A Low Iron Diet Protects from Steatohepatitis in a Mouse Model

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Experimentation
2.2. Study Approval
2.3. Cell Culture
2.4. Commercial Assays
2.5. MIP OES and LC-MS/MS
2.6. RNA Sequencing
2.7. Reverse Transcription and Real-time PCR Quantification
2.8. Seahorse Mito Stress Test
2.9. Statistics
3. Results
3.1. Metabolic Phenotyping of Mice on Diet for 3–4 Months
3.2. Iron Indices Are Not Altered by the FF Diet, and Hepatic Fat Content Is Not Affected by Dietary Iron
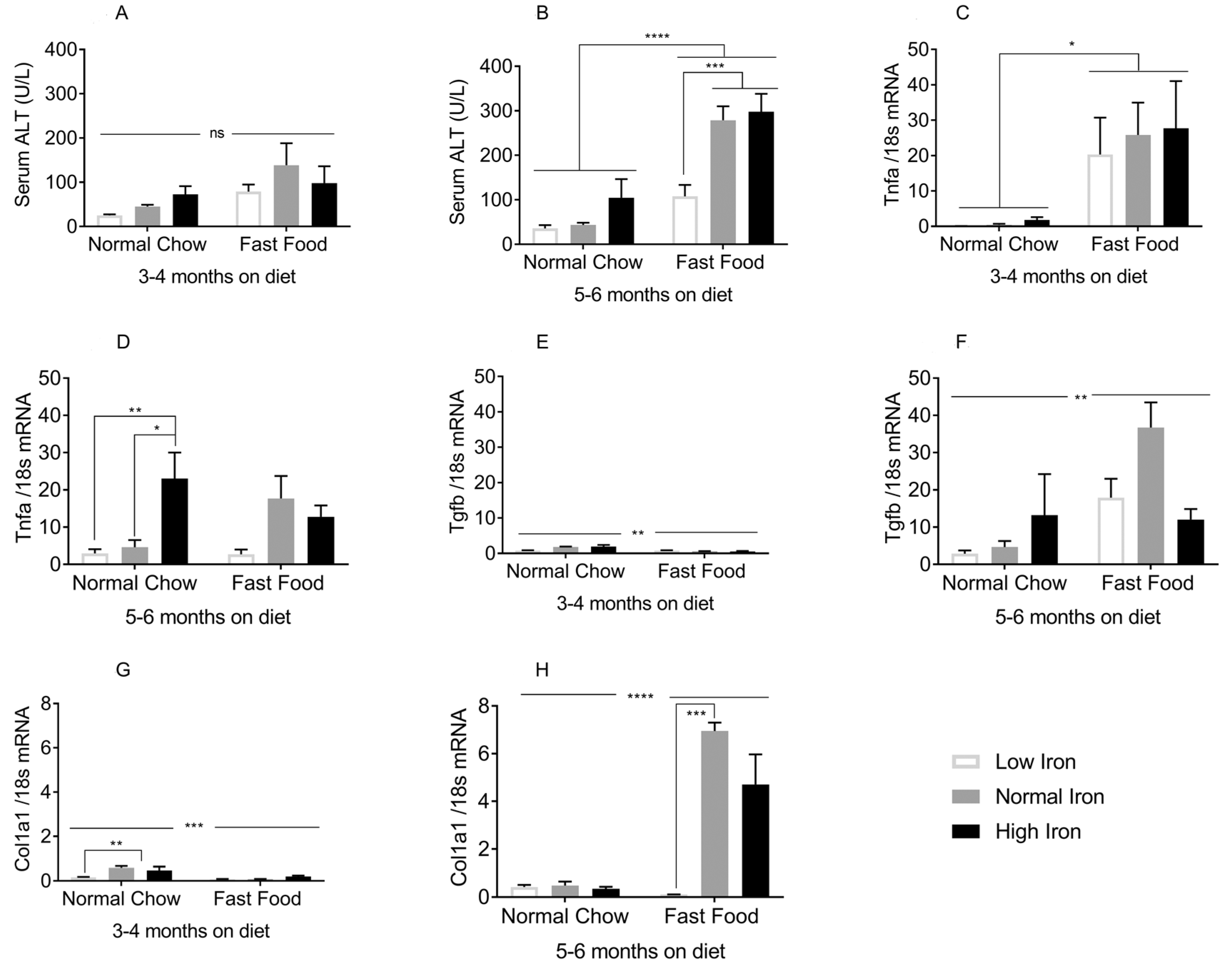
3.3. Iron Restriction Limits Progression of NAFLD to NASH
3.4. Iron Transcriptionally Regulates the TGF-Β Signaling Pathway
3.5. Hepatocyte–Stellate Cell Interaction is Necessary for Iron- and Fat-Induced Fibrogenesis
3.6. Oxidative Stress and Other Previously Identified Mediators Are Not Predominant Early Drivers of NASH in This Model
3.7. Iron Does Not Affect Mitochondrial Function Prior to the Initiation of Fibrosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2017, 15, 11. [Google Scholar] [CrossRef] [PubMed]
- Arab, J.P.; Arrese, M.; Trauner, M. Recent Insights into the Pathogenesis of Nonalcoholic Fatty Liver Disease. Annu. Rev. Pathol. Mech. Disease 2018, 13, 321–350. [Google Scholar] [CrossRef] [PubMed]
- Benedict, M.; Zhang, X. Non-alcoholic fatty liver disease: An expanded review. World J. Hepatol. 2017, 9, 715–732. [Google Scholar] [CrossRef] [PubMed]
- Pickett-Blakely, O.; Young, K.; Carr, R.M. Micronutrients in Nonalcoholic Fatty Liver Disease Pathogenesis. Cell. Mol. Gastroenterol. Hepatol. 2018, 6, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Bulaj, Z.J.; Ajioka, R.S.; Phillips, J.D.; LaSalle, B.A.; Jorde, L.B.; Griffen, L.M.; Edwards, C.Q.; Kushner, J.P. Disease-related conditions in relatives of patients with hemochromatosis. N. Engl. J. Med. 2000, 343, 1529–1535. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Valenti, L. A Nutrigenomic Approach to Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2017, 18, 1534. [Google Scholar] [CrossRef] [PubMed]
- Simcox, J.A.; McClain, D.A. Iron and diabetes risk. Cell Metab. 2013, 17, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Real, J.M.; McClain, D.; Manco, M. Mechanisms Linking Glucose Homeostasis and Iron Metabolism Toward the Onset and Progression of Type 2 Diabetes. Diabetes Care 2015, 38, 2169–2176. [Google Scholar] [CrossRef] [PubMed]
- Chitturi, S.; George, J. Interaction of iron, insulin resistance, and nonalcoholic steatohepatitis. Curr. Gastroenterol. Rep. 2003, 5, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Charlton, M.; Krishnan, A.; Viker, K.; Sanderson, S.; Cazanave, S.; McConico, A.; Masuoko, H.; Gores, G. Fast food diet mouse: Novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am. J. Physiol.-Gastrointest. Liver Physiol. 2011, 301, 825–834. [Google Scholar] [CrossRef]
- Nam, H.; Jones, D.; Cooksey, R.C.; Gao, Y.; Sink, S.; Cox, J.; McClain, D.A. Synergistic Inhibitory Effects of Hypoxia and Iron Deficiency on Hepatic Glucose Response in Mouse Liver. Diabetes 2016, 65, 1521–1533. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Kir, J.; Liu, D.; Bryant, D.; Guo, Y.; Stephens, R.; Baseler, M.W.; Lane, H.C.; et al. DAVID Bioinformatics Resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007, 35, W169–W175. [Google Scholar] [CrossRef] [PubMed]
- Kramer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]
- Bharadwaj, M.S.; Tyrrell, D.J.; Lyles, M.F.; Demons, J.L.; Rogers, G.W.; Molina, A.J.A. Preparation and respirometric assessment of mitochondria isolated from skeletal muscle tissue obtained by percutaneous needle biopsy. J. Vis. Exp. JoVE 2015, 98, e52350. [Google Scholar] [CrossRef]
- Britton, L.; Bridle, K.; Reiling, J.; Santrampurwala, N.; Wockner, L.; Ching, H.; Stuart, K.; Subramaniam, V.N.; Jeffrey, G.; St Pierre, T.; et al. Hepatic iron concentration correlates with insulin sensitivity in nonalcoholic fatty liver disease. Hepatol. Commun. 2018, 2, 644–653. [Google Scholar] [CrossRef]
- Huang, J.; Simcox, J.; Mitchell, T.C.; Jones, D.; Cox, J.; Luo, B.; Cooksey, R.C.; Boros, L.G.; McClain, D.A. Iron regulates glucose homeostasis in liver and muscle via AMP-activated protein kinase in mice. FASEB J. 2013, 27, 2845–2854. [Google Scholar] [CrossRef]
- Teufel, A.; Itzel, T.; Erhart, W.; Brosch, M.; Wang, X.Y.; Kim, Y.O.; von Schonfels, W.; Herrmann, A.; Bruckner, S.; Stickel, F.; et al. Comparison of Gene Expression Patterns Between Mouse Models of Nonalcoholic Fatty Liver Disease and Liver Tissues from Patients. Gastroenterology 2016, 151, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Van Koppen, A.; Verschuren, L.; van den Hoek, A.M.; Verheij, J.; Morrison, M.C.; Li, K.; Nagabukuro, H.; Costessi, A.; Caspers, M.P.M.; van den Broek, T.J.; et al. Uncovering a Predictive Molecular Signature for the Onset of NASH-Related Fibrosis in a Translational NASH Mouse Model. Cell. Mol. Gastroenterol. Hepatol. 2017, 5, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Cooksey, R.C.; Jouihan, H.A.; Ajioka, R.S.; Hazel, M.W.; Jones, D.L.; Kushner, J.P.; McClain, D.A. Oxidative stress, beta-cell apoptosis, and decreased insulin secretory capacity in mouse models of hemochromatosis. Endocrinology 2004, 145, 5305–5312. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Fillebeen, C.; Haliotis, T.; Mui, J.; Vali, H.; Pantopoulos, K. Mouse models of hereditary hemochromatosis do not develop early liver fibrosis in response to a high fat diet. PLoS ONE 2019, 14, e0221455. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.C.H.; Crawford, D.H.G.; Jaskowski, L.A.; Murphy, T.M.; Heritage, M.L.; Subramaniam, V.N.; Clouston, A.D.; Anderson, G.J.; Fletcher, L.M. Altered lipid metabolism in Hfe-knockout mice promotes severe NAFLD and early fibrosis. Am. J. Physiol.-Gastrointest. Liver Physiol. 2011, 301, 865–876. [Google Scholar] [CrossRef]
- Hansen, H.H.; Feigh, M.; Veidal, S.S.; Rigbolt, K.T.; Vrang, N.; Fosgerau, K. Mouse models of nonalcoholic steatohepatitis in preclinical drug development. Drug Discov. Today 2017, 22, 1707–1718. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.-W.; Nie, Y.-Z.; Taniguchi, H. Cellular reprogramming and hepatocellular carcinoma development. World J. Gastroenterol. 2013, 19, 8850–8860. [Google Scholar] [CrossRef]
- Herrera, B.; Addante, A.; Sánchez, A. BMP Signalling at the Crossroad of Liver Fibrosis and Regeneration. Int. J. Mol. Sci. 2017, 19, 39. [Google Scholar] [CrossRef]
- Khan, R.S.; Newsome, P.N. Novel insights into mechanisms of disease progression. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 71. [Google Scholar] [CrossRef]
- Mehta, K.J.; Farnaud, S.J.; Sharp, P.A. Iron and liver fibrosis: Mechanistic and clinical aspects. World J. Gastroenterol. 2019, 25, 521–538. [Google Scholar] [CrossRef]
- Sharma, R.S.; Harrison, D.J.; Kisielewski, D.; Cassidy, D.M.; McNeilly, A.D.; Gallagher, J.R.; Walsh, S.V.; Honda, T.; McCrimmon, R.J.; Dinkova-Kostova, A.T.; et al. Experimental Nonalcoholic Steatohepatitis and Liver Fibrosis Are Ameliorated by Pharmacologic Activation of Nrf2 (NF-E2 p45-Related Factor 2). Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 367–398. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.K.; Kolhe, R.; Black, S.M.; Keller, J.R.; Mivechi, N.F.; Satyanarayana, A. Inhibitor of differentiation 1 transcription factor promotes metabolic reprogramming in hepatocellular carcinoma cells. FASEB J. 2016, 30, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Strong, N.; Millena, A.C.; Walker, L.; Chaudhary, J.; Khan, S.A. Inhibitor of differentiation 1 (Id1) and Id3 proteins play different roles in TGFbeta effects on cell proliferation and migration in prostate cancer cells. Prostate 2013, 73, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Bauckman, K.A.; Haller, E.; Flores, I.; Nanjundan, M. Iron modulates cell survival in a Ras- and MAPK-dependent manner in ovarian cells. Cell Death Dis. 2013, 4, e592. [Google Scholar] [CrossRef] [PubMed]
- Lai, D.; Teng, F.; Hammad, S.; Werle, J.; Maas, T.; Teufel, A.; Muckenthaler, M.U.; Dooley, S.; Vujic Spasic, M. Hepatic Smad7 overexpression causes severe iron overload in mice. Blood 2018, 131, 581–585. [Google Scholar] [CrossRef] [PubMed]
- An, P.; Wang, H.; Wu, Q.; Wang, J.; Xia, Z.; He, X.; Wang, X.; Chen, Y.; Min, J.; Wang, F. Smad7 deficiency decreases iron and haemoglobin through hepcidin up-regulation by multilayer compensatory mechanisms. J. Cell. Mol. Med. 2018, 22, 3035–3044. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Wang, L.; Wang, X.; Luo, X.; Yang, L.; Zhang, R.; Yin, H.; Xie, D.; Pan, Y.; Chen, Y. Hepatic deletion of Smad7 in mouse leads to spontaneous liver dysfunction and aggravates alcoholic liver injury. PLoS ONE 2011, 6, e17415. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Li, J.; Xiang, W.; Cui, Y.; Xie, B.; Wang, X.; Xu, Z.; Gan, L. Metformin increases hepatic leptin receptor and decreases steatosis in mice. J. Endocrinol. 2016, 230, 227–237. [Google Scholar] [CrossRef]
- Murali, A.R.; Gupta, A.; Brown, K. Systematic review and meta-analysis to determine the impact of iron depletion in dysmetabolic iron overload syndrome and non-alcoholic fatty liver disease. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2018, 48, 30–41. [Google Scholar] [CrossRef]
- Adolph, T.E.; Grander, C.; Grabherr, F.; Tilg, H. Adipokines and Non-Alcoholic Fatty Liver Disease: Multiple Interactions. Int. J. Mol. Sci. 2017, 18, 1649. [Google Scholar] [CrossRef]
- Gao, Y.; Li, Z.; Gabrielsen, J.S.; Simcox, J.A.; Lee, S.H.; Jones, D.; Cooksey, B.; Stoddard, G.; Cefalu, W.T.; McClain, D.A. Adipocyte iron regulates leptin and food intake. J. Clin. Investig. 2015, 125, 3681–3691. [Google Scholar] [CrossRef] [PubMed]
- Gabrielsen, J.S.; Gao, Y.; Simcox, J.A.; Huang, J.; Thorup, D.; Jones, D.; Cooksey, R.C.; Gabrielsen, D.; Adams, T.D.; Hunt, S.C.; et al. Adipocyte iron regulates adiponectin and insulin sensitivity. J. Clin. Investig. 2012, 122, 3529–3540. [Google Scholar] [CrossRef] [PubMed]
- Schroder, T.; Kucharczyk, D.; Bar, F.; Pagel, R.; Derer, S.; Jendrek, S.T.; Sunderhauf, A.; Brethack, A.K.; Hirose, M.; Moller, S.; et al. Mitochondrial gene polymorphisms alter hepatic cellular energy metabolism and aggravate diet-induced non-alcoholic steatohepatitis. Mol. Metab. 2016, 5, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, P.; Kowdley, K.V. The Metabolic Syndrome and Its Influence on Nonalcoholic Steatohepatitis. Clin. Liver. Dis. 2016, 20, 225–243. [Google Scholar] [CrossRef] [PubMed]
- Miranda, M.A.; St Pierre, C.L.; Macias-Velasco, J.F.; Nguyen, H.A.; Schmidt, H.; Agnello, L.T.; Wayhart, J.P.; Lawson, H.A. Dietary iron interacts with genetic background to influence glucose homeostasis. Nutr. Metab. 2019, 16, 13. [Google Scholar] [CrossRef]
- Elpek, G.O. Cellular and molecular mechanisms in the pathogenesis of liver fibrosis: An update. World J. Gastroenterol. 2014, 20, 7260–7276. [Google Scholar] [CrossRef]
- Saeed, A.; Dullaart, R.P.F.; Schreuder, T.C.M.A.; Blokzijl, H.; Faber, K.N. Disturbed Vitamin A Metabolism in Non-Alcoholic Fatty Liver Disease (NAFLD). Nutrients 2017, 10, 29. [Google Scholar] [CrossRef]
- Adams, L.A.; Crawford, D.H.; Stuart, K.; House, M.J.; St Pierre, T.G.; Webb, M.; Ching, H.L.; Kava, J.; Bynevelt, M.; MacQuillan, G.C.; et al. The impact of phlebotomy in nonalcoholic fatty liver disease: A prospective, randomized, controlled trial. Hepatology 2015, 61, 1555–1564. [Google Scholar] [CrossRef]
- Valenti, L.; Fracanzani, A.L.; Dongiovanni, P.; Rovida, S.; Rametta, R.; Fatta, E.; Pulixi, E.A.; Maggioni, M.; Fargion, S. A randomized trial of iron depletion in patients with nonalcoholic fatty liver disease and hyperferritinemia. World J. Gastroenterol. 2014, 20, 3002–3010. [Google Scholar] [CrossRef]
- Iwasa, M.; Hara, N.; Iwata, K.; Ishidome, M.; Sugimoto, R.; Tanaka, H.; Fujita, N.; Kobayashi, Y.; Takei, Y. Restriction of calorie and iron intake results in reduction of visceral fat and serum alanine aminotransferase and ferritin levels in patients with chronic liver disease. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2010, 40, 1188–1194. [Google Scholar] [CrossRef]
- Laine, F.; Ruivard, M.; Loustaud-Ratti, V.; Bonnet, F.; Cales, P.; Bardou-Jacquet, E.; Sacher-Huvelin, S.; Causse, X.; Beusnel, C.; Renault, A.; et al. Metabolic and hepatic effects of bloodletting in dysmetabolic iron overload syndrome: A randomized controlled study in 274 patients. Hepatology 2017, 65, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Beaton, M.D.; Chakrabarti, S.; Levstik, M.; Speechley, M.; Marotta, P.; Adams, P. Phase II clinical trial of phlebotomy for non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2013, 37, 720–729. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salaye, L.; Bychkova, I.; Sink, S.; Kovalic, A.J.; Bharadwaj, M.S.; Lorenzo, F.; Jain, S.; Harrison, A.V.; Davis, A.T.; Turnbull, K.; et al. A Low Iron Diet Protects from Steatohepatitis in a Mouse Model. Nutrients 2019, 11, 2172. https://doi.org/10.3390/nu11092172
Salaye L, Bychkova I, Sink S, Kovalic AJ, Bharadwaj MS, Lorenzo F, Jain S, Harrison AV, Davis AT, Turnbull K, et al. A Low Iron Diet Protects from Steatohepatitis in a Mouse Model. Nutrients. 2019; 11(9):2172. https://doi.org/10.3390/nu11092172
Chicago/Turabian StyleSalaye, Lipika, Ielizaveta Bychkova, Sandy Sink, Alexander J. Kovalic, Manish S. Bharadwaj, Felipe Lorenzo, Shalini Jain, Alexandria V. Harrison, Ashley T. Davis, Katherine Turnbull, and et al. 2019. "A Low Iron Diet Protects from Steatohepatitis in a Mouse Model" Nutrients 11, no. 9: 2172. https://doi.org/10.3390/nu11092172
APA StyleSalaye, L., Bychkova, I., Sink, S., Kovalic, A. J., Bharadwaj, M. S., Lorenzo, F., Jain, S., Harrison, A. V., Davis, A. T., Turnbull, K., Meegalla, N. T., Lee, S.-h., Cooksey, R., Donati, G. L., Kavanagh, K., Bonkovsky, H. L., & McClain, D. A. (2019). A Low Iron Diet Protects from Steatohepatitis in a Mouse Model. Nutrients, 11(9), 2172. https://doi.org/10.3390/nu11092172