Consumption of the Total Western Diet Promotes Colitis and Inflammation-Associated Colorectal Cancer in Mice

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Animals and Experimental Diets
2.3. Experiment Designs
2.3.1. Experiment A: AOM + DSS Model of CAC with the TWD
2.3.2. Experiment B: Restoration of Calcium and Vitamin D or Methyl Donor Micronutrients in TWD to Amounts in the AIN Diet
2.3.3. Experiment C: ApcMin/+ Model of Intestinal Tumorigenesis with TWD and DIO Diets
2.3.4. Experiment D: Longitudinal Effects of TWD on Colitis and Recovery from Gut Injury
2.3.5. Experiment E: Impact of TWD on DSS-induced Colitis and Expression of Immune- and Cancer-Related Genes in the Colon
2.4. Assessment of Tumors of the Colon or Small Intestine
2.5. Histopathology and Immunohistochemistry
2.6. Analysis of Transcript Abundance Data for Colon Mucosa
2.7. General Data Analysis
3. Results
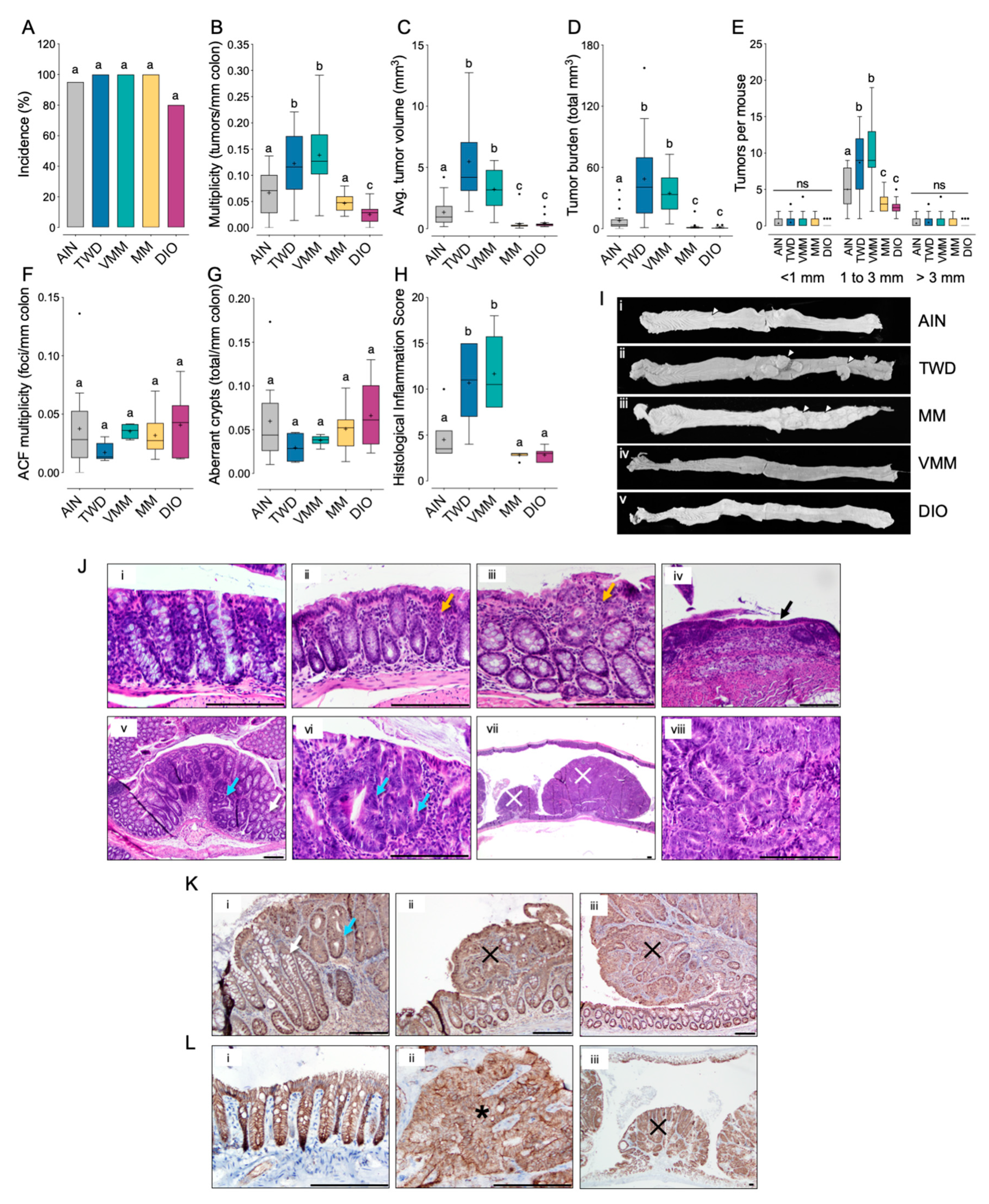
3.1. Experiment A: AOM + DSS Model of CAC with the TWD
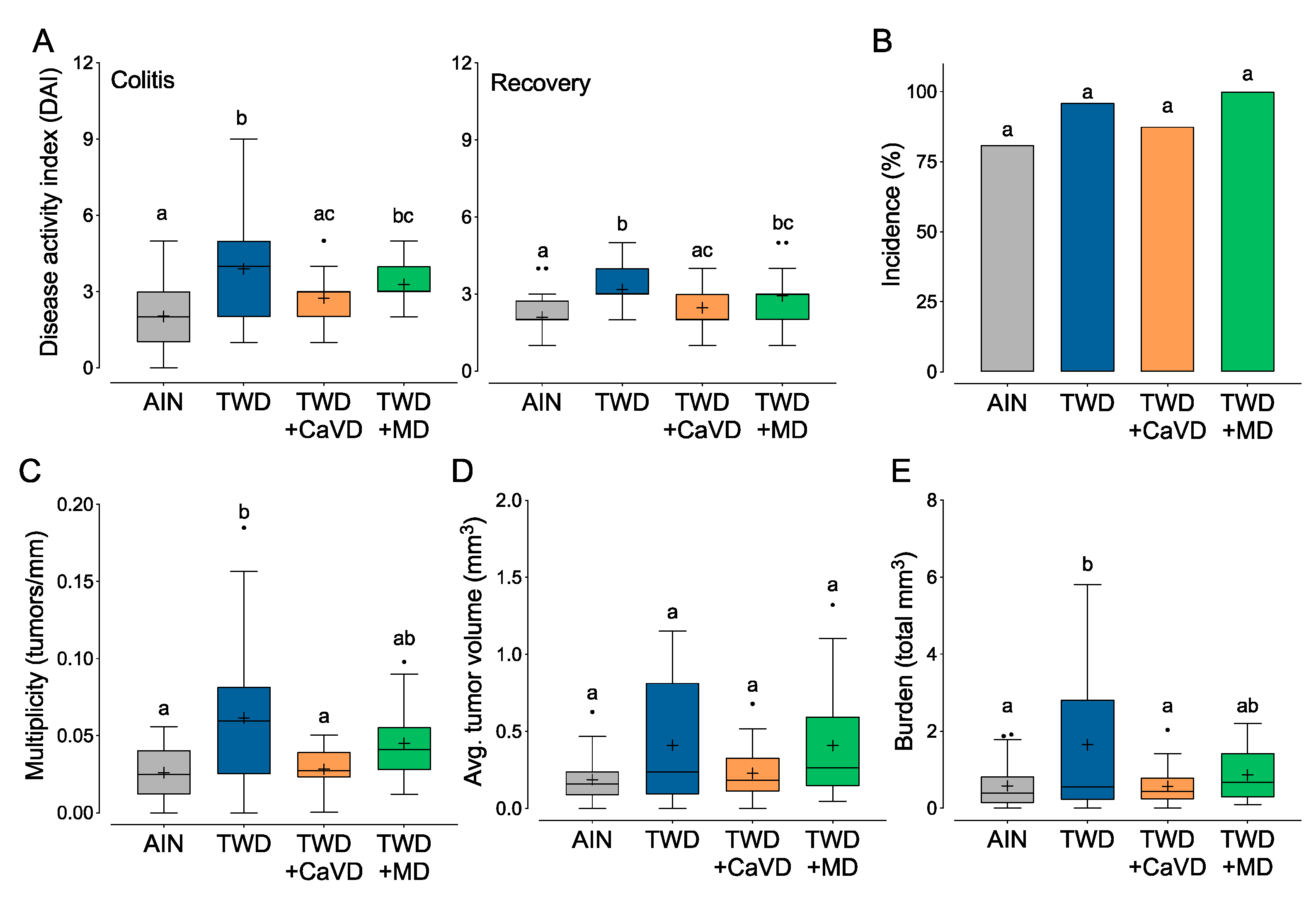
3.2. Experiment B: Restoration of Calcium and Vitamin D or Methyl Donor Micronutrients in TWD to Amounts in the AIN Diet
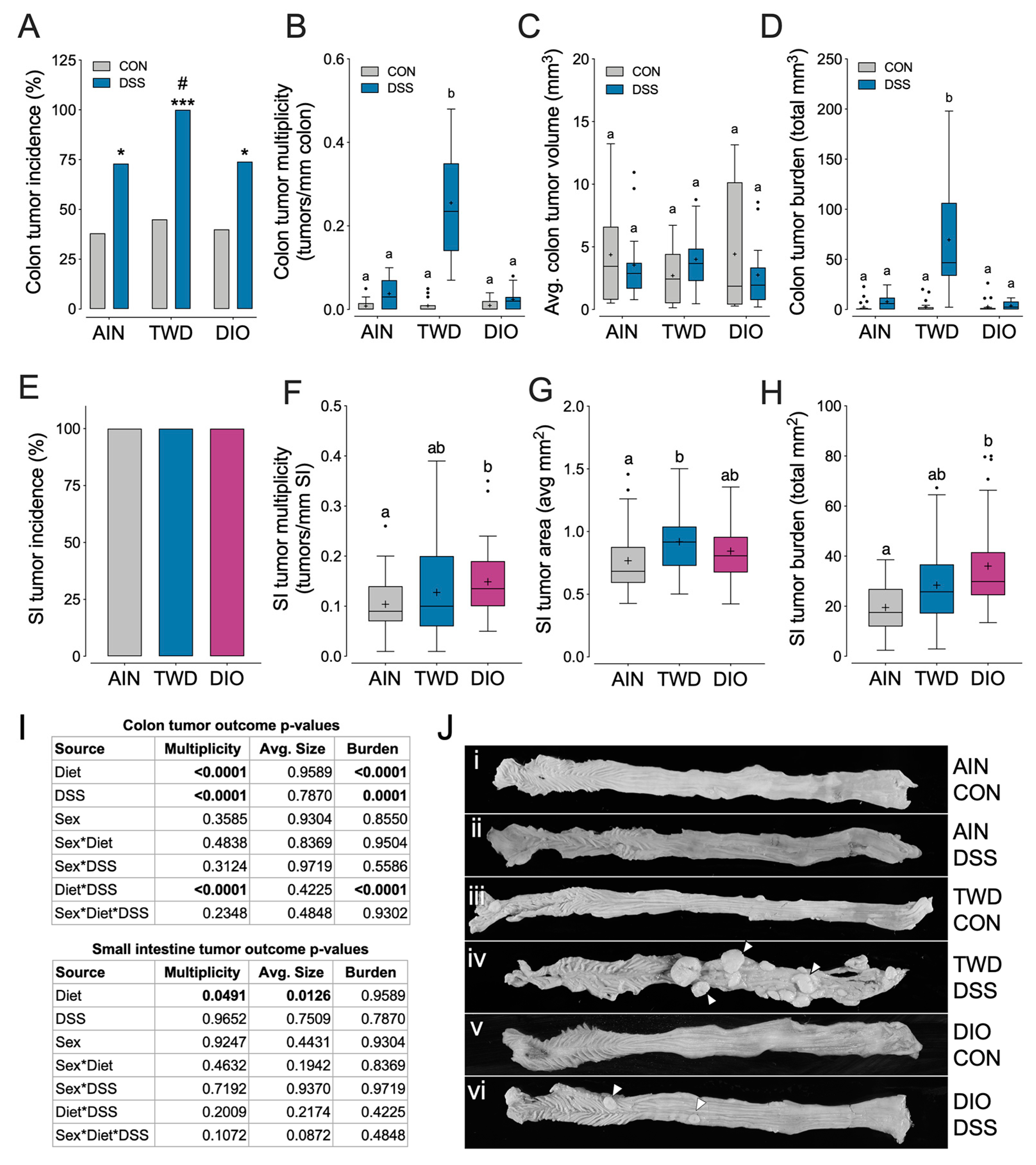
3.3. Experiment C: ApcMin/+ Model of Colorectal Carcinogenesis with TWD and DIO Diets
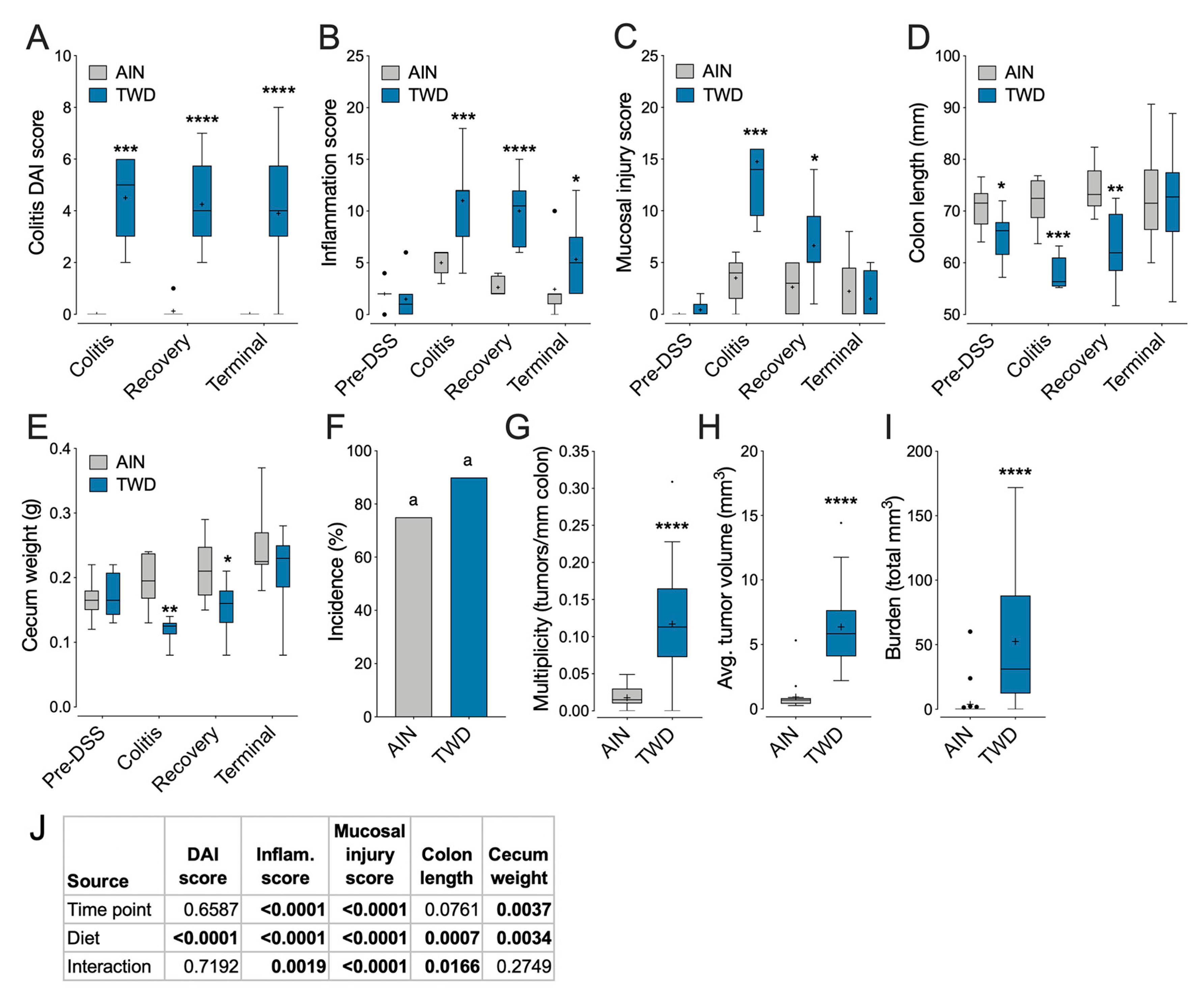
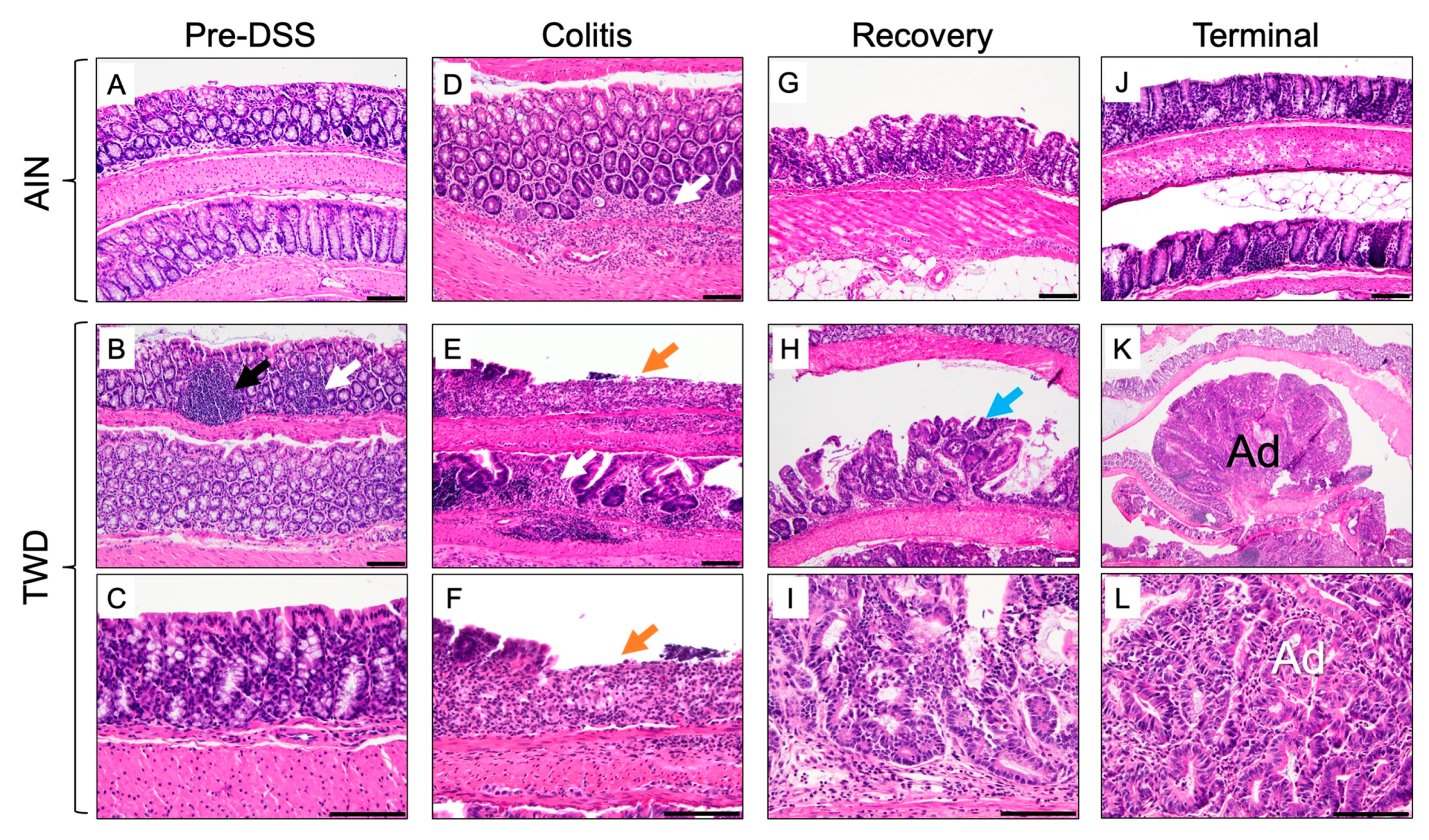
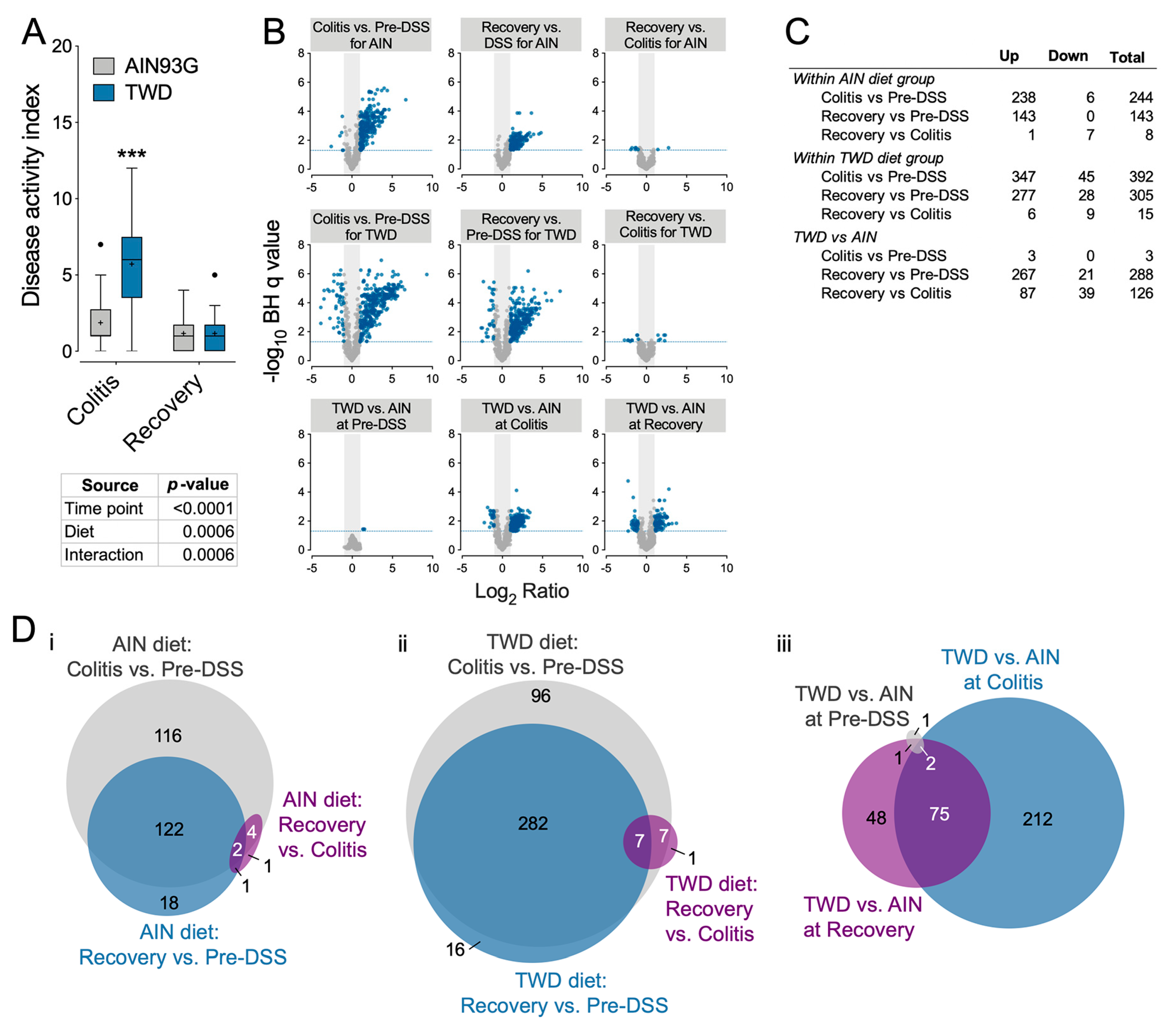
3.4. Experiment D: Longitudinal Effects of TWD on Colitis and Recovery from Gut Injury
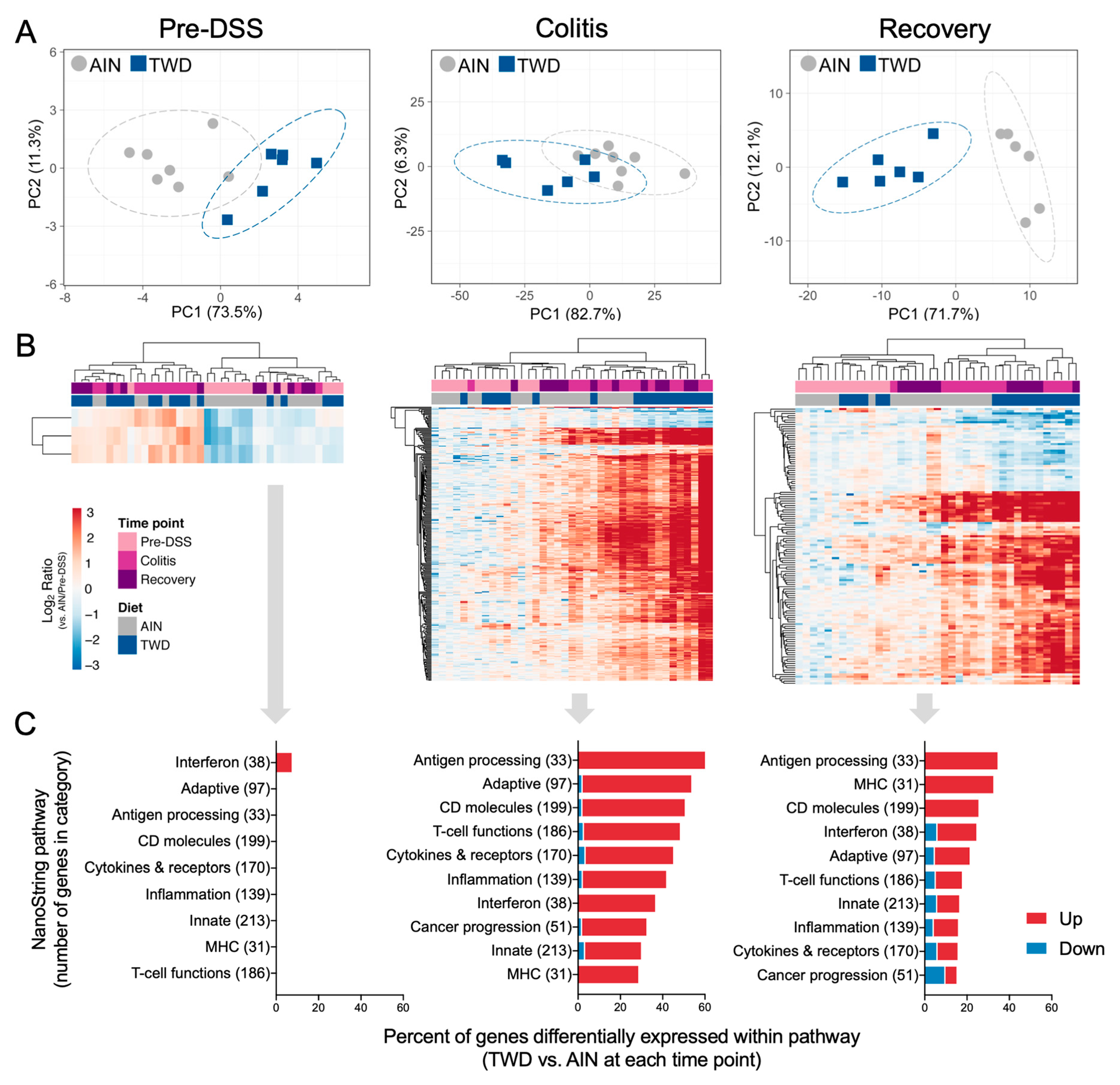
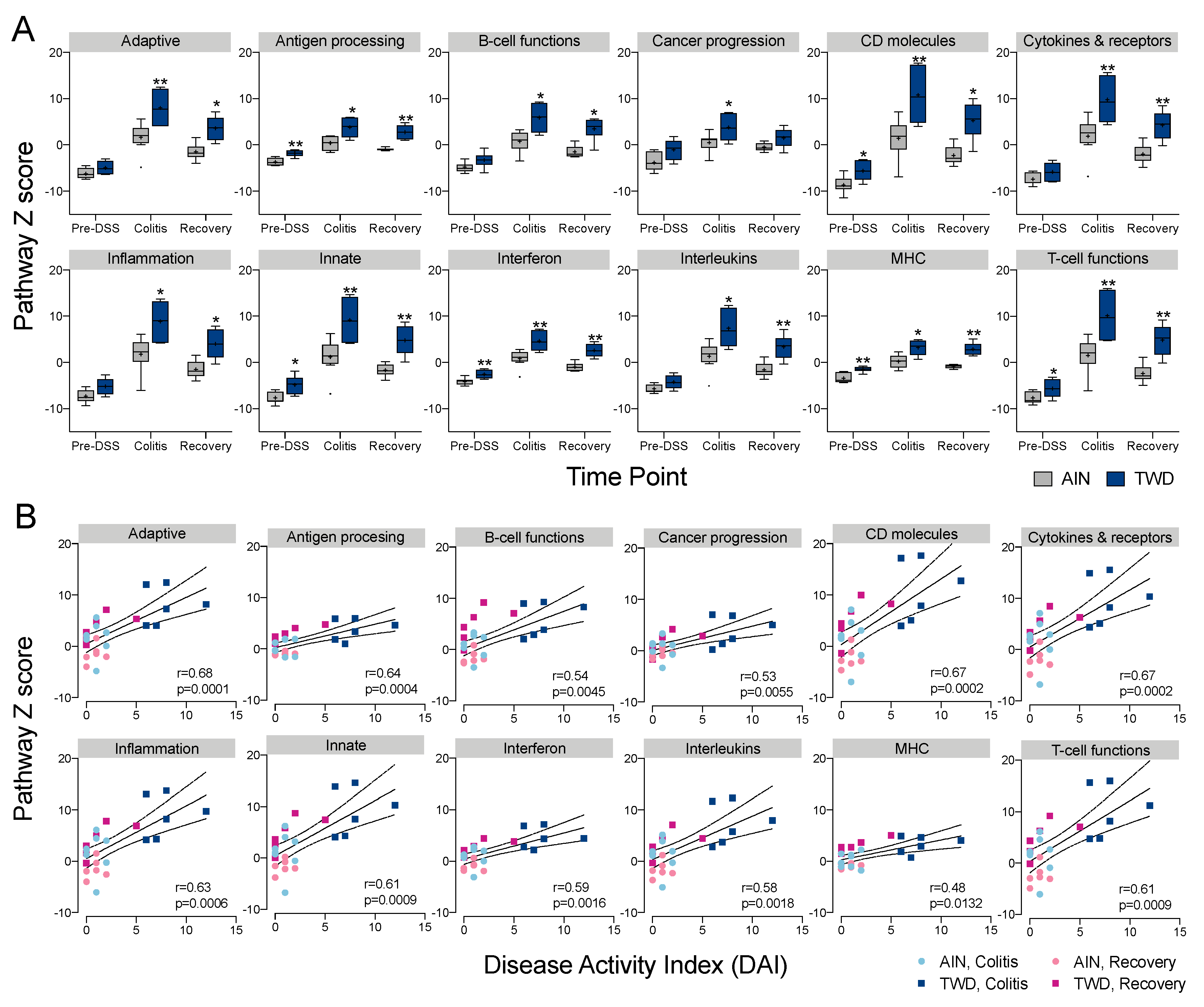
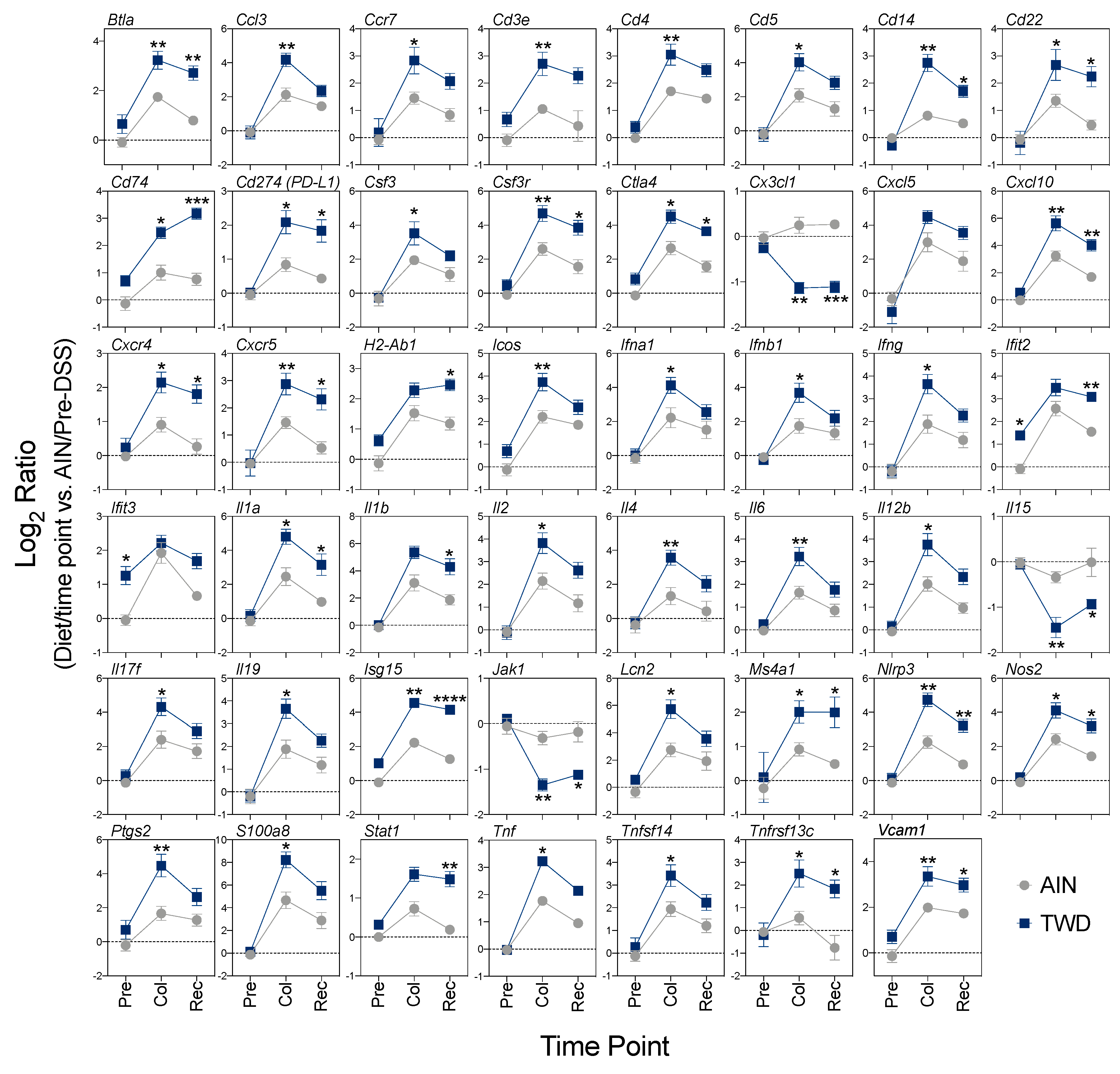
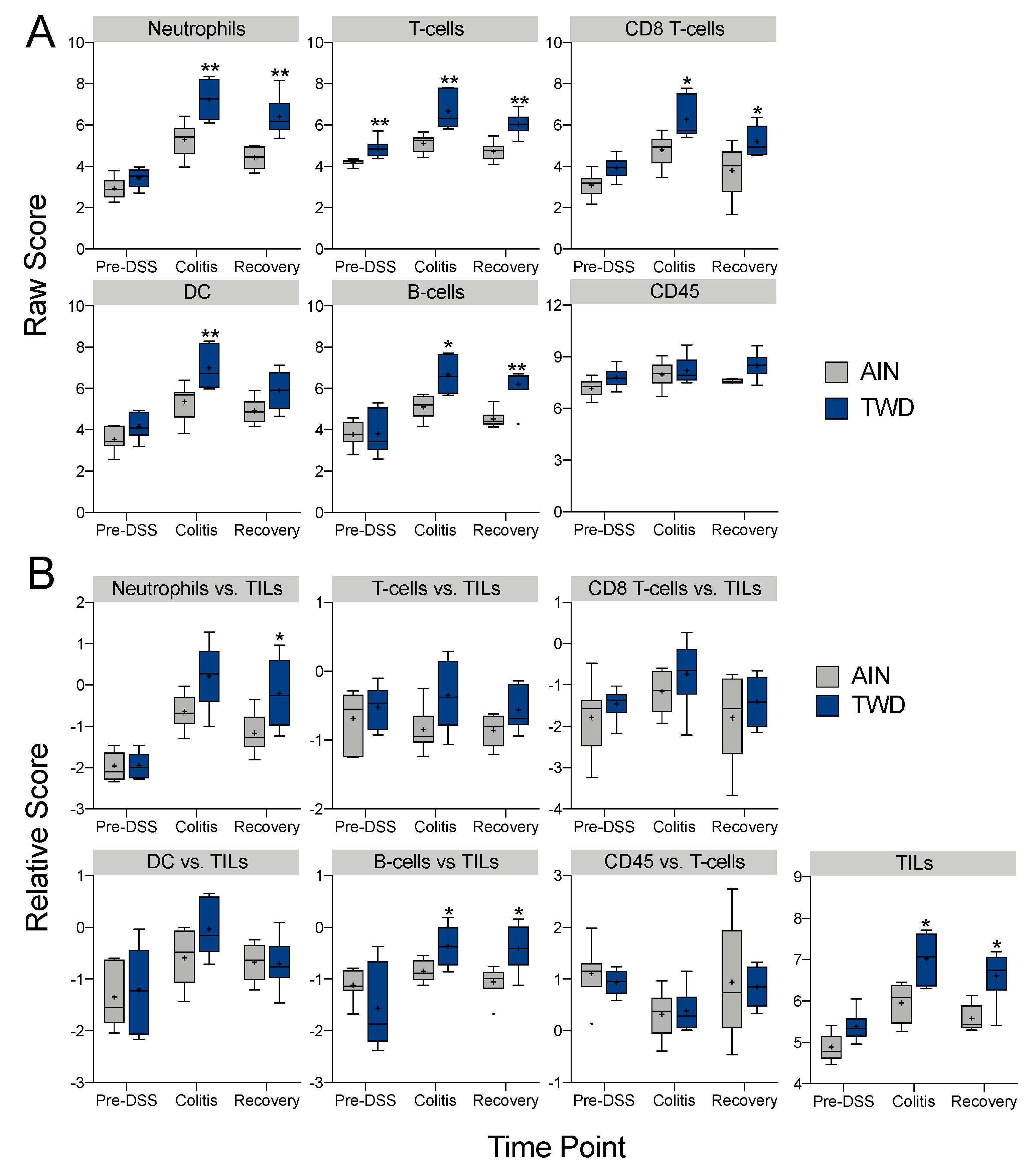
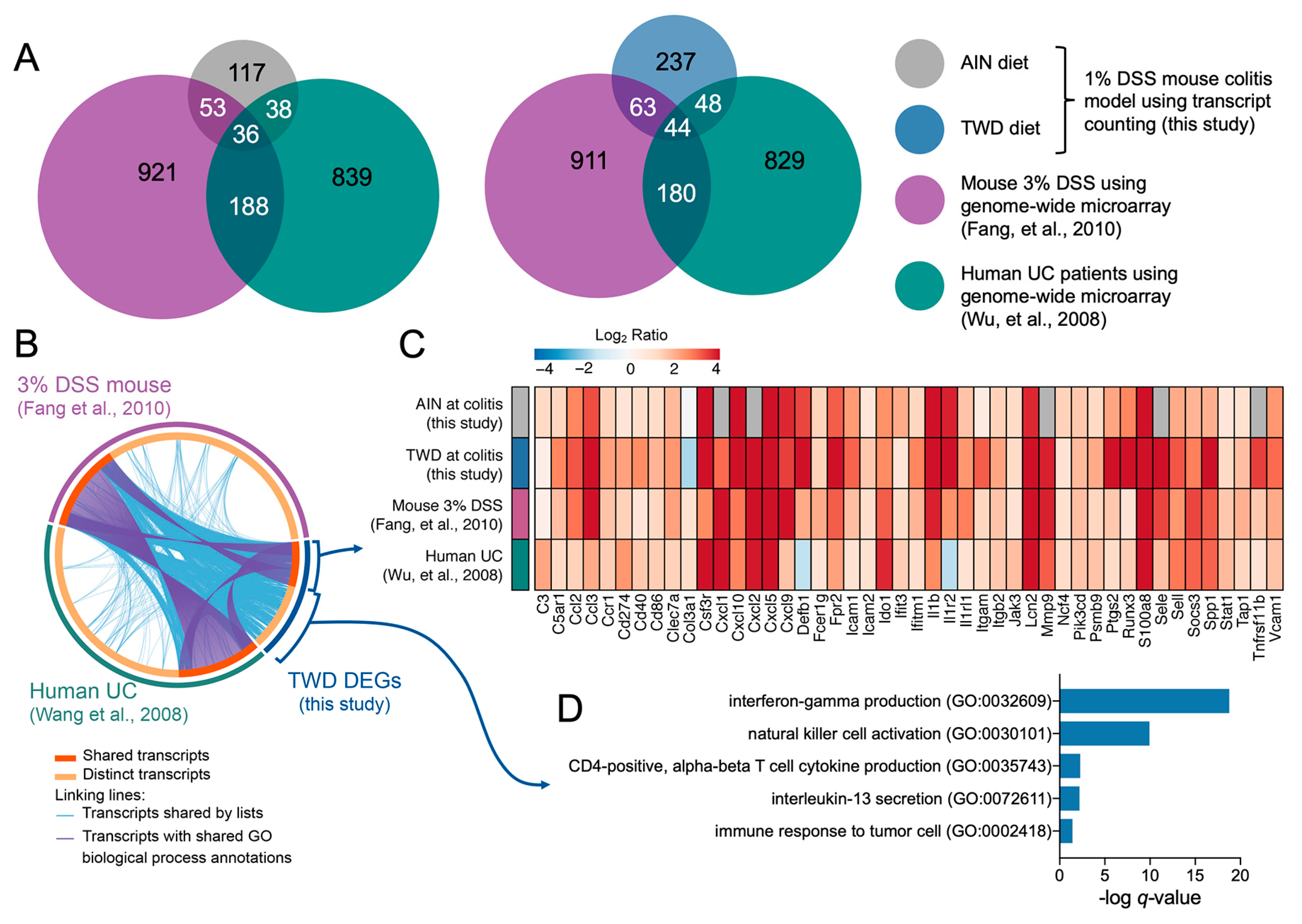
3.5. Experiment E: Immune- and Cancer-Related Gene Expression and cell Types in Colon Mucosa of TWD-fed Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ward, E.; Sherman, R.L.; Henley, S.J.; Jemal, A.; Siegel, D.A.; Feuer, E.J.; Firth, A.U.; Kohler, B.A.; Scott, S.; Ma, J.; et al. Annual report to the nation on the status of cancer, 1999-2015, featuring cancer in men and women ages 20–49. J. Natl. Cancer Inst. 2019. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, C.N.; Blanchard, J.F.; Kliewer, E.; Wajda, A. Cancer risk in patients with inflammatory bowel disease: A population-based study. Cancer 2001, 91, 854–862. [Google Scholar] [CrossRef]
- Canavan, C.; Abrams, K.R.; Mayberry, J. Meta-analysis: Colorectal and small bowel cancer risk in patients with Crohn’s disease. Aliment. Pharmacol. Ther. 2006, 23, 1097–1104. [Google Scholar] [CrossRef]
- Munkholm, P. Review article: The incidence and prevalence of colorectal cancer in inflammatory bowel disease. Aliment. Pharmacol. Ther. 2003, 18, 1–5. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Antoni, L.; Nuding, S.; Wehkamp, J.; Stange, E.F. Intestinal barrier in inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 1165–1179. [Google Scholar] [CrossRef]
- McCole, D.F. IBD candidate genes and intestinal barrier regulation. Inflamm. Bowel Dis. 2014, 20, 1829–1849. [Google Scholar] [CrossRef] [Green Version]
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef]
- Triantafillidis, J.K.; Nasioulas, G.; Kosmidis, P.A. Colorectal cancer and inflammatory bowel disease: Epidemiology, risk factors, mechanisms of carcinogenesis and prevention strategies. Anticancer Res. 2009, 29, 2727–2737. [Google Scholar]
- Tanaka, T.; Kohno, H.; Suzuki, R.; Yamada, Y.; Sugie, S.; Mori, H. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci. 2003, 94, 965–973. [Google Scholar] [CrossRef]
- Clapper, M.L.; Cooper, H.S.; Chang, W.C. Dextran sulfate sodium-induced colitis-associated neoplasia: A promising model for the development of chemopreventive interventions. Acta Pharmacol. Sin. 2007, 28, 1450–1459. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, M.; Wakabayashi, K. Gene mutations and altered gene expression in azoxymethane-induced colon carcinogenesis in rodents. Cancer Sci. 2004, 95, 475–480. [Google Scholar] [CrossRef]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA-Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [Green Version]
- Cordain, L.; Eaton, S.B.; Sebastian, A.; Mann, N.; Lindeberg, S.; Watkins, B.A.; O’Keefe, J.H.; Brand-Miller, J. Origins and evolution of the Western diet: Health implications for the 21st century. Am. J. Clin. Nutr. 2005, 81, 341–354. [Google Scholar] [CrossRef]
- Astrup, A.; Dyerberg, J.; Selleck, M.; Stender, S. Nutrition transition and its relationship to the development of obesity and related chronic diseases. Obes. Rev. 2008, 9, 48–52. [Google Scholar] [CrossRef]
- Carrera-Bastos, P.; Fontes-Villalba, M.; O’Keefe, J.; Lindeberg, S.; Cordain, L. The western diet and lifestyle and diseases of civilization. Res. Rep. Clin. Oncol. 2011, 2, 15–35. [Google Scholar] [CrossRef] [Green Version]
- Reeves, P.G.; Nielsen, F.H.; Fahey, G.C., Jr. AIN-93 purified diets for laboratory rodents: Final report of the American Institute of Nutrition ad hoc writing committee on the reformulation of the AIN-76A rodent diet. J. Nutr. 1993, 123, 1939–1951. [Google Scholar] [CrossRef]
- Newmark, H.L.; Lipkin, M.; Maheshwari, N. Colonic hyperplasia and hyperproliferation induced by a nutritional stress diet with four components of western-style diet. J. Natl. Cancer Inst. 1990, 82, 491–496. [Google Scholar] [CrossRef]
- Newmark, H.L.; Yang, K.; Kurihara, N.; Fan, K.; Augenlicht, L.H.; Lipkin, M. Western-style diet-induced colonic tumors and their modulation by calcium and vitamin D in C57Bl/6 mice: A preclinical model for human sporadic colon cancer. Carcinogenesis 2009, 30, 88–92. [Google Scholar] [CrossRef] [Green Version]
- Newmark, H.L.; Yang, K.; Lipkin, M.; Kopelovich, L.; Liu, Y.; Fan, K.; Shinozaki, H. A Western-style diet induces benign and malignant neoplasms in the colon of normal C57Bl/6 mice. Carcinogenesis 2001, 22, 1871–1875. [Google Scholar] [CrossRef] [Green Version]
- Envigo. Envigo Custom Research Diets: Diet Induced Obesity. Available online: https://www.envigo.com/p/teklad/laboratory-animal-diets/custom-research/diet-induced-obesity/ (accessed on 3 January 2020).
- Jawien, J.; Nastalek, P.; Korbut, R. Mouse models of experimental atherosclerosis. J. Physiol. Pharmacol. 2004, 55, 503–517. [Google Scholar] [PubMed]
- Hintze, K.J.; Benninghoff, A.D.; Ward, R.E. Formulation of the Total Western Diet (TWD) as a Basal Diet for Rodent Cancer Studies. J. Agric. Food Chem. 2012, 60, 6736–6742. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.E.; Benninghoff, A.D.; Healy, B.J.; Li, M.; Vagu, B.; Hintze, K.J. Consumption of the total Western diet differentially affects the response to green tea in rodent models of chronic disease compared to the AIN93G diet. Mol. Nutr. Food Res. 2017, 61, 1600720. [Google Scholar] [CrossRef]
- Su, L.K.; Kinzler, K.W.; Vogelstein, B.; Preisinger, A.C.; Moser, A.R.; Luongo, C.; Gould, K.A.; Dove, W.F. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 1992, 256, 668–670. [Google Scholar] [CrossRef]
- Truett, G.E.; Heeger, P.; Mynatt, R.L.; Truett, A.A.; Walker, J.A.; Warman, M.L. Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT). Biotechniques 2000, 29, 52–54. [Google Scholar] [CrossRef]
- Rosenberg, D.W.; Giardina, C.; Tanaka, T. Mouse models for the study of colon carcinogenesis. Carcinogenesis 2009, 30, 183–196. [Google Scholar] [CrossRef] [Green Version]
- Papanikolaou, A.; Wang, Q.S.; Delker, D.A.; Rosenberg, D.W. Azoxymethane-induced colon tumors and aberrant crypt foci in mice of different genetic susceptibility. Cancer Lett. 1998, 130, 29–34. [Google Scholar] [CrossRef]
- Boivin, G.P.; Washington, K.; Yang, K.; Ward, J.M.; Pretlow, T.P.; Russell, R.; Besselsen, D.G.; Godfrey, V.L.; Doetschman, T.; Dove, W.; et al. Pathology of mouse models of intestinal cancer: Consensus report and recommendations. Gastroenterology 2003, 124, 762–777. [Google Scholar] [CrossRef] [Green Version]
- Washington, M.K.; Powell, A.E.; Sullivan, R.; Sundberg, J.P.; Wright, N.; Coffey, R.J.; Dove, W.F. Pathology of rodent models of intestinal cancer: Progress report and recommendations. Gastroenterology 2013, 144, 705–717. [Google Scholar] [CrossRef] [Green Version]
- Bates, M.A.; Benninghoff, A.D.; Gilley, K.N.; Holian, A.; Harkema, J.R.; Pestka, J.J. Mapping of dynamic transcriptome changes associated with silica-triggered autoimmune pathogenesis in the lupus-prone NZBWF1 mouse. Front. Immunol. 2019, 10, 632. [Google Scholar] [CrossRef]
- Hulsen, T.; de Vlieg, J.; Alkema, W. BioVenn—A web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genom. 2008, 9, 488. [Google Scholar] [CrossRef] [Green Version]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Danaher, P.; Warren, S.; Dennis, L.; D’Amico, L.; White, A.; Disis, M.L.; Geller, M.A.; Odunsi, K.; Beechem, J.; Fling, S.P. Gene expression markers of tumor infiltrating leukocytes. J. Immunother. Cancer 2017, 5, 18. [Google Scholar] [CrossRef] [Green Version]
- Monsanto, S.P.; Hintze, K.J.; Ward, R.E.; Larson, D.P.; Lefevre, M.; Benninghoff, A.D. The new total Western diet for rodents does not induce an overweight phenotype or alter parameters of metabolic syndrome in mice. Nutr. Res. 2016, 36, 1031–1044. [Google Scholar] [CrossRef]
- Morotomi, M.; Sakaitani, Y.; Satou, M.; Takahashi, T.; Takagi, A.; Onoue, M. Effects of a high-fat diet on azoxymethane-induced aberrant crypt foci and fecal biochemistry and microbial activity in rats. Nutr. Cancer 1997, 27, 84–91. [Google Scholar] [CrossRef]
- Bull, A.W.; Soullier, B.K.; Wilson, P.S.; Hayden, M.T.; Nigro, N.D. Promotion of azoxymethane-induced intestinal cancer by high-fat diet in rats. Cancer Res. 1979, 39, 4956–4959. [Google Scholar]
- Kune, G.; Watson, L. Colorectal cancer protective effects and the dietary micronutrients folate, methionine, vitamins B6, B12, C, E, selenium, and lycopene. Nutr. Cancer 2006, 56, 11–21. [Google Scholar] [CrossRef]
- Hara, A.; Sasazuki, S.; Inoue, M.; Iwasaki, M.; Shimazu, T.; Sawada, N.; Yamaji, T.; Takachi, R.; Tsugane, S. Zinc and heme iron intakes and risk of colorectal cancer: A population-based prospective cohort study in Japan. Am. J. Clin. Nutr. 2012, 96, 864–873. [Google Scholar] [CrossRef]
- Gupta, D.; Lis, C.G.; Granick, J.; Grutsch, J.F.; Vashi, P.G.; Lammersfeld, C.A. Malnutrition was associated with poor quality of life in colorectal cancer: A retrospective analysis. J. Clin. Epidemiol. 2006, 59, 704–709. [Google Scholar] [CrossRef]
- Zhang, X.; Giovannucci, E. Calcium, vitamin D and colorectal cancer chemoprevention. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 485–494. [Google Scholar] [CrossRef]
- Wactawski-Wende, J.; Kotchen, J.M.; Anderson, G.L.; Assaf, A.R.; Brunner, R.L.; O’Sullivan, M.J.; Margolis, K.L.; Ockene, J.K.; Phillips, L.; Pottern, L.; et al. Calcium plus vitamin D supplementation and the risk of colorectal cancer. N. Engl. J. Med. 2006, 354, 684–696. [Google Scholar] [CrossRef] [Green Version]
- Bolland, M.J.; Grey, A.; Gamble, G.D.; Reid, I.R. Calcium and vitamin D supplements and health outcomes: A reanalysis of the Women’s Health Initiative (WHI) limited-access data set. Am. J. Clin. Nutr. 2011, 94, 1144–1149. [Google Scholar] [CrossRef]
- Yang, K.; Lamprecht, S.A.; Shinozaki, H.; Fan, K.; Yang, W.; Newmark, H.L.; Kopelovich, L.; Edelmann, W.; Jin, B.; Gravaghi, C.; et al. Dietary calcium and cholecalciferol modulate cyclin D1 expression, apoptosis, and tumorigenesis in intestine of adenomatous polyposis coli1638n/+ mice. J. Nutr. 2008, 138, 1658–1663. [Google Scholar] [CrossRef] [Green Version]
- Erdelyi, I.; Levenkova, N.; Lin, E.Y.; Pinto, J.T.; Lipkin, M.; Quimby, F.W.; Holt, P.R. Western-style diets induce oxidative stress and dysregulate immune responses in the colon in a mouse model of sporadic colon cancer. J. Nutr. 2009, 139, 2072–2078. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, D.R.; Holmstrom, S.R.; Fon Tacer, K.; Bookout, A.L.; Kliewer, S.A.; Mangelsdorf, D.J. Regulation of bile acid synthesis by fat-soluble vitamins A and D. J. Biol. Chem. 2010, 285, 14486–14494. [Google Scholar] [CrossRef] [Green Version]
- Makishima, M.; Lu, T.T.; Xie, W.; Whitfield, G.K.; Domoto, H.; Evans, R.M.; Haussler, M.R.; Mangelsdorf, D.J. Vitamin D receptor as an intestinal bile acid sensor. Science 2002, 296, 1313–1316. [Google Scholar] [CrossRef] [Green Version]
- Lapre, J.A.; De Vries, H.T.; Van der Meer, R. Cytotoxicity of fecal water is dependent on the type of dietary fat and is reduced by supplemental calcium phosphate in rats. J. Nutr. 1993, 123, 578–585. [Google Scholar] [CrossRef]
- Govers, M.J.; Van der Meet, R. Effects of dietary calcium and phosphate on the intestinal interactions between calcium, phosphate, fatty acids, and bile acids. Gut 1993, 34, 365–370. [Google Scholar] [CrossRef] [Green Version]
- Payne, C.M.; Bernstein, C.; Dvorak, K.; Bernstein, H. Hydrophobic bile acids, genomic instability, Darwinian selection, and colon carcinogenesis. Clin. Exp. Gastroenterol. 2008, 1, 19–47. [Google Scholar] [CrossRef] [Green Version]
- Barrasa, J.I.; Olmo, N.; Lizarbe, M.A.; Turnay, J. Bile acids in the colon, from healthy to cytotoxic molecules. Toxicol Vitr. 2013, 27, 964–977. [Google Scholar] [CrossRef]
- Lim, W.C.; Hanauer, S.B.; Li, Y.C. Mechanisms of disease: Vitamin D and inflammatory bowel disease. Nat. Clin. Pract. Gastroenterol. Hepatol. 2005, 2, 308–315. [Google Scholar] [CrossRef]
- Loftus, E.V., Jr. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology 2004, 126, 1504–1517. [Google Scholar] [CrossRef]
- Parizadeh, S.M.; Jafarzadeh-Esfehani, R.; Hassanian, S.M.; Mottaghi-Moghaddam, A.; Ghazaghi, A.; Ghandehari, M.; Alizade-Noghani, M.; Khazaei, M.; Ghayour-Mobarhan, M.; Ferns, G.A.; et al. Vitamin D in inflammatory bowel disease: From biology to clinical implications. Complement. Ther. Med. 2019, 47, 102189. [Google Scholar] [CrossRef]
- Gubatan, J.; Chou, N.D.; Nielsen, O.H.; Moss, A.C. Systematic review with meta-analysis: Association of vitamin D status with clinical outcomes in adult patients with inflammatory bowel disease. Aliment. Pharmacol. Ther. 2019, 50, 1146–1158. [Google Scholar] [CrossRef]
- Fletcher, J.; Cooper, S.C.; Ghosh, S.; Hewison, M. The role of vitamin D in inflammatory nowel disease: Mechanism to management. Nutrients 2019, 11, 1019. [Google Scholar] [CrossRef] [Green Version]
- Niculescu, M.D.; Zeisel, S.H. Diet, methyl donors and DNA methylation: Interactions between dietary folate, methionine and choline. J. Nutr. 2002, 132, 2333S–2335S. [Google Scholar] [CrossRef]
- Stefanska, B.; Karlic, H.; Varga, F.; Fabianowska-Majewska, K.; Haslberger, A. Epigenetic mechanisms in anti-cancer actions of bioactive food components--the implications in cancer prevention. Br. J. Pharmacol. 2012, 167, 279–297. [Google Scholar] [CrossRef] [Green Version]
- Marchand, L.L.; White, K.K.; Nomura, A.M.Y.; Wilkens, L.R.; Selhub, J.S.; Tiirikainen, M.; Goodman, M.T.; Murphy, S.P.; Henderson, B.E.; Kolonel, L.N. Plasma levels of B vitamins and colorectal cancer risk: The multiethnic cohort study. Cancer Epidemiol. Biomark. Prev. 2009, 18, 2195–2201. [Google Scholar] [CrossRef] [Green Version]
- Sanjoaquin, M.A.; Allen, N.; Couto, E.; Roddam, A.W.; Key, T.J. Folate intake and colorectal cancer risk: A meta-analytical approach. Int. J. Cancer 2005, 113, 825–828. [Google Scholar] [CrossRef]
- Je, Y.; Lee, J.E.; Ma, J.; Zhang, X.; Cho, E.; Rosner, B.; Selhub, J.; Fuchs, C.S.; Meyerhardt, J.; Giovannucci, E. Prediagnostic plasma vitamin B6 (pyridoxal 5’-phosphate) and survival in patients with colorectal cancer. Cancer Cause Control. 2013, 24, 719–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindzon, G.M.; Medline, A.; Sohn, K.-J.; Depeint, F.; Croxford, R.; Kim, Y.-I. Effect of folic acid supplementation on the progression of colorectal aberrant crypt foci. Carcinogenesis 2009, 30, 1536–1543. [Google Scholar] [CrossRef] [PubMed]
- Otani, T.; Iwasaki, M.; Hanaoka, T.; Kobayashi, M.; Ishihara, J.; Natsukawa, S.; Shaura, K.; Koizumi, Y.; Kasuga, Y.; Yoshimura, K.; et al. Folate, vitamin B6, vitamin B12, and vitamin B2 intake, genetic polymorphisms of related enzymes, and risk of colorectal cancer in a hospital-based case-control study in Japan. Nutr. Cancer 2005, 53, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Bergomas, F.; Grizzi, F.; Doni, A.; Pesce, S.; Laghi, L.; Allavena, P.; Mantovani, A.; Marchesi, F. Tertiary intratumor lymphoid tissue in colo-rectal cancer. Cancers 2011, 4, 1–10. [Google Scholar] [CrossRef]
- Fang, K.; Bruce, M.; Pattillo, C.B.; Zhang, S.; Stone, R., 2nd; Clifford, J.; Kevil, C.G. Temporal genomewide expression profiling of DSS colitis reveals novel inflammatory and angiogenesis genes similar to ulcerative colitis. Physiol. Genom. 2011, 43, 43–56. [Google Scholar] [CrossRef]
- Holgersen, K.; Kutlu, B.; Fox, B.; Serikawa, K.; Lord, J.; Hansen, A.K.; Holm, T.L. High-resolution gene expression profiling using RNA sequencing in patients with inflammatory bowel disease and in mouse models of colitis. J. Crohns Colitis 2015, 9, 492–506. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Zikusoka, M.; Trindade, A.; Dassopoulos, T.; Harris, M.L.; Bayless, T.M.; Brant, S.R.; Chakravarti, S.; Kwon, J.H. MicroRNAs are differentially expressed in ulcerative colitis and alter expression of macrophage inflammatory peptide-2 alpha. Gastroenterology 2008, 135, 1624–1635. [Google Scholar] [CrossRef]
- Chassaing, B.; Aitken, J.D.; Malleshappa, M.; Vijay-Kumar, M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr. Protoc. Immunol. 2014, 104. [Google Scholar] [CrossRef]
- Keubler, L.M.; Buettner, M.; Hager, C.; Bleich, A. A multihit model: Colitis lessons from the interleukin-10-deficient mouse. Inflamm. Bowel Dis. 2015, 21, 1967–1975. [Google Scholar] [CrossRef]
- Hintze, K.J.; Benninghoff, A.D.; Cho, C.E.; Ward, R.E. Modeling the Western diet for preclinical investigations. Adv. Nutr. 2018, 9, 263–271. [Google Scholar] [CrossRef] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AIN93G (AIN) | TWD | DIO | VMM | MM | TWD + CaVD | TWD + MD | |
|---|---|---|---|---|---|---|---|
| Energy density (kcal/g) | 3.8 | 4.4 | 4.6 | 3.7 | 4.4 | 4.4 | 4.4 |
| Macronutrients | |||||||
| Carbohydrates (g/kg diet) | |||||||
| Corn starch | 398 | 230 | 85 | 398 | 230 | 230 | 230 |
| Maltodextrin | 132 | 70 | 115 | 132 | 70 | 70 | 70 |
| Sucrose | 100 | 261 | 200 | 100 | 261 | 261 | 261 |
| Cellulose | 50 | 30 | 58 | 50 | 30 | 30 | 30 |
| kcal (% of total) | 60.1% | 54.5% | 36.2% | 60.1% | 54.5% | 54.5% | 54.5% |
| Proteins (g/kg) | |||||||
| Casein | 200 | 190 | 245 | 200 | 190 | 190 | 190 |
| L-cystine | 3 | 2.8 | 3.5 | 3 | 2.8 | 2.8 | 2.8 |
| kcal (% of total) | 18.8% | 15.4% | 19.0% | 18.8% | 15.4% | 15.4% | 15.4% |
| Fats (g/kg) | |||||||
| Soybean oil | 70 | 31.4 | 30 | 70 | 31.4 | 31.4 | 31.4 |
| Anhydrous milk fat | 0 | 36.3 | 0 | 0 | 36.3 | 36.3 | 36.3 |
| Olive oil | 0 | 28.0 | 0 | 0 | 28.0 | 28.0 | 28.0 |
| Lard | 0 | 28.0 | 195 | 0 | 28.0 | 28.0 | 28.0 |
| Beef tallow | 0 | 24.8 | 0 | 0 | 24.8 | 24.8 | 24.8 |
| Corn oil | 0 | 16.5 | 0 | 0 | 16.5 | 16.5 | 16.5 |
| Cholesterol | 0 | 0.4 | 0 | 0 | 0.4 | 0.4 | 0.4 |
| kcal (% of total) | 17.2% | 34.5% | 44.8% | 17.2% | 34.5% | 34.5% | 34.5% |
| Micronutrients | |||||||
| Minerals (mg/kg) | |||||||
| Calcium | 5000 | 2011 | 5000 | 2011 | 5000 | 5000 | 2011 |
| Phosphorus | 3000 | 2757 | 3000 | 2757 | 3000 | 2757 | 2757 |
| Sodium | 1019 | 7078 | 1019 | 7078 | 1019 | 7078 | 7078 |
| Potassium | 3600 | 5333 | 3600 | 5333 | 3600 | 5333 | 5333 |
| Magnesium | 507 | 589 | 507 | 589 | 507 | 589 | 589 |
| Iron | 35 | 31 | 35 | 31 | 35 | 31 | 31 |
| Zinc | 30 | 25 | 30 | 25 | 30 | 25 | 25 |
| Copper | 6 | 2.6 | 6 | 2.6 | 6 | 2.6 | 2.6 |
| Selenium | 0.15 | 0.2 | 0.15 | 0.2 | 0.15 | 0.2 | 0.2 |
| Vitamins (unit/kg) | |||||||
| Thiamin (mg) | 5 | 3.5 | 5 | 3.5 | 5 | 3.5 | 3.5 |
| Riboflavin (B2) (mg) | 6 | 4.4 | 6 | 4.4 | 6 | 4.4 | 6 |
| Niacin (mg) | 30 | 50.6 | 30 | 50.6 | 30 | 50.6 | 50.6 |
| Pyridoxine (B6) (mg) | 6 | 3.9 | 6 | 3.9 | 6 | 3.9 | 6 |
| Folate (mg) | 2 | 1.3 | 2 | 1.3 | 2 | 1.3 | 2 |
| Vitamin B12 (μg) | 25 | 11 | 25 | 11 | 25 | 11 | 25 |
| Vitamin A (IU) | 4000 | 4300 | 4000 | 4300 | 4000 | 4300 | 4300 |
| Vitamin D (IU) | 1000 | 391 | 1000 | 391 | 1000 | 1000 | 391 |
| Vitamin E (IU) | 75 | 24.6 | 75 | 24.6 | 75 | 24.6 | 24.6 |
| Vitamin K (μg) | 750 | 189 | 750 | 189 | 750 | 189 | 189 |
| Choline (mg) | 1027 | 648 | 1027 | 648 | 1027 | 648 | 1027 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benninghoff, A.D.; Hintze, K.J.; Monsanto, S.P.; Rodriguez, D.M.; Hunter, A.H.; Phatak, S.; Pestka, J.J.; Van Wettere, A.J.; Ward, R.E. Consumption of the Total Western Diet Promotes Colitis and Inflammation-Associated Colorectal Cancer in Mice. Nutrients 2020, 12, 544. https://doi.org/10.3390/nu12020544
Benninghoff AD, Hintze KJ, Monsanto SP, Rodriguez DM, Hunter AH, Phatak S, Pestka JJ, Van Wettere AJ, Ward RE. Consumption of the Total Western Diet Promotes Colitis and Inflammation-Associated Colorectal Cancer in Mice. Nutrients. 2020; 12(2):544. https://doi.org/10.3390/nu12020544
Chicago/Turabian StyleBenninghoff, Abby D., Korry J. Hintze, Stephany P. Monsanto, Daphne M. Rodriguez, Ashli H. Hunter, Sumira Phatak, James J. Pestka, Arnaud J. Van Wettere, and Robert E. Ward. 2020. "Consumption of the Total Western Diet Promotes Colitis and Inflammation-Associated Colorectal Cancer in Mice" Nutrients 12, no. 2: 544. https://doi.org/10.3390/nu12020544