Gut Microbiota-Derived Components and Metabolites in the Progression of Non-Alcoholic Fatty Liver Disease (NAFLD)




{kind=link}
Abstract
:1. Introduction
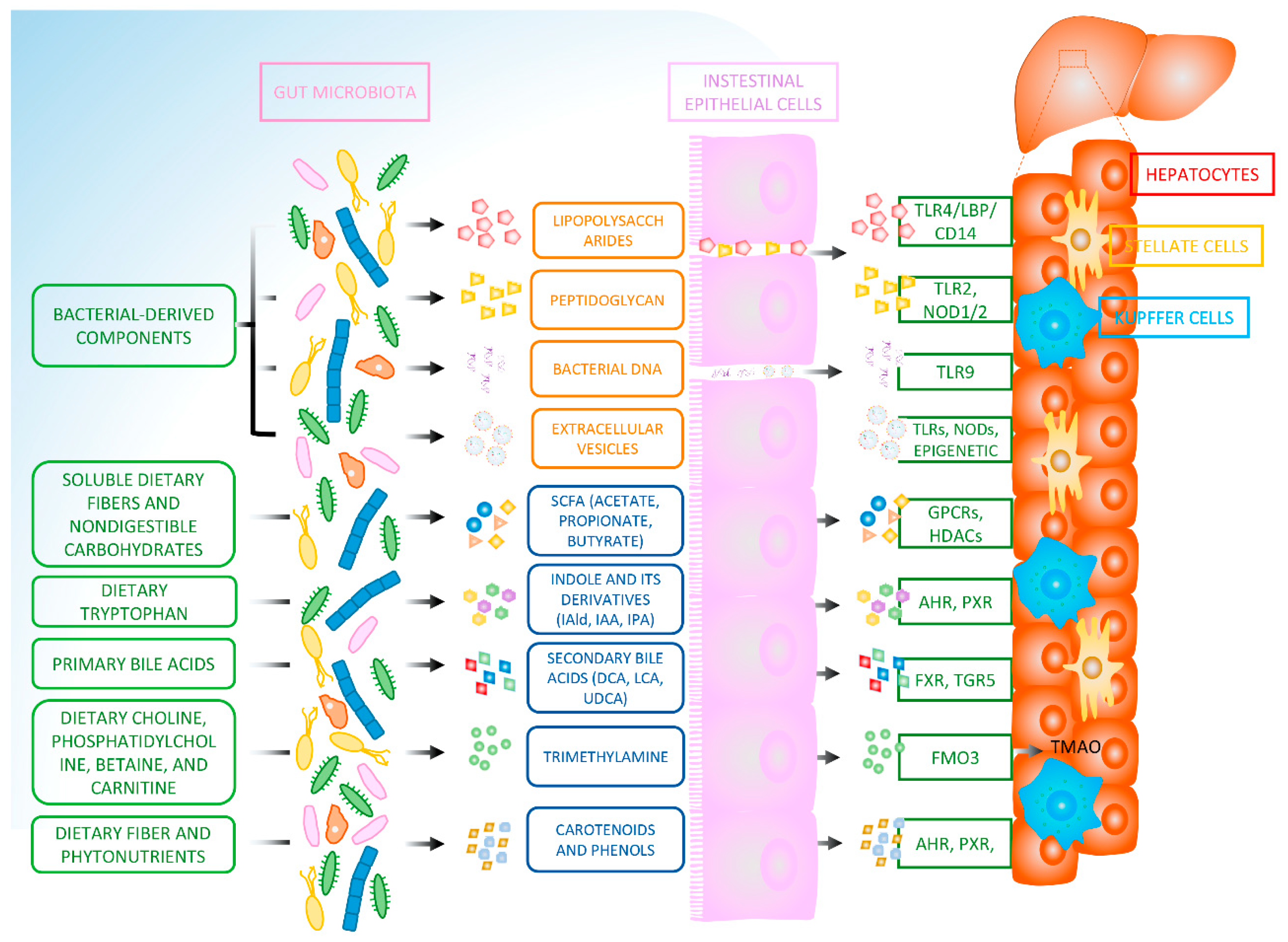
2. Lipopolysaccharides
3. Peptidoglycan
4. Bacterial DNA
5. Extracellular Vesicles
6. Short-Chain Fatty Acids
7. Indole and Its Derivatives
8. Bile Acids
9. Trimethylamine
10. Carotenoids and Phenolic Compounds
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AhR | Aryl hydrocarbon receptor |
| EV | Extracellular vesicle |
| FXR | Farnesoid X receptor |
| GPCR | G-protein coupling receptor |
| HDAC | Histone deacetylase |
| HFD | high fat diet |
| IAA | Indole-3-acetic acid |
| IAld | Indole-3-aldehyde |
| IPA | Indole-3-propionic acid |
| LBP | LPS binding protein |
| LPS | Lipopolysaccharide |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| NOD1 | Nucleotide Binding Oligomerization Domain Containing 1 |
| PGN | Peptidoglycan |
| PXR | Pregnane X receptor |
| SCFA | Short-chain fatty acid |
| TGR5 | Takeda-G-protein-receptor-5 |
| TLR | Toll-like receptor |
| TMA | Trimethylamine |
| TMAO | Trimethylamine-N-oxide |
References
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Touros, A.; Kim, W.R. Nonalcoholic Fatty Liver Disease and Metabolic Syndrome. Clin. Liver Dis. 2018, 22, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Baker, R.D.; Baker, S.S. Gut microbiome and nonalcoholic fatty liver diseases. Pediatr. Res. 2015, 77, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Son, G.; Kremer, M.; Hines, I.N. Contribution of gut bacteria to liver pathobiology. Gastroenterol. Res. Pract. 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Pappo, I.; Bercovier, H.; Berry, E.M.; Haviv, Y.; Gallily, R.; Freund, H.R. Polymyxin B reduces total parenteral nutrition-associated hepatic steatosis by its antibacterial activity and by blocking deleterious effects of lipopolysaccharide. JPEN J. Parenter Enteral. Nutr. 1992, 16, 529–532. [Google Scholar] [CrossRef]
- Harte, A.L.; da Silva, N.F.; Creely, S.J.; McGee, K.C.; Billyard, T.; Youssef-Elabd, E.M.; Tripathi, G.; Ashour, E.; Abdalla, M.S.; Sharada, H.M.; et al. Elevated endotoxin levels in non-alcoholic fatty liver disease. J. Inflamm. 2010, 7, 15. [Google Scholar] [CrossRef]
- Kim, K.A.; Gu, W.; Lee, I.A.; Joh, E.H.; Kim, D.H. High fat diet-induced gut microbiota exacerbates inflammation and obesity in mice via the TLR4 signaling pathway. PLoS ONE 2012, 7, e47713. [Google Scholar] [CrossRef]
- Sharifnia, T.; Antoun, J.; Verriere, T.G.; Suarez, G.; Wattacheril, J.; Wilson, K.T.; Peek, R.M., Jr.; Abumrad, N.N.; Flynn, C.R. Hepatic TLR4 signaling in obese NAFLD. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G270–G278. [Google Scholar] [CrossRef] [Green Version]
- Fukunishi, S.; Sujishi, T.; Takeshita, A.; Ohama, H.; Tsuchimoto, Y.; Asai, A.; Tsuda, Y.; Higuchi, K. Lipopolysaccharides accelerate hepatic steatosis in the development of nonalcoholic fatty liver disease in Zucker rats. J. Clin. Biochem. Nutr. 2014, 54, 39–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceccarelli, S.; Panera, N.; Mina, M.; Gnani, D.; De Stefanis, C.; Crudele, A.; Rychlicki, C.; Petrini, S.; Bruscalupi, G.; Agostinelli, L.; et al. LPS-induced TNF-alpha factor mediates pro-inflammatory and pro-fibrogenic pattern in non-alcoholic fatty liver disease. Oncotarget 2015, 6, 41434–41452. [Google Scholar] [CrossRef] [PubMed]
- Poggi, M.; Bastelica, D.; Gual, P.; Iglesias, M.A.; Gremeaux, T.; Knauf, C.; Peiretti, F.; Verdier, M.; Juhan-Vague, I.; Tanti, J.F.; et al. C3H/HeJ mice carrying a toll-like receptor 4 mutation are protected against the development of insulin resistance in white adipose tissue in response to a high-fat diet. Diabetologia 2007, 50, 1267–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Y.; Freeswick, P.D.; Khemlani, L.S.; Kispert, P.H.; Wang, S.C.; Su, G.L.; Billiar, T.R. Role of lipopolysaccharide (LPS), interleukin-1, interleukin-6, tumor necrosis factor, and dexamethasone in regulation of LPS-binding protein expression in normal hepatocytes and hepatocytes from LPS-treated rats. Infect. Immun. 1995, 63, 2435–2442. [Google Scholar] [Green Version]
- Pang, J.; Xu, W.; Zhang, X.; Wong, G.L.; Chan, A.W.; Chan, H.Y.; Tse, C.H.; Shu, S.S.; Choi, P.C.; Chan, H.L.; et al. Significant positive association of endotoxemia with histological severity in 237 patients with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2017, 46, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.; Wong, G.L.; Chan, H.Y.; Yeung, D.K.; Chan, R.S.; Chim, A.M.; Chan, C.K.; Tse, Y.K.; Woo, J.; Chu, W.C.; et al. Bacterial endotoxin and non-alcoholic fatty liver disease in the general population: A prospective cohort study. Aliment. Pharmacol. Ther. 2015, 42, 731–740. [Google Scholar] [CrossRef]
- Jin, C.J.; Engstler, A.J.; Ziegenhardt, D.; Bischoff, S.C.; Trautwein, C.; Bergheim, I. Loss of lipopolysaccharide-binding protein attenuates the development of diet-induced non-alcoholic fatty liver disease in mice. J. Gastroenterol. Hepatol. 2017, 32, 708–715. [Google Scholar] [CrossRef]
- Levels, J.H.; Marquart, J.A.; Abraham, P.R.; van den Ende, A.E.; Molhuizen, H.O.; van Deventer, S.J.; Meijers, J.C. Lipopolysaccharide is transferred from high-density to low-density lipoproteins by lipopolysaccharide-binding protein and phospholipid transfer protein. Infect. Immun. 2005, 73, 2321–2326. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Takeuchi, S.; Kubota, K.; Kobayashi, Y.; Kozakai, S.; Ukai, I.; Shichiku, A.; Okubo, M.; Numasaki, M.; Kanemitsu, Y.; et al. Lipopolysaccharide (LPS)-binding protein stimulates CD14-dependent Toll-like receptor 4 internalization and LPS-induced TBK1-IKK-IRF3 axis activation. J. Biol. Chem. 2018, 293, 10186–10201. [Google Scholar] [CrossRef]
- Hamann, L.; Alexander, C.; Stamme, C.; Zahringer, U.; Schumann, R.R. Acute-phase concentrations of lipopolysaccharide (LPS)-binding protein inhibit innate immune cell activation by different LPS chemotypes via different mechanisms. Infect. Immun. 2005, 73, 193–200. [Google Scholar] [CrossRef]
- Chenevier-Gobeaux, C.; Borderie, D.; Weiss, N.; Mallet-Coste, T.; Claessens, Y.E. Presepsin (sCD14-ST), an innate immune response marker in sepsis. Clin. Chim. Acta 2015, 450, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Roncon-Albuquerque, R., Jr.; Moreira-Rodrigues, M.; Faria, B.; Ferreira, A.P.; Cerqueira, C.; Lourenco, A.P.; Pestana, M.; von Hafe, P.; Leite-Moreira, A.F. Attenuation of the cardiovascular and metabolic complications of obesity in CD14 knockout mice. Life Sci. 2008, 83, 502–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, Y.; Imajo, K.; Yoneda, M.; Kessoku, T.; Tomeno, W.; Shinohara, Y.; Kato, S.; Mawatari, H.; Nozaki, Y.; Fujita, K.; et al. Soluble CD14 levels reflect liver inflammation in patients with nonalcoholic steatohepatitis. PLoS ONE 2013, 8, e65211. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Al-Sadi, R.; Said, H.M.; Ma, T.Y. Lipopolysaccharide causes an increase in intestinal tight junction permeability in vitro and in vivo by inducing enterocyte membrane expression and localization of TLR-4 and CD14. Am. J. Pathol. 2013, 182, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Nighot, M.; Al-Sadi, R.; Guo, S.; Rawat, M.; Nighot, P.; Watterson, M.D.; Ma, T.Y. Lipopolysaccharide-Induced Increase in Intestinal Epithelial Tight Permeability Is Mediated by Toll-Like Receptor 4/Myeloid Differentiation Primary Response 88 (MyD88) Activation of Myosin Light Chain Kinase Expression. Am. J. Pathol. 2017, 187, 2698–2710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S.; Nighot, M.; Al-Sadi, R.; Alhmoud, T.; Nighot, P.; Ma, T.Y. Lipopolysaccharide Regulation of Intestinal Tight Junction Permeability Is Mediated by TLR4 Signal Transduction Pathway Activation of FAK and MyD88. J. Immunol. 2015, 195, 4999–5010. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Xu, C.; Yu, C.; Li, Y. Role of NLRP3 Inflammasome in the Progression of NAFLD to NASH. Can. J. Gastroenterol. Hepatol. 2016, 2016, 6489012. [Google Scholar] [CrossRef] [PubMed]
- Humann, J.; Lenz, L.L. Bacterial peptidoglycan degrading enzymes and their impact on host muropeptide detection. J. Innate. Immun. 2009, 1, 88–97. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.; Inohara, N.; Nunez, G. Peptidoglycan signaling in innate immunity and inflammatory disease. J. Biol. Chem. 2005, 280, 20177–20180. [Google Scholar] [CrossRef]
- Miura, K.; Ohnishi, H. Role of gut microbiota and Toll-like receptors in nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 7381–7391. [Google Scholar] [CrossRef]
- Cengiz, M.; Ozenirler, S.; Elbeg, S. Role of serum toll-like receptors 2 and 4 in non-alcoholic steatohepatitis and liver fibrosis. J. Gastroenterol. Hepatol. 2015, 30, 1190–1196. [Google Scholar] [CrossRef] [PubMed]
- Ehses, J.A.; Meier, D.T.; Wueest, S.; Rytka, J.; Boller, S.; Wielinga, P.Y.; Schraenen, A.; Lemaire, K.; Debray, S.; Van Lommel, L.; et al. Toll-like receptor 2-deficient mice are protected from insulin resistance and beta cell dysfunction induced by a high-fat diet. Diabetologia 2010, 53, 1795–1806. [Google Scholar] [CrossRef] [PubMed]
- Himes, R.W.; Smith, C.W. Tlr2 is critical for diet-induced metabolic syndrome in a murine model. FASEB J. 2010, 24, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Wan, Z.; Durrer, C.; Mah, D.; Simtchouk, S.; Little, J.P. One-week high-fat diet leads to reduced toll-like receptor 2 expression and function in young healthy men. Nutr. Res. 2014, 34, 1045–1051. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, A.; Karasudani, Y.; Otsuka, Y.; Hasegawa, M.; Inohara, N.; Fujimoto, Y.; Fukase, K. Synthesis of diaminopimelic acid containing peptidoglycan fragments and tracheal cytotoxin (TCT) and investigation of their biological functions. Chemistry 2008, 14, 10318–10330. [Google Scholar] [CrossRef] [PubMed]
- Girardin, S.E.; Boneca, I.G.; Viala, J.; Chamaillard, M.; Labigne, A.; Thomas, G.; Philpott, D.J.; Sansonetti, P.J. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J. Biol. Chem. 2003, 278, 8869–8872. [Google Scholar] [CrossRef] [PubMed]
- Martinic, M.M.; Caminschi, I.; O’Keeffe, M.; Thinnes, T.C.; Grumont, R.; Gerondakis, S.; McKay, D.B.; Nemazee, D.; Gavin, A.L. The Bacterial Peptidoglycan-Sensing Molecules NOD1 and NOD2 Promote CD8(+) Thymocyte Selection. J. Immunol. 2017, 198, 2649–2660. [Google Scholar] [CrossRef]
- Meli, R.; Mattace Raso, G.; Calignano, A. Role of innate immune response in non-alcoholic Fatty liver disease: Metabolic complications and therapeutic tools. Front. Immunol. 2014, 5, 177. [Google Scholar] [CrossRef] [PubMed]
- Schertzer, J.D.; Tamrakar, A.K.; Magalhaes, J.G.; Pereira, S.; Bilan, P.J.; Fullerton, M.D.; Liu, Z.; Steinberg, G.R.; Giacca, A.; Philpott, D.J.; et al. NOD1 activators link innate immunity to insulin resistance. Diabetes 2011, 60, 2206–2215. [Google Scholar] [CrossRef]
- Nonogaki, K.; Moser, A.H.; Pan, X.M.; Staprans, I.; Grunfeld, C.; Feingold, K.R. Lipoteichoic acid stimulates lipolysis and hepatic triglyceride secretion in rats in vivo. J. Lipid Res. 1995, 36, 1987–1995. [Google Scholar]
- Osawa, Y.; Iho, S.; Takauji, R.; Takatsuka, H.; Yamamoto, S.; Takahashi, T.; Horiguchi, S.; Urasaki, Y.; Matsuki, T.; Fujieda, S. Collaborative action of NF-kappaB and p38 MAPK is involved in CpG DNA-induced IFN-alpha and chemokine production in human plasmacytoid dendritic cells. J. Immunol. 2006, 177, 4841–4852. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.T.; Campos, P.C.; Pereira Gde, S.; Bartholomeu, D.C.; Splitter, G.; Oliveira, S.C. TLR9 is required for MAPK/NF-kappaB activation but does not cooperate with TLR2 or TLR6 to induce host resistance to Brucella abortus. J. Leukoc. Biol. 2016, 99, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Minton, K. LC3 anchors TLR9 signalling. Nat. Rev. Immunol. 2018, 18, 418–419. [Google Scholar] [CrossRef]
- Hayashi, K.; Taura, M.; Iwasaki, A. The interaction between IKKalpha and LC3 promotes type I interferon production through the TLR9-containing LAPosome. Sci. Signal. 2018, 11. [Google Scholar] [CrossRef]
- Bauer, S.; Kirschning, C.J.; Hacker, H.; Redecke, V.; Hausmann, S.; Akira, S.; Wagner, H.; Lipford, G.B. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc. Natl. Acad. Sci. USA 2001, 98, 9237–9242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mridha, A.R.; Haczeyni, F.; Yeh, M.M.; Haigh, W.G.; Ioannou, G.N.; Barn, V.; Ajamieh, H.; Adams, L.; Hamdorf, J.M.; Teoh, N.C.; et al. TLR9 is up-regulated in human and murine NASH: Pivotal role in inflammatory recruitment and cell survival. Clin. Sci. 2017, 131, 2145–2159. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Martinez, I.; Santoro, N.; Chen, Y.; Hoque, R.; Ouyang, X.; Caprio, S.; Shlomchik, M.J.; Coffman, R.L.; Candia, A.; Mehal, W.Z. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J. Clin. Investig. 2016, 126, 859–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, K.; Kodama, Y.; Inokuchi, S.; Schnabl, B.; Aoyama, T.; Ohnishi, H.; Olefsky, J.M.; Brenner, D.A.; Seki, E. Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice. Gastroenterology 2010, 139, 323–334.e7. [Google Scholar] [CrossRef]
- Yanez-Mo, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borras, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [Green Version]
- Anand, D.; Chaudhuri, A. Bacterial outer membrane vesicles: New insights and applications. Mol. Membr. Biol. 2016, 33, 125–137. [Google Scholar] [CrossRef] [Green Version]
- Tulkens, J.; Vergauwen, G.; Van Deun, J.; Geeurickx, E.; Dhondt, B.; Lippens, L.; De Scheerder, M.A.; Miinalainen, I.; Rappu, P.; De Geest, B.G.; et al. Increased levels of systemic LPS-positive bacterial extracellular vesicles in patients with intestinal barrier dysfunction. Gut 2018. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Meng, R.; Tang, Y.; Zhao, K.; Liang, F.; Zhang, R.; Xue, Q.; Chen, F.; Xiao, X.; Wang, H.; et al. Toll-Like Receptor 4 Signaling Licenses the Cytosolic Transport of Lipopolysaccharide From Bacterial Outer Membrane Vesicles. Shock 2019, 51, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Bielig, H.; Rompikuntal, P.K.; Dongre, M.; Zurek, B.; Lindmark, B.; Ramstedt, M.; Wai, S.N.; Kufer, T.A. NOD-like receptor activation by outer membrane vesicles from Vibrio cholerae non-O1 non-O139 strains is modulated by the quorum-sensing regulator HapR. Infect. Immun. 2011, 79, 1418–1427. [Google Scholar] [CrossRef] [PubMed]
- Prados-Rosales, R.; Baena, A.; Martinez, L.R.; Luque-Garcia, J.; Kalscheuer, R.; Veeraraghavan, U.; Camara, C.; Nosanchuk, J.D.; Besra, G.S.; Chen, B.; et al. Mycobacteria release active membrane vesicles that modulate immune responses in a TLR2-dependent manner in mice. J. Clin. Investig. 2011, 121, 1471–1483. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Kim, S.I.; Ryu, S.; Yoon, H. Identification and characterization of outer membrane vesicle-associated proteins in Salmonella enterica serovar Typhimurium. Infect. Immun. 2014, 82, 4001–4010. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.S.; Kim, D.K.; Choi, S.J.; Lee, J.; Choi, J.P.; Rho, S.; Park, S.H.; Kim, Y.K.; Hwang, D.; Gho, Y.S. Proteomic analysis of outer membrane vesicles derived from Pseudomonas aeruginosa. Proteomics 2011, 11, 3424–3429. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, K.; Hampton, T.H.; Jarek, M.; Scharfe, M.; Gerber, S.A.; Mielcarz, D.W.; Demers, E.G.; Dolben, E.L.; Hammond, J.H.; Hogan, D.A.; et al. A Novel Mechanism of Host-Pathogen Interaction through sRNA in Bacterial Outer Membrane Vesicles. PLoS Pathog. 2016, 12, e1005672. [Google Scholar] [CrossRef]
- Den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborti, C.K. New-found link between microbiota and obesity. World J. Gastrointest. Pathophysiol. 2015, 6, 110–119. [Google Scholar] [CrossRef]
- Kimura, I.; Inoue, D.; Hirano, K.; Tsujimoto, G. The SCFA Receptor GPR43 and Energy Metabolism. Front. Endocrinol. 2014, 5, 85. [Google Scholar] [CrossRef] [Green Version]
- Inoue, D.; Tsujimoto, G.; Kimura, I. Regulation of Energy Homeostasis by GPR41. Front. Endocrinol. 2014, 5, 81. [Google Scholar] [CrossRef] [PubMed]
- Ang, Z.; Ding, J.L. GPR41 and GPR43 in Obesity and Inflammation-Protective or Causative? Front. Immunol. 2016, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Cornall, L.M.; Mathai, M.L.; Hryciw, D.H.; McAinch, A.J. Diet-induced obesity up-regulates the abundance of GPR43 and GPR120 in a tissue specific manner. Cell Physiol. Biochem. 2011, 28, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, N.; Pluznick, J.L. Olfaction in the kidney: “Smelling” gut microbial metabolites. Exp. Physiol. 2016, 101, 478–481. [Google Scholar] [CrossRef] [PubMed]
- Fleischer, J.; Bumbalo, R.; Bautze, V.; Strotmann, J.; Breer, H. Expression of odorant receptor Olfr78 in enteroendocrine cells of the colon. Cell Tissue Res. 2015, 361, 697–710. [Google Scholar] [CrossRef] [PubMed]
- Bellahcene, M.; O’Dowd, J.F.; Wargent, E.T.; Zaibi, M.S.; Hislop, D.C.; Ngala, R.A.; Smith, D.M.; Cawthorne, M.A.; Stocker, C.J.; Arch, J.R. Male mice that lack the G-protein-coupled receptor GPR41 have low energy expenditure and increased body fat content. Br. J. Nutr. 2013, 109, 1755–1764. [Google Scholar] [CrossRef] [PubMed]
- Bjursell, M.; Admyre, T.; Goransson, M.; Marley, A.E.; Smith, D.M.; Oscarsson, J.; Bohlooly, Y.M. Improved glucose control and reduced body fat mass in free fatty acid receptor 2-deficient mice fed a high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E211–E220. [Google Scholar] [CrossRef] [Green Version]
- Kimura, I.; Ozawa, K.; Inoue, D.; Imamura, T.; Kimura, K.; Maeda, T.; Terasawa, K.; Kashihara, D.; Hirano, K.; Tani, T.; et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat. Commun. 2013, 4, 1829. [Google Scholar] [CrossRef]
- Rau, M.; Rehman, A.; Dittrich, M.; Groen, A.K.; Hermanns, H.M.; Seyfried, F.; Beyersdorf, N.; Dandekar, T.; Rosenstiel, P.; Geier, A. Fecal SCFAs and SCFA-producing bacteria in gut microbiome of human NAFLD as a putative link to systemic T-cell activation and advanced disease. United Eur. Gastroenterol. J 2018, 6, 1496–1507. [Google Scholar] [CrossRef]
- Nishina, P.M.; Freedland, R.A. Effects of propionate on lipid biosynthesis in isolated rat hepatocytes. J. Nutr. 1990, 120, 668–673. [Google Scholar] [CrossRef]
- Tedelind, S.; Westberg, F.; Kjerrulf, M.; Vidal, A. Anti-inflammatory properties of the short-chain fatty acids acetate and propionate: A study with relevance to inflammatory bowel disease. World J. Gastroenterol. 2007, 13, 2826–2832. [Google Scholar] [CrossRef] [PubMed]
- Sahuri-Arisoylu, M.; Brody, L.P.; Parkinson, J.R.; Parkes, H.; Navaratnam, N.; Miller, A.D.; Thomas, E.L.; Frost, G.; Bell, J.D. Reprogramming of hepatic fat accumulation and ‘browning’ of adipose tissue by the short-chain fatty acid acetate. Int. J. Obes. 2016, 40, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Pan, Q.; Xin, F.Z.; Zhang, R.N.; He, C.X.; Chen, G.Y.; Liu, C.; Chen, Y.W.; Fan, J.G. Sodium butyrate attenuates high-fat diet-induced steatohepatitis in mice by improving gut microbiota and gastrointestinal barrier. World J. Gastroenterol. 2017, 23, 60–75. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Chen, Y.W.; Zhao, Z.H.; Yang, R.X.; Xin, F.Z.; Liu, X.L.; Pan, Q.; Zhou, H.; Fan, J.G. Sodium butyrate reduces high-fat diet-induced non-alcoholic steatohepatitis through upregulation of hepatic GLP-1R expression. Exp. Mol. Med. 2018, 50, 157. [Google Scholar] [CrossRef]
- Jin, C.J.; Sellmann, C.; Engstler, A.J.; Ziegenhardt, D.; Bergheim, I. Supplementation of sodium butyrate protects mice from the development of non-alcoholic steatohepatitis (NASH). Br. J. Nutr. 2015, 114, 1745–1755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Lv, L.; Wu, W.; Li, Y.; Shi, D.; Fang, D.; Guo, F.; Jiang, H.; Yan, R.; Ye, W.; et al. Butyrate Protects Mice Against Methionine-Choline-Deficient Diet-Induced Non-alcoholic Steatohepatitis by Improving Gut Barrier Function, Attenuating Inflammation and Reducing Endotoxin Levels. Front Microbiol. 2018, 9, 1967. [Google Scholar] [CrossRef]
- Verdone, L.; Caserta, M.; Di Mauro, E. Role of histone acetylation in the control of gene expression. Biochem. Cell Biol. 2005, 83, 344–353. [Google Scholar] [CrossRef]
- Sasaki-Imamura, T.; Yoshida, Y.; Suwabe, K.; Yoshimura, F.; Kato, H. Molecular basis of indole production catalyzed by tryptophanase in the genus Prevotella. FEMS Microbiol. Lett. 2011, 322, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Devlin, A.S.; Marcobal, A.; Dodd, D.; Nayfach, S.; Plummer, N.; Meyer, T.; Pollard, K.S.; Sonnenburg, J.L.; Fischbach, M.A. Modulation of a Circulating Uremic Solute via Rational Genetic Manipulation of the Gut Microbiota. Cell Host Microbe 2016, 20, 709–715. [Google Scholar] [CrossRef] [Green Version]
- Sasaki-Imamura, T.; Yano, A.; Yoshida, Y. Production of indole from L-tryptophan and effects of these compounds on biofilm formation by Fusobacterium nucleatum ATCC 25586. Appl. Environ. Microbiol. 2010, 76, 4260–4268. [Google Scholar] [CrossRef]
- Chu, W.; Zere, T.R.; Weber, M.M.; Wood, T.K.; Whiteley, M.; Hidalgo-Romano, B.; Valenzuela, E., Jr.; McLean, R.J. Indole production promotes Escherichia coli mixed-culture growth with Pseudomonas aeruginosa by inhibiting quorum signaling. Appl. Environ. Microbiol. 2012, 78, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Bansal, T.; Alaniz, R.C.; Wood, T.K.; Jayaraman, A. The bacterial signal indole increases epithelial-cell tight-junction resistance and attenuates indicators of inflammation. Proc. Natl. Acad. Sci. USA 2010, 107, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Whitfield-Cargile, C.M.; Cohen, N.D.; Chapkin, R.S.; Weeks, B.R.; Davidson, L.A.; Goldsby, J.S.; Hunt, C.L.; Steinmeyer, S.H.; Menon, R.; Suchodolski, J.S.; et al. The microbiota-derived metabolite indole decreases mucosal inflammation and injury in a murine model of NSAID enteropathy. Gut Microbes 2016, 7, 246–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chimerel, C.; Emery, E.; Summers, D.K.; Keyser, U.; Gribble, F.M.; Reimann, F. Bacterial metabolite indole modulates incretin secretion from intestinal enteroendocrine L cells. Cell Rep. 2014, 9, 1202–1208. [Google Scholar] [CrossRef] [PubMed]
- Beaumont, M.; Neyrinck, A.M.; Olivares, M.; Rodriguez, J.; de Rocca Serra, A.; Roumain, M.; Bindels, L.B.; Cani, P.D.; Evenepoel, P.; Muccioli, G.G.; et al. The gut microbiota metabolite indole alleviates liver inflammation in mice. FASEB J. 2018. [Google Scholar] [CrossRef] [PubMed]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Alexeev, E.E.; Lanis, J.M.; Kao, D.J.; Campbell, E.L.; Kelly, C.J.; Battista, K.D.; Gerich, M.E.; Jenkins, B.R.; Walk, S.T.; Kominsky, D.J.; et al. Microbiota-Derived Indole Metabolites Promote Human and Murine Intestinal Homeostasis through Regulation of Interleukin-10 Receptor. Am. J. Pathol. 2018, 188, 1183–1194. [Google Scholar] [CrossRef] [Green Version]
- Natividad, J.M.; Agus, A.; Planchais, J.; Lamas, B.; Jarry, A.C.; Martin, R.; Michel, M.L.; Chong-Nguyen, C.; Roussel, R.; Straube, M.; et al. Impaired Aryl Hydrocarbon Receptor Ligand Production by the Gut Microbiota Is a Key Factor in Metabolic Syndrome. Cell Metab. 2018, 28, 737–749. [Google Scholar] [CrossRef]
- Krishnan, S.; Ding, Y.; Saedi, N.; Choi, M.; Sridharan, G.V.; Sherr, D.H.; Yarmush, M.L.; Alaniz, R.C.; Jayaraman, A.; Lee, K. Gut Microbiota-Derived Tryptophan Metabolites Modulate Inflammatory Response in Hepatocytes and Macrophages. Cell Rep. 2018, 23, 1099–1111. [Google Scholar] [CrossRef]
- Venkatesh, M.; Mukherjee, S.; Wang, H.; Li, H.; Sun, K.; Benechet, A.P.; Qiu, Z.; Maher, L.; Redinbo, M.R.; Phillips, R.S.; et al. Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity 2014, 41, 296–310. [Google Scholar] [CrossRef]
- Poeggeler, B.; Pappolla, M.A.; Hardeland, R.; Rassoulpour, A.; Hodgkins, P.S.; Guidetti, P.; Schwarcz, R. Indole-3-propionate: A potent hydroxyl radical scavenger in rat brain. Brain Res. 1999, 815, 382–388. [Google Scholar] [CrossRef]
- Hwang, I.K.; Yoo, K.Y.; Li, H.; Park, O.K.; Lee, C.H.; Choi, J.H.; Jeong, Y.G.; Lee, Y.L.; Kim, Y.M.; Kwon, Y.G.; et al. Indole-3-propionic acid attenuates neuronal damage and oxidative stress in the ischemic hippocampus. J. Neurosci. Res. 2009, 87, 2126–2137. [Google Scholar] [CrossRef] [PubMed]
- Karbownik, M.; Reiter, R.J.; Garcia, J.J.; Cabrera, J.; Burkhardt, S.; Osuna, C.; Lewinski, A. Indole-3-propionic acid, a melatonin-related molecule, protects hepatic microsomal membranes from iron-induced oxidative damage: Relevance to cancer reduction. J. Cell Biochem. 2001, 81, 507–513. [Google Scholar] [CrossRef]
- Abildgaard, A.; Elfving, B.; Hokland, M.; Wegener, G.; Lund, S. The microbial metabolite indole-3-propionic acid improves glucose metabolism in rats, but does not affect behaviour. Arch. Physiol. Biochem. 2018, 124, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Taoka, H.; Yokoyama, Y.; Morimoto, K.; Kitamura, N.; Tanigaki, T.; Takashina, Y.; Tsubota, K.; Watanabe, M. Role of bile acids in the regulation of the metabolic pathways. World J. Diabetes 2016, 7, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Chow, M.D.; Lee, Y.H.; Guo, G.L. The role of bile acids in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Mol. Aspects Med. 2017, 56, 34–44. [Google Scholar] [CrossRef] [PubMed]
- McMillin, M.; DeMorrow, S. Effects of bile acids on neurological function and disease. FASEB J. 2016, 30, 3658–3668. [Google Scholar] [CrossRef] [Green Version]
- Keren, N.; Konikoff, F.M.; Paitan, Y.; Gabay, G.; Reshef, L.; Naftali, T.; Gophna, U. Interactions between the intestinal microbiota and bile acids in gallstones patients. Environ. Microbiol. Rep. 2015, 7, 874–880. [Google Scholar] [CrossRef]
- Yang, H.; Duan, Z. Bile Acids and the Potential Role in Primary Biliary Cirrhosis. Digestion 2016, 94, 145–153. [Google Scholar] [CrossRef]
- Muili, K.A.; Wang, D.; Orabi, A.I.; Sarwar, S.; Luo, Y.; Javed, T.A.; Eisses, J.F.; Mahmood, S.M.; Jin, S.; Singh, V.P.; et al. Bile acids induce pancreatic acinar cell injury and pancreatitis by activating calcineurin. J. Biol. Chem. 2013, 288, 570–580. [Google Scholar] [CrossRef]
- Tiratterra, E.; Franco, P.; Porru, E.; Katsanos, K.H.; Christodoulou, D.K.; Roda, G. Role of bile acids in inflammatory bowel disease. Ann. Gastroenterol. 2018, 31, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Wahlstrom, A.; Sayin, S.I.; Marschall, H.U.; Backhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tremblay, S.; Romain, G.; Roux, M.; Chen, X.L.; Brown, K.; Gibson, D.L.; Ramanathan, S.; Menendez, A. Bile Acid Administration Elicits an Intestinal Antimicrobial Program and Reduces the Bacterial Burden in Two Mouse Models of Enteric Infection. Infect. Immun. 2017, 85, e00942-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, A.F.; Eckmann, L. How bile acids confer gut mucosal protection against bacteria. Proc. Natl. Acad. Sci. USA 2006, 103, 4333–4334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Xie, C.; Nichols, R.G.; Chan, S.H.; Jiang, C.; Hao, R.; Smith, P.B.; Cai, J.; Simons, M.N.; Hatzakis, E.; et al. Farnesoid X Receptor Signaling Shapes the Gut Microbiota and Controls Hepatic Lipid Metabolism. MSystems 2016, 1, e00070-16. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.X.; Shen, W.; Sun, H. Effects of nuclear receptor FXR on the regulation of liver lipid metabolism in patients with non-alcoholic fatty liver disease. Hepatol. Int. 2010, 4, 741–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, K.; Saha, P.K.; Chan, L.; Moore, D.D. Farnesoid X receptor is essential for normal glucose homeostasis. J. Clin. Investig. 2006, 116, 1102–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cariou, B.; Duran-Sandoval, D.; Kuipers, F.; Staels, B. Farnesoid X receptor: A new player in glucose metabolism? Endocrinology 2005, 146, 981–983. [Google Scholar] [CrossRef] [PubMed]
- Lou, G.; Ma, X.; Fu, X.; Meng, Z.; Zhang, W.; Wang, Y.D.; Van Ness, C.; Yu, D.; Xu, R.; Huang, W. GPBAR1/TGR5 mediates bile acid-induced cytokine expression in murine Kupffer cells. PLoS ONE 2014, 9, e93567. [Google Scholar] [CrossRef]
- Thomas, C.; Gioiello, A.; Noriega, L.; Strehle, A.; Oury, J.; Rizzo, G.; Macchiarulo, A.; Yamamoto, H.; Mataki, C.; Pruzanski, M.; et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009, 10, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Houten, S.M.; Mataki, C.; Christoffolete, M.A.; Kim, B.W.; Sato, H.; Messaddeq, N.; Harney, J.W.; Ezaki, O.; Kodama, T.; et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 2006, 439, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Fennema, D.; Phillips, I.R.; Shephard, E.A. Trimethylamine and Trimethylamine N-Oxide, a Flavin-Containing Monooxygenase 3 (FMO3)-Mediated Host-Microbiome Metabolic Axis Implicated in Health and Disease. Drug Metab. Dispos. 2016, 44, 1839–1850. [Google Scholar] [CrossRef] [PubMed]
- Barrea, L.; Annunziata, G.; Muscogiuri, G.; Di Somma, C.; Laudisio, D.; Maisto, M.; de Alteriis, G.; Tenore, G.C.; Colao, A.; Savastano, S. Trimethylamine-N-oxide (TMAO) as Novel Potential Biomarker of Early Predictors of Metabolic Syndrome. Nutrients 2018, 10, 1971. [Google Scholar] [CrossRef] [PubMed]
- Fon Tacer, K.; Rozman, D. Nonalcoholic Fatty liver disease: Focus on lipoprotein and lipid deregulation. J. Lipids 2011, 2011, 783976. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.-l.; Tan, X.-Y.; Liu, Y.; Chen, X.-L. Increased circulating trimethylamine N-oxide, a gut-flora-dependent metabolite of choline and betaine in nonalcoholic fatty liver disease is associated with high serum bile acids. FASEB J. 2016, 30, 272-2. [Google Scholar]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Warrier, M.; Shih, D.M.; Burrows, A.C.; Ferguson, D.; Gromovsky, A.D.; Brown, A.L.; Marshall, S.; McDaniel, A.; Schugar, R.C.; Wang, Z.; et al. The TMAO-Generating Enzyme Flavin Monooxygenase 3 Is a Central Regulator of Cholesterol Balance. Cell Rep. 2015, 10, 326–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, D.M.; Wang, Z.; Lee, R.; Meng, Y.; Che, N.; Charugundla, S.; Qi, H.; Wu, J.; Pan, C.; Brown, J.M.; et al. Flavin containing monooxygenase 3 exerts broad effects on glucose and lipid metabolism and atherosclerosis. J. Lipid Res. 2015, 56, 22–37. [Google Scholar] [CrossRef] [Green Version]
- Oellgaard, J.; Winther, S.A.; Hansen, T.S.; Rossing, P.; von Scholten, B.J. Trimethylamine N-oxide (TMAO) as a New Potential Therapeutic Target for Insulin Resistance and Cancer. Curr. Pharm. Des. 2017, 23, 3699–3712. [Google Scholar] [CrossRef] [PubMed]
- Christensen, K.; Lawler, T.; Mares, J. Dietary Carotenoids and Non-Alcoholic Fatty Liver Disease among US Adults, NHANES 2003(-)2014. Nutrients 2019, 11, 1101. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, B.; Sahin, K.; Bilen, H.; Bahcecioglu, I.H.; Bilir, B.; Ashraf, S.; Halazun, K.J.; Kucuk, O. Carotenoids and non-alcoholic fatty liver disease. Hepatobiliary Surg. Nutr. 2015, 4, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Nagashimada, M.; Zhuge, F.; Zhan, L.; Nagata, N.; Tsutsui, A.; Nakanuma, Y.; Kaneko, S.; Ota, T. Astaxanthin prevents and reverses diet-induced insulin resistance and steatohepatitis in mice: A comparison with vitamin E. Sci. Rep. 2015, 5, 17192. [Google Scholar] [CrossRef] [PubMed]
- Eo, H.; Jeon, Y.J.; Lee, M.; Lim, Y. Brown Alga Ecklonia cava polyphenol extract ameliorates hepatic lipogenesis, oxidative stress, and inflammation by activation of AMPK and SIRT1 in high-fat diet-induced obese mice. J. Agric. Food Chem. 2015, 63, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Anhe, F.F.; Nachbar, R.T.; Varin, T.V.; Vilela, V.; Dudonne, S.; Pilon, G.; Fournier, M.; Lecours, M.A.; Desjardins, Y.; Roy, D.; et al. A polyphenol-rich cranberry extract reverses insulin resistance and hepatic steatosis independently of body weight loss. Mol. Metab. 2017, 6, 1563–1573. [Google Scholar] [CrossRef] [PubMed]
- Abenavoli, L.; Milic, N.; Luzza, F.; Boccuto, L.; De Lorenzo, A. Polyphenols Treatment in Patients with Nonalcoholic Fatty Liver Disease. J. Transl. Int. Med. 2017, 5, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Mezzomo, N.; Ferreira, S.R.S. Carotenoids Functionality, Sources, and Processing by Supercritical Technology: A Review. J. Chem.-Ny 2016. [Google Scholar] [CrossRef]
- Pandey, K.B.; Rizvi, S.I. Plant polyphenols as dietary antioxidants in human health and disease. Oxid. Med. Cell. Longev. 2009, 2, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Palafox-Carlos, H.; Ayala-Zavala, J.F.; Gonzalez-Aguilar, G.A. The role of dietary fiber in the bioaccessibility and bioavailability of fruit and vegetable antioxidants. J. Food Sci. 2011, 76, R6–R15. [Google Scholar] [CrossRef] [PubMed]
- Djuric, Z.; Bassis, C.; Ren, J.W.; Chan, R.; Kato, I.; Ruffin, M.T.; Sen, A.; Turgeon, D.K. Predictors of Carotenoid Concentrations in Human Serum: Role of the Intestinal Microbiome. Faseb J. 2017, 31, 168-7. [Google Scholar]
- Klassen, J.L. Phylogenetic and evolutionary patterns in microbial carotenoid biosynthesis are revealed by comparative genomics. PLoS ONE 2010, 5, e11257. [Google Scholar] [CrossRef]
- Nordlund, E.; Aura, A.M.; Mattila, I.; Kosso, T.; Rouau, X.; Poutanen, K. Formation of Phenolic Microbial Metabolites and Short-Chain Fatty Acids from Rye, Wheat, and Oat Bran and Their Fractions in the Metabolical in Vitro Colon Model. J. Agric. Food Chem. 2012, 60, 8134–8145. [Google Scholar] [CrossRef] [PubMed]
- Amakura, Y.; Tsutsumi, T.; Sasaki, K.; Nakamura, M.; Yoshida, T.; Maitani, T. Influence of food polyphenols on aryl hydrocarbon receptor-signaling pathway estimated by in vitro bioassay. Phytochemistry 2008, 69, 3117–3130. [Google Scholar] [CrossRef] [PubMed]
- Busbee, P.B.; Rouse, M.; Nagarkatti, M.; Nagarkatti, P.S. Use of natural AhR ligands as potential therapeutic modalities against inflammatory disorders. Nutr. Rev. 2013, 71, 353–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, I.A.; Perdew, G.H. Ligand activation of the Ah receptor contributes to gastrointestinal homeostasis. Curr. Opin. Toxicol. 2017, 2, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruhl, R. Induction of PXR-mediated metabolism by beta-carotene. Biochim. Biophys. Acta 2005, 1740, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, S.; Chen, M.; Wang, J.; Xie, B.; Sun, Z. (-)-Epigallocatechin-3-gallate (EGCG) inhibits starch digestion and improves glucose homeostasis through direct or indirect activation of PXR/CAR-mediated phase II metabolism in diabetic mice. Food Funct. 2018, 9, 4651–4663. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, Y.; Yin, Y.; Li, Z.; Zhang, W. Gut Microbiota-Derived Components and Metabolites in the Progression of Non-Alcoholic Fatty Liver Disease (NAFLD). Nutrients 2019, 11, 1712. https://doi.org/10.3390/nu11081712
Ji Y, Yin Y, Li Z, Zhang W. Gut Microbiota-Derived Components and Metabolites in the Progression of Non-Alcoholic Fatty Liver Disease (NAFLD). Nutrients. 2019; 11(8):1712. https://doi.org/10.3390/nu11081712
Chicago/Turabian StyleJi, Yun, Yue Yin, Ziru Li, and Weizhen Zhang. 2019. "Gut Microbiota-Derived Components and Metabolites in the Progression of Non-Alcoholic Fatty Liver Disease (NAFLD)" Nutrients 11, no. 8: 1712. https://doi.org/10.3390/nu11081712
APA StyleJi, Y., Yin, Y., Li, Z., & Zhang, W. (2019). Gut Microbiota-Derived Components and Metabolites in the Progression of Non-Alcoholic Fatty Liver Disease (NAFLD). Nutrients, 11(8), 1712. https://doi.org/10.3390/nu11081712