Preserved Skeletal Muscle Mitochondrial Function, Redox State, Inflammation and Mass in Obese Mice with Chronic Heart Failure

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Methods
2.1. Study Protocol
2.2. Blood Glucose
2.3. Mitochondrial Function
2.4. Superoxide Generation and Redox State
2.5. Protein Expression and Phosphorylation
2.6. Statistical Analysis
3. Results
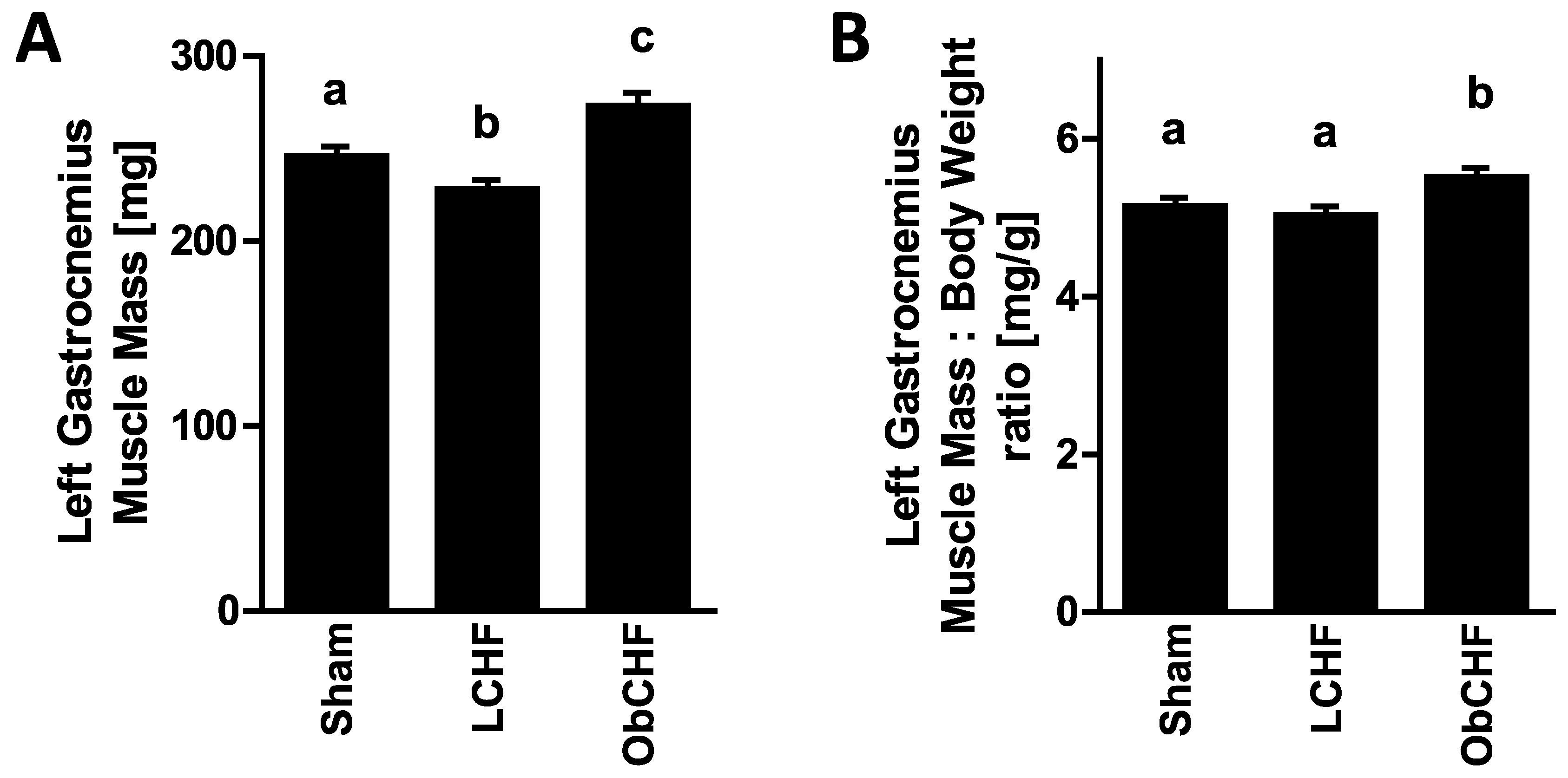
3.1. Calorie Intake, Body and Tissue Weight and Plasma Glucose
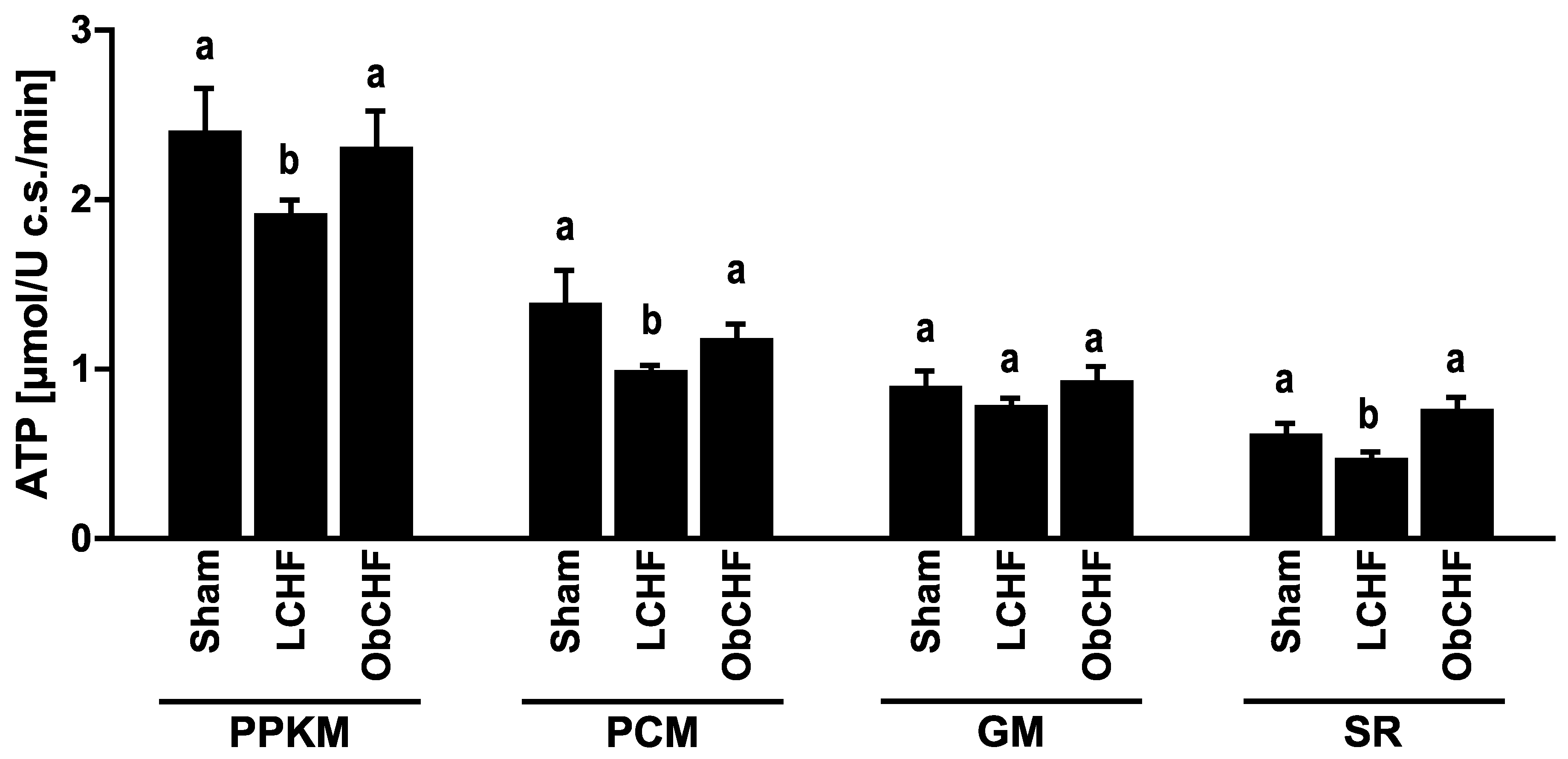
3.2. Skeletal Muscle Mitochondrial ATP and ROS Production
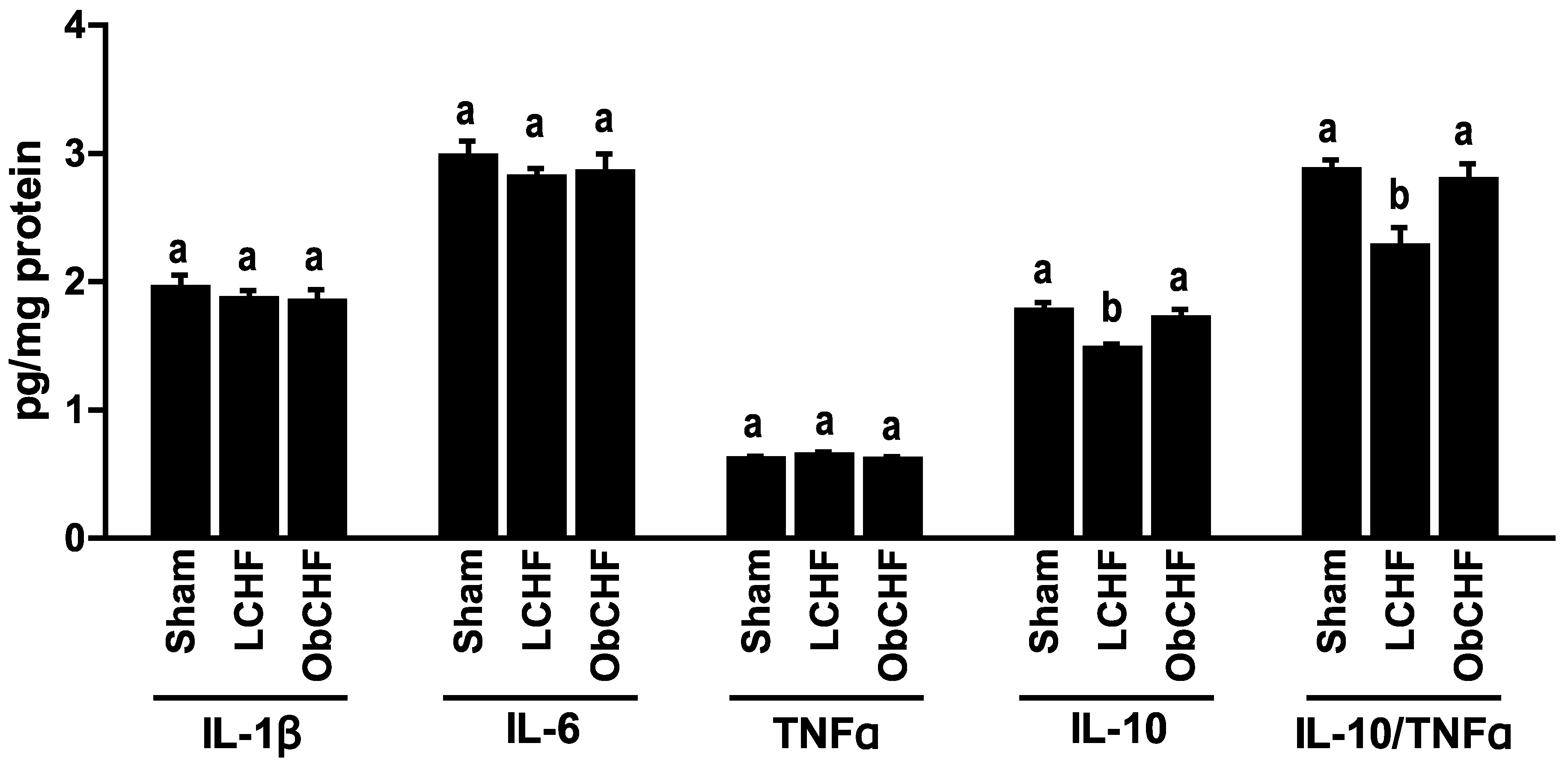
3.3. Skeletal Muscle Cytokine Profile
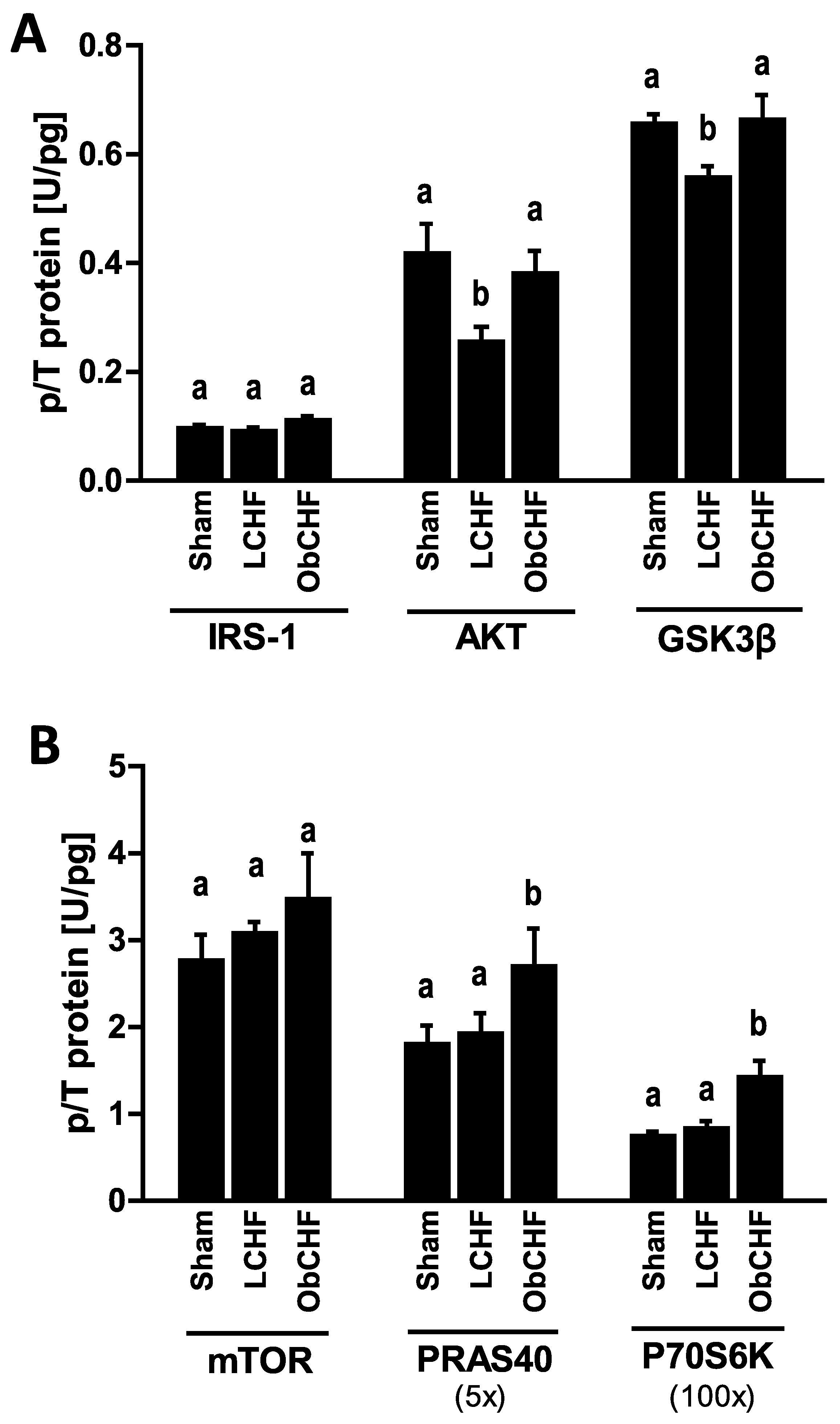
3.4. Skeletal Muscle Insulin Signalling
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- von Haehling, S.; Anker, M.S.; Anker, S.D. Prevalence and clinical impact of cachexia in chronic illness in Europe, USA, and Japan: Facts and numbers update 2016. J. Cachexia Sarcopenia Muscle 2016, 7, 507–509. [Google Scholar] [CrossRef]
- Tomasoni, D.; Adamo, M.; Lombardi, C.M.; Metra, M. Highlights in heart failure. ESC Heart Fail. 2019, 6, 1105–1127. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, A.M.; Lucas, R.; Cowie, M.R. Assessing health-related quality of life in heart failure patients attending an outpatient clinic: A pragmatic approach. ESC Heart Fail. 2019, 6, 3–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbone, S.; Billingsley, H.E.; Rodriguez-Miguelez, P.; Kirkman, D.L.; Garten, R.; Franco, R.L.; Lee, D.-C.; Lavie, C.J. Lean Mass Abnormalities in Heart Failure: The Role of Sarcopenia, Sarcopenic Obesity, and Cachexia. Curr. Probl. Cardiol. 2019, 100417. [Google Scholar] [CrossRef] [PubMed]
- Fulster, S.; Tacke, M.; Sandek, A.; Ebner, N.; Tschope, C.; Doehner, W.; Anker, S.D.; von Haehling, S. Muscle wasting in patients with chronic heart failure: Results from the studies investigating co-morbidities aggravating heart failure (SICA-HF). Eur. Heart J. 2013, 34, 512–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Palus, S.; Springer, J. Skeletal muscle wasting in chronic heart failure. ESC Heart Fail. 2018, 5, 1099–1107. [Google Scholar] [CrossRef] [PubMed]
- von Haehling, S. Muscle wasting and sarcopenia in heart failure: A brief overview of the current literature. ESC Heart Fail. 2018, 5, 1074–1082. [Google Scholar] [CrossRef] [Green Version]
- Emami, A.; Saitoh, M.; Valentova, M.; Sandek, A.; Evertz, R.; Ebner, N.; Loncar, G.; Springer, J.; Doehner, W.; Lainscak, M.; et al. Comparison of sarcopenia and cachexia in men with chronic heart failure: Results from the Studies Investigating Co-morbidities Aggravating Heart Failure (SICA-HF). Eur. J. Heart Fail. 2018, 20, 1580–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ventura, H.O.; Carbone, S.; Lavie, C.J. Muscling up to improve heart failure prognosis. Eur. J. Heart Fail. 2018, 20, 1588–1590. [Google Scholar] [CrossRef] [Green Version]
- Mangner, N.; Weikert, B.; Bowen, T.S.; Sandri, M.; Hollriegel, R.; Erbs, S.; Hambrecht, R.; Schuler, G.; Linke, A.; Gielen, S.; et al. Skeletal muscle alterations in chronic heart failure: Differential effects on quadriceps and diaphragm. J. Cachexia Sarcopenia Muscle 2015, 6, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Tromp, J.; Westenbrink, B.D.; Ouwerkerk, W.; van Veldhuisen, D.J.; Samani, N.J.; Ponikowski, P.; Metra, M.; Anker, S.D.; Cleland, J.G.; Dickstein, K.; et al. Identifying Pathophysiological Mechanisms in Heart Failure WithReduced Versus Preserved EjectionFraction. J. Am. Coll. Cardiol. 2018, 72, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Z.; Marcinek, D.J. Skeletal muscle bioenergetics in aging and heart failure. Heart Fail. Rev. 2017, 22, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Lu, X.; Qian, Z.; Xu, W.; Zhou, X. New insights into the pathogenesis and treatment of sarcopenia in chronic heart failure. Theranostics 2019, 9, 4019–4029. [Google Scholar] [CrossRef] [PubMed]
- Bowen, T.S.; Rolim, N.P.L.; Fischer, T.; Baekkerud, F.H.; Medeiros, A.; Werner, S.; Bronstad, E.; Rognmo, O.; Mangner, N.; Linke, A.; et al. Heart failure with preserved ejection fraction induces molecular, mitochondrial, histological, and functional alterations in rat respiratory and limb skeletal muscle. Eur. J. Heart Fail. 2015, 17, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Lena, A.; Coats, A.J.S.; Anker, M.S. Metabolic disorders in heart failure and cancer. ESC Heart Fail. 2018, 5, 1092–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tkaczyszyn, M.; Drozd, M.; Wegrzynowska-Teodorczyk, K.; Flinta, I.; Kobak, K.; Banasiak, W.; Ponikowski, P.; Jankowska, E.A. Depleted iron stores are associated with inspiratory muscle weakness independently of skeletal muscle mass in men with systolic chronic heart failure. J. Cachexia Sarcopenia Muscle 2018, 9, 547–556. [Google Scholar] [CrossRef]
- Garnham, J.O.; Roberts, L.D.; Caspi, T.; Al-Owais, M.M.; Bullock, M.; Swoboda, P.P.; Koshy, A.; Gierula, J.; Paton, M.F.; Cubbon, R.M.; et al. Divergent skeletal muscle mitochondrial phenotype between male and female patients with chronic heart failure. J. Cachexia Sarcopenia Muscle 2020, 11, 79–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garnier, A.; Fortin, D.; Zoll, J.; N’Guessan, B.; Mettauer, B.; Lampert, E.; Veksler, V.; Ventura-Clapier, R. Coordinated changes in mitochondrial function and biogenesis in healthy and diseased human skeletal muscle. FASEB J. 2005, 19, 43–52. [Google Scholar] [CrossRef]
- Kumar, A.A.; Kelly, D.P.; Chirinos, J.A. Mitochondrial Dysfunction in Heart Failure with Preserved Ejection Fraction. Circulation 2019, 139, 1435–1450. [Google Scholar] [CrossRef]
- Ohta, Y.; Kinugawa, S.; Matsushima, S.; Ono, T.; Sobirin, M.A.; Inoue, N.; Yokota, T.; Hirabayashi, K.; Tsutsui, H. Oxidative stress impairs insulin signal in skeletal muscle and causes insulin resistance in postinfarct heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H1637–H1644. [Google Scholar] [CrossRef] [PubMed]
- Martins, T.; Vitorino, R.; Moreira-Goncalves, D.; Amado, F.; Duarte, J.A.; Ferreira, R. Recent insights on the molecular mechanisms and therapeutic approaches for cardiac cachexia. Clin. Biochem. 2014, 47, 8–15. [Google Scholar] [CrossRef]
- Markousis-Mavrogenis, G.; Tromp, J.; Ouwerkerk, W.; Devalaraja, M.; Anker, S.D.; Cleland, J.G.; Dickstein, K.; Filippatos, G.S.; van der Harst, P.; Lang, C.C.; et al. The clinical significance of interleukin-6 in heart failure: Results from the BIOSTAT-CHF study. Eur. J. Heart Fail. 2019, 21, 965–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating oxidative stress in heart failure: Past, present and future. Eur. J. Heart Fail. 2019, 21, 425–435. [Google Scholar] [CrossRef]
- Tsuda, M.; Fukushima, A.; Matsumoto, J.; Takada, S.; Kakutani, N.; Nambu, H.; Yamanashi, K.; Furihata, T.; Yokota, T.; Okita, K.; et al. Protein acetylation in skeletal muscle mitochondria is involved in impaired fatty acid oxidation and exercise intolerance in heart failure. J. Cachexia Sarcopenia Muscle 2018, 9, 844–859. [Google Scholar] [CrossRef] [PubMed]
- Seiler, M.; Bowen, T.S.; Rolim, N.; Dieterlen, M.-T.; Werner, S.; Hoshi, T.; Fischer, T.; Mangner, N.; Linke, A.; Schuler, G.; et al. Skeletal Muscle Alterations Are Exacerbated in Heart Failure with Reduced Compared with Preserved Ejection Fraction: Mediated by Circulating Cytokines? Circ. Heart Fail. 2016, 9, e003027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barazzoni, R.; Gortan Cappellari, G.; Palus, S.; Vinci, P.; Ruozi, G.; Zanetti, M.; Semolic, A.; Ebner, N.; von Haehling, S.; Sinagra, G.; et al. Acylated ghrelin treatment normalizes skeletal muscle mitochondrial oxidative capacity and AKT phosphorylation in rat chronic heart failure. J. Cachexia Sarcopenia Muscle 2017, 8, 991–998. [Google Scholar] [CrossRef]
- Gortan Cappellari, G.; Semolic, A.; Ruozi, G.; Vinci, P.; Guarnieri, G.; Bortolotti, F.; Barbetta, D.; Zanetti, M.; Giacca, M.; Barazzoni, R. Unacylated ghrelin normalizes skeletal muscle oxidative stress and prevents muscle catabolism by enhancing tissue mitophagy in experimental chronic kidney disease. FASEB J. 2017, 31, 5159–5171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gortan Cappellari, G.; Zanetti, M.; Semolic, A.; Vinci, P.; Ruozi, G.; Falcione, A.; Filigheddu, N.; Guarnieri, G.; Graziani, A.; Giacca, M.; et al. Unacylated Ghrelin Reduces Skeletal Muscle Reactive Oxygen Species Generation and Inflammation and Prevents High-Fat Diet-Induced Hyperglycemia and Whole-Body Insulin Resistance in Rodents. Diabetes 2016, 65, 874–886. [Google Scholar] [CrossRef] [Green Version]
- Barazzoni, R.; Zanetti, M.; Gortan Cappellari, G.; Semolic, A.; Boschelle, M.; Codarin, E.; Pirulli, A.; Cattin, L.; Guarnieri, G. Fatty acids acutely enhance insulin-induced oxidative stress and cause insulin resistance by increasing mitochondrial reactive oxygen species (ROS) generation and nuclear factor-kappaB inhibitor (IkappaB)-nuclear factor-kappaB (NFkappaB) activation in rat muscle, in the absence of mitochondrial dysfunction. Diabetologia 2012, 55, 773–782. [Google Scholar] [PubMed] [Green Version]
- Barazzoni, R. Skeletal muscle mitochondrial protein metabolism and function in ageing and type 2 diabetes. Curr. Opin. Clin. Nutr. Metab. Care 2004, 7, 97–102. [Google Scholar] [CrossRef]
- Qaisar, R.; Bhaskaran, S.; Premkumar, P.; Ranjit, R.; Natarajan, K.S.; Ahn, B.; Riddle, K.; Claflin, D.R.; Richardson, A.; Brooks, S.V.; et al. Oxidative stress-induced dysregulation of excitation-contraction coupling contributes to muscle weakness. J. Cachexia Sarcopenia Muscle 2018, 9, 1003–1017. [Google Scholar] [CrossRef]
- Garnham, J.O.; Roberts, L.D.; Espino-Gonzalez, E.; Whitehead, A.; Swoboda, P.P.; Koshy, A.; Gierula, J.; Paton, M.F.; Cubbon, R.M.; Kearney, M.T.; et al. Chronic heart failure with diabetes mellitus is characterized by a severe skeletal muscle pathology. J. Cachexia Sarcopenia Muscle 2020, 11, 394–404. [Google Scholar] [CrossRef]
- Oreopoulos, A.; Padwal, R.; Kalantar-Zadeh, K.; Fonarow, G.C.; Norris, C.M.; McAlister, F.A. Body mass index and mortality in heart failure: A meta-analysis. Am. Heart J. 2008, 156, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Wawrzenczyk, A.; Anaszewicz, M.; Wawrzenczyk, A.; Budzynski, J. Clinical significance of nutritional status in patients with chronic heart failure-a systematic review. Heart Fail. Rev. 2019, 24, 671–700. [Google Scholar] [CrossRef]
- Fonseca, G.; Dos Santos, M.R.; de Souza, F.R.; Takayama, L.; Rodrigues Pereira, R.M.; Negrao, C.E.; Alves, M.N.N. Discriminating sarcopenia in overweight/obese male patients with heart failure: The influence of body mass index. ESC Heart Fail. 2019. [Google Scholar] [CrossRef]
- Barazzoni, R.; Palmisano, S.; Gortan Cappellari, G.; Giuricin, M.; Moretti, E.; Vinci, P.; Semolic, A.; Guarnieri, G.; Zanetti, M.; Manzini, N.D. Gastric bypass-induced weight loss alters obesity-associated patterns of plasma pentraxin-3 and systemic inflammatory markers. Surg. Obes. Relat. Dis. 2016, 12, 23–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barazzoni, R.; Gortan Cappellari, G.; Ragni, M.; Nisoli, E. Insulin resistance in obesity: An overview of fundamental alterations. Eat. Weight Disord. 2018, 23, 149–157. [Google Scholar] [CrossRef]
- Holloszy, J.O. Skeletal muscle “mitochondrial deficiency” does not mediate insulin resistance. Am. J. Clin. Nutr. 2009, 89, 463S–466S. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Roves, P.; Huss, J.M.; Han, D.H.; Hancock, C.R.; Iglesias-Gutierrez, E.; Chen, M.; Holloszy, J.O. Raising plasma fatty acid concentration induces increased biogenesis of mitochondria in skeletal muscle. Proc. Natl. Acad. Sci. USA 2007, 104, 10709–10713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, N.; Kowalski, G.M.; Leslie, S.J.; Risis, S.; Yang, C.; Lee-Young, R.S.; Babb, J.R.; Meikle, P.J.; Lancaster, G.I.; Henstridge, D.C.; et al. Distinct patterns of tissue-specific lipid accumulation during the induction of insulin resistance in mice by high-fat feeding. Diabetologia 2013, 56, 1638–1648. [Google Scholar] [CrossRef]
- Barazzoni, R.; Zanetti, M.; Semolic, A.; Cattin, M.R.; Pirulli, A.; Cattin, L.; Guarnieri, G. High-fat diet with acyl-ghrelin treatment leads to weight gain with low inflammation, high oxidative capacity and normal triglycerides in rat muscle. PLoS ONE 2011, 6, e26224. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.-Y.; Hong, T.; Wen, G.-B.; Han, J.; Zuo, D.; Liu, Z.; Cao, W. Increased basal level of Akt-dependent insulin signaling may be responsible for the development of insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E898–E906. [Google Scholar] [CrossRef] [Green Version]
- Forbes, G.B.; Welle, S.L. Lean body mass in obesity. Int. J. Obes. 1983, 7, 99–107. [Google Scholar]
- Eulalio, A.; Mano, M.; Dal Ferro, M.; Zentilin, L.; Sinagra, G.; Zacchigna, S.; Giacca, M. Functional screening identifies miRNAs inducing cardiac regeneration. Nature 2012, 492, 376–381. [Google Scholar] [CrossRef]
- Ruozi, G.; Bortolotti, F.; Falcione, A.; Dal Ferro, M.; Ukovich, L.; Macedo, A.; Zentilin, L.; Filigheddu, N.; Gortan Cappellari, G.; Baldini, G.; et al. AAV-mediated in vivo functional selection of tissue-protective factors against ischaemia. Nat. Commun. 2015, 6, 7388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gortan Cappellari, G.; Barazzoni, R.; Cattin, L.; Muro, A.F.; Zanetti, M. Lack of Fibronectin Extra Domain A Alternative Splicing Exacerbates Endothelial Dysfunction in Diabetes. Sci. Rep. 2016, 6, 37965. [Google Scholar] [CrossRef] [Green Version]
- Gortan Cappellari, G.; Losurdo, P.; Mazzucco, S.; Panizon, E.; Jevnicar, M.; Macaluso, L.; Fabris, B.; Barazzoni, R.; Biolo, G.; Carretta, R.; et al. Treatment with n-3 polyunsaturated fatty acids reverses endothelial dysfunction and oxidative stress in experimental menopause. J. Nutr. Biochem. 2013, 24, 371–379. [Google Scholar] [CrossRef]
- Zanetti, M.; Gortan Cappellari, G.; Graziani, A.; Barazzoni, R. Unacylated Ghrelin Improves Vascular Dysfunction and Attenuates Atherosclerosis during High-Fat Diet Consumption in Rodents. Int. J. Mol. Sci. 2019, 20, 499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayner, J.J.; Neubauer, S.; Rider, O.J. The paradox of obesity cardiomyopathy and the potential for weight loss as a therapy. Obes. Rev. 2015, 16, 679–690. [Google Scholar] [CrossRef]
- Kokkinos, P.; Faselis, C.; Franklin, B.; Lavie, C.J.; Sidossis, L.; Moore, H.; Karasik, P.; Myers, J. Cardiorespiratory fitness, body mass index and heart failure incidence. Eur. J. Heart Fail. 2019, 21, 436–444. [Google Scholar] [CrossRef]
- Packer, M. Do most patients with obesity or type 2 diabetes, and atrial fibrillation, also have undiagnosed heart failure? A critical conceptual framework for understanding mechanisms and improving diagnosis and treatment. Eur. J. Heart Fail. 2020, 22, 214–227. [Google Scholar] [CrossRef]
- Wang, S.; Ren, J. Obesity Paradox in Aging: From Prevalence to Pathophysiology. Prog. Cardiovasc. Dis. 2018, 61, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Karpe, F.; Dickmann, J.R.; Frayn, K.N. Fatty acids, obesity, and insulin resistance: Time for a reevaluation. Diabetes 2011, 60, 2441–2449. [Google Scholar] [CrossRef] [Green Version]
- Jheng, H.-F.; Tsai, P.-J.; Guo, S.-M.; Kuo, L.-H.; Chang, C.-S.; Su, I.-J.; Chang, C.-R.; Tsai, Y.-S. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol. Cell. Biol. 2012, 32, 309–319. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, H.; Zheng, A.; Yang, L.; Liu, J.; Chen, C.; Tang, Y.; Zou, X.; Li, Y.; Long, J.; et al. Mitochondrial dysfunction-associated OPA1 cleavage contributes to muscle degeneration: Preventative effect of hydroxytyrosol acetate. Cell Death Dis. 2014, 5, e1521. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Wang, Z.; Qin, X.; Xu, J.; Hou, Z.; Yang, H.; Mao, X.; Xing, W.; Li, X.; Zhang, X.; et al. Enhancing fatty acid utilization ameliorates mitochondrial fragmentation and cardiac dysfunction via rebalancing optic atrophy 1 processing in the failing heart. Cardiovasc. Res. 2018, 114, 979–991. [Google Scholar] [CrossRef] [Green Version]
- Wai, T.; Garcia-Prieto, J.; Baker, M.J.; Merkwirth, C.; Benit, P.; Rustin, P.; Ruperez, F.J.; Barbas, C.; Ibanez, B.; Langer, T. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 2015, 350, aad0116. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, E.; Sestili, P. Reactive oxygen species in skeletal muscle signaling. J. Signal Transduct. 2012, 2012, 982794. [Google Scholar] [CrossRef] [Green Version]
- Van Remmen, H.; Salvador, C.; Yang, H.; Huang, T.T.; Epstein, C.J.; Richardson, A. Characterization of the antioxidant status of the heterozygous manganese superoxide dismutase knockout mouse. Arch. Biochem. Biophys. 1999, 363, 91–97. [Google Scholar] [CrossRef]
- Powers, S.K.; Morton, A.B.; Ahn, B.; Smuder, A.J. Redox control of skeletal muscle atrophy. Free Radic. Biol. Med. 2016, 98, 208–217. [Google Scholar] [CrossRef] [Green Version]
- Anderson, E.J.; Lustig, M.E.; Boyle, K.E.; Woodlief, T.L.; Kane, D.A.; Lin, C.T.; Price, J.W., 3rd; Kang, L.; Rabinovitch, P.S.; Szeto, H.H.; et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Investig. 2009, 119, 573–581. [Google Scholar] [CrossRef]
- Dagdeviren, S.; Jung, D.Y.; Lee, E.; Friedline, R.H.; Noh, H.L.; Kim, J.H.; Patel, P.R.; Tsitsilianos, N.; Tsitsilianos, A.V.; Tran, D.A.; et al. Altered Interleukin-10 Signaling in Skeletal Muscle Regulates Obesity-Mediated Inflammation and Insulin Resistance. Mol. Cell. Biol. 2016, 36, 2956–2966. [Google Scholar] [CrossRef] [Green Version]
- Baar, K.; Esser, K. Phosphorylation of p70(S6k) correlates with increased skeletal muscle mass following resistance exercise. Am. J. Physiol. 1999, 276, C120–C127. [Google Scholar] [CrossRef]
- Yoon, M.S. mTOR as a Key Regulator in Maintaining Skeletal Muscle Mass. Front. Physiol. 2017, 8, 788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, A. Muscle wasting is associated with reduced exercise capacity and advanced disease in patients with chronic heart failure. Future Cardiol. 2013, 9, 767–770. [Google Scholar] [CrossRef]
- Kovesdy, C.P.; Kalantar-Zadeh, K. Why is protein-energy wasting associated with mortality in chronic kidney disease? Semin. Nephrol. 2009, 29, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Shachar, S.S.; Williams, G.R.; Muss, H.B.; Nishijima, T.F. Prognostic value of sarcopenia in adults with solid tumours: A meta-analysis and systematic review. Eur. J. Cancer 2016, 57, 58–67. [Google Scholar] [CrossRef]
- Donini, L.M.; Busetto, L.; Bauer, J.M.; Bischoff, S.; Boirie, Y.; Cederholm, T.; Cruz-Jentoft, A.J.; Dicker, D.; Fruhbeck, G.; Giustina, A.; et al. Critical appraisal of definitions and diagnostic criteria for sarcopenic obesity based on a systematic review. Clin. Nutr. 2019, 39, 2368–2388. [Google Scholar] [CrossRef]
- Eckel, R.H.; Jakicic, J.M.; Ard, J.D.; de Jesus, J.M.; Houston Miller, N.; Hubbard, V.S.; Lee, I.M.; Lichtenstein, A.H.; Loria, C.M.; Millen, B.E.; et al. 2013 AHA/ACC guideline on lifestyle management to reduce cardiovascular risk: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014, 129, S76–S99. [Google Scholar] [CrossRef] [Green Version]
- Skinner, J.S.; Cooper, A.; Feder, G.S.; Guideline Development, G. Secondary prevention for patients following a myocardial infarction: Summary of NICE guidance. Heart 2007, 93, 862–864. [Google Scholar] [CrossRef]
- Nichols, S.; McGregor, G.; Al-Mohammad, A.; Ali, A.N.; Tew, G.; O’Doherty, A.F. The effect of protein and essential amino acid supplementation on muscle strength and performance in patients with chronic heart failure: A systematic review. Eur. J. Nutr. 2020, 59, 1785–1801. [Google Scholar] [CrossRef] [Green Version]
- Moreau, J.; Ordan, M.A.; Barbe, C.; Mazza, C.; Perrier, M.; Botsen, D.; Brasseur, M.; Portefaix, C.; Renard, Y.; Talliere, B.; et al. Correlation between muscle mass and handgrip strength in digestive cancer patients undergoing chemotherapy. Cancer Med. 2019, 8, 3677–3684. [Google Scholar] [CrossRef] [Green Version]
- Zanetti, M.; Gortan Cappellari, G.; Barazzoni, R.; Sanson, G. The Impact of Protein Supplementation Targeted at Improving Muscle Mass on Strength in Cancer Patients: A Scoping Review. Nutrients 2020, 12, 2099. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sham | LCHF | ObCHF | ||
|---|---|---|---|---|
| Body Weight [g] | T0 | 42.0 ± 1.5 a | 42.4 ± 0.9 a | 41.9 ± 1.6 a |
| Body Weight [g] | T12 | 44.1 ± 1.2 a | 43.1 ± 1.3 a | 53.0 ± 0.9 b,* |
| Body Weight [g] | T20 | 46.9 ± 1.3 a,* | 43.9 ± 1.1 b | 53.6 ± 1.4 c,* |
| Average daily caloric intake (kcal/d) | T0-T12 | 7.7 ± 0.6 a | 7.8 ± 0.3 a | 25.5 ± 4.1 b |
| Average daily caloric intake (kcal/d) | T13-T20 | 7.7 ± 0.9 a | 8.1 ± 0.4 a | 8.9 ± 2.1 a,* |
| Left Retroperitoneal Fat Pad mass [mg] | T20 | 294 ± 70 a | 249 ± 58 b | 493 ± 72 c |
| Left Epididimal Fat Pad mass [mg] | T20 | 1031 ± 161 a | 675 ± 162 b | 1351 ± 126 c |
| Plasma Glucose [mg/dl] | T20 | 131 ± 4 a | 140 ± 3 a | 155 ± 3 b |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gortan Cappellari, G.; Aleksova, A.; Dal Ferro, M.; Cannatà, A.; Semolic, A.; Zanetti, M.; Springer, J.; Anker, S.D.; Giacca, M.; Sinagra, G.; et al. Preserved Skeletal Muscle Mitochondrial Function, Redox State, Inflammation and Mass in Obese Mice with Chronic Heart Failure. Nutrients 2020, 12, 3393. https://doi.org/10.3390/nu12113393
Gortan Cappellari G, Aleksova A, Dal Ferro M, Cannatà A, Semolic A, Zanetti M, Springer J, Anker SD, Giacca M, Sinagra G, et al. Preserved Skeletal Muscle Mitochondrial Function, Redox State, Inflammation and Mass in Obese Mice with Chronic Heart Failure. Nutrients. 2020; 12(11):3393. https://doi.org/10.3390/nu12113393
Chicago/Turabian StyleGortan Cappellari, Gianluca, Aneta Aleksova, Matteo Dal Ferro, Antonio Cannatà, Annamaria Semolic, Michela Zanetti, Jochen Springer, Stefan D. Anker, Mauro Giacca, Gianfranco Sinagra, and et al. 2020. "Preserved Skeletal Muscle Mitochondrial Function, Redox State, Inflammation and Mass in Obese Mice with Chronic Heart Failure" Nutrients 12, no. 11: 3393. https://doi.org/10.3390/nu12113393