Dietary Fat-Accelerating Leptin Signaling Promotes Protumorigenic Gastric Environment in Mice

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals, Diets, and Chemical
2.2. Histological Analysis
2.3. Plasma Assay
2.4. qPCR for Bacterial 16S rRNA Gene and Cytokine mRNA
2.5. Statistical Analysis
3. Results
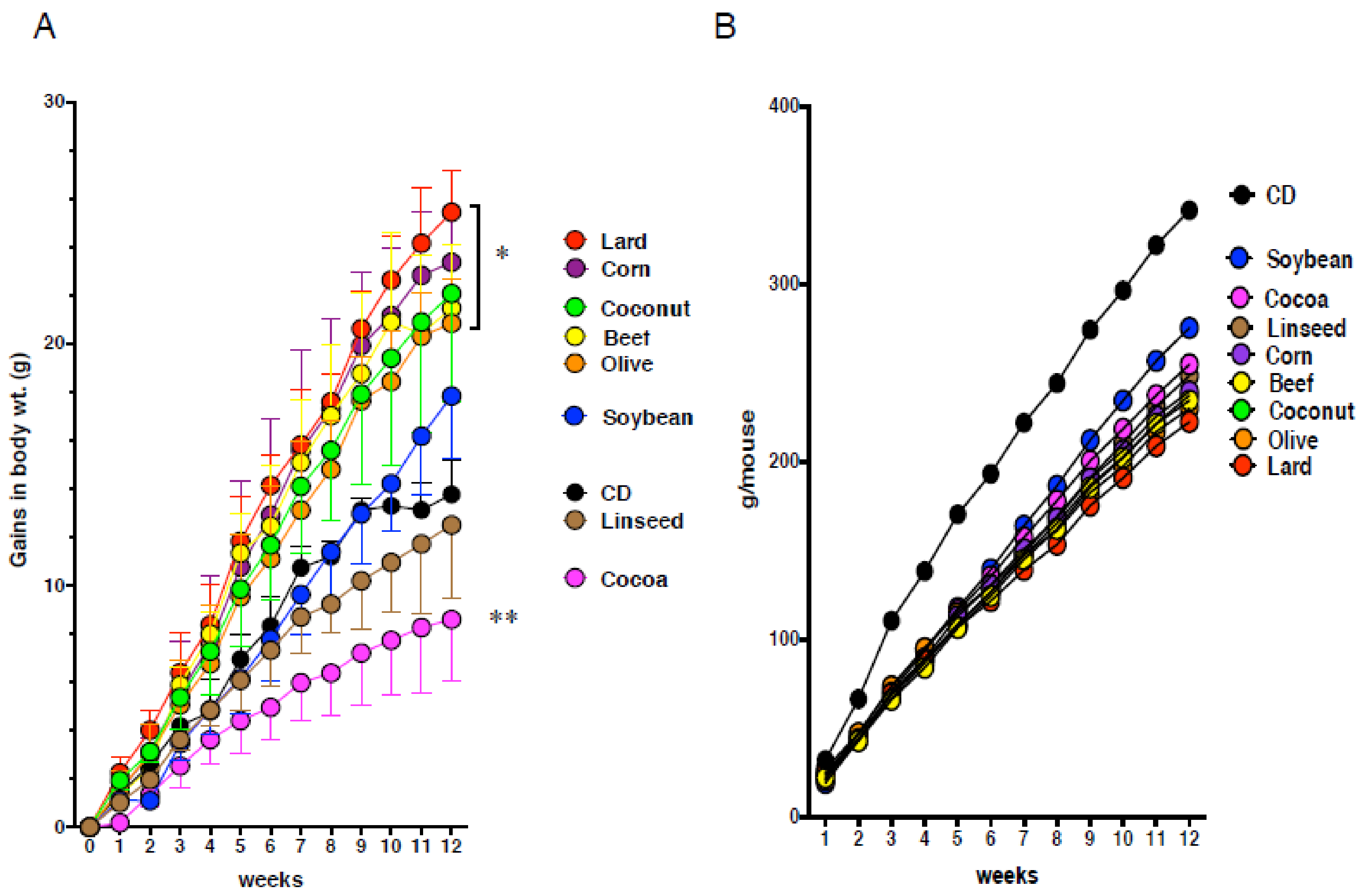
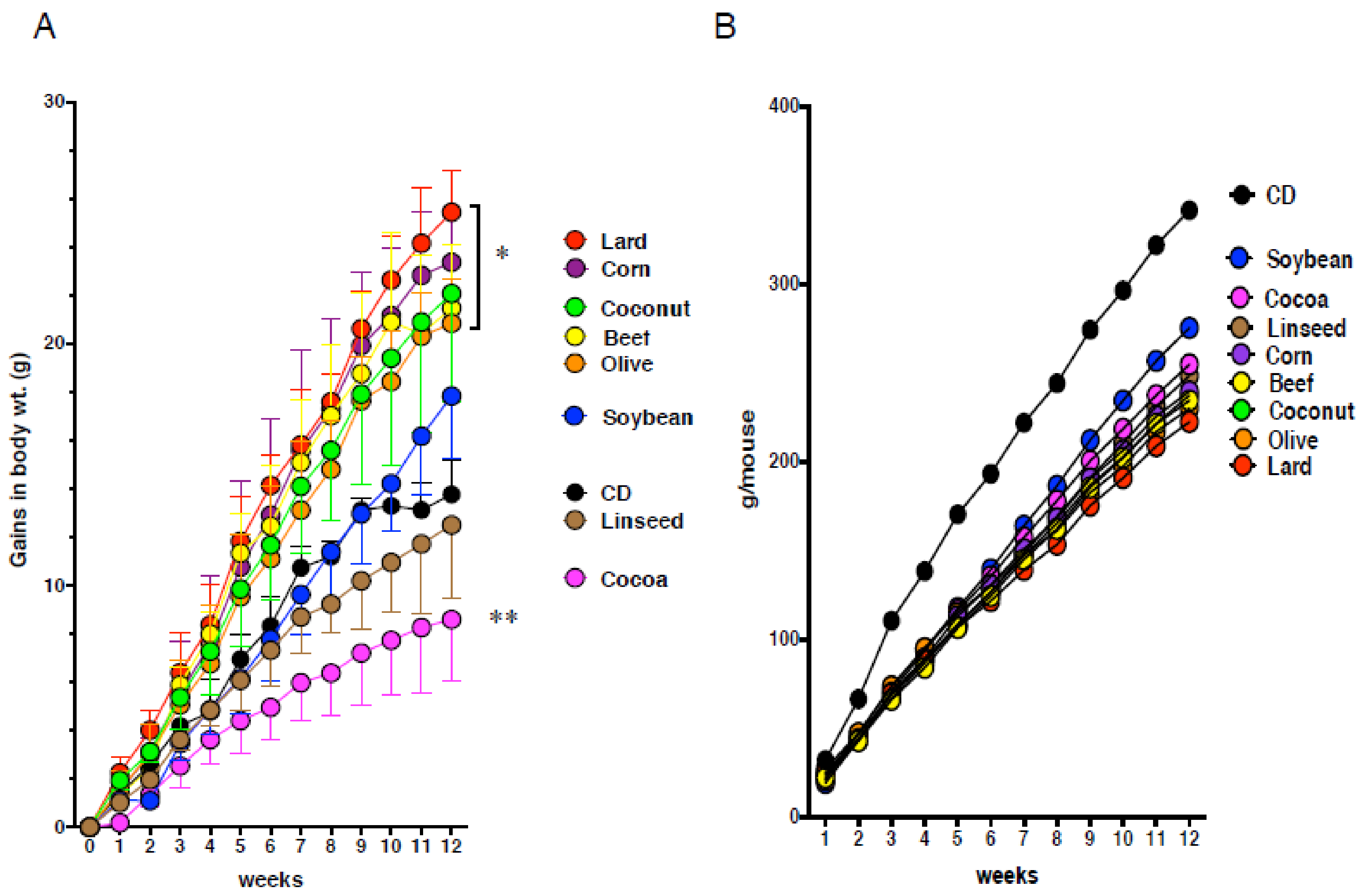
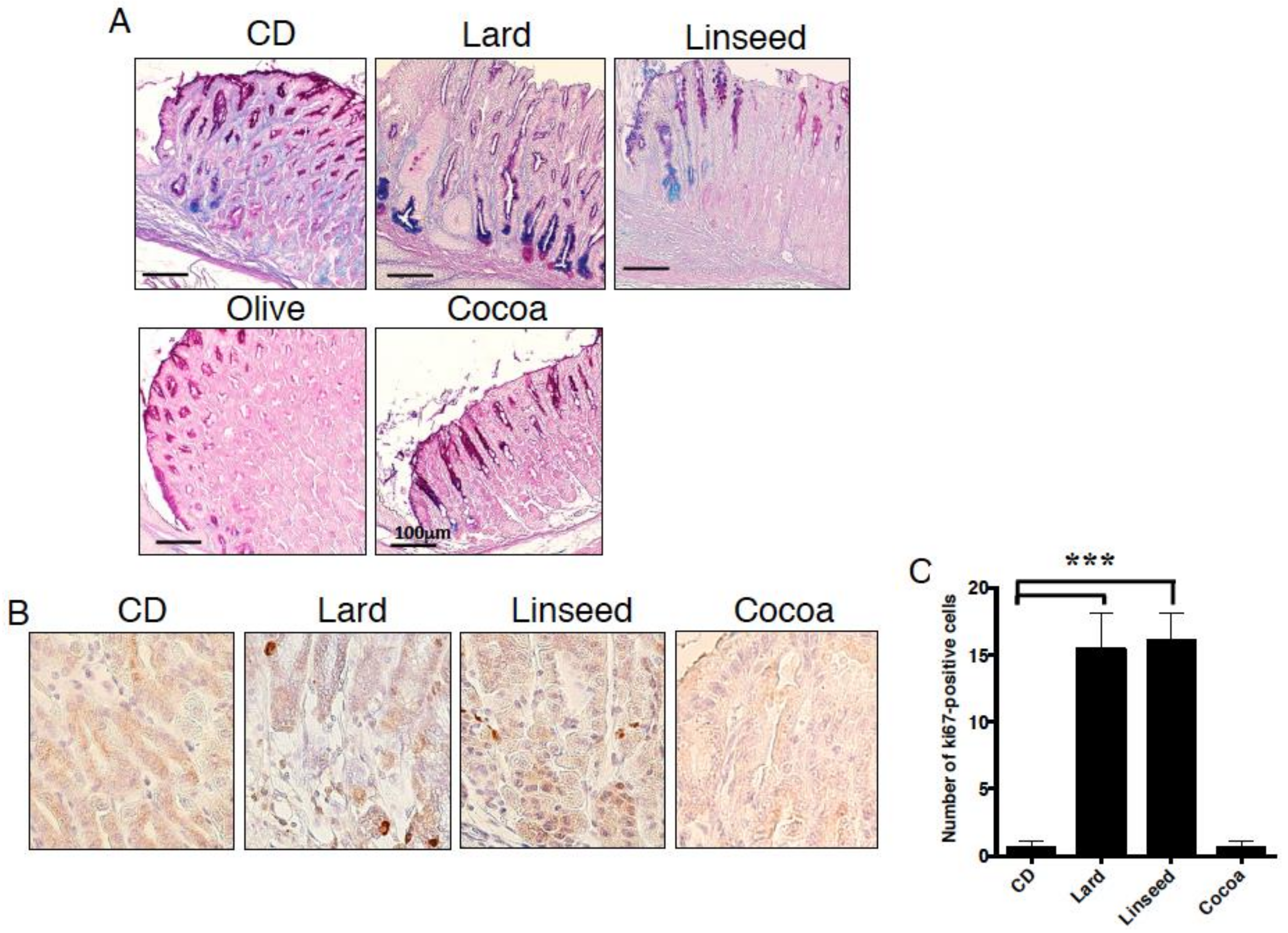
3.1. Obesity Does Not Necessarily Cause Intestinal Metaplasia in the Gastric Mucosa
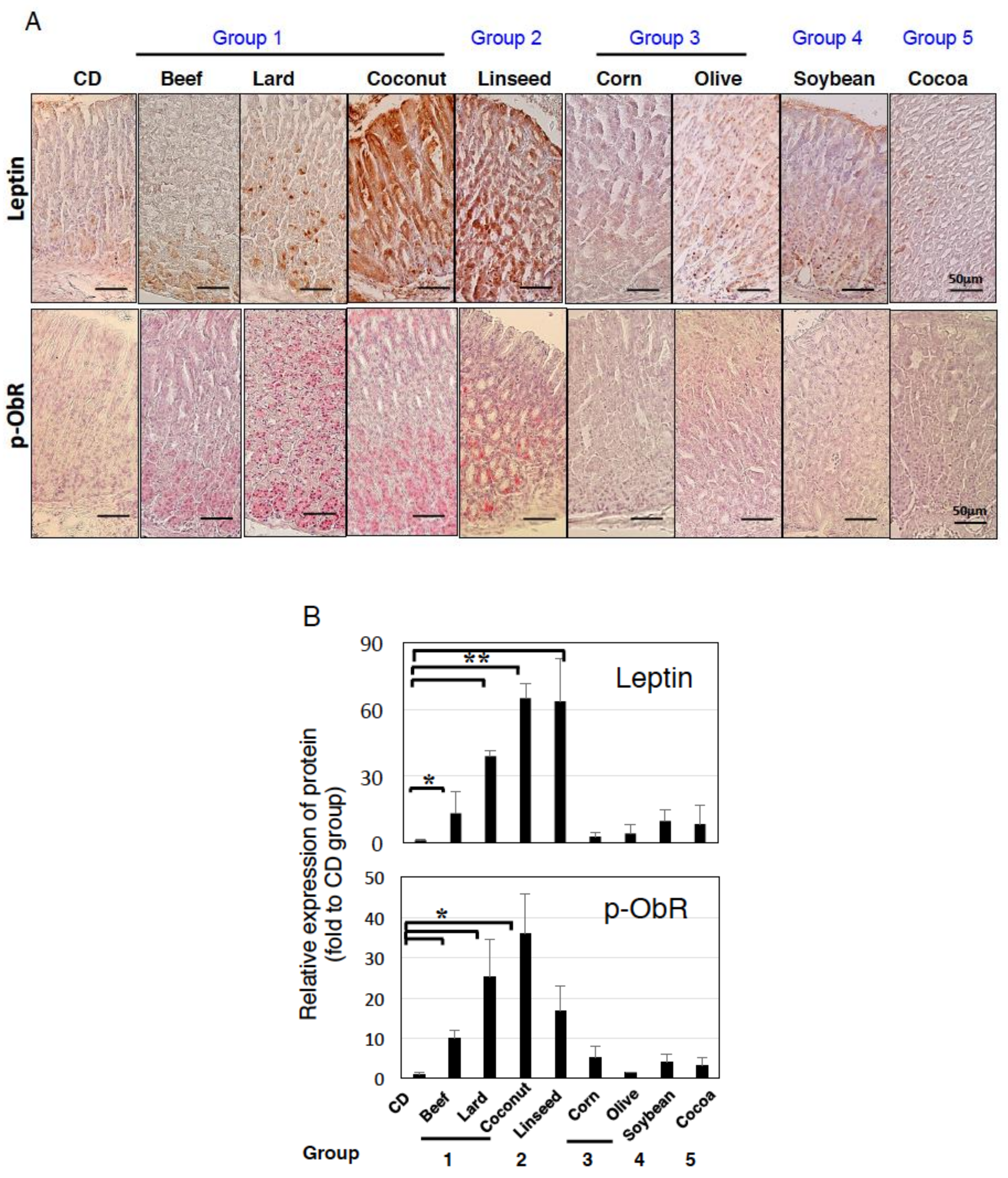
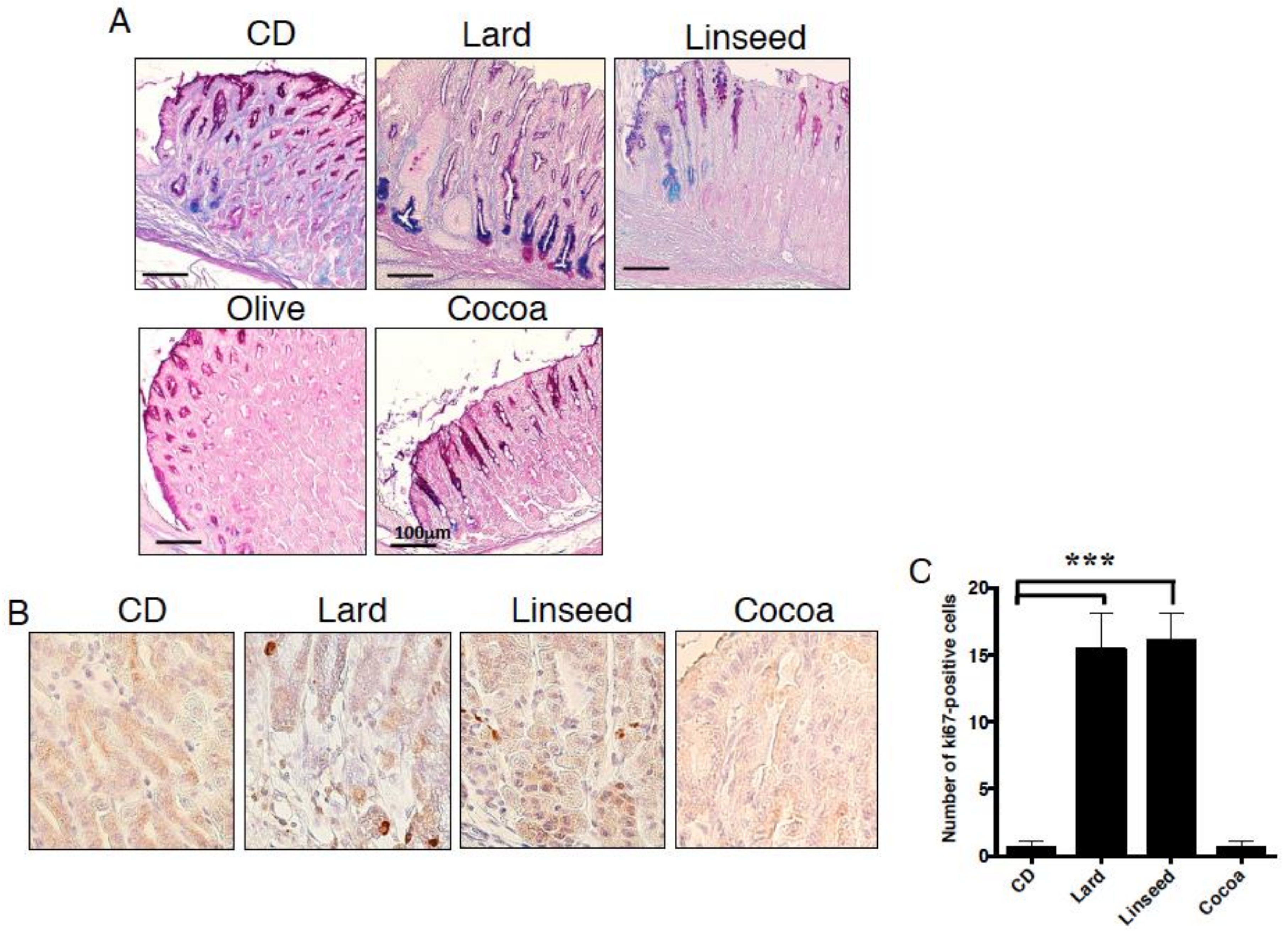
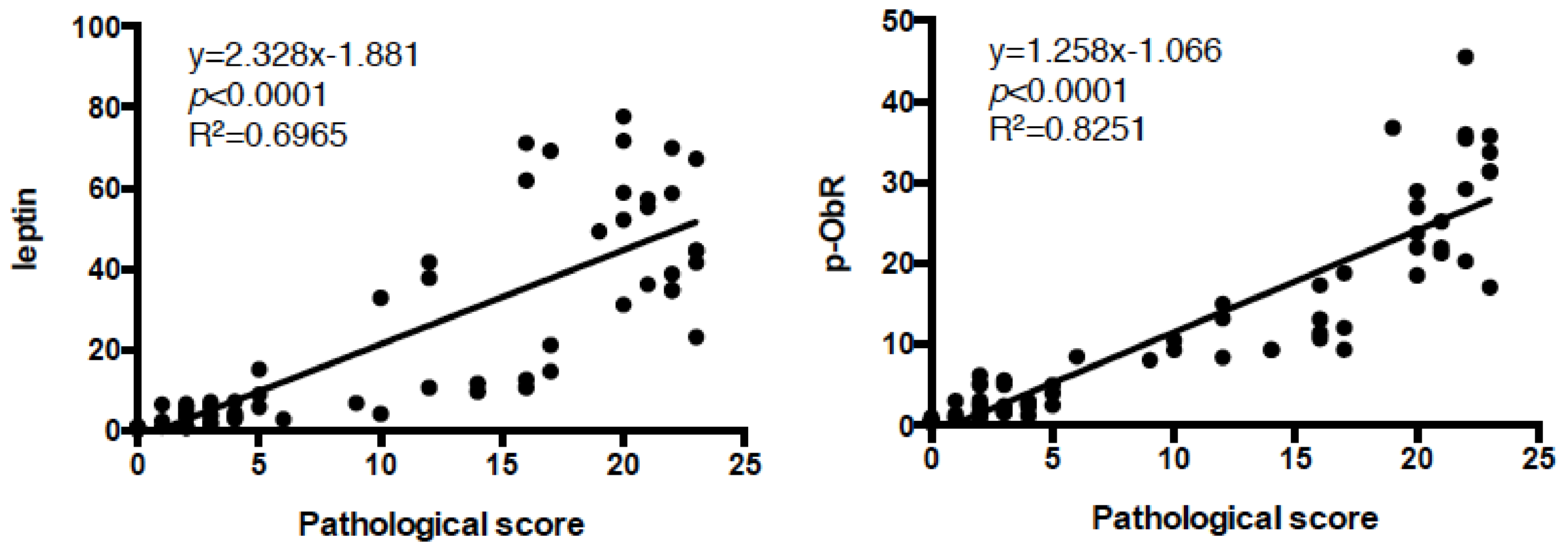
3.2. Intestinal Metaplasia in the Stomach Is Developed by HFDs that Induced Gastric Leptin Production
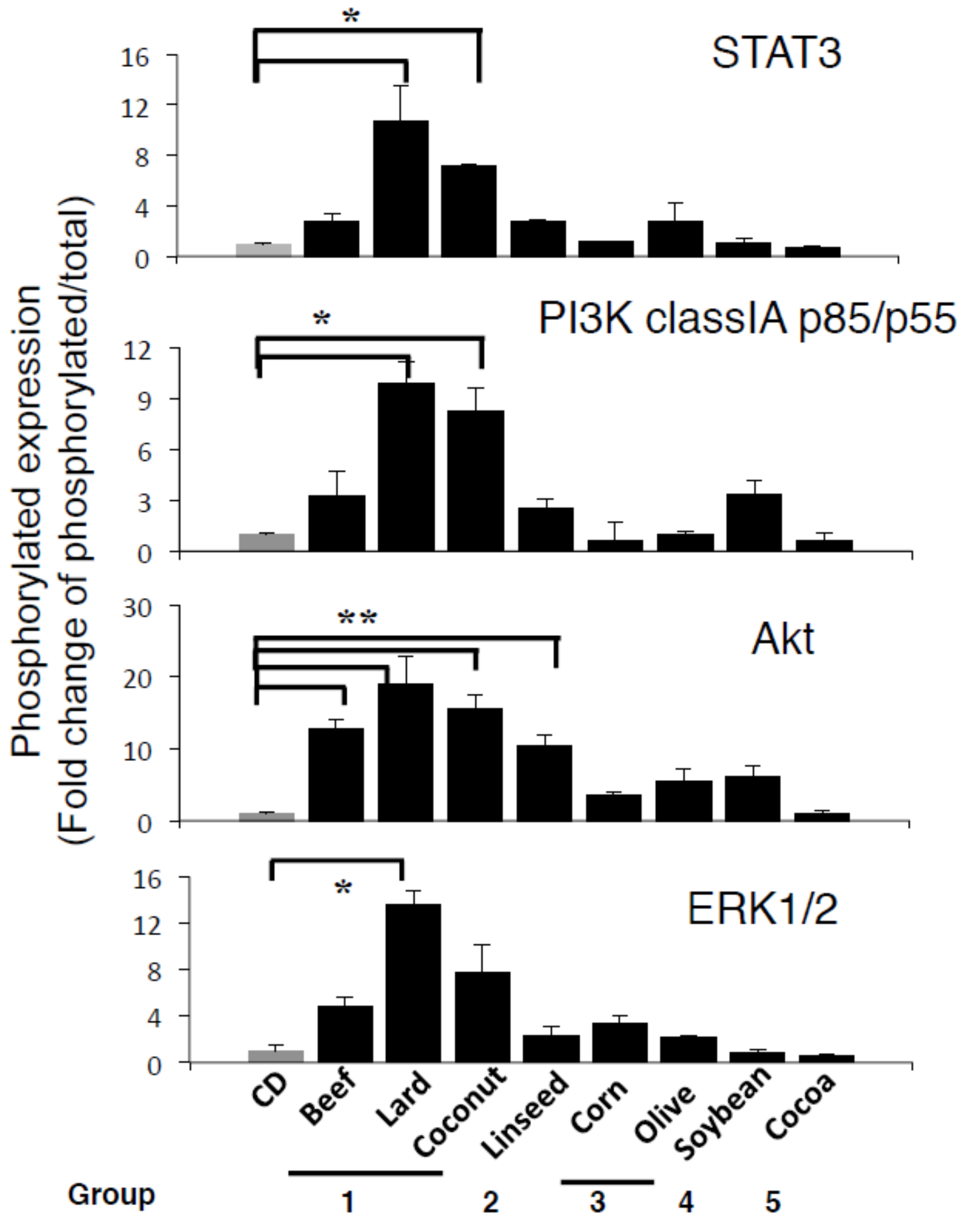
3.3. β- Catenin Signaling Is Increased in the Gastric Mucosa of Mice Fed HFDs Accelerating Gastric Leptin Production
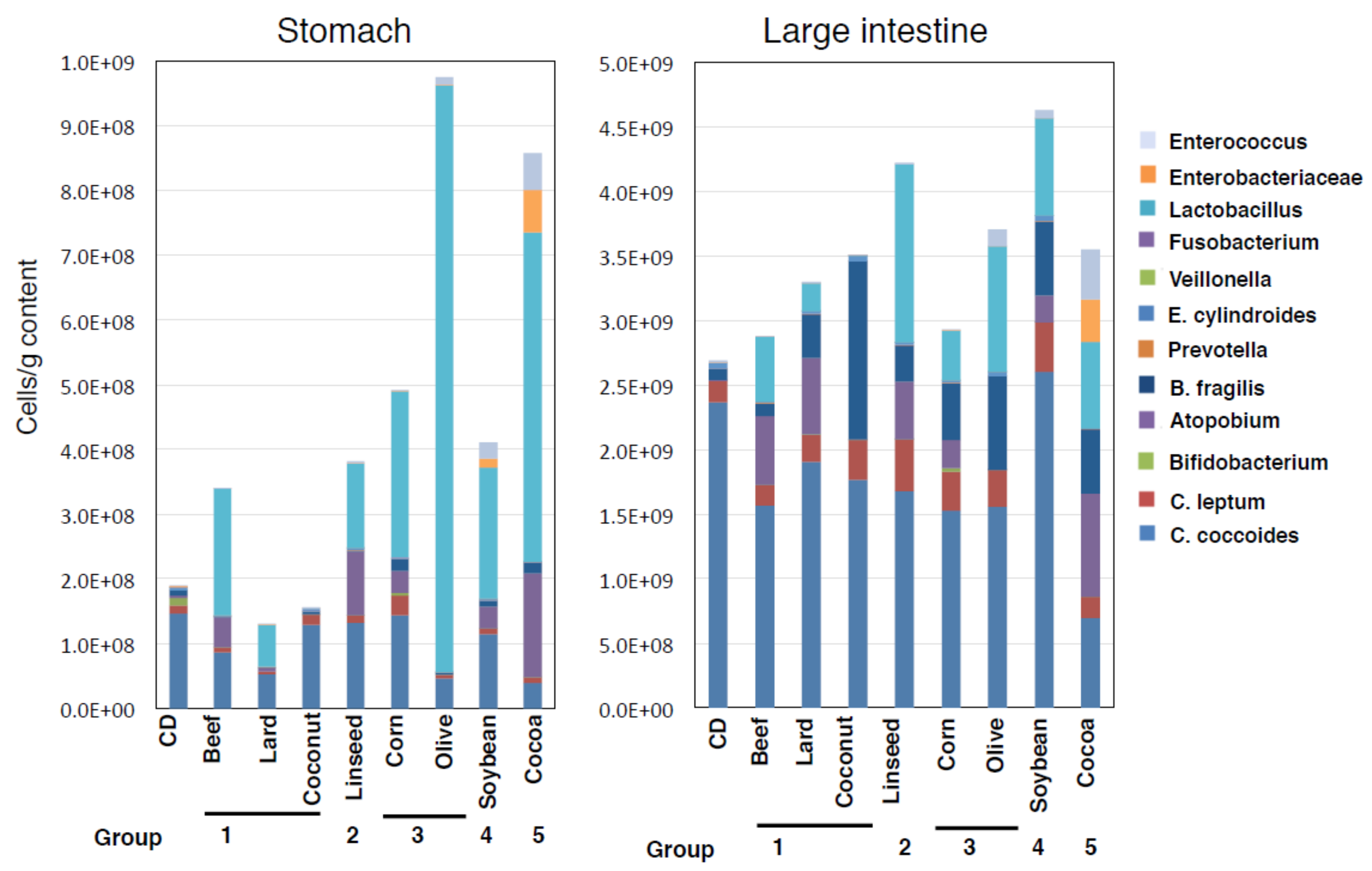
3.4. Alteration of Microbial Component Induced by HFDs Is Irrespective of Body Mass
3.5. HFDs-Accelerating Gastric Leptin Production Enhance Dysplasia in Mice Administered MNNG
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [Green Version]
- Bhandari, A.; Crowe, S.E. Helicobacter pylori in gastric malignancies. Curr. Gastroenterol. Rep. 2012, 14, 489–496. [Google Scholar] [CrossRef]
- Helicobacter and Cancer Collaborative Group. Gastric cancer and Helicobacter pylori: A combined analysis of 12 case control studies nested within prospective cohorts. Gut 2001, 49, 347–353. [Google Scholar] [CrossRef]
- Peek, R.M., Jr.; Crabtree, J.E. Helicobacter infection and gastric neoplasia. J. Pathol. 2006, 208, 233–248. [Google Scholar] [CrossRef]
- Wroblewski, L.E.; Peek, R.M., Jr.; Wilson, K.T. Helicobacter pylori and gastric cancer: Factors that modulate disease risk. Clin. Microbiol. Rev. 2010, 23, 713–739. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, L.; Wang, X.; Wang, J.; Yan, Z.; Cheng, J.; Gong, G.; Li, G. Body mass index and risk of gastric cancer: A meta-analysis of a population with more than ten million from 24 prospective studies. Cancer Epidemiol. Biomark. Prev. 2013, 22, 1395–1408. [Google Scholar] [CrossRef]
- Dutta, S.K.; Arora, M.; Kireet, A.; Bashandy, H.; Gandsas, A. Upper gastrointestinal symptoms and associated disorders in morbidly obese patients: A prospective study. Dig. Dis. Sci. 2009, 54, 1243–1246. [Google Scholar] [CrossRef]
- O’Keefe, S.J. Diet, microorganisms and their metabolites, and colon cancer. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 691–706. [Google Scholar] [CrossRef]
- Butler, L.M.; Wang, R.; Koh, W.P.; Stern, M.C.; Yuan, J.M.; Yu, M.C. Marine n-3 and saturated fatty acids in relation to risk of colorectal cancer in Singapore Chinese: A prospective study. Int. J. Cancer 2009, 124, 678–686. [Google Scholar] [CrossRef]
- Liu, L.; Zhuang, W.; Wang, R.Q.; Mukherjee, R.; Xiao, S.M.; Chen, Z.; Wu, X.T.; Zhou, Y.; Zhang, H.Y. Is dietary fat associated with the risk of colorectal cancer? A meta-analysis of 13 prospective cohort studies. Eur. J. Nutr. 2011, 50, 173–184. [Google Scholar] [CrossRef]
- O’Doherty, M.G.; Freedman, N.D.; Hollenbeck, A.R.; Schatzkin, A.; Murray, L.J.; Cantwell, M.M.; Abnet, C.C. Association of dietary fat intakes with risk of esophageal and gastric cancer in the NIH-AARP diet and health study. Int. J. Cancer 2012, 131, 1376–1387. [Google Scholar] [CrossRef]
- Graham, S.; Haughey, B.; Marshall, J.; Brasure, J.; Zielezny, M.; Freudenheim, J.; West, D.; Nolan, J.; Wilkinson, G. Diet in the epidemiology of gastric cancer. Nutr. Cancer 1990, 13, 19–34. [Google Scholar] [CrossRef]
- Hu, J.; La Vecchia, C.; Negri, E.; de Groh, M.; Morrison, H.; Mery, L.; Canadian Cancer Registries Epidemiology Research, G. Macronutrient intake and stomach cancer. Cancer Causes Control. 2015, 26, 839–847. [Google Scholar] [CrossRef]
- Han, J.; Jiang, Y.; Liu, X.; Meng, Q.; Xi, Q.; Zhuang, Q.; Han, Y.; Gao, Y.; Ding, Q.; Wu, G. Dietary Fat Intake and Risk of Gastric Cancer: A Meta-Analysis of Observational Studies. PLoS ONE 2015, 10, e0138580. [Google Scholar] [CrossRef]
- Inagaki-Ohara, K.; Okamoto, S.; Takagi, K.; Saito, K.; Arita, S.; Tang, L.; Hori, T.; Kataoka, H.; Matsumoto, S.; Minokoshi, Y. Leptin receptor signaling is required for high-fat diet-induced atrophic gastritis in mice. Nutr. Metab. 2016, 13, 7. [Google Scholar] [CrossRef]
- Arita, S.; Kinoshita, Y.; Ushida, K.; Enomoto, A.; Inagaki-Ohara, K. High-fat diet feeding promotes stemness and precancerous changes in murine gastric mucosa mediated by leptin receptor signaling pathway. Arch. Biochem. Biophys. 2016, 610, 16–24. [Google Scholar] [CrossRef]
- Ericksen, R.E.; Rose, S.; Westphalen, C.B.; Shibata, W.; Muthupalani, S.; Tailor, Y.; Friedman, R.A.; Han, W.; Fox, J.G.; Ferrante, A.W., Jr.; et al. Obesity accelerates Helicobacter felis-induced gastric carcinogenesis by enhancing immature myeloid cell trafficking and TH17 response. Gut 2014, 63, 385–394. [Google Scholar] [CrossRef]
- Castello, A.; Fernandez de Larrea, N.; Martin, V.; Davila-Batista, V.; Boldo, E.; Guevara, M.; Moreno, V.; Castano-Vinyals, G.; Gomez-Acebo, I.; Fernandez-Tardon, G.; et al. High adherence to the Western, Prudent, and Mediterranean dietary patterns and risk of gastric adenocarcinoma: MCC-Spain study. Gastric Cancer 2018, 21, 372–382. [Google Scholar] [CrossRef]
- Gu, Y.; Yu, S.; Lambert, J.D. Dietary cocoa ameliorates obesity-related inflammation in high fat-fed mice. Eur. J. Nutr. 2014, 53, 149–158. [Google Scholar] [CrossRef]
- Sittipo, P.; Lobionda, S.; Lee, Y.K.; Maynard, C.L. Intestinal microbiota and the immune system in metabolic diseases. J. Microbiol. 2018, 56, 154–162. [Google Scholar] [CrossRef]
- Ley, R.E.; Backhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef] [Green Version]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015, 26, 26191. [Google Scholar] [CrossRef]
- Arita, S.; Inagaki-Ohara, K. High-fat diet-induced modulations of leptin signaling and gastric microbiota drive precancerous lesions in the stomach. Nutrition 2019, in press. [Google Scholar] [CrossRef]
- Caesar, R.; Tremaroli, V.; Kovatcheva-Datchary, P.; Cani, P.D.; Backhed, F. Crosstalk between Gut Microbiota and Dietary Lipids Aggravates WAT Inflammation through TLR Signaling. Cell Metab. 2015, 22, 658–668. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.M.; Halaas, J.L. Leptin and the regulation of body weight in mammals. Nature 1998, 395, 763–770. [Google Scholar] [CrossRef]
- Bado, A.; Levasseur, S.; Attoub, S.; Kermorgant, S.; Laigneau, J.P.; Bortoluzzi, M.N.; Moizo, L.; Lehy, T.; Guerre-Millo, M.; Le Marchand-Brustel, Y.; et al. The stomach is a source of leptin. Nature 1998, 394, 790–793. [Google Scholar] [CrossRef]
- Howard, J.M.; Pidgeon, G.P.; Reynolds, J.V. Leptin and gastro-intestinal malignancies. Obes. Rev. 2010, 11, 863–874. [Google Scholar] [CrossRef]
- Inagaki-Ohara, K. Gastric Leptin and Tumorigenesis: Beyond Obesity. Int. J. Mol. Sci. 2019, 20, 2622. [Google Scholar] [CrossRef]
- Lovatt, M.; Bijlmakers, M.J. Stabilisation of beta-catenin downstream of T cell receptor signalling. PLoS ONE 2010, 5, e12794. [Google Scholar] [CrossRef]
- Miller, D.M.; Thomas, S.D.; Islam, A.; Muench, D.; Sedoris, K. c-Myc and cancer metabolism. Clin. Cancer Res. 2012, 18, 5546–5553. [Google Scholar] [CrossRef]
- Huang, L.; Qi, D.J.; He, W.; Xu, A.M. Omeprazole promotes carcinogenesis of fore-stomach in mice with co-stimulation of nitrosamine. Oncotarget 2017, 8, 70332–70344. [Google Scholar] [CrossRef] [Green Version]
- Chen, I.S.; Subramaniam, S.; Vahouny, G.V.; Cassidy, M.M.; Ikeda, I.; Kritchevsky, D. A comparison of the digestion and absorption of cocoa butter and palm kernel oil and their effects on cholesterol absorption in rats. J. Nutr. 1989, 119, 1569–1573. [Google Scholar] [CrossRef]
- Apgar, J.L.; Shively, C.A.; Tarka, S.M., Jr. Digestibility of cocoa butter and corn oil and their influence on fatty acid distribution in rats. J. Nutr. 1987, 117, 660–665. [Google Scholar] [CrossRef]
- Zeng, L.; Wu, G.Z.; Goh, K.J.; Lee, Y.M.; Ng, C.C.; You, A.B.; Wang, J.; Jia, D.; Hao, A.; Yu, Q.; et al. Saturated fatty acids modulate cell response to DNA damage: Implication for their role in tumorigenesis. PLoS ONE 2008, 3, e2329. [Google Scholar] [CrossRef]
- Camarda, R.; Zhou, A.Y.; Kohnz, R.A.; Balakrishnan, S.; Mahieu, C.; Anderton, B.; Eyob, H.; Kajimura, S.; Tward, A.; Krings, G.; et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat. Med. 2016, 22, 427–432. [Google Scholar] [CrossRef]
- Hashimoto, S.; Mikami, S.; Sugino, H.; Yoshikawa, A.; Hashimoto, A.; Onodera, Y.; Furukawa, S.; Handa, H.; Oikawa, T.; Okada, Y.; et al. Lysophosphatidic acid activates Arf6 to promote the mesenchymal malignancy of renal cancer. Nat. Commun. 2016, 7, 10656. [Google Scholar] [CrossRef] [Green Version]
- Jeon, S.M.; Chandel, N.S.; Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 2012, 485, 661–665. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Dai, Q.; Zhang, T.; Li, C. Palmitate acid promotes gastric cancer metastasis via FABP5/SP1/UCA1 pathway. Cancer Cell Int. 2019, 19, 69. [Google Scholar] [CrossRef]
- Xiang, F.; Wu, K.; Liu, Y.; Shi, L.; Wang, D.; Li, G.; Tao, K.; Wang, G. Omental adipocytes enhance the invasiveness of gastric cancer cells by oleic acid-induced activation of the PI3K-Akt signaling pathway. Int. J. Biochem. Cell Biol. 2017, 84, 14–21. [Google Scholar] [CrossRef]
- Mocellin, M.C.; Fernandes, R.; Chagas, T.R.; Trindade, E. A meta-analysis of n-3 polyunsaturated fatty acids effects on circulating acute-phase protein and cytokines in gastric cancer. Clin. Nutr. 2018, 37, 840–850. [Google Scholar] [CrossRef]
- Ali Abd El-Aal, Y.; Mohamed Abdel-Fattah, D.; El-Dawy Ahmed, K. Some biochemical studies on trans fatty acid-containing diet. Diabetes Metab. Syndr. 2019, 13, 1753–1757. [Google Scholar] [CrossRef]
- Saadatdoust, Z.; Pandurangan, A.K.; Ananda Sadagopan, S.K.; Mohd Esa, N.; Ismail, A.; Mustafa, M.R. Dietary cocoa inhibits colitis associated cancer: A crucial involvement of the IL-6/STAT3 pathway. J. Nutr. Biochem. 2015, 26, 1547–1558. [Google Scholar] [CrossRef]
- Blanpain, C.; Fuchs, E. Epidermal homeostasis: A balancing act of stem cells in the skin. Nat. Rev. Mol. Cell Biol. 2009, 10, 207–217. [Google Scholar] [CrossRef]
- Clevers, H.; Nusse, R. Wnt/beta-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef]
- Zhong, W.; Li, Q.; Xie, G.; Sun, X.; Tan, X.; Sun, X.; Jia, W.; Zhou, Z. Dietary fat sources differentially modulate intestinal barrier and hepatic inflammation in alcohol-induced liver injury in rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G919–G932. [Google Scholar] [CrossRef]
- Vivarelli, S.; Salemi, R.; Candido, S.; Falzone, L.; Santagati, M.; Stefani, S.; Torino, F.; Banna, G.L.; Tonini, G.; Libra, M. Gut Microbiota and Cancer: From Pathogenesis to Therapy. Cancers 2019, 11, 38. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef]
- Ferrer, M.; Ruiz, A.; Lanza, F.; Haange, S.B.; Oberbach, A.; Till, H.; Bargiela, R.; Campoy, C.; Segura, M.T.; Richter, M.; et al. Microbiota from the distal guts of lean and obese adolescents exhibit partial functional redundancy besides clear differences in community structure. Environ. Microbiol. 2013, 15, 211–226. [Google Scholar] [CrossRef]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef]
- Patil, D.P.; Dhotre, D.P.; Chavan, S.G.; Sultan, A.; Jain, D.S.; Lanjekar, V.B.; Gangawani, J.; Shah, P.S.; Todkar, J.S.; Shah, S.; et al. Molecular analysis of gut microbiota in obesity among Indian individuals. J. Biosci. 2012, 37, 647–657. [Google Scholar] [CrossRef]
- Ferreira, R.M.; Pereira-Marques, J.; Pinto-Ribeiro, I.; Costa, J.L.; Carneiro, F.; Machado, J.C.; Figueiredo, C. Gastric microbial community profiling reveals a dysbiotic cancer-associated microbiota. Gut 2018, 67, 226–236. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arita, S.; Ogawa, T.; Murakami, Y.; Kinoshita, Y.; Okazaki, M.; Inagaki-Ohara, K. Dietary Fat-Accelerating Leptin Signaling Promotes Protumorigenic Gastric Environment in Mice. Nutrients 2019, 11, 2127. https://doi.org/10.3390/nu11092127
Arita S, Ogawa T, Murakami Y, Kinoshita Y, Okazaki M, Inagaki-Ohara K. Dietary Fat-Accelerating Leptin Signaling Promotes Protumorigenic Gastric Environment in Mice. Nutrients. 2019; 11(9):2127. https://doi.org/10.3390/nu11092127
Chicago/Turabian StyleArita, Seiya, Takumi Ogawa, Yuta Murakami, Yuta Kinoshita, Masaharu Okazaki, and Kyoko Inagaki-Ohara. 2019. "Dietary Fat-Accelerating Leptin Signaling Promotes Protumorigenic Gastric Environment in Mice" Nutrients 11, no. 9: 2127. https://doi.org/10.3390/nu11092127
APA StyleArita, S., Ogawa, T., Murakami, Y., Kinoshita, Y., Okazaki, M., & Inagaki-Ohara, K. (2019). Dietary Fat-Accelerating Leptin Signaling Promotes Protumorigenic Gastric Environment in Mice. Nutrients, 11(9), 2127. https://doi.org/10.3390/nu11092127