Exp. Lung Res. 1991, 17, 17–23. [Google Scholar] [CrossRef]
- Papoff, P.; Fiorucci, P.; Ottaviano, C.; Bucci, G. Interleukin-8: A potent neutrophil chemotactic factor. Arch. Dis. Child. Fetal Neonatal Ed. 1995, 73, F54. [Google Scholar]
- Grenier, A. Presence of a mobilizable intracellular pool of hepatocyte growth factor in human polymorphonuclear neutrophils. Blood 2002, 99, 2997–3004. [Google Scholar] [CrossRef]
- Taieb, J.; de larche, C.; Ethuin, F.; Selloum, S.; Poynard, T.; Gougerot-Pocidalo, M.A.; Chollet-Martin, S. Alcohol-induced inhibition of cytokine release and protein degranulation in human neutrophils. J. Leukoc. Biol. 2002, 72, 1142–1147. [Google Scholar]
- Jonsson, H.; Allen, P.; Peng, S.L. Inflammatory arthritis requires foxo3a to prevent fas ligand-induced neutrophil apoptosis. Nat. Med. 2005, 11, 666–671. [Google Scholar] [CrossRef]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 1997, 385, 729–733. [Google Scholar]
- Moss, M.L.; Sklair-Tavron, L.; Nudelman, R. Drug insight: Tumor necrosis factor-converting enzyme as a pharmaceutical target for rheumatoid arthritis. Nat. Clin. Pract. Rheumatol. 2008, 4, 300–309. [Google Scholar] [CrossRef]
- Aggarwal, B.B. Tumour necrosis factors receptor associated signalling molecules and their role in activation of apoptosis, JNK and NF-kappaB. Ann. Rheum. Dis. 2000, 59 (Suppl. 1), i6–i16. [Google Scholar] [CrossRef]
- Nelson, S.; Bagby, G.I.; Bainton, B.G.; Summer, W.R. The effects of acute and chronic alcoholism on tumor necrosis factor and the inflammatory response. J. Infect. Dis. 1989, 160, 422–429. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
McCaskill, M.L.; Romberger, D.J.; DeVasure, J.; Boten, J.; Sisson, J.H.; Bailey, K.L.; Poole, J.A.; Wyatt, T.A. Alcohol Exposure Alters Mouse Lung Inflammation in Response to Inhaled Dust. Nutrients 2012, 4, 695-710. https://doi.org/10.3390/nu4070695
McCaskill ML, Romberger DJ, DeVasure J, Boten J, Sisson JH, Bailey KL, Poole JA, Wyatt TA. Alcohol Exposure Alters Mouse Lung Inflammation in Response to Inhaled Dust. Nutrients. 2012; 4(7):695-710. https://doi.org/10.3390/nu4070695
Chicago/Turabian StyleMcCaskill, Michael L., Debra J. Romberger, Jane DeVasure, Jessica Boten, Joseph H. Sisson, Kristina L. Bailey, Jill A. Poole, and Todd A. Wyatt. 2012. "Alcohol Exposure Alters Mouse Lung Inflammation in Response to Inhaled Dust" Nutrients 4, no. 7: 695-710. https://doi.org/10.3390/nu4070695