Molecular Insight into the Interaction between Epigenetics and Leptin in Metabolic Disorders

 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Leptin
2.1. Structure
2.2. Secretion
2.3. Functions
3. Mechanisms of Epigenetic Modifications
4. Epigenetic Regulation of Leptin’s Expression in Metabolic Disorders
4.1. DNA Methylation
4.2. Postranscriptional Histone Modifications
4.3. miRNAs
5. Leptin-Induced Epigenetic Modifications in Metabolic Disorders
5.1. DNA Methylation
5.2. Postranscriptional Histone Modifications
5.3. miRNAs
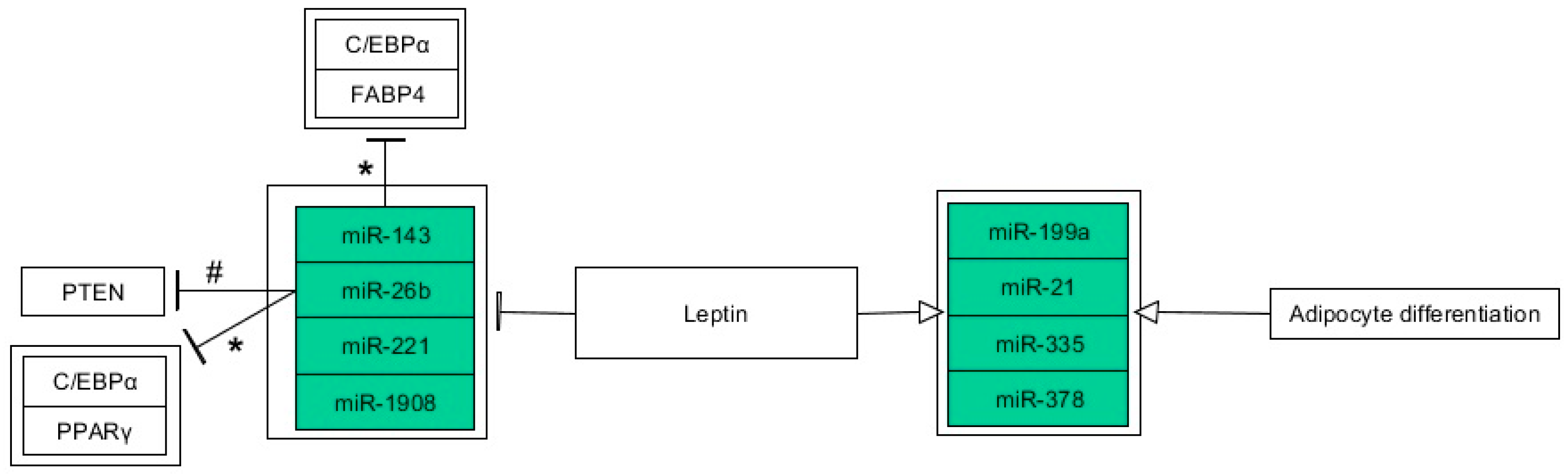
6. Epigenetics of Leptin in Adipogenesis
6.1. DNA Methylation
6.2. Postranscriptional Histone Modifications
6.3. miRNAs
7. Epigenetics of Leptin in Fetal Programming
7.1. DNA Methylation
7.2. Postranscriptional Histone Modifications
7.3. miRNAs
8. Epigenetics of Leptin in Early Postnatal Programming
9. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- O’Neill, S.; O’Driscoll, L. Metabolic syndrome: A closer look at the growing epidemic and its associated pathologies. Obes. Rev. 2015, 16, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Nolan, P.B.; Carrick-Ranson, G.; Stinear, J.W.; Reading, S.A.; Dalleck, L.C. Prevalence of metabolic syndrome and metabolic syndrome components in young adults: A pooled analysis. Prev. Med. Rep. 2017, 7, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Szymczak-Pajor, I.; Sliwinska, A. Analysis of Association between Vitamin D Deficiency and Insulin Resistance. Nutrients 2019, 11, 794. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, R.; Ravindran, R.; Dhanasekaran, S. Emerging Role of Adipocytokines in Type 2 Diabetes as Mediators of Insulin Resistance and Cardiovascular Disease. Can. J. Diabetes 2018, 42, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-H.; Liu, Y.-C.; Wang, J.-H.; Lee, C.-J.; Hsu, B.-G. Serum leptin level positively correlates with metabolic syndrome among elderly Taiwanese. Tzu-chi Med. J. 2017, 29, 159–164. [Google Scholar]
- Sainz, N.; Barrenetxe, J.; Moreno-Aliaga, M.J.; Martinez, J.A. Leptin resistance and diet-induced obesity: Central and peripheral actions of leptin. Metabolism 2015, 64, 35–46. [Google Scholar] [CrossRef]
- Reinehr, T.; Woelfle, J.; Wiegand, S.; Karges, B.; Meissner, T.; Nagl, K.; Holl, R.W. Leptin but not adiponectin is related to type 2 diabetes mellitus in obese adolescents. Pediatr. Diabetes 2016, 17, 281–288. [Google Scholar] [CrossRef]
- Abdelgadir, M.; Elbagir, M.; Eltom, M.; Berne, C.; Ahren, B. Reduced leptin concentrations in subjects with type 2 diabetes mellitus in Sudan. Metabolism 2002, 51, 304–306. [Google Scholar] [CrossRef]
- Kasinska, M.A.; Drzewoski, J.; Sliwinska, A. Epigenetic modifications in adipose tissue—relation to obesity and diabetes. Arch. Med. Sci. 2016, 12, 1293–1301. [Google Scholar] [CrossRef]
- Munzberg, H.; Morrison, C.D. Structure, production and signaling of leptin. Metabolism 2015, 64, 13–23. [Google Scholar] [CrossRef]
- LEP Leptin [Homo Sapiens (Human)]—Gene—NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene?Db=gene&Cmd=DetailsSearch&Term=3952 (accessed on 17 July 2019).
- Fruhbeck, G. Intracellular signalling pathways activated by leptin. Biochem. J. 2006, 393, 7–20. [Google Scholar] [CrossRef]
- Kwon, H.; Pessin, J.E. Adipokines mediate inflammation and insulin resistance. Front. Endocrinol. (Lausanne) 2013, 4, 71. [Google Scholar] [CrossRef]
- Gogga, P.; Karbowska, J.; Meissner, W.; Kochan, Z. [Role of leptin in the regulation of lipid and carbohydrate metabolism]. Postepy Hig. Med. Dosw. (Online) 2011, 65, 255–262. [Google Scholar]
- Kershaw, E.E.; Flier, J.S. Adipose tissue as an endocrine organ. J. Clin. Endocrinol. Metab. 2004, 89, 2548–2556. [Google Scholar] [CrossRef]
- Zhou, Y.; Rui, L. Leptin signaling and leptin resistance. Front. Med. 2013, 7, 207–222. [Google Scholar] [CrossRef]
- Aragones, G.; Ardid-Ruiz, A.; Ibars, M.; Suarez, M.; Blade, C. Modulation of leptin resistance by food compounds. Mol. Nutr. Food Res. 2016, 60, 1789–1803. [Google Scholar] [CrossRef]
- Loscalzo, J.; Handy, D.E. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease (2013 Grover Conference series). Pulm. Circ. 2014, 4, 169–174. [Google Scholar] [CrossRef]
- Carey, N.; Marques, C.J.; Reik, W. DNA demethylases: A new epigenetic frontier in drug discovery. Drug Discov. Today 2011, 16, 683–690. [Google Scholar] [CrossRef]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef]
- Becker, J.S.; Nicetto, D.; Zaret, K.S. H3K9me3-Dependent Heterochromatin: Barrier to Cell Fate Changes. Trends Genet. 2016, 32, 29–41. [Google Scholar] [CrossRef]
- Farooq, Z.; Banday, S.; Pandita, T.K.; Altaf, M. The many faces of histone H3K79 methylation. Mutat. Res. Rev. Mutat. Res. 2016, 768, 46–52. [Google Scholar] [CrossRef]
- Catalanotto, C.; Cogoni, C.; Zardo, G. MicroRNA in Control of Gene Expression: An Overview of Nuclear Functions. Int. J. Mol. Sci. 2016, 17, 1712. [Google Scholar] [CrossRef]
- Mongan, N.P.; Emes, R.D.; Archer, N. Detection and analysis of RNA methylation. F1000Research 2019, 8. [Google Scholar] [CrossRef]
- Sun, T.; Wu, R.; Ming, L. The role of m6A RNA methylation in cancer. Biomed. Pharmacother. 2019, 112, 108613. [Google Scholar] [CrossRef]
- Hofmann, N.R. Epitranscriptomics and Flowering: mRNA Methylation/Demethylation Regulates Flowering Time. Plant Cell 2017, 29, 2949–2950. [Google Scholar] [CrossRef]
- Yao, Q.; Chen, Y.; Zhou, X. The roles of microRNAs in epigenetic regulation. Curr. Opin. Chem. Biol. 2019, 51, 11–17. [Google Scholar] [CrossRef]
- Raghuraman, S.; Donkin, I.; Versteyhe, S.; Barres, R.; Simar, D. The Emerging Role of Epigenetics in Inflammation and Immunometabolism. Trends Endocrinol. Metab. 2016, 27, 782–795. [Google Scholar] [CrossRef]
- Franzago, M.; Fraticelli, F.; Stuppia, L.; Vitacolonna, E. Nutrigenetics, epigenetics and gestational diabetes: Consequences in mother and child. Epigenetics 2019, 14, 215–235. [Google Scholar] [CrossRef]
- Ling, C.; Ronn, T. Epigenetics in Human Obesity and Type 2 Diabetes. Cell Metab. 2019, 29, 1028–1044. [Google Scholar] [CrossRef]
- de Castro Barbosa, T.; Ingerslev, L.R.; Alm, P.S.; Versteyhe, S.; Massart, J.; Rasmussen, M.; Donkin, I.; Sjogren, R.; Mudry, J.M.; Vetterli, L.; et al. High-fat diet reprograms the epigenome of rat spermatozoa and transgenerationally affects metabolism of the offspring. Mol. Metab. 2016, 5, 184–197. [Google Scholar] [CrossRef]
- de Castro Barbosa, T.; Alm, P.S.; Krook, A.; Barres, R.; Zierath, J.R. Paternal high-fat diet transgenerationally impacts hepatic immunometabolism. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 6269–6280. [Google Scholar] [CrossRef]
- Lecoutre, S.; Petrus, P.; Ryden, M.; Breton, C. Transgenerational Epigenetic Mechanisms in Adipose Tissue Development. Trends Endocrinol. Metab. 2018, 29, 675–685. [Google Scholar] [CrossRef]
- Carson, C.; Lawson, H.A. Epigenetics of metabolic syndrome. Physiol. Genom. 2018, 50, 947–955. [Google Scholar] [CrossRef]
- Dunstan, J.; Bressler, J.P.; Moran, T.H.; Pollak, J.S.; Hirsch, A.G.; Bailey-Davis, L.; Glass, T.A.; Schwartz, B.S. Associations of LEP, CRH, ICAM-1, and LINE-1 methylation, measured in saliva, with waist circumference, body mass index, and percent body fat in mid-childhood. Clin. Epigenetics 2017, 9, 29. [Google Scholar] [CrossRef]
- Garcia-Cardona, M.C.; Huang, F.; Garcia-Vivas, J.M.; Lopez-Camarillo, C.; Del Rio Navarro, B.E.; Navarro Olivos, E.; Hong-Chong, E.; Bolanos-Jimenez, F.; Marchat, L.A. DNA methylation of leptin and adiponectin promoters in children is reduced by the combined presence of obesity and insulin resistance. Int. J. Obes. (Lond.) 2014, 38, 1457–1465. [Google Scholar] [CrossRef]
- El Sayed, S.; Khairy, E.; Basheer, A.R.; Zaki, W.S.; Ahmad, G.F.; Kassim, S.K. Evaluation of leptin and MMP2 genes methylation in childhood obesity. Gene Rep. 2018, 11, 79–86. [Google Scholar] [CrossRef]
- Houde, A.-A.; Legare, C.; Hould, F.-S.; Lebel, S.; Marceau, P.; Tchernof, A.; Vohl, M.-C.; Hivert, M.-F.; Bouchard, L. Cross-tissue comparisons of leptin and adiponectin: DNA methylation profiles. Adipocyte 2014, 3, 132–140. [Google Scholar] [CrossRef]
- Houde, A.-A.; Legare, C.; Biron, S.; Lescelleur, O.; Biertho, L.; Marceau, S.; Tchernof, A.; Vohl, M.-C.; Hivert, M.-F.; Bouchard, L. Leptin and adiponectin DNA methylation levels in adipose tissues and blood cells are associated with BMI, waist girth and LDL-cholesterol levels in severely obese men and women. BMC Med. Genet. 2015, 16, 29. [Google Scholar] [CrossRef]
- Marchi, M.; Lisi, S.; Curcio, M.; Barbuti, S.; Piaggi, P.; Ceccarini, G.; Nannipieri, M.; Anselmino, M.; Di Salvo, C.; Vitti, P.; et al. Human leptin tissue distribution, but not weight loss-dependent change in expression, is associated with methylation of its promoter. Epigenetics 2011, 6, 1198–1206. [Google Scholar] [CrossRef]
- Cordero, P.; Campion, J.; Milagro, F.I.; Goyenechea, E.; Steemburgo, T.; Javierre, B.M.; Martinez, J.A. Leptin and TNF-alpha promoter methylation levels measured by MSP could predict the response to a low-calorie diet. J. Physiol. Biochem. 2011, 67, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Castellano-Castillo, D.; Moreno-Indias, I.; Sanchez-Alcoholado, L.; Ramos-Molina, B.; Alcaide-Torres, J.; Morcillo, S.; Ocana-Wilhelmi, L.; Tinahones, F.; Queipo-Ortuno, M.I.; Cardona, F. Altered Adipose Tissue DNA Methylation Status in Metabolic Syndrome: Relationships Between Global DNA Methylation and Specific Methylation at Adipogenic, Lipid Metabolism and Inflammatory Candidate Genes and Metabolic Variables. J. Clin. Med. 2019, 8, 87. [Google Scholar] [CrossRef] [PubMed]
- Milagro, F.I.; Campion, J.; Garcia-Diaz, D.F.; Goyenechea, E.; Paternain, L.; Martinez, J.A. High fat diet-induced obesity modifies the methylation pattern of leptin promoter in rats. J. Physiol. Biochem. 2009, 65, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zwamborn, R.A.J.; Slieker, R.C.; Mulder, P.C.A.; Zoetemelk, I.; Verschuren, L.; Suchiman, H.E.D.; Toet, K.H.; Droog, S.; Slagboom, P.E.; Kooistra, T.; et al. Prolonged high-fat diet induces gradual and fat depot-specific DNA methylation changes in adult mice. Sci. Rep. 2017, 7, 43261. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Wang, C.; Lu, Y.; Fan, C.; Ding, X.; Fu, H.; Qi, K. Time-specific changes in DNA methyltransferases associated with the leptin promoter during the development of obesity. Nutr. Hosp. 2014, 30, 1248–1255. [Google Scholar] [PubMed]
- Okada, Y.; Sakaue, H.; Nagare, T.; Kasuga, M. Diet-induced up-regulation of gene expression in adipocytes without changes in DNA methylation. Kobe J. Med. Sci. 2009, 54, E241–E249. [Google Scholar] [PubMed]
- Shen, W.; Wang, C.; Xia, L.; Fan, C.; Dong, H.; Deckelbaum, R.J.; Qi, K. Epigenetic modification of the leptin promoter in diet-induced obese mice and the effects of N-3 polyunsaturated fatty acids. Sci. Rep. 2014, 4, 5282. [Google Scholar] [CrossRef] [PubMed]
- Virtue, A.; Johnson, C.; Lopez-Pastrana, J.; Shao, Y.; Fu, H.; Li, X.; Li, Y.-F.; Yin, Y.; Mai, J.; Rizzo, V.; et al. MicroRNA-155 Deficiency Leads to Decreased Atherosclerosis, Increased White Adipose Tissue Obesity, and Non-alcoholic Fatty Liver Disease: A novel mouse model of obesity paradox. J. Biol. Chem. 2017, 292, 1267–1287. [Google Scholar] [CrossRef]
- Kang, M.; Liu, X.; Fu, Y.; Timothy Garvey, W. Improved systemic metabolism and adipocyte biology in miR-150 knockout mice. Metabolism 2018, 83, 139–148. [Google Scholar] [CrossRef]
- Zhang, J.; Ma, W.; He, Y.; Dawar, F.U.; Xiong, S.; Mei, J. Potential Contributions of miR-200a/-200b and Their Target Gene-Leptin to the Sexual Size Dimorphism in Yellow Catfish. Front. Physiol. 2017, 8, 970. [Google Scholar] [CrossRef]
- Zhou, B.; Li, H.; Shi, J. miR27 inhibits the NF-kappaB signaling pathway by targeting leptin in osteoarthritic chondrocytes. Int. J. Mol. Med. 2017, 40, 523–530. [Google Scholar] [CrossRef]
- van Iersel, M.P.; Kelder, T.; Pico, A.R.; Hanspers, K.; Coort, S.; Conklin, B.R.; Evelo, C. Presenting and exploring biological pathways with PathVisio. BMC Bioinform. 2008, 9, 399. [Google Scholar] [CrossRef] [PubMed]
- Sonne, S.B.; Yadav, R.; Yin, G.; Dalgaard, M.D.; Myrmel, L.S.; Gupta, R.; Wang, J.; Madsen, L.; Kajimura, S.; Kristiansen, K. Obesity is associated with depot-specific alterations in adipocyte DNA methylation and gene expression. Adipocyte 2017, 6, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Kamei, Y.; Suganami, T.; Ehara, T.; Kanai, S.; Hayashi, K.; Yamamoto, Y.; Miura, S.; Ezaki, O.; Okano, M.; Ogawa, Y. Increased expression of DNA methyltransferase 3a in obese adipose tissue: Studies with transgenic mice. Obesity (Silver Spring) 2010, 18, 314–321. [Google Scholar] [CrossRef]
- You, D.; Nilsson, E.; Tenen, D.E.; Lyubetskaya, A.; Lo, J.C.; Jiang, R.; Deng, J.; Dawes, B.A.; Vaag, A.; Ling, C.; et al. Dnmt3a is an epigenetic mediator of adipose insulin resistance. Elife 2017, 6, e30766. [Google Scholar] [CrossRef] [PubMed]
- Kabra, D.G.; Pfuhlmann, K.; Garcia-Caceres, C.; Schriever, S.C.; Casquero Garcia, V.; Kebede, A.F.; Fuente-Martin, E.; Trivedi, C.; Heppner, K.; Uhlenhaut, N.H.; et al. Hypothalamic leptin action is mediated by histone deacetylase 5. Nat. Commun. 2016, 7, 10782. [Google Scholar] [CrossRef]
- Song, N.-Y.; Lee, Y.-H.; Na, H.-K.; Baek, J.-H.; Surh, Y.-J. Leptin induces SIRT1 expression through activation of NF-E2-related factor 2: Implications for obesity-associated colon carcinogenesis. Biochem. Pharmacol. 2018, 153, 282–291. [Google Scholar] [CrossRef]
- Fiedor, E.; Zajda, K.; Gregoraszczuk, E.L. Leptin Receptor Antagonists’ Action on HDAC Expression Eliminating the Negative Effects of Leptin in Ovarian Cancer. Cancer Genom. Proteom. 2018, 15, 329–336. [Google Scholar] [CrossRef]
- Tchio, C.M.; Harbuzariu, A.; Harmon, T.; Beech, D.; Gonzalez-Perez, R. Leptin modulation of PCSC, HDAC, and microRNA in pancreatic adenocarcinoma. In Proceedings of the AACR 107th Annual Meeting 2016; American Association for Cancer Research, New Orleans, LA, USA, 16–20 April 2016; Volume 76, p. 1901. [Google Scholar]
- Benoit, C.; Ould-Hamouda, H.; Crepin, D.; Gertler, A.; Amar, L.; Taouis, M. Early leptin blockade predisposes fat-fed rats to overweight and modifies hypothalamic microRNAs. J. Endocrinol. 2013, 218, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Sangiao-Alvarellos, S.; Pena-Bello, L.; Manfredi-Lozano, M.; Tena-Sempere, M.; Cordido, F. Perturbation of hypothalamic microRNA expression patterns in male rats after metabolic distress: Impact of obesity and conditions of negative energy balance. Endocrinology 2014, 155, 1838–1850. [Google Scholar] [CrossRef] [PubMed]
- Dhar, M.; Zhu, M.; Impey, S.; Lambert, T.J.; Bland, T.; Karatsoreos, I.N.; Nakazawa, T.; Appleyard, S.M.; Wayman, G.A. Leptin induces hippocampal synaptogenesis via CREB-regulated microRNA-132 suppression of p250GAP. Mol. Endocrinol. 2014, 28, 1073–1087. [Google Scholar] [CrossRef]
- Derghal, A.; Djelloul, M.; Airault, C.; Pierre, C.; Dallaporta, M.; Troadec, J.-D.; Tillement, V.; Tardivel, C.; Bariohay, B.; Trouslard, J.; et al. Leptin is required for hypothalamic regulation of miRNAs targeting POMC 3′UTR. Front. Cell. Neurosci. 2015, 9, 172. [Google Scholar] [CrossRef]
- Crepin, D.; Benomar, Y.; Riffault, L.; Amine, H.; Gertler, A.; Taouis, M. The over-expression of miR-200a in the hypothalamus of ob/ob mice is linked to leptin and insulin signaling impairment. Mol. Cell. Endocrinol. 2014, 384, 1–11. [Google Scholar] [CrossRef]
- Derghal, A.; Djelloul, M.; Azzarelli, M.; Degonon, S.; Tourniaire, F.; Landrier, J.-F.; Mounien, L. MicroRNAs are involved in the hypothalamic leptin sensitivity. Epigenetics 2018, 13, 1127–1140. [Google Scholar] [CrossRef]
- Alisi, A.; Da Sacco, L.; Bruscalupi, G.; Piemonte, F.; Panera, N.; De Vito, R.; Leoni, S.; Bottazzo, G.F.; Masotti, A.; Nobili, V. Mirnome analysis reveals novel molecular determinants in the pathogenesis of diet-induced nonalcoholic fatty liver disease. Lab. Investig. 2011, 91, 283–293. [Google Scholar] [CrossRef]
- Li, Z.; Ji, L.; Su, S.; Zhu, X.; Cheng, F.; Jia, X.; Zhou, Q.; Zhou, Y. Leptin up-regulates microRNA-27a/b-3p level in hepatic stellate cells. Exp. Cell Res. 2018, 366, 63–70. [Google Scholar] [CrossRef]
- Dattaroy, D.; Pourhoseini, S.; Das, S.; Alhasson, F.; Seth, R.K.; Nagarkatti, M.; Michelotti, G.A.; Diehl, A.M.; Chatterjee, S. Micro-RNA 21 inhibition of SMAD7 enhances fibrogenesis via leptin-mediated NADPH oxidase in experimental and human nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G298–G312. [Google Scholar] [CrossRef]
- Alhasson, F.; Seth, R.K.; Sarkar, S.; Kimono, D.A.; Albadrani, M.S.; Dattaroy, D.; Chandrashekaran, V.; Scott, G.I.; Raychoudhury, S.; Nagarkatti, M.; et al. High circulatory leptin mediated NOX-2-peroxynitrite-miR21 axis activate mesangial cells and promotes renal inflammatory pathology in nonalcoholic fatty liver disease. Redox Biol. 2018, 17, 1–15. [Google Scholar] [CrossRef]
- Zhai, X.; Cheng, F.; Ji, L.; Zhu, X.; Cao, Q.; Zhang, Y.; Jia, X.; Zhou, Q.; Guan, W.; Zhou, Y. Leptin reduces microRNA-122 level in hepatic stellate cells in vitro and in vivo. Mol. Immunol. 2017, 92, 68–75. [Google Scholar] [CrossRef]
- Cao, Q.; Zhu, X.; Zhai, X.; Ji, L.; Cheng, F.; Zhu, Y.; Yu, P.; Zhou, Y. Leptin suppresses microRNA-122 promoter activity by phosphorylation of foxO1 in hepatic stellate cell contributing to leptin promotion of mouse liver fibrosis. Toxicol. Appl. Pharmacol. 2018, 339, 143–150. [Google Scholar] [CrossRef]
- Nakanishi, N.; Nakagawa, Y.; Tokushige, N.; Aoki, N.; Matsuzaka, T.; Ishii, K.; Yahagi, N.; Kobayashi, K.; Yatoh, S.; Takahashi, A.; et al. The up-regulation of microRNA-335 is associated with lipid metabolism in liver and white adipose tissue of genetically obese mice. Biochem. Biophys. Res. Commun. 2009, 385, 492–496. [Google Scholar] [CrossRef]
- Zhu, L.; Chen, L.; Shi, C.-M.; Xu, G.-F.; Xu, L.-L.; Zhu, L.-L.; Guo, X.-R.; Ni, Y.; Cui, Y.; Ji, C. MiR-335, an adipogenesis-related microRNA, is involved in adipose tissue inflammation. Cell Biochem. Biophys. 2014, 68, 283–290. [Google Scholar] [CrossRef]
- Xu, X.; Dong, Z.; Li, Y.; Yang, Y.; Yuan, Z.; Qu, X.; Kong, B. The upregulation of signal transducer and activator of transcription 5-dependent microRNA-182 and microRNA-96 promotes ovarian cancer cell proliferation by targeting forkhead box O3 upon leptin stimulation. Int. J. Biochem. Cell Biol. 2013, 45, 536–545. [Google Scholar] [CrossRef]
- Yang, W.-H.; Chang, A.-C.; Wang, S.-W.; Wang, S.-J.; Chang, Y.-S.; Chang, T.-M.; Hsu, S.-K.; Fong, Y.-C.; Tang, C.-H. Leptin promotes VEGF-C production and induces lymphangiogenesis by suppressing miR-27b in human chondrosarcoma cells. Sci. Rep. 2016, 6, 28647. [Google Scholar] [CrossRef]
- Avtanski, D.B.; Nagalingam, A.; Kuppusamy, P.; Bonner, M.Y.; Arbiser, J.L.; Saxena, N.K.; Sharma, D. Honokiol abrogates leptin-induced tumor progression by inhibiting Wnt1-MTA1-beta-catenin signaling axis in a microRNA-34a dependent manner. Oncotarget 2015, 6, 16396–16410. [Google Scholar] [CrossRef]
- Avtanski, D.B.; Nagalingam, A.; Bonner, M.Y.; Arbiser, J.L.; Saxena, N.K.; Sharma, D. Honokiol activates LKB1-miR-34a axis and antagonizes the oncogenic actions of leptin in breast cancer. Oncotarget 2015, 6, 29947–29962. [Google Scholar] [CrossRef]
- Meerson, A.; Yehuda, H. Leptin and insulin up-regulate miR-4443 to suppress NCOA1 and TRAF4, and decrease the invasiveness of human colon cancer cells. BMC Cancer 2016, 16, 882. [Google Scholar] [CrossRef]
- Jasinski-Bergner, S.; Kielstein, H. Adipokines Regulate the Expression of Tumor-Relevant MicroRNAs. Obes. Facts 2019, 12, 211–225. [Google Scholar] [CrossRef]
- Kloting, N.; Berthold, S.; Kovacs, P.; Schon, M.R.; Fasshauer, M.; Ruschke, K.; Stumvoll, M.; Bluher, M. MicroRNA expression in human omental and subcutaneous adipose tissue. PLoS ONE 2009, 4, e4699. [Google Scholar] [CrossRef]
- Al-Rawaf, H.A. Circulating microRNAs and adipokines as markers of metabolic syndrome in adolescents with obesity. Clin. Nutr. 2018, in press. [Google Scholar] [CrossRef]
- Catalioto, R.-M.; Maggi, C.A.; Giuliani, S. Chemically distinct HDAC inhibitors prevent adipose conversion of subcutaneous human white preadipocytes at an early stage of the differentiation program. Exp. Cell Res. 2009, 315, 3267–3280. [Google Scholar] [CrossRef]
- Ali, A.T.; Hochfeld, W.E.; Myburgh, R.; Pepper, M.S. Adipocyte and adipogenesis. Eur. J. Cell Biol. 2013, 92, 229–236. [Google Scholar] [CrossRef]
- Melzner, I.; Scott, V.; Dorsch, K.; Fischer, P.; Wabitsch, M.; Brüderlein, S.; Hasel, C.; Möller, P. Leptin gene expression in human preadipocytes is switched on by maturation-induced demethylation of distinct CpGs in its proximal promoter. J. Biol. Chem. 2002, 277, 45420–45427. [Google Scholar] [CrossRef]
- Yokomori, N.; Tawata, M.; Onaya, T. DNA demethylation modulates mouse leptin promoter activity during the differentiation of 3T3-L1 cells. Diabetologia 2002, 45, 140–148. [Google Scholar] [CrossRef]
- Noer, A.; Sørensen, A.L.; Boquest, A.C.; Collas, P. Stable CpG hypomethylation of adipogenic promoters in freshly isolated, cultured, and differentiated mesenchymal stem cells from adipose tissue. Mol. Biol. Cell 2006, 17, 3543–3556. [Google Scholar] [CrossRef]
- Noer, A.; Boquest, A.C.; Collas, P. Dynamics of adipogenic promoter DNA methylation during clonal culture of human adipose stem cells to senescence. BMC Cell Biol. 2007, 8, 18. [Google Scholar] [CrossRef]
- Musri, M.M.; Corominola, H.; Casamitjana, R.; Gomis, R.; Parrizas, M. Histone H3 lysine 4 dimethylation signals the transcriptional competence of the adiponectin promoter in preadipocytes. J. Biol. Chem. 2006, 281, 17180–17188. [Google Scholar] [CrossRef]
- Lagace, D.C.; McLeod, R.S.; Nachtigal, M.W. Valproic acid inhibits leptin secretion and reduces leptin messenger ribonucleic acid levels in adipocytes. Endocrinology 2004, 145, 5493–5503. [Google Scholar] [CrossRef]
- Yoo, E.J.; Chung, J.-J.; Choe, S.S.; Kim, K.H.; Kim, J.B. Down-regulation of histone deacetylases stimulates adipocyte differentiation. J. Biol. Chem. 2006, 281, 6608–6615. [Google Scholar] [CrossRef]
- Gu, N.; You, L.; Shi, C.; Yang, L.; Pang, L.; Cui, X.; Ji, C.; Zheng, W.; Guo, X. Expression of miR-199a-3p in human adipocytes is regulated by free fatty acids and adipokines. Mol. Med. Rep. 2016, 14, 1180–1186. [Google Scholar] [CrossRef]
- Jiang, X.; Xue, M.; Fu, Z.; Ji, C.; Guo, X.; Zhu, L.; Xu, L.; Pang, L.; Xu, M.; Qu, H. Insight into the Effects of Adipose Tissue Inflammation Factors on miR-378 Expression and the Underlying Mechanism. Cell. Physiol. Biochem. 2014, 33, 1778–1788. [Google Scholar] [CrossRef]
- Zhang, N.; Zhang, N.; Song, L.; Xie, H.; Zhao, C.; Li, S.; Zhao, W.; Zhao, Y.; Gao, C.; Xu, G. Adipokines and free fatty acids regulate insulin sensitivity by increasing microRNA-21 expression in human mature adipocytes. Mol. Med. Rep. 2017, 16, 2254–2258. [Google Scholar] [CrossRef]
- Jiang, X.; Yang, L.; Pang, L.; Chen, L.; Guo, X.; Ji, C.; Shi, C.; Ni, Y. Expression of obesityrelated miR1908 in human adipocytes is regulated by adipokines, free fatty acids and hormones. Mol. Med. Rep. 2014, 10, 1164–1169. [Google Scholar] [CrossRef]
- Meerson, A.; Traurig, M.; Ossowski, V.; Fleming, J.M.; Mullins, M.; Baier, L.J. Human adipose microRNA-221 is upregulated in obesity and affects fat metabolism downstream of leptin and TNF-alpha. Diabetologia 2013, 56, 1971–1979. [Google Scholar] [CrossRef]
- Kim, Y.J.; Hwang, S.J.; Bae, Y.C.; Jung, J.S. MiR-21 regulates adipogenic differentiation through the modulation of TGF-beta signaling in mesenchymal stem cells derived from human adipose tissue. Stem Cells 2009, 27, 3093–3102. [Google Scholar]
- Xu, G.; Ji, C.; Shi, C.; Fu, H.; Zhu, L.; Zhu, L.; Xu, L.; Chen, L.; Feng, Y.; Zhao, Y.; et al. Modulation of hsa-miR-26b levels following adipokine stimulation. Mol. Biol. Rep. 2013, 40, 3577–3582. [Google Scholar] [CrossRef]
- Song, G.; Xu, G.; Ji, C.; Shi, C.; Shen, Y.; Chen, L.; Zhu, L.; Yang, L.; Zhao, Y.; Guo, X. The role of microRNA-26b in human adipocyte differentiation and proliferation. Gene 2014, 533, 481–487. [Google Scholar] [CrossRef]
- Kajimoto, K.; Naraba, H.; Iwai, N. MicroRNA and 3T3-L1 pre-adipocyte differentiation. RNA 2006, 12, 1626–1632. [Google Scholar] [CrossRef]
- Li, G.; Ning, C.; Ma, Y.; Jin, L.; Tang, Q.; Li, X.; Li, M.; Liu, H. miR-26b Promotes 3T3-L1 Adipocyte Differentiation Through Targeting PTEN. DNA Cell Biol. 2017, 36, 672–681. [Google Scholar] [CrossRef]
- Liang, Y.-Z.; Li, J.-J.-H.; Xiao, H.-B.; He, Y.; Zhang, L.; Yan, Y.-X. Identification of stress-related microRNA biomarkers in type 2 diabetes mellitus: A systematic review and meta-analysis. J. Diabetes 2018. [Google Scholar] [CrossRef]
- Zhu, L.; Shi, C.; Ji, C.; Xu, G.; Chen, L.; Yang, L.; Fu, Z.; Cui, X.; Lu, Y.; Guo, X. FFAs and adipokine-mediated regulation of hsa-miR-143 expression in human adipocytes. Mol. Biol. Rep. 2013, 40, 5669–5675. [Google Scholar] [CrossRef]
- Esau, C.; Kang, X.; Peralta, E.; Hanson, E.; Marcusson, E.G.; Ravichandran, L.V.; Sun, Y.; Koo, S.; Perera, R.J.; Jain, R.; et al. MicroRNA-143 regulates adipocyte differentiation. J. Biol. Chem. 2004, 279, 52361–52365. [Google Scholar] [CrossRef]
- Li, H.; Zhang, Z.; Zhou, X.; Wang, Z.; Wang, G.; Han, Z. Effects of microRNA-143 in the differentiation and proliferation of bovine intramuscular preadipocytes. Mol. Biol. Rep. 2011, 38, 4273–4280. [Google Scholar] [CrossRef]
- Jordan, S.D.; Krüger, M.; Willmes, D.M.; Redemann, N.; Wunderlich, F.T.; Brönneke, H.S.; Merkwirth, C.; Kashkar, H.; Olkkonen, V.M.; Böttger, T.; et al. Obesity-induced overexpression of miRNA-143 inhibits insulin-stimulated AKT activation and impairs glucose metabolism. Nat. Cell Biol. 2011, 13, 434. [Google Scholar] [CrossRef]
- Ruchat, S.-M.; Hivert, M.-F.; Bouchard, L. Epigenetic programming of obesity and diabetes by in utero exposure to gestational diabetes mellitus. Nutr. Rev. 2013, 71 (Suppl. 1), S88–S94. [Google Scholar] [CrossRef]
- Nugent, B.M.; Bale, T.L. The omniscient placenta: Metabolic and epigenetic regulation of fetal programming. Front. Neuroendocrinol. 2015, 39, 28–37. [Google Scholar] [CrossRef]
- Saad, A.F.; Dickerson, J.; Kechichian, T.B.; Yin, H.; Gamble, P.; Salazar, A.; Patrikeev, I.; Motamedi, M.; Saade, G.R.; Costantine, M.M. High-fructose diet in pregnancy leads to fetal programming of hypertension, insulin resistance, and obesity in adult offspring. Am. J. Obstet. Gynecol. 2016, 215, e1–e6. [Google Scholar] [CrossRef]
- Fall, C.H.D.; Kumaran, K. Metabolic programming in early life in humans. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2019, 374, 20180123. [Google Scholar] [CrossRef]
- Hur, S.S.J.; Cropley, J.E.; Suter, C.M. Paternal epigenetic programming: Evolving metabolic disease risk. J. Mol. Endocrinol. 2017, 58, R159–R168. [Google Scholar] [CrossRef]
- Lesseur, C.; Armstrong, D.A.; Paquette, A.G.; Koestler, D.C.; Padbury, J.F.; Marsit, C.J. Tissue-specific Leptin promoter DNA methylation is associated with maternal and infant perinatal factors. Mol. Cell. Endocrinol. 2013, 381, 160–167. [Google Scholar] [CrossRef]
- Tobi, E.W.; Heijmans, B.T.; Kremer, D.; Putter, H.; Delemarre-van de Waal, H.A.; Finken, M.J.J.; Wit, J.M.; Slagboom, P.E. DNA methylation of IGF2, GNASAS, INSIGF and LEP and being born small for gestational age. Epigenetics 2011, 6, 171–176. [Google Scholar] [CrossRef]
- Broholm, C.; Olsson, A.H.; Perfilyev, A.; Hansen, N.S.; Schrolkamp, M.; Strasko, K.S.; Scheele, C.; Ribel-Madsen, R.; Mortensen, B.; Jorgensen, S.W.; et al. Epigenetic programming of adipose-derived stem cells in low birthweight individuals. Diabetologia 2016, 59, 2664–2673. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Xu, X.-X.; Sun, H.; Han, Y.; Lei, Z.-F.; Wang, Y.-C.; Yan, H.-T.; Yang, X.-J. Cord blood leptin DNA methylation levels are associated with macrosomia during normal pregnancy. Pediatr. Res. 2019. [Google Scholar] [CrossRef]
- Hjort, L.; Jørgensen, S.W.; Gillberg, L.; Hall, E.; Brøns, C.; Frystyk, J.; Vaag, A.A.; Ling, C. 36 h fasting of young men influences adipose tissue DNA methylation of LEP and ADIPOQ in a birth weight-dependent manner. Clin. Epigenetics 2017, 9, 40. [Google Scholar] [CrossRef]
- Kadakia, R.; Zheng, Y.; Zhang, Z.; Zhang, W.; Hou, L.; Josefson, J.L. Maternal pre-pregnancy BMI downregulates neonatal cord blood LEP methylation. Pediatr. Obes. 2017, 12 (Suppl. 1), 57–64. [Google Scholar] [CrossRef]
- Nogues, P.; Dos Santos, E.; Jammes, H.; Berveiller, P.; Arnould, L.; Vialard, F.; Dieudonne, M.-N. Maternal obesity influences expression and DNA methylation of the adiponectin and leptin systems in human third-trimester placenta. Clin. Epigenetics 2019, 11, 20. [Google Scholar] [CrossRef]
- Lesseur, C.; Armstrong, D.A.; Paquette, A.G.; Li, Z.; Padbury, J.F.; Marsit, C.J. Maternal obesity and gestational diabetes are associated with placental leptin DNA methylation. Am. J. Obstet. Gynecol. 2014, 211, e1–e9. [Google Scholar] [CrossRef]
- Bouchard, L.; Thibault, S.; Guay, S.-P.; Santure, M.; Monpetit, A.; St-Pierre, J.; Perron, P.; Brisson, D. Leptin gene epigenetic adaptation to impaired glucose metabolism during pregnancy. Diabetes Care 2010, 33, 2436–2441. [Google Scholar] [CrossRef]
- Allard, C.; Desgagné, V.; Patenaude, J.; Lacroix, M.; Guillemette, L.; Battista, M.C.; Doyon, M.; Ménard, J.; Ardilouze, J.L.; Perron, P.; et al. Mendelian randomization supports causality between maternal hyperglycemia and epigenetic regulation of leptin gene in newborns. Epigenetics 2015, 10, 342–351. [Google Scholar] [CrossRef]
- Houshmand-Oeregaard, A.; Hansen, N.S.; Hjort, L.; Kelstrup, L.; Broholm, C.; Mathiesen, E.R.; Clausen, T.D.; Damm, P.; Vaag, A. Differential adipokine DNA methylation and gene expression in subcutaneous adipose tissue from adult offspring of women with diabetes in pregnancy. Clin. Epigenetics 2017, 9, 37. [Google Scholar] [CrossRef]
- Lecoutre, S.; Oger, F.; Pourpe, C.; Butruille, L.; Marousez, L.; Dickes-Coopman, A.; Laborie, C.; Guinez, C.; Lesage, J.; Vieau, D.; et al. Maternal obesity programs increased leptin gene expression in rat male offspring via epigenetic modifications in a depot-specific manner. Mol. Metab. 2017, 6, 922–930. [Google Scholar] [CrossRef]
- Jousse, C.; Parry, L.; Lambert-Langlais, S.; Maurin, A.-C.; Averous, J.; Bruhat, A.; Carraro, V.; Tost, J.; Letteron, P.; Chen, P.; et al. Perinatal undernutrition affects the methylation and expression of the leptin gene in adults: Implication for the understanding of metabolic syndrome. FASEB J. 2011, 25, 3271–3278. [Google Scholar] [CrossRef]
- Tobi, E.W.; Lumey, L.H.; Talens, R.P.; Kremer, D.; Putter, H.; Stein, A.D.; Slagboom, P.E.; Heijmans, B.T. DNA methylation differences after exposure to prenatal famine are common and timing- and sex-specific. Hum. Mol. Genet. 2009, 18, 4046–4053. [Google Scholar] [CrossRef]
- Tian, F.-Y.; Rifas-Shiman, S.L.; Cardenas, A.; Baccarelli, A.A.; DeMeo, D.L.; Litonjua, A.A.; Rich-Edwards, J.W.; Gillman, M.W.; Oken, E.; Hivert, M.-F. Maternal corticotropin-releasing hormone is associated with LEP DNA methylation at birth and in childhood: An epigenome-wide study in Project Viva. Int. J. Obes. (Lond.) 2019, 43, 1244–1255. [Google Scholar] [CrossRef]
- Pauwels, S.; Ghosh, M.; Duca, R.C.; Bekaert, B.; Freson, K.; Huybrechts, I.; Langie, S.A.S.; Koppen, G.; Devlieger, R.; Godderis, L. Maternal intake of methyl-group donors affects DNA methylation of metabolic genes in infants. Clin. Epigenetics 2017, 9, 16. [Google Scholar] [CrossRef]
- Tsai, C.-C.; Lin, Y.-J.; Yu, H.-R.; Sheen, J.-M.; Lin, I.-C.; Lai, Y.-J.; Tain, Y.-L.; Huang, L.-T.; Tiao, M.-M. Regulation of Leptin Methylation Not via Apoptosis by Melatonin in the Rescue of Chronic Programming Liver Steatosis. Int. J. Mol. Sci. 2018, 19, 3565. [Google Scholar] [CrossRef]
- Masuyama, H.; Hiramatsu, Y. Effects of a high-fat diet exposure in utero on the metabolic syndrome-like phenomenon in mouse offspring through epigenetic changes in adipocytokine gene expression. Endocrinology 2012, 153, 2823–2830. [Google Scholar] [CrossRef]
- Masuyama, H.; Mitsui, T.; Nobumoto, E.; Hiramatsu, Y. The Effects of High-Fat Diet Exposure in Utero on the Obesogenic and Diabetogenic Traits through Epigenetic Changes in Adiponectin and Leptin Gene Expression for Multiple Generations in Female Mice. Endocrinology 2015, 156, 2482–2491. [Google Scholar] [CrossRef]
- Masuyama, H.; Mitsui, T.; Eguchi, T.; Tamada, S.; Hiramatsu, Y. The effects of paternal high-fat diet exposure on offspring metabolism with epigenetic changes in the mouse adiponectin and leptin gene promoters. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E236–E245. [Google Scholar] [CrossRef]
- Sinha, N.; Biswas, A.; Nave, O.; Seger, C.; Sen, A. Endocrinology Gestational Diabetes Epigenetically Reprograms the Cart Promoter in Fetal Ovary Causing Sub-fertility in Adult Life Gestational Diabetes Epigenetically Reprograms the Cart Promoter in Fetal Ovary. Endocrinology 2019, 160, 1684–1700. [Google Scholar] [CrossRef]
- Ma, X.; Hayes, E.; Prizant, H.; Srivastava, R.K.; Hammes, S.R.; Sen, A. Leptin-Induced CART (Cocaine- and Amphetamine-Regulated Transcript) Is a Novel Intraovarian Mediator of Obesity-Related Infertility in Females. Endocrinology 2016, 157, 1248–1257. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, R.; Zhang, Y.; Li, J.; Grossmann, R.; Zhao, R. In ovo leptin administration affects hepatic lipid metabolism and microRNA expression in newly hatched broiler chickens. J. Anim. Sci. Biotechnol. 2012, 3, 16. [Google Scholar] [CrossRef]
- Pico, C.; Jilkova, Z.M.; Kus, V.; Palou, A.; Kopecky, J. Perinatal programming of body weight control by leptin: Putative roles of AMP kinase and muscle thermogenesis. Am. J. Clin. Nutr. 2011, 94, 1830S–1837S. [Google Scholar] [CrossRef]
- Palou, M.; Pico, C.; Palou, A. Leptin as a breast milk component for the prevention of obesity. Nutr. Rev. 2018, 76, 875–892. [Google Scholar] [CrossRef]
- Obermann-Borst, S.A.; Eilers, P.H.C.; Tobi, E.W.; de Jong, F.H.; Slagboom, P.E.; Heijmans, B.T.; Steegers-Theunissen, R.P.M. Duration of breastfeeding and gender are associated with methylation of the LEPTIN gene in very young children. Pediatr. Res. 2013, 74, 344–349. [Google Scholar] [CrossRef]
- Palou, M.; Pico, C.; McKay, J.A.; Sanchez, J.; Priego, T.; Mathers, J.C.; Palou, A. Protective effects of leptin during the suckling period against later obesity may be associated with changes in promoter methylation of the hypothalamic pro-opiomelanocortin gene. Br. J. Nutr. 2011, 106, 769–778. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wróblewski, A.; Strycharz, J.; Świderska, E.; Drewniak, K.; Drzewoski, J.; Szemraj, J.; Kasznicki, J.; Śliwińska, A. Molecular Insight into the Interaction between Epigenetics and Leptin in Metabolic Disorders. Nutrients 2019, 11, 1872. https://doi.org/10.3390/nu11081872
Wróblewski A, Strycharz J, Świderska E, Drewniak K, Drzewoski J, Szemraj J, Kasznicki J, Śliwińska A. Molecular Insight into the Interaction between Epigenetics and Leptin in Metabolic Disorders. Nutrients. 2019; 11(8):1872. https://doi.org/10.3390/nu11081872
Chicago/Turabian StyleWróblewski, Adam, Justyna Strycharz, Ewa Świderska, Karolina Drewniak, Józef Drzewoski, Janusz Szemraj, Jacek Kasznicki, and Agnieszka Śliwińska. 2019. "Molecular Insight into the Interaction between Epigenetics and Leptin in Metabolic Disorders" Nutrients 11, no. 8: 1872. https://doi.org/10.3390/nu11081872
APA StyleWróblewski, A., Strycharz, J., Świderska, E., Drewniak, K., Drzewoski, J., Szemraj, J., Kasznicki, J., & Śliwińska, A. (2019). Molecular Insight into the Interaction between Epigenetics and Leptin in Metabolic Disorders. Nutrients, 11(8), 1872. https://doi.org/10.3390/nu11081872