
Analyzing Type 2 Diabetes Associations with the Gut Microbiome in Individuals from Two Ethnic Backgrounds Living in the Same Geographic Area

 , , ,
, , ,
Abstract
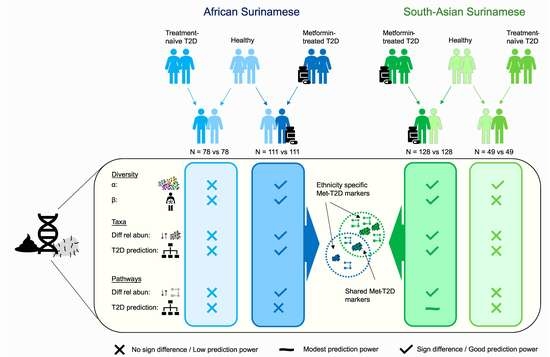
:
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Baseline Data Collection
2.3. Profiling of Fecal Microbiota Composition
2.4. Bioinformatics Pipeline
2.5. Ascertainment of T2D and Controls
2.6. Statistical Analysis
2.7. Machine Learning
3. Results
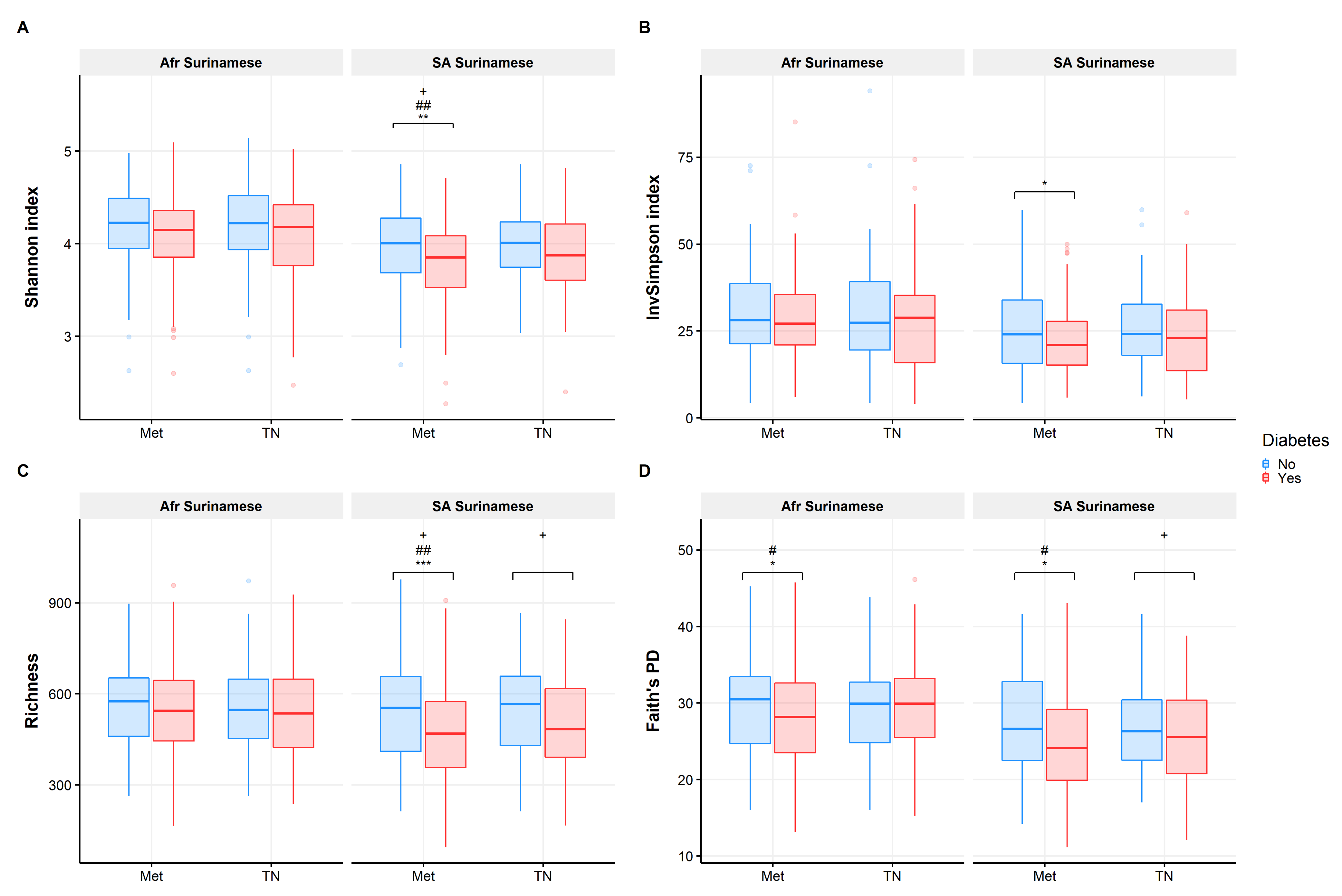
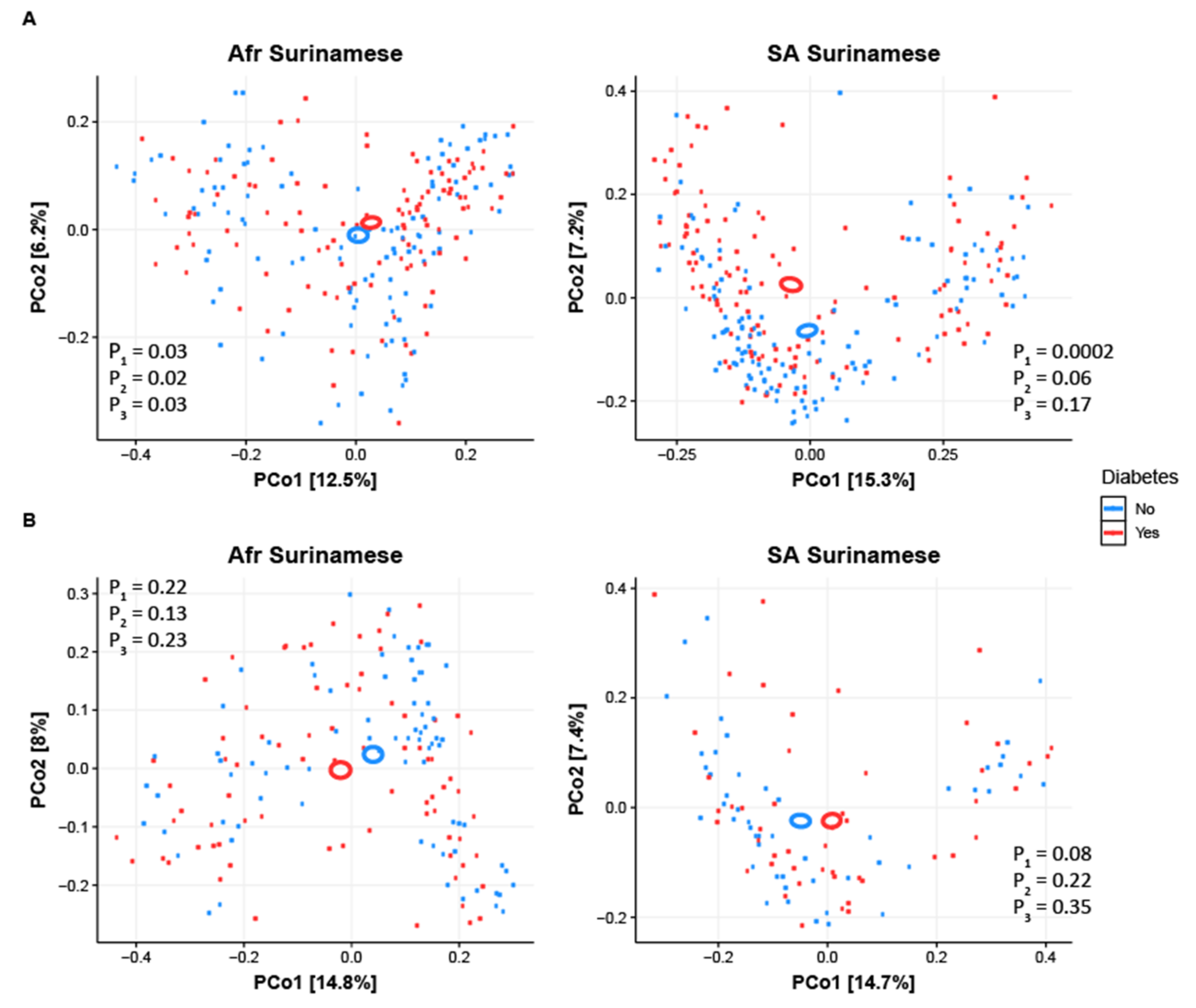
3.1. Gut Microbiome Diversity Analysis
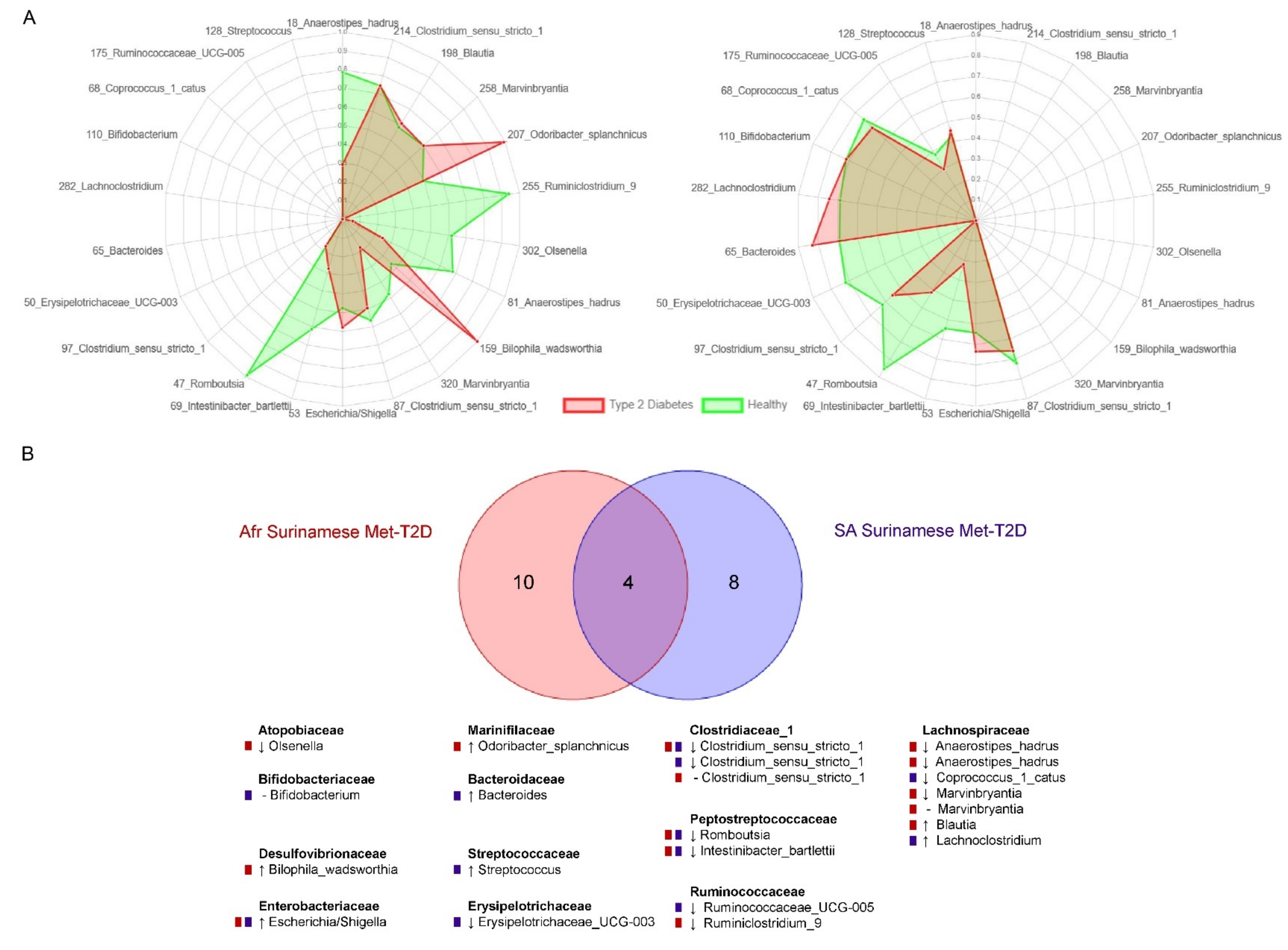
3.2. Analysis of Biomarkers for T2D
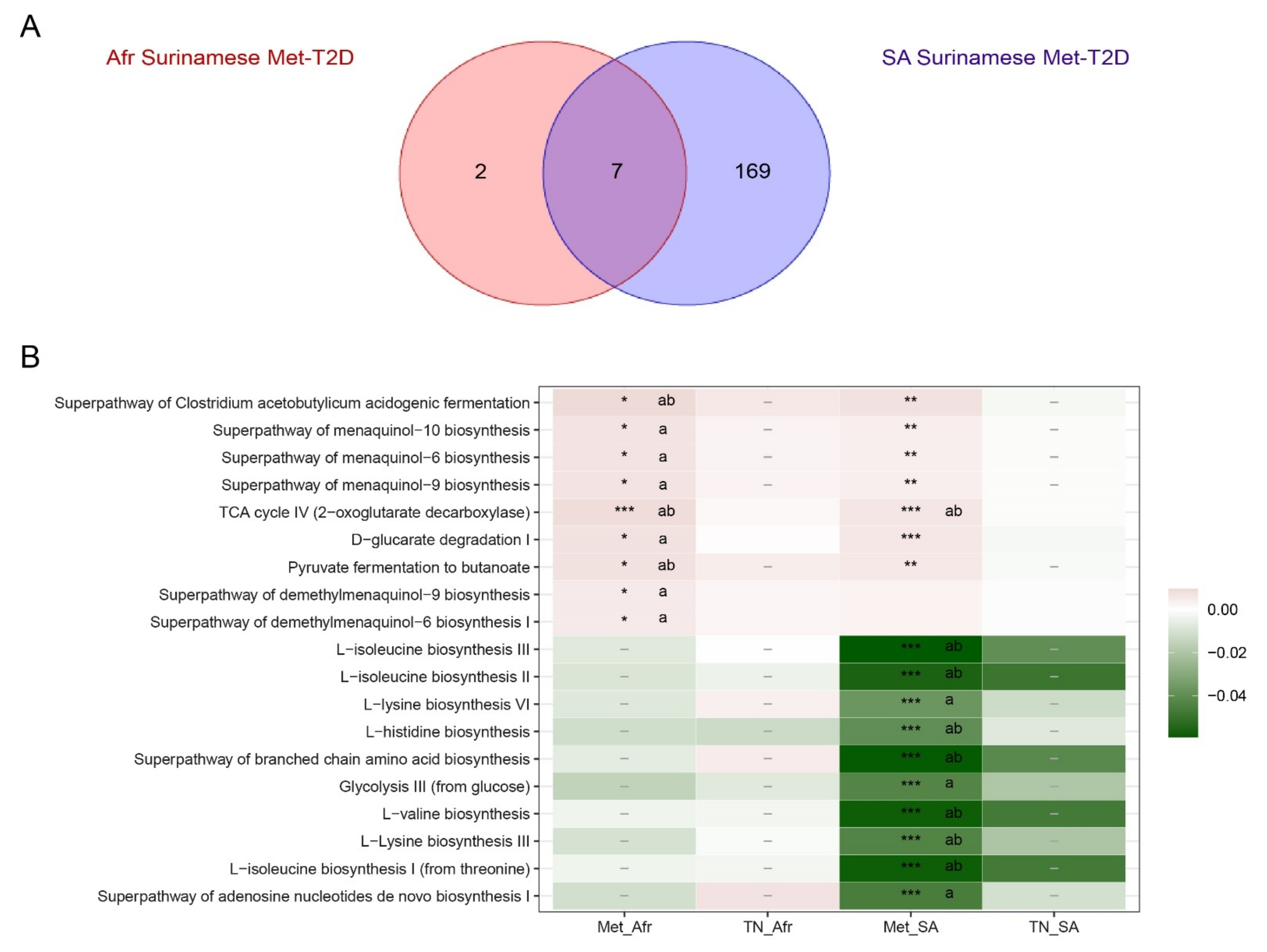
3.3. Functional Analysis
4. Discussion
4.1. Gut Microbiota Composition Is Comparable in Treatment Naïve Diabetic Cases and Controls
4.2. Overlapping Biomarkers between Ethnicities Were Previously Related to Metformin Use
4.3. Biomarkers Related to Cardiometabolic Indicators Mostly Represented by Different ASVs across Ethnicities
4.4. Alterations in Functional Potential of the Gut Microbiome Were More Frequent in South-Asian Surinamese
4.5. Relation to Results in Other Cohorts with the Same Genetic Background
4.6. Strengths and Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DeFronzo, R.A.; Ferrannini, E.; Groop, L.; Henry, R.R.; Herman, W.H.; Holst, J.J.; Hu, F.B.; Kahn, C.R.; Raz, I.; Shulman, G.I.; et al. Type 2 diabetes mellitus. Nat. Rev. Dis. Primers 2015, 1, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Meeks, K.A.C.; Freitas-Da-Silva, D.; Adeyemo, A.; Beune, E.J.A.J.; Modesti, P.A.; Stronks, K.; Zafarmand, M.H.; Agyemang, C. Disparities in type 2 diabetes preva-lence among ethnic minority groups resident in Europe: A systematic review and meta-analysis. Intern. Emerg. Med. 2016, 11, 327–340. [Google Scholar] [CrossRef]
- Snijder, M.B.; Agyemang, C.; Peters, R.J.; Stronks, K.; Ujcic-Voortman, J.K.; van Valkengoed, I.G.M. Case Finding and Medical Treatment of Type 2 Diabetes among Different Ethnic Minority Groups: The HELIUS Study. J. Diabetes Res. 2017, 2017, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tillin, T.; Hughes, A.D.; Godsland, I.F.; Whincup, P.; Forouhi, N.G.; Welsh, P.; Sattar, N.; McKeigue, P.M.; Chaturvedi, N. Insulin Resistance and Truncal Obesity as Important Determinants of the Greater Incidence of Diabetes in Indian Asians and African Caribbeans Compared with Europeans: The Southall And Brent REvisited (SABRE) cohort. Diabetes Care 2012, 36, 383–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muilwijk, M.; Nieuwdorp, M.; Snijder, M.B.; Hof, M.H.P.; Stronks, K.; van Valkengoed, I. The high risk for type 2 diabetes among ethnic minority populations is not explained by low-grade inflammation. Sci. Rep. 2019, 9, 19871–19878. [Google Scholar] [CrossRef] [PubMed]
- ID Federation. IDF Diabetes Atlas, 9th ed.; ID Federation: Brussels, Belgium, 2019. [Google Scholar]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergström, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Bäckhed, F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef]
- Forslund, K.; Hildebrand, F.; Nielsen, T.; Falony, G.; Le Chatelier, E.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundsdottir, V.; Pedersen, H.K.; et al. Disentangling the effects of type 2 diabetes and metformin on the human gut microbiota. Nature 2015, 528, 262–266. [Google Scholar] [CrossRef]
- Wu, H.; Tremaroli, V.; Schmidt, C.; Lundqvist, A.; Olsson, L.M.; Krämer, M.; Gummesson, A.; Perkins, R.; Bergström, G.; Bäckhed, F. The Gut Microbiota in Prediabetes and Diabetes: A Population-Based Cross-Sectional Study. Cell Metab. 2020, 32, 379–390.e3. [Google Scholar] [CrossRef]
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [Green Version]
- Gérard, C.; Vidal, H. Impact of Gut Microbiota on Host Glycemic Control. Front. Endocrinol. 2019, 10, 29. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, H.K.; Gudmundsdottir, V.; Nielsen, H.B.; Hyotylainen, T.; Nielsen, T.; Jensen, B.A.H.; Forslund, K.; Hildebrand, F.; Prifti, E.; Falony, G.; et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature 2016, 535, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Alemida, M.; Arumugam, M.; Batto, J.-M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Koh, A.; Molinaro, A.; Ståhlman, M.; Khan, M.T.; Schmidt, C.; Mannerås-Holm, L.; Wu, H.; Carreras, A.; Jeong, H.; Olofsson, L.E.; et al. Microbially Produced Imidazole Propionate Impairs Insulin Signaling through mTORC1. Cell 2018, 175, 947–961.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaike, A.H.; Paul, D.; Bhute, S.; Dhotre, D.P.; Pande, P.; Upadhyaya, S.; Reddy, Y.; Sampath, R.; Ghosh, D.; Chandraprabha, D.; et al. The Gut Microbial Diversity of Newly Diagnosed Diabetics but Not of Prediabetics Is Significantly Different from That of Healthy Nondiabetics. mSystems 2020, 5, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Bhute, S.; Suryavanshi, M.V.; Joshi, S.M.; Yajnik, C.S.; Shouche, Y.S.; Ghaskadbi, S.S. Gut Microbial Diversity Assessment of Indian Type-2-Diabetics Reveals Alterations in Eubacteria, Archaea, and Eukaryotes. Front. Microbiol. 2017, 8, 214. [Google Scholar] [CrossRef]
- De La Cuesta-Zuluaga, J.; Mueller, N.T.; Corrales-Agudelo, V.; Velásquez-Mejía, E.P.; Carmona, J.A.; Abad, J.M.; Escobar, J.S. Metformin is associated with higher relative abundance of mucin-degrading akkermansia muciniphila and several short-chain fatty ac-id-producing microbiota in the gut. Diabetes Care 2017, 40, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Zhong, H.; Ren, H.; Lu, Y.; Fang, C.; Hou, G.; Yang, Z.; Chen, B.; Yang, F.; Zhao, Y.; Shi, Z.; et al. Distinct gut metagenomics and metaproteomics signatures in prediabetics and treatment-naïve type 2 diabetics. EBioMedicine 2019, 47, 373–383. [Google Scholar] [CrossRef]
- Allin, K.H.; Tremaroli, V.; Caesar, R.; Jensen, B.; Damgaard, M.T.F.; Bahl, M.I.; Licht, T.R.; Hansen, T.; Nielsen, T.; Dantoft, T.M.; et al. Aberrant intestinal microbiota in individuals with prediabetes. Diabetologia 2018, 61, 810–820. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Silva, C.; Kashani, A.; Hansen, T.H.; Pinna, N.K.; Anjana, R.M.; Dutta, A.; Saxena, S.; Støy, J.; Kampmann, U.; Nielsen, T.; et al. Trans-ethnic gut microbiota signatures of type 2 diabetes in Denmark and India. Genome Med. 2021, 13, 1–13. [Google Scholar] [CrossRef]
- Wu, H.; Esteve, E.; Tremaroli, V.; Khan, M.T.; Caesar, R.; Mannerås-Holm, L.; Ståhlman, M.; Olsson, L.M.; Serino, M.; Planas-Fèlix, M.; et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat. Med. 2017, 23, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Bryrup, T.; Thomsen, C.W.; Kern, T.; Allin, K.H.; Brandslund, I.; Jørgensen, N.R.; Vestergaard, H.; Hansen, T.H.; Hansen, T.; Pedersen, O.; et al. Metformin-induced changes of the gut microbiota in healthy young men: Results of a non-blinded, one-armed intervention study. Diabetologia 2019, 62, 1024–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbere, I.; Kalnina, I.; Silamikelis, I.; Konrade, I.L.; Zaharenko, L.; Sekace, K.; Radovica-Spalvina, I.; Fridmanis, D.; Gudra, D.; Pirags, V.; et al. Association of metformin administration with gut microbiome dysbiosis in healthy volunteers. PLoS ONE 2018, 13, e0204317. [Google Scholar] [CrossRef]
- Wang, Y.; Luo, X.; Mao, X.; Tao, Y.; Ran, X.; Zhao, H.; Xiong, J.; Li, L. Gut microbiome analysis of type 2 diabetic patients from the Chinese minority ethnic groups the Uygurs and Kazaks. PLoS ONE 2017, 12, e0172774. [Google Scholar] [CrossRef] [Green Version]
- Deschasaux, M.; Bouter, K.E.; Prodan, A.; Levin, E.; Groen, A.K.; Herrema, H.; Tremaroli, V.; Bakker, G.J.; Attaye, I.; Pinto-Sietsma, S.-J.; et al. Depicting the composition of gut microbiota in a population with varied ethnic origins but shared geography. Nat. Med. 2018, 24, 1526–1531. [Google Scholar] [CrossRef]
- Vangay, P.; Johnson, A.; Ward, T.L.; Al-Ghalith, G.A.; Shields-Cutler, R.R.; Hillmann, B.M.; Lucas, S.K.; Beura, L.K.; Thompson, E.A.; Till, L.M.; et al. US Immigration Westernizes the Human Gut Microbiome. Cell 2018, 175, 962–972.e10. [Google Scholar] [CrossRef] [Green Version]
- Keohane, D.M.; Ghosh, T.; Jeffery, I.B.; Molloy, M.G.; O’Toole, P.W.; Shanahan, F. Microbiome and health implications for ethnic minorities after enforced lifestyle changes. Nat. Med. 2020, 26, 1089–1095. [Google Scholar] [CrossRef]
- Stronks, K.; Snijder, M.B.; Peters, R.J.G.; Prins, M.; Schene, A.H.; Zwinderman, A.H. Unravelling the impact of ethnicity on health in Europe: The HELIUS study. BMC Public Health 2013, 13, 402. [Google Scholar] [CrossRef] [Green Version]
- Snijder, M.B.; Galenkamp, H.; Prins, M.; Derks, E.; Peters, R.J.G.; Zwinderman, A.H.; Stronks, K. Cohort profile: The Healthy Life in an Urban Setting (HELIUS) study in Amsterdam, The Netherlands. BMJ Open 2017, 7, e017873. [Google Scholar] [CrossRef] [Green Version]
- Beukers, M.; Dekker, L.; de Boer, E.; Perenboom, C.W.M.; Meijboom, S.; Nicolaou, M.; de Vries, J.H.M.; Brants, H.A.M. Development of the HELIUS food frequency questionnaires: Ethnic-specific questionnaires to assess the diet of a multiethnic population in The Netherlands. Eur. J. Clin. Nutr. 2015, 69, 579–584. [Google Scholar] [CrossRef]
- Dekker, L.H.; Snijder, M.B.; Beukers, M.H.; De Vries, J.H.M.; Brants, H.A.; De Boer, E.J.; Van Dam, R.M.; Stronks, K.; Nicolaou, M. A prospective cohort study of dietary patterns of non-western migrants in the Netherlands in relation to risk factors for cardiovascular diseases: HELIUS-Dietary Patterns. BMC Public Health 2011, 11, 441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mobini, R.; Tremaroli, V.; Ståhlman, M.; Karlsson, F.; Levin, M.; Ljungberg, M.; Sohlin, M.; Forslund, H.B.; Perkins, R.; Bäckhed, F.; et al. Metabolic effects ofLactobacillus reuteriDSM 17938 in people with type 2 diabetes: A randomized controlled trial. Diabetes Obes. Metab. 2017, 19, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a Dual-Index Sequencing Strategy and Curation Pipeline for Analyzing Amplicon Sequence Data on the MiSeq Illumina Sequencing Platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLOS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package, R Package Version 2.5–7. 2019. Available online: https://CRAN.R-project.org/package=vegan (accessed on 18 September 2021).
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.E.; Imai, K.; King, G.; Stuart, E. MatchIt: Nonparametric Preprocessing for Parametric Causal Inference. J. Stat. Softw. 2011, 42, 1–28. [Google Scholar] [CrossRef] [Green Version]
- Pearce, N. Analysis of matched case-control studies. BMJ 2016, 352, i969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revelle, W. Psych: Procedures for Personality and Psychological Research, R Package Version 1.8.10. Available online: https://CRAN.R-project.org/package=psych (accessed on 18 September 2021).
- Kembel, S.W.; Cowan, P.D.; Helmus, M.; Cornwell, W.K.; Morlon, H.; Ackerly, D.D.; Blomberg, S.P.; Webb, C.O. Picante: R tools for integrating phylogenies and ecology. Bioinformatics 2010, 26, 1463–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozupone, C.; Knight, R. UniFrac: A New Phylogenetic Method for Comparing Microbial Communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2018, 35, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.R-project.org (accessed on 18 September 2021).
- Meinshausen, N.; Bühlmann, P. Stability selection. J. R. Stat. Soc. Ser. B Stat. Methodol. 2010, 72, 417–473. [Google Scholar] [CrossRef]
- Geurts, P.; Ernst, D.; Wehenkel, L. Extremely randomized trees. Mach. Learn. 2006, 63, 3–42. [Google Scholar] [CrossRef] [Green Version]
- Fisher, A.; Rudin, C.; Dominici, F. Model class reliance: Variable importance measures for any machine learning model class, from the “rashomon” perspective. arXiv 2018, arXiv:1801.01489. [Google Scholar]
- Lundberg, S.M.; Erion, G.; Chen, H.; DeGrave, A.; Prutkin, J.M.; Nair, B.; Katz, R.; Himmelfarb, J.; Bansal, N.; Lee, S.-I. From local explanations to global understanding with explainable AI for trees. Nat. Mach. Intell. 2020, 2, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, W.; Wang, C.; Wang, L.; He, T.; Hu, H.; Song, J.; Cui, C.; Qiao, J.; Qing, L.; et al. Enterotype Bacteroides Is Associated with a High Risk in Patients with Diabetes: A Pilot Study. J. Diabetes Res. 2020, 2020, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vujkovic-Cvijin, I.; Sklar, J.; Jiang, L.; Natarajan, L.; Knight, R.; Belkaid, Y. Host variables confound gut microbiota studies of human disease. Nat. Cell Biol. 2020, 587, 448–454. [Google Scholar] [CrossRef]
- Falony, G.; Joossens, M.; Vieira-Silva, S.; Wang, J.; Darzi, Y.; Faust, K.; Kurilshikov, A.; Bonder, M.J.; Valles-Colomer, M.; Vandeputte, D.; et al. Population-level analysis of gut microbiome variation. Science 2016, 352, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Xie, C.; Wang, G.; Wu, Y.; Wu, Q.; Wang, X.; Liu, J.; Deng, Y.; Xia, J.; Chen, B.; et al. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat. Med. 2018, 24, 1919–1929. [Google Scholar] [CrossRef]
- Elbere, I.; Silamikelis, I.; Dindune, I.I.; Kalnina, I.; Ustinova, M.; Zaharenko, L.; Silamikele, L.; Rovite, V.; Gudra, D.; Konrade, I.; et al. Baseline gut microbiome composition predicts metformin therapy short-term efficacy in newly diagnosed type 2 diabetes patients. PLoS ONE 2020, 15, e0241338. [Google Scholar] [CrossRef]
- Therdtatha, P.; Song, Y.; Tanaka, M.; Mariyatun, M.; Almunifah, M.; Manurung, N.; Indriarsih, S.; Lu, Y.; Nagata, K.; Fukami, K.; et al. Gut Microbiome of Indonesian Adults Associated with Obesity and Type 2 Diabetes: A Cross-Sectional Study in an Asian City, Yogyakarta. Microorganisms 2021, 9, 897. [Google Scholar] [CrossRef]
- Li, R.; Yao, Y.; Gao, P.; Bu, S. The Therapeutic Efficacy of Curcumin vs. Metformin in Modulating the Gut Microbiota in NAFLD Rats: A Comparative Study. Front. Microbiol. 2021, 11, 1–15. [Google Scholar] [CrossRef]
- Magruder, M.; Edusei, E.; Zhang, L.; Albakry, S.; Satlin, M.J.; Westblade, L.F.; Malha, L.; Sze, C.; Lubetzky, M.; Dadhania, D.M.; et al. Gut commensal microbiota and decreased risk for Enterobacteriaceae bacteriuria and urinary tract infection. Gut Microbes 2020, 12, 1805281. [Google Scholar] [CrossRef]
- Vich Vila, A.; Collij, V.; Sanna, S.; Sinha, T.; Imhann, F.; Bourgonje, A.; Mujagic, Z.; Jonkers, D.M.A.E.; Masclee, A.A.M.; Fu, J.; et al. Impact of commonly used drugs on the composition and metabolic function of the gut microbiota. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Krautkramer, K.A.; Fan, J.; Bäckhed, F. Gut microbial metabolites as multi-kingdom intermediates. Nat. Rev. Genet. 2021, 19, 77–94. [Google Scholar] [CrossRef]
- Zeevi, D.; Korem, T.; Godneva, A.; Bar, N.; Kurilshikov, A.; Lotan-Pompan, M.; Weinberger, A.; Fu, J.; Wijmenga, C.; Zhernakova, A.; et al. Structural variation in the gut microbiome associates with host health. Nature 2019, 568, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Verhaar, B.J.H.; Collard, D.; Prodan, A.; Levels, J.H.M.; Zwinderman, A.H.; Bäckhed, F.; Vogt, L.; Peters, M.J.L.; Muller, M.; Nieuwdorp, M.; et al. Associations between gut microbiota, faecal short-chain fatty acids, and blood pressure across ethnic groups: The HELIUS study. Eur. Heart J. 2020, 41, 4259–4267. [Google Scholar] [CrossRef] [PubMed]
- Vojinovic, D.; Radjabzadeh, D.; Kurilshikov, A.; Amin, N.; Wijmenga, C.; Franke, L.; Ikram, M.A.; Uitterlinden, A.G.; Zhernakova, A.; Fu, J.; et al. Relationship between Gut Microbiota and Circulating Metabolites in Population-Based Cohorts. Nat. Commun. 2019, 10, 5813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doumatey, A.P.; Adeyemo, A.; Zhou, J.; Lei, L.; Adebamowo, S.N.; Adebamowo, C.; Rotimi, C.N. Gut Microbiome Profiles Are Associated with Type 2 Diabetes in Urban Africans. Front. Cell. Infect. Microbiol. 2020, 10, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afolayan, A.; Adebusoye, L.; Cadmus, E.; Ayeni, F. Insights into the gut microbiota of Nigerian elderly with type 2 diabetes and non-diabetic elderly persons. Heliyon 2020, 6, e03971. [Google Scholar] [CrossRef]
- Pushpanathan, P.; Srikanth, P.; Seshadri, K.G.; Selvarajan, S.; Pitani, R.S.; Kumar, T.D.; Janarthanan, R. Gut Microbiota in Type 2 Diabetes Individuals and Correlation with Monocyte Chemoattractant Protein1 and Interferon Gamma from Patients Attending a Tertiary Care Centre in Chennai, India. Indian J. Endocrinol. Metab. 2016, 20, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Das, T.; Jayasudha, R.; Chakravarthy, S.; Prashanthi, G.S.; Bhargava, A.; Tyagi, M.; Rani, P.K.; Pappuru, R.R.; Sharma, S.; Shivaji, S. Alterations in the gut bacterial microbiome in people with type 2 diabetes mellitus and diabetic retinopathy. Sci. Rep. 2021, 11, 1–15. [Google Scholar] [CrossRef]
- Vieira-Silva, S.; Falony, G.; Belda, E.; Nielsen, T.; Aron-Wisnewsky, J.; Chakaroun, R.; Forslund, S.K.; Assmann, K.; Valles-Colomer, M.; Nguyen, T.T.D.; et al. Statin Therapy Is Associated with Lower Prevalence of Gut Microbiota Dysbiosis. Nature 2020, 581, 310–315. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| African Surinamese | South-Asian Surinamese | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Met-T2D vs. Controls | TN-T2D vs. Controls | Met-T2D vs. Controls | TN-T2D vs. Controls | |||||||||
| Healthy | T2D | p | Healthy | T2D | p | Healthy | T2D | p | Healthy | T2D | p | |
| n | 111 | 111 | 78 | 78 | 128 | 128 | 49 | 49 | ||||
| Sex male | 41 (36.9%) | 38 (34.2%) | 0.78 (*) | 46 (59.0%) | 41 (52.6%) | 0.52 (*) | 47 (36.7%) | 75 (58.6%) | 0.00073 (*) | 22 (44.9%) | 23 (46.9%) | 1 (*) |
| Age (in years) | 56.5 ± 6.8 | 58.7 ± 6.5 | 0.013 (+) | 57 (53–61) | 57 (52–62.75) | 0.65 (&) | 45 (33.85–51.3) | 58 (54.8–64) | <2.2 × 10−16 (&) | 53 (49–59) | 58 (50–62) | 0.047 (&) |
| BMI (in kg/m2) | 29.2 ± 4.8 | 30.3 ± 4.8 | 0.087 (+) | 28.95 ± 5.1 | 31.33 ± 6.5 | 0.012 (+) | 24.1 (22.0–26.5) | 27.7 (25.3–30.7) | 2.0 × 10−14 (&) | 25.8 (23.6–28.0) | 26.5 (24.1–30.4) | 0.11 (&) |
| n with diet information | 33 (29.7%) | 27 (24.3%) | 0.45 (*) | 24 (30.8%) | 20 (25.6%) | 0.59 | 44 (34.4%) | 57 (44.5%) | 0.12 (*) | 15 (30.6%) | 16 (32.7%) | 1 (*) |
| PCDiet 1 | −0.12 (−0.53–0.11) | −0.12 (−0.43–0.10) | 0.79 (&) | −0.12 (−0.58–0.32) | −0.12 (−0.33–0.01) | 0.90 (&) | −0.16 (−0.41–0.08) | −0.04 (−0.37–0.31) | 0.16 (&) | −0.20 (−0.37− -0.05) | −0.24 (−0.45–0.19) | 0.89 (&) |
| PCDiet 2 | −0.44 (−0.72–0.06) | −0.36 (−0.74–0.03) | 0.82 (&) | −0.42 (−0.73–0.14) | −0.30 (−0.77–0.23) | 0.86 (&) | −0.11 (−0.52–0.48) | −0.34 (−0.63–0.38) | 0.26 (&) | −0.39 (−0.69– -0.11) | −0.17 (−0.49–0.90) | 0.14 (&) |
| Fasting glucose (mmol/L) | 5.0 (4.8–5.3) | 7.2 (6.4–8.4) | <2.2 × 10−16 (&) | 5.1 (4.8–5.3) | 7.0 (6.1–7.9) | <2.2 × 10−16 (&) | 5 (4.8–5.2) | 7.6 (6.4–8.6) | <2.2 × 10−16 (&) | 5.1 (4.9–5.3) | 6.9 (6–7.5) | 1.2 × 10−14 (&) |
| HbA1c (mmol/mol) | 36 (33–38) | 51 (46–60) | <2.2 × 10−16 (&) | 36 (32.3–37.8) | 50 (48–53) | <2.2 × 10−16 (&) | 36 (34–37) | 53 (49–64) | <2.2 × 10−16 (&) | 37 (35–38) | 50 (48–53) | <2.2 × 10−16 (&) |
| MetSyn Yes | 0 (0%) | 104 (93.7%) | <2.2 × 10−16 ($) | 0 (0%) | 63 (80.8%) | <2.2 × 10−16 ($) | 0 (0%) | 123 (96.1%) | <2.2 × 10−16 ($) | 0 (0%) | 40 (81.6%) | <2.2 × 10−16 ($) |
| Central obesity Yes | 86 (77.5%) | 101 (91.0%) | 0.0099(*) | 51 (65.4%) | 64 (82.1%) | 0.029 (*) | 77 (60.2%) | 122 (95.3%) | 3.8 × 10−11 (*) | 38 (77.6%) | 40 (81.6%) | 0.80 (*) |
| High blood pressure Yes | 80 (72.1%) | 102 (91.9%) | 0.00025 (*) | 56 (71.2%) | 67 (85.9%) | 0.050 (*) | 34 (26.6%) | 116 (90.6%) | <2.2 × 10−16 (*) | 21 (42.9%) | 35 (71.4%) | 0.0080 (*) |
| Low HDL Yes | 2 (1.8%) | 79 (71.2%) | <2.2 × 10−16 (*) | 3 (3.8%) | 39 (50%) | 2.7 × 10−10 (*) | 20 (15.6%) | 107 (83.6%) | <2.2 × 10−16 (*) | 4 (8.2%) | 35 (71.4%) | 6.0 × 10−10 (*) |
| High Triglycerides Yes | 0 (0%) | 71 (64.0%) | <2.2 × 10−16 ($) | 0 (0%) | 29 (37.2%) | 1.5 × 10−10 ($) | 7 (5.5%) | 104 (81.3%) | <2.2 × 10−16 (*) | 7 (14.3%) | 25 (51.0%) | 0.00025 (*) |
| High glucose Yes | 0 (0%) | 111 (100%) | <2.2 × 10−16 ($) | 0 (0%) | 73 (93.6%) | <2.2 × 10−16 ($) | 0 (0%) | 128 (100%) | <2.2 × 10−16 ($) | 0 (0%) | 42 (85.7%) | <2.2 × 10−16 ($) |
| Insulin use Yes | 0 (0%) | 25 (22.5%) | 1.3 × 10−8 ($) | 0 (0%) | 0 (0%) | 1 ($) | 0 (0%) | 25 (19.5%) | 1.6 × 10−8 ($) | 0 (0%) | 0 (0%) | 1 ($) |
| PPI use Yes | 12 (10.8%) | 22 (19.8%) | 0.093 (*) | 5 (6.4%) | 12 (15.4%) | 0.12 (*) | 4 (3.1%) | 34 (26.6%) | 3.4 × 10−7 (*) | 3 (6.1%) | 6 (12.2%) | 0.49 ($) |
| Statin use Yes | 0 (0%) | 59 (53.2%) | <2.2 × 10−16 ($) | 0 (0%) | 13 (16.7%) | 0.00014 ($) | 0 (0%) | 90 (70.3%) | <2.2 × 10−16 ($) | 0 (0%) | 17 (34.7%) | 2.8 × 10−6 ($) |
| Laxative use Yes | 4 (3.6%) | 0 (0%) | 0.12 ($) | 1 (1.3%) | 1 (1.3%) | 1 ($) | 1 (0.8%) | 4 (3.1%) | 0.37 ($) | 0 (0%) | 4 (8.2%) | 0.12 ($) |
| Beta blockers Yes | 9 (8.1%) | 27 (24.3%) | 0.0020 (*) | 7 (9.0%) | 13 (16.7%) | 0.23 (*) | 3 (2.3%) | 39 (30.5%) | 3.5 × 10−9 (*) | 2 (4.1%) | 9 (18.4%) | 0.055 |
| Renin-angiotensin use Yes | 21 (18.9%) | 65 (58.6%) | 3.1 × 10−9 (*) | 14 (17.9%) | 24 (30.8%) | 0.093 (*) | 7 (5.5%) | 71 (55.5%) | <2.2 × 10−16 (*) | 4 (8.2%) | 12 (24.5%) | 0.056 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balvers, M.; Deschasaux, M.; van den Born, B.-J.; Zwinderman, K.; Nieuwdorp, M.; Levin, E. Analyzing Type 2 Diabetes Associations with the Gut Microbiome in Individuals from Two Ethnic Backgrounds Living in the Same Geographic Area. Nutrients 2021, 13, 3289. https://doi.org/10.3390/nu13093289
Balvers M, Deschasaux M, van den Born B-J, Zwinderman K, Nieuwdorp M, Levin E. Analyzing Type 2 Diabetes Associations with the Gut Microbiome in Individuals from Two Ethnic Backgrounds Living in the Same Geographic Area. Nutrients. 2021; 13(9):3289. https://doi.org/10.3390/nu13093289
Chicago/Turabian StyleBalvers, Manon, Mélanie Deschasaux, Bert-Jan van den Born, Koos Zwinderman, Max Nieuwdorp, and Evgeni Levin. 2021. "Analyzing Type 2 Diabetes Associations with the Gut Microbiome in Individuals from Two Ethnic Backgrounds Living in the Same Geographic Area" Nutrients 13, no. 9: 3289. https://doi.org/10.3390/nu13093289