Supplementary Nitric Oxide Donors and Exercise as Potential Means to Improve Vascular Health in People with Type 1 Diabetes: Yes to NO?


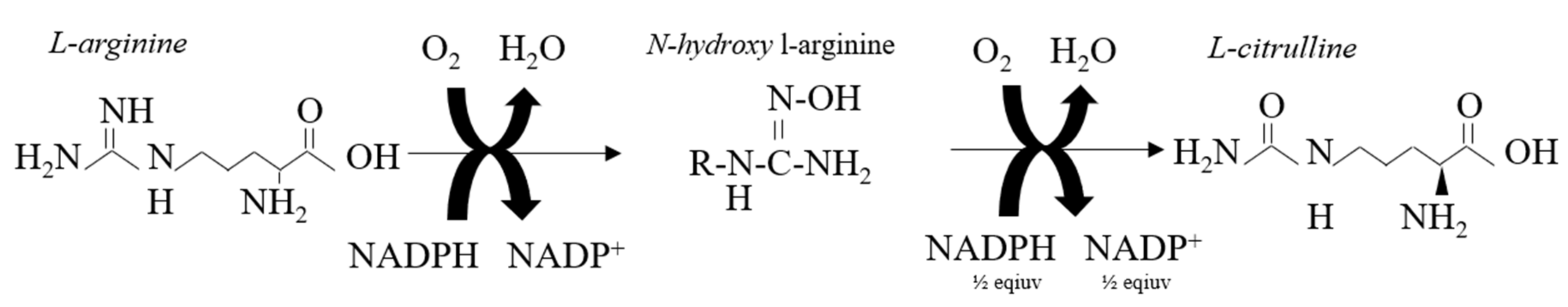{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Nitric Oxide Synthesis and Biochemical Formation in The Vascular Endothelium
3. Potential Pathogenic Mediators of Endothelial Damage in Type 1 Diabetes
3.1. Abnormal NO Production
3.2. Hyperglycaemia
4. The Energetic and Therapeutic Potential of Exercise-Induced Nitric Oxide Synthase
5. Potential Therapeutic Mediators of Endothelial Vitality in Type 1 Diabetes
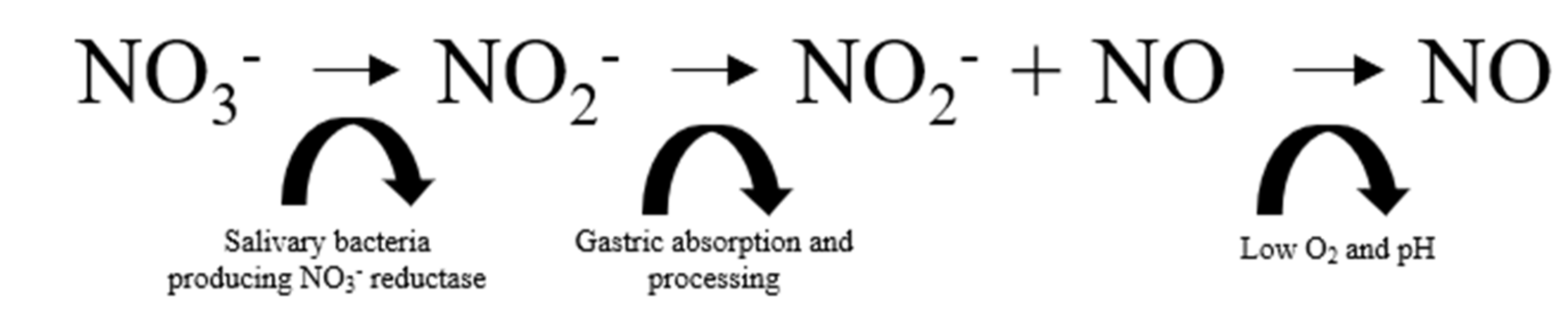
5.1. NO Donor 1: Dietary Nitrate
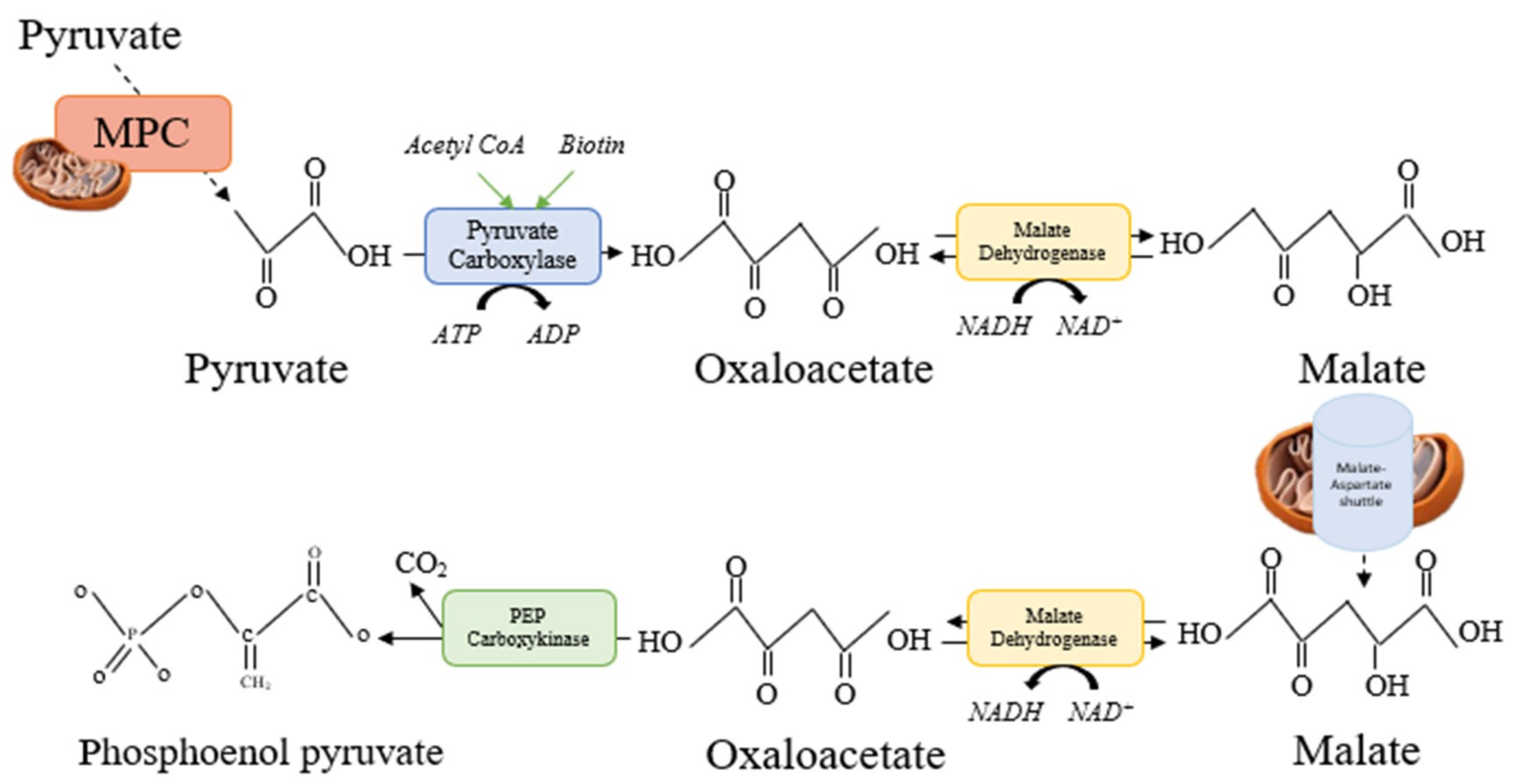
5.2. NO Donor 2: Citrulline Malate
6. Directions for Future Research
7. Conclusions
Funding
Conflicts of Interest
References
- Atkinson, M.A.; Eisenbarth, G.S. Type 1 diabetes: New perspectives on disease pathogenesis and treatment. Lancet (Lond. Engl.) 2001, 358, 221–229. [Google Scholar] [CrossRef]
- Soedamah-Muthu, S.S.; Fuller, J.H.; Mulnier, H.E.; Raleigh, V.S.; Lawrenson, R.A.; Colhoun, H.M. High risk of cardiovascular disease in patients with type 1 diabetes in the U.K.: A cohort study using the general practice research database. Diabetes Care 2006, 29, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Sousa, G.R.; Pober, D.; Galderisi, A.; Lv, H.; Yu, L.; Pereira, A.C.; Doria, A.; Kosiborod, M.; Lipes, M.A. Glycemic Control, Cardiac Autoimmunity, and Long-Term Risk of Cardiovascular Disease in Type 1 Diabetes Mellitus: A DCCT/EDIC Cohort-Based Study. Circulation 2018. [Google Scholar] [CrossRef] [PubMed]
- Gregg, E.W.; Li, Y.; Wang, J.; Rios Burrows, N.; Ali, M.K.; Rolka, D.; Geiss, L.; Williams, D.E. Changes in Diabetes-Related Complications in the United States, 1990–2010. N. Engl. J. Med. 2014, 370, 1514–1523. [Google Scholar] [CrossRef] [PubMed]
- Pambianco, G.; Costacou, T.; Ellis, D.; Becker, D.J.; Klein, R.; Orchard, T.J. The 30-year natural history of type 1 diabetes complications: The Pittsburgh Epidemiology of Diabetes Complications Study experience. Diabetes 2006, 55, 1463–1469. [Google Scholar] [CrossRef] [PubMed]
- Waden, J.; Forsblom, C.; Thorn, L.M.; Saraheimo, M.; Rosengård-Bärlund, M.; Heikkilä, O.; Hietala, K.; Ong, K.; Wareham, N.; Groop, P.H.; et al. Adult Stature and Diabetes Complications in Patients with Type 1 Diabetes: The FinnDiane Study and the Diabetes Control and Complications Trial. Diabetes 2009, 58, 1914–1920. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Hiuge, A.; Makino, H.; Nagumo, A.; Takaki, H.; Konishi, H.; Goto, Y.; Yoshimasa, Y.; Miyamoto, Y. Effect of exercise intervention on endothelial function and incidence of cardiovascular disease in patients with type 2 diabetes. J. Atheroscler. Thromb. 2010, 17, 828–833. [Google Scholar] [CrossRef]
- Rubanyi, G.M. The role of endothelium in cardiovascular homeostasis and diseases. J. Cardiovasc. Pharmacol. 1993, 22 (Suppl. 4), S1–S14. [Google Scholar] [CrossRef]
- Newman, P.; Berndt, M.; Gorski, J.; White, G.C.; Lyman, S.; Paddock, C.; Muller, W.A. PECAM-1 (CD31) cloning and relation to adhesion molecules of the immunoglobulin gene superfamily. Science 1990, 247, 1219–1222. [Google Scholar] [CrossRef]
- Sibal, L.; Aldibbiat, A.; Agarwal, S.C.; Mitchell, G.; Oates, C.; Razvi, S.; Weaver, J.U.; Shaw, J.A.; Home, P.D. Circulating endothelial progenitor cells, endothelial function, carotid intima–media thickness and circulating markers of endothelial dysfunction in people with type 1 diabetes without macrovascular disease or microalbuminuria. Diabetologia 2009, 52, 1464–1473. [Google Scholar] [CrossRef]
- Glowinska-Olszewska, B.; Moniuszko, M.; Hryniewicz, A.; Jeznach, M.; Rusak, M.; Dąbrowska, M.; Łuczyński, W.; Bodzenta-Łukaszyk, A.; Bossowski, A. Relationship between circulating endothelial progenitor cells and endothelial dysfunction in children with type 1 diabetes: A novel paradigm of early atherosclerosis in high-risk young patients. Eur. J. Endocrinol. 2013, 168, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, S.L.; Gong, J.H.; Chen, L.; Wu, I.H.; Sun, J.K.; Keenan, H.A.; King, G.L. Characterization of circulating and endothelial progenitor cells in patients with extreme-duration type 1 diabetes. Diabetes Care 2014, 37, 2193–2201. [Google Scholar] [CrossRef] [PubMed]
- Liebner, S.; Cavallaro, U.; Dejana, E. ATVB In Focus Novel Mediators and Mechanisms in Angiogenesis and Vasculogenesis The Multiple Languages of Endothelial Cell-to-Cell Communication. Arterioscler. Thromb. Vasc. Biol. 2006. [Google Scholar] [CrossRef] [PubMed]
- Creager, M.A.; Lüscher, T.F.; Cosentino, F.; Beckman, J.A. Diabetes and Vascular Disease. Circulation 2003. [Google Scholar] [CrossRef] [PubMed]
- Livingstone, S.J.; Looker, H.C.; Hothersall, E.J.; Wild, S.H.; Lindsay, R.S.; Chalmers, J.; Cleland, S.; Leese, G.P.; McKnight, J.; Morris, A.D.; et al. Risk of Cardiovascular Disease and Total Mortality in Adults with Type 1 Diabetes: Scottish Registry Linkage Study. PLoS Med. 2012, 9, e1001321. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, B.; Weber, M.M.; Kreisselmeier, H.P.; Kugler, M.; Ries, C.; Pfützner, A.; Kann, P.H.; Forst, T. Endothelial function and arterial stiffness in uncomplicated type 1 diabetes and healthy controls and the impact of insulin on these parameters during an euglycemic clamp. J. Diabetes Sci. Technol. 2007, 1, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Järvisalo, M.J.; Raitakari, M.; Toikka, J.O.; Putto-Laurila, A.; Rontu, R.; Laine, S.; Lehtimäki, T.; Rönnemaa, T.; Viikari, J.; Raitakari, O.T. Endothelial Dysfunction and Increased Arterial Intima-Media Thickness in Children with Type 1 Diabetes. Circulation 2004, 109, 1750–1755. [Google Scholar] [CrossRef]
- Stamler, J.S.; Meissner, G. Physiology of Nitric Oxide in Skeletal Muscle. Physiol. Rev. 2001, 81, 209–237. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, G.M.; Nishida, C.R.; Mooney, S.D.; Ortiz de Montellano, P.R. Nitric-oxide synthase (NOS) reductase domain models suggest a new control element in endothelial NOS that attenuates calmodulin-dependent activity. J. Biol. Chem. 2003, 278, 31814–31824. [Google Scholar] [CrossRef]
- Arnal, J.-F.; Dinh-Xuan, A.-T.; Pueyo, M.; Darblade, B.; Rami, J. Endothelium-Derived Nitric Oxide and Vascular Physiology and Pathology. Cell Mol. Life Sci. 1999, 55, 1078–1087. Available online: https://search.proquest.com/docview/884634397?accountid=14680&rfr_id=info%3Axri%2Fsid%3Aprimo (accessed on 24 April 2019). [CrossRef]
- Lundberg, J.O.; Gladwin, M.T.; Ahluwalia, A.; Benjamin, N.; Bryan, N.S.; Butler, A.; Cabrales, P.; Fago, A.; Feelisch, M.; Ford, P.C.; et al. Nitrate and nitrite in biology, nutrition and therapeutics. Nat. Chem. Biol. 2009, 5, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.M. Dietary nitrate supplementation and exercise performance. Sports Med. 2014, 44 (Suppl. 1), S35–S45. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.A.; Creager, M.A.; Libby, P. Diabetes and Atherosclerosis: Epidemiology, Pathophysiology, and Management. JAMA 2002, 287, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-H.; Lin, S.-J.; Lin, F.-Y.; Wu, T.C.; Tsao, C.R.; Huang, P.H.; Liu, P.L.; Chen, Y.L.; Chen, J.W. High Glucose Impairs Early and Late Endothelial Progenitor Cells by Modifying Nitric Oxide-Related but Not Oxidative Stress-Mediated Mechanisms. Diabetes 2007, 56, 1559–1568. [Google Scholar] [CrossRef] [PubMed]
- Pitocco, D.; Zaccardi, F.; Di Stasio, E.; Romitelli, F.; Martini, F.; Scaglione, G.L.; Speranza, D.; Santini, S.; Zuppi, C.; Ghirlanda, G. Role of asymmetric-dimethyl-l-arginine (ADMA) and nitrite/nitrate (NOx) in the pathogenesis of oxidative stress in female subjects with uncomplicated type 1 diabetes mellitus. Diabetes Res. Clin. Pract. 2009, 86, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Pitocco, D.; Di Stasio, E.; Romitelli, F.; Zaccardi, F.; Tavazzi, B.; Manto, A.; Caputo, S.; Musella, T.; Zuppi, C.; Santini, S.A.; et al. Hypouricemia linked to an overproduction of nitric oxide is an early marker of oxidative stress in female subjects with type 1 diabetes. Diabetes Metab. Res. Rev. 2008, 24, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Diabetes Control and Complications Trial (DCCT)/Epidemiology of Diabetes Interventions and Complications (EDIC) Study Research Group. Intensive Diabetes Treatment and Cardiovascular Outcomes in Type 1 Diabetes: The DCCT/EDIC Study 30-Year Follow-up. Diabetes Care 2016, 39, 686–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef]
- Lee, A.Y.W.; Chung, S.S.M. Contributions of polyol pathway to oxidative stress in diabetic cataract. FASEB J. 1999, 13, 23–30. [Google Scholar] [CrossRef] [Green Version]
- Musicki, B.; Kramer, M.F.; Becker, R.E.; Burnett, A.L. Inactivation of phosphorylated endothelial nitric oxide synthase (Ser-1177) by O-GlcNAc in diabetes-associated erectile dysfunction. Proc. Natl. Acad. Sci. USA 2005, 102, 11870–11875. [Google Scholar] [CrossRef]
- Seftel, A.D.; Vaziri, N.D.; Ni, Z.; Razmjouei, K.; Fogarty, J.; Hampel, N.; Polak, J.; Wang, R.Z.; Ferguson, K.; Block, C.; et al. Advanced glycation end products in human penis: Elevation in diabetic tissue, site of deposition, and possible effect through inos or enos. Urology 1997, 50, 1016–1026. [Google Scholar] [CrossRef]
- Wagenmakers, A.J.M. The Biochemical Basis of the Health Effects of Exercise; Portland Press: London, UK, 2006. [Google Scholar]
- Ganz, M.B.; Seftel, A. Glucose-induced changes in protein kinase C and nitric oxide are prevented by vitamin E. Am. J. Physiol. Metab. 2000, 278, E146–E152. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A.; Esposito, K.; Ihnat, M.; Thorpe, J.; Giugliano, D. Long-Term Glycemic Control Influences the Long-Lasting Effect of Hyperglycemia on Endothelial Function in Type 1 Diabetes. J. Clin. Endocrinol. Metab. 2009, 94, 2751–2756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, M.S.K.; Ng, W.K.; Chan, J.K.Y. Concise Review: Endothelial Progenitor Cells in Regenerative Medicine: Applications and Challenges. Stem Cells Transl. Med. 2016, 5, 530–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loomans, C.J.M.; de Koning, E.J.P.; Staal, F.J.T.; Rookmaaker, M.B.; Verseyden, C.; de Boer, H.C.; Verhaar, M.C.; Braam, B.; Rabelink, T.J.; van Zonneveld, A.J. Endothelial progenitor cell dysfunction: A novel concept in the pathogenesis of vascular complications of type 1 diabetes. Diabetes 2004, 53, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Waclawovsky, G.; Umpierre, D.; Figueira, F.R.; De lima, E.S.; Alegretti, A.P.; Schneider, L.; Matte, U.S.; Rodrigues, T.C.; Schaan, B.D. Exercise on Progenitor Cells in Healthy Subjects and Patients with Type 1 Diabetes. Am. Coll. Sport Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- West, D.J.; Campbell, M.D.; Gonzalez, J.T.; Walker, M.; Stevenson, E.J.; Ahmed, F.W.; Wijaya, S.; Shaw, J.A.; Weaver, J.U. The inflammation, vascular repair and injury responses to exercise in fit males with and without Type 1 diabetes: An observational study. Cardiovasc. Diabetol. 2015, 14, 71. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A.; De Nigris, V.; Pujadas, G.; La Sala, L.; Bonfigli, A.R.; Testa, R.; Genovese, S.; Uccellatore, A. The simultaneous control of hyperglycemia and GLP-1 infusion normalize endothelial function in type 1 diabetes. Diabetes Res. Clin. Pract. 2016, 114, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A.; Novials, A.; Ortega, E.; Canivell, S.; La Sala, L.; Pujadas, G.; Esposito, K.; Giugliano, D.; Genovese, S. Glucagon-like peptide 1 reduces endothelial dysfunction, inflammation, and oxidative stress induced by both hyperglycemia and hypoglycemia in type 1 diabetes. Diabetes Care 2013, 36, 2346–2350. [Google Scholar] [CrossRef]
- Writing Group for the DCCT/EDIC Research Group TWG for the DR. Coprogression of Cardiovascular Risk Factors in Type 1 Diabetes During 30 Years of Follow-up in the DCCT/EDIC Study. Diabetes Care 2016, 39, 1621–1630. [Google Scholar] [CrossRef] [Green Version]
- Lind, M.; Bounias, I.; Olsson, M.; Gudbjörnsdottir, S.; Svensson, A.-M.; Rosengren, A. Glycaemic control and incidence of heart failure in 20,985 patients with type 1 diabetes: An observational study. Lancet 2011, 378, 140–146. [Google Scholar] [CrossRef]
- Nathan, D.M. DCCT/EDIC Research Group for the DR. The diabetes control and complications trial/epidemiology of diabetes interventions and complications study at 30 years: Overview. Diabetes Care 2014, 37, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Moser, O.; Eckstein, M.L.; McCarthy, O.; Deere, R.; Bain, S.C.; Haahr, H.L.; Zijlstra, E.; Bracken, R.M. Poor glycaemic control is associated with reduced exercise performance and oxygen economy during cardio-pulmonary exercise testing in people with type 1 diabetes. Diabetol. Metab. Syndr. 2017, 9, 93. [Google Scholar] [CrossRef] [PubMed]
- Strength and Conditioning: Biological Principles and Practical Applications; Wiley: Hoboken, NJ, USA, 2013.
- Egan, B.; Zierath, J.R. Exercise Metabolism and the Molecular Regulation of Skeletal Muscle Adaptation. Cell Metab. 2013, 17, 162–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balon, T.W.; Nadler, J.L. Evidence that nitric oxide increases glucose transport in skeletal muscle. J. Appl Physiol. 1997, 82, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Kindig, C.A.; Richardson, T.E.; Poole, D.C. Skeletal muscle capillary hemodynamics from rest to contractions: Implications for oxygen transfer. J. Appl Physiol. 2002, 92, 2513–2520. [Google Scholar] [CrossRef] [PubMed]
- Joyner, M.J.; Wilkins, B.W. Exercise hyperaemia: Is anything obligatory but the hyperaemia? J. Physiol. 2007, 583, 855–860. [Google Scholar] [CrossRef]
- Kusters, Y.H.A.M.; Barrett, E.J. Muscle microvasculature’s structural and functional specializations facilitate muscle metabolism. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E379–E387. [Google Scholar] [CrossRef]
- Williams, B.K.; Guelfi, K.J.; Jones, T.W.; Davis, E.A. Lower cardiorespiratory fitness in children with Type 1 diabetes. Diabet Med. 2011, 28, 1005–1007. [Google Scholar] [CrossRef]
- Moser, O.; Eckstein, M.L.; McCarthy, O.; Deere, R.; Bain, S.C.; Haahr, H.L.; Zijlstra, E.; Heise, T.; Bracken, R.M. Heart rate dynamics during cardio-pulmonary exercise testing are associated with glycemic control in individuals with type 1 diabetes. PLoS ONE 2018, 13, e0194750. [Google Scholar] [CrossRef]
- Irace, C.; Messiniti, V.; Tassone, B.; Cortese, C.; Barrett, E.J.; Gnasso, A. Evidence for congruent impairment in micro and macrovascular function in type 1 diabetes. PLoS ONE 2017, 12, e0187525. [Google Scholar] [CrossRef] [PubMed]
- Baldi, J.C.; Cassuto, N.A.; Foxx-Lupo, W.T.; Wheatley, C.M.; Snyder, E.M. Glycemic Status Affects Cardiopulmonary Exercise Response in Athletes with Type I Diabetes. Med. Sci. Sport Exerc. 2010, 42, 1454–1459. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.M.; Funk, M.; Grey, M. Cardiovascular health in adults with type 1 diabetes. Prev. Med. (Baltim) 2016, 91, 138–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craig, M.E.; Jones, T.W.; Silink, M.; Ping, Y.J. Diabetes care, glycemic control, and complications in children with type 1 diabetes from Asia and the Western Pacific Region. J. Diabetes Complicat. 2007, 21, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Cardwell, C.R.; Patterson, C.C.; Allen, M.; Carson, D.J. Northern Ireland Paediatric Diabetes Study Group. Diabetes care provision and glycaemic control in Northern Ireland: A UK regional audit. Arch. Dis. Child. 2005, 90, 468–473. [Google Scholar] [CrossRef] [PubMed]
- Danne, T.; Mortensen, H.B.; Hougaard, P.; Lynggaard, H.; Aanstoot, H.J.; Chiarelli, F.; Daneman, D.; Dorchy, H.; Garandeau, P.; Greene, S.A.; et al. Persistent differences among centers over 3 years in glycemic control and hypoglycemia in a study of 3805 children and adolescents with type 1 diabetes from the Hvidøre Study Group. Diabetes Care 2001, 24, 1342–1347. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.R.; Brekke, M.; Bergengen, L.; Sandvik, L.; Arnesen, H.; Hanssen, K.F.; Dahl-Jorgensen, K. Mean HbA1c over 18 years predicts carotid intima media thickness in women with type 1 diabetes. Diabetologia 2005, 48, 776–779. [Google Scholar] [CrossRef] [Green Version]
- How to Assess Endothelial Function for Detection of Pre-Clinical Atherosclerosis. Available online: https://www.escardio.org/Journals/E-Journal-of-Cardiology-Practice/Volume-10/How-to-assess-endothelial-function-for-detection-of-pre-clinical-atherosclerosis (accessed on 26 September 2017).
- Dabelea, D.; Mayer-Davis, E.J.; Saydah, S.; Imperatore, G.; Linder, B.; Divers, J.; Bell, R.; Badaru, A.; Talton, J.W.; Crume, T.; et al. Prevalence of Type 1 and Type 2 Diabetes Among Children and Adolescents From 2001 to 2009. JAMA 2014, 311, 1778. [Google Scholar] [CrossRef]
- Van Craenenbroeck, E.M.F.; Vrints, C.J.; Haine, S.E.; Vermeulen, K.; Goovaerts, I.; Van Tendeloo, V.F.I.; Hoymans, V.Y.; Conraads, V.M.A. A maximal exercise bout increases the number of circulating CD34+/KDR+ endothelial progenitor cells in healthy subjects. Relation with lipid profile. J. Appl. Physiol. 2008, 104, 1006–1013. [Google Scholar] [CrossRef] [Green Version]
- Naylor, L.H.; Davis, E.A.; Kalic, R.J.; Paramalingam, N.; Abraham, M.B.; Jones, T.W.; Green, D.J. Exercise training improves vascular function in adolescents with type 2 diabetes. Physiol. Rep. 2016, 4. [Google Scholar] [CrossRef]
- Gavin, T.P.; Drew, J.L.; Kubik, C.J.; Pofahl, W.E.; Hickner, R.C. Acute resistance exercise increases skeletal muscle angiogenic growth factor expression. Acta Physiol. 2007, 191, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Niemiro, G.M.; Parel, J.; Beals, J.; van Vliet, S.; Paluska, S.A.; Moore, D.R.; Burd, N.A.; De Lisio, M. Kinetics of circulating progenitor cell mobilization during submaximal exercise. J. Appl. Physiol. 2017, 122, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Rehman, J.; Li, J.; Parvathaneni, L.; Karlsson, G.; Panchal, V.R.; Temm, C.J.; Mahenthiran, J.; March, K.L. Exercise acutely increases circulating endothelial progenitor cells and monocyte-/macrophage-derived angiogenic cells. J. Am. Coll. Cardiol. 2004, 43, 2314–2318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashor, A.W.; Lara, J.; Siervo, M.; Celis-Morales, C.; Oggioni, C.; Jakovljevic, D.G.; Mathers, J.C. Exercise Modalities and Endothelial Function: A Systematic Review and Dose–Response Meta-Analysis of Randomized Controlled Trials. Sport Med. 2015, 45, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.M.; De Lisio, M.; Parise, G. Endurance exercise training promotes medullary hematopoiesis. FASEB J. 2011, 25, 4348–4357. [Google Scholar] [CrossRef] [PubMed]
- Emmons, R.; Niemiro, G.M.; De Lisio, M. Exercise as an Adjuvant Therapy for Hematopoietic Stem Cell Mobilization. Stem Cells Int. 2016, 2016, 7131359. [Google Scholar] [CrossRef] [PubMed]
- Marycz, K.; Mierzejewska, K.; Śmieszek, A.; Suszynska, E.; Malicka, I.; Kucia, M.; Ratajczak, M.Z. Endurance Exercise Mobilizes Developmentally Early Stem Cells into Peripheral Blood and Increases Their Number in Bone Marrow: Implications for Tissue Regeneration. Stem Cells Int. 2016, 2016, 5756901. [Google Scholar] [CrossRef]
- Morici, G.; Zangla, D.; Santoro, A.; Pelosi, E.; Petrucci, E.; Gioia, M.; Bonanno, A.; Profita, M.; Bellia, V.; Testa, U.; et al. Supramaximal exercise mobilizes hematopoietic progenitors and reticulocytes in athletes. AJP Regul. Integr. Comp. Physiol. 2005, 289, R1496–R1503. [Google Scholar] [CrossRef]
- Boff, W.; da Silva, A.M.; Farinha, J.B.; Rodrigues-Krause, J.; Reischak-Oliveira, A.; Tschiedel, B.; Puñales, M.; Bertoluci, M.C. Superior Effects of High-Intensity Interval vs. Moderate-Intensity Continuous Training on Endothelial Function and Cardiorespiratory Fitness in Patients with Type 1 Diabetes: A Randomized Controlled Trial. Front. Physiol. 2019, 10, 450. [Google Scholar] [CrossRef]
- Stellos, K.; Gawaz, M. Platelets and Stromal Cell-Derived Factor-1 in Progenitor Cell Recruitment. Semin. Thromb. Hemost. 2007, 33, 159–164. [Google Scholar] [CrossRef]
- Asahara, T.; Takahashi, T.; Masuda, H.; Kalka, C.; Chen, D.; Iwaguro, H.; Inai, Y.; Silver, M.; Isner, J.M. VEGF contributes to postnatal neovascularization by mobilizing bone marrow-derived endothelial progenitor cells. EMBO J. 1999, 18, 3964–3972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, M.D.; Wekesa, A.L.; Phelan, J.P.; Harrison, M. Resistance exercise increases endothelial progenitor cells and angiogenic factors. Med. Sci. Sports Exerc. 2014, 46, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; McDonald, S.P.; Coates, P.T.H.; Bonder, C.S. Endothelial progenitor cells: Novel biomarker and promising cell therapy for cardiovascular disease. Clin. Sci. 2011, 120, 263–283. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, J.O.; Weitzberg, E. NO generation from inorganic nitrate and nitrite: Role in physiology, nutrition and therapeutics. Arch. Pharm. Res. 2009, 32, 1119–1126. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.; Sampaio-Pinto, V.; Andrade, S.; Rodrigues, I.; Costa, R.; Guerreiro, S.; Carvalho, E.; Pinto-do-Ó, P.; Nascimento, D.S.; Soares, R. Establishing a Link Between Endothelial Cell Metabolism and Vascular Behaviour in a Type 1 Diabetes Mouse Model. Cell. Physiol. Biochem. 2019, 52, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Kleinbongard, P.; Dejam, A.; Lauer, T.; Jax, T.; Kerber, S.; Gharini, P.; Balzer, J.; Zotz, R.B.; Scharf, R.E.; Willers, R.; et al. Plasma nitrite concentrations reflect the degree of endothelial dysfunction in humans. Free Radic. Biol. Med. 2006, 40, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Rassaf, T.; Heiss, C.; Hendgen-Cotta, U.; Balzer, J.; Matern, S.; Kleinbongard, P.; Lee, A.; Lauer, T.; Kelm, M. Plasma nitrite reserve and endothelial function in the human forearm circulation. Free Radic. Biol. Med. 2006, 41, 295–301. [Google Scholar] [CrossRef]
- Kroll, J.L.; Werchan, C.A.; Rosenfield, D.; Ritz, T. Acute ingestion of beetroot juice increases exhaled nitric oxide in healthy individuals. PLoS ONE 2018, 13, e0191030. [Google Scholar] [CrossRef]
- Neha, P.; Sk, J.; Nk, J.; Hk, J.; Hk, M. Chemical and functional properties of Beetroot (Beta vulgaris L.) for product development: A review. Int. J. Chem. Stud. 2018, 6, 3190–3194. [Google Scholar]
- Raubenheimer, K.; Hickey, D.; Leveritt, M.; Fassett, R.; Ortiz de Zevallos Munoz, J.; Allen, J.D.; Briskey, D.; Parker, T.J.; Kerr, G.; Peake, J.M.; et al. Acute Effects of Nitrate-Rich Beetroot Juice on Blood Pressure, Hemostasis and Vascular Inflammation Markers in Healthy Older Adults: A Randomized, Placebo-Controlled Crossover Study. Nutrients 2017, 9, 1270. [Google Scholar] [CrossRef]
- Velmurugan, S.; Gan, J.M.; Rathod, K.S.; Khambata, R.S.; Ghosh, S.M.; Hartley, A.; Van Eijl, S.; Sagi-Kiss, V.; Chowdhury, T.A.; Curtis, M.; et al. Dietary nitrate improves vascular function in patients with hypercholesterolemia: A randomized, double-blind, placebo-controlled study. Am. J. Clin. Nutr. 2016, 103, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Webb, A.J.; Patel, N.; Loukogeorgakis, S.; Okorie, M.; Aboud, Z.; Misra, S.; Rashid, R.; Miall, P.; Deanfield, J.; Benjamin, N.; et al. Acute blood pressure lowering, vasoprotective, and antiplatelet properties of dietary nitrate via bioconversion to nitrite. Hypertens 2008, 51, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Shannon, O.M.; Duckworth, L.; Barlow, M.J.; Woods, D.; Lara, J.; Siervo, M.; O’Hara, J.P. Dietary nitrate supplementation enhances high-intensity running performance in moderate normobaric hypoxia, independent of aerobic fitness. Nitric Oxide 2016, 59, 63–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, K.G.; Turner, L.; Prichard, J.; Dodd, F.; Kennedy, D.O.; Haskell, C.; Blackwell, J.R.; Jones, A.M. Influence of dietary nitrate supplementation on physiological and cognitive responses to incremental cycle exercise. Respir. Physiol. Neurobiol. 2014, 193, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Muggeridge, D.J.; Howe, C.C.F.; Spendiff, O.; Pedlar, C.; James, P.E.; Easton, C. A Single Dose of Beetroot Juice Enhances Cycling Performance in Simulated Altitude. Med. Sci. Sport Exerc. 2014, 46, 143–150. [Google Scholar] [CrossRef]
- Patrician, A.; Schagatay, E. Dietary nitrate enhances arterial oxygen saturation after dynamic apnea. Scand. J. Med. Sci. Sports 2017, 27, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.; Wylie, L.J.; Fulford, J.; Kelly, J.; Black, M.I.; McDonagh, S.T.; Jeukendrup, A.E.; Vanhatalo, A.; Jones, A.M. Dietary nitrate improves sprint performance and cognitive function during prolonged intermittent exercise. Eur. J. Appl. Physiol. 2015, 115, 1825–1834. [Google Scholar] [CrossRef] [PubMed]
- Peeling, P.; Cox, G.R.; Bullock, N.; Burke, L.M. Beetroot Juice Improves On-Water 500 M Time-Trial Performance, and Laboratory-Based Paddling Economy in National and International-Level Kayak Athletes. Int. J. Sport Nutr. Exerc. Metab. 2015, 25, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Pinna, M.; Roberto, S.; Milia, R.; Marongiu, E.; Olla, S.; Loi, A.; Migliaccio, G.M.; Padulo, J.; Orlandi, C.; Tocco, F.; et al. Effect of Beetroot Juice Supplementation on Aerobic Response during Swimming. Nutrients 2014, 6, 605–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wylie, L.J.; Mohr, M.; Krustrup, P.; Jackman, S.R.; Ermιdis, G.; Kelly, J.; Black, M.I.; Bailey, S.J.; Vanhatalo, A.; Jones, A.M. Dietary nitrate supplementation improves team sport-specific intense intermittent exercise performance. Eur. J. Appl. Physiol. 2013, 113, 1673–1684. [Google Scholar] [CrossRef]
- Mosher, S.L.; Sparks, S.A.; Williams, E.L.; Bentley, D.J.; Mc Naughton, L.R. Ingestion of a Nitric Oxide Enhancing Supplement Improves Resistance Exercise Performance. J. Strength Cond. Res. 2016, 30, 3520–3524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haider, G.; Follad, J.P. Nitrate Supplementation Enhances the Contractile Properties of Human Skeletal Muscle. Med. Sci. Sport Exerc. 2014, 46, 2234–2243. [Google Scholar] [CrossRef] [PubMed]
- Kuennen, M.; Jansen, L.; Gillum, T.; Granados, J.; Castillo, W.; Nabiyar, A.; Christmas, K. Dietary nitrate reduces the O2 cost of desert marching but elevates the rise in core temperature. Eur. J. Appl. Physiol. 2015, 115, 2557–2569. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.M.; Ferguson, S.K.; Bailey, S.J.; Vanhatalo, A.; Poole, D.C. Fiber Type-Specific Effects of Dietary Nitrate. Exerc. Sport Sci. Rev. 2016, 44, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Cosby, K.; Partovi, K.S.; Crawford, J.H.; Patel, R.P.; Reiter, C.D.; Martyr, S.; Yang, B.K.; Waclawiw, M.A.; Zalos, G.; Xu, X.; et al. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat. Med. 2003, 9, 1498–1505. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, S.K.; Holdsworth, C.T.; Wright, J.L.; Fees, A.J.; Allen, J.D.; Jones, A.M.; Musch, T.I.; Poole, D.C. Microvascular oxygen pressures in muscles comprised of different fiber types: Impact of dietary nitrate supplementation. Nitric Oxide Biol. Chem. 2015, 48, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Crowther, G.J.; Milstein, J.M.; Jubrias, S.A.; Kushmerick, M.J.; Gronka, R.K.; Conley, K.E. Altered energetic properties in skeletal muscle of men with well-controlled insulin-dependent (type 1) diabetes. Am. J. Physiol. Metab. 2003, 284, E655–E662. [Google Scholar] [CrossRef] [Green Version]
- Cree-Green, M.; Newcomer, B.R.; Brown, M.S.; Baumgartner, A.D.; Bergman, B.; Drew, B.; Regensteiner, J.G.; Pyle, L.; Reusch, J.E.; Nadeau, K.J. Delayed skeletal muscle mitochondrial ADP recovery in youth with type 1 diabetes relates to muscle insulin resistance. Diabetes 2015, 64, 383–392. [Google Scholar] [CrossRef]
- Orlando, G.; Balducci, S.; Bazzucchi, I.; Pugliese, G.; Sacchetti, M. The impact of type 1 diabetes and diabetic polyneuropathy on muscle strength and fatigability. Acta Diabetol. 2017, 54, 543–550. [Google Scholar] [CrossRef]
- Kelly, J.; Vanhatalo, A.; Bailey, S.J.; Wylie, L.J.; Tucker, C.; List, S.; Winyard, P.G.; Jones, A.M. Dietary nitrate supplementation: Effects on plasma nitrite and pulmonary O2 uptake dynamics during exercise in hypoxia and normoxia. Am. J. Physiol. Integr. Comp. Physiol. 2014, 307, R920–R930. [Google Scholar] [CrossRef]
- Masschelein, E.; Van Thienen, R.; Wang, X.; Van Schepdael, A.; Thomis, M.; Hespel, P. Dietary nitrate improves muscle but not cerebral oxygenation status during exercise in hypoxia. J. Appl. Physiol. 2012, 113, 736–745. [Google Scholar] [CrossRef] [Green Version]
- Allen, J.D.; Stabler, T.; Kenjale, A.; Ham, K.L.; Robbins, J.L.; Duscha, B.D.; Dobrosielski, D.A.; Annex, B.H. Plasma nitrite flux predicts exercise performance in peripheral arterial disease after 3months of exercise training. Free Radic. Biol. Med. 2010, 49, 1138–1144. [Google Scholar] [CrossRef] [Green Version]
- Curis, E.; Nicolis, I.; Moinard, C.; Osowska, S.; Zerrouk, N.; Bénazeth, S.; Cynober, L. Almost all about citrulline in mammals. Amino Acids 2005, 29, 177–205. [Google Scholar] [CrossRef]
- Pérez-Guisado, J.; Jakeman, P.M. Citrulline Malate Enhances Athletic Anaerobic Performance and Relieves Muscle Soreness. J. Strength Cond. Res. 2010, 24, 1215–1222. [Google Scholar] [CrossRef]
- Schwedhelm, E.; Maas, R.; Freese, R.; Jung, D.; Lukacs, Z.; Jambrecina, A.; Spickler, W.; Schulze, F.; Böger, R.H. Pharmacokinetic and pharmacodynamic properties of oral L-citrulline and L-arginine: Impact on nitric oxide metabolism. Br. J. Clin. Pharmacol. 2008, 65, 51–59. [Google Scholar] [CrossRef]
- Elwafi, F.; Curis, E.; Zerrouk, N.; Neveux, N.; Chaumeil, J.C.; Arnaud, P.; Cynober, L.; Moinard, C. Endotoxemia affects citrulline, arginine and glutamine bioavailability. Eur. J. Clin. Investig. 2012, 42, 282–289. [Google Scholar] [CrossRef]
- Wijnands, K.A.P.; Vink, H.; Briedé, J.J.; van Faassen, E.E.; Lamers, W.H.; Buurman, W.A.; Poeze, M. Citrulline a More Suitable Substrate than Arginine to Restore NO Production and the Microcirculation during Endotoxemia. PLoS ONE 2012, 7, e37439. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Hsu, J.W.; Emrick, L.T.; Wong, L.J.; Craigen, W.J.; Jahoor, F.; Scaglia, F. Restoration of impaired nitric oxide production in, M.E.L.A.; S syndrome with citrulline and arginine supplementation. Mol. Genet. Metab. 2012, 105, 607–614. [Google Scholar] [CrossRef]
- Wijnands, K.A.P.; Castermans, T.M.R.; Hommen, M.P.J.; Meesters, D.M.; Poeze, M. Arginine and citrulline and the immune response in sepsis. Nutrients 2015, 7, 1426–1463. [Google Scholar] [CrossRef]
- Persson, P.; Fasching, A.; Teerlink, T.; Hansell, P.; Palm, F. L-Citrulline, But Not l-Arginine, Prevents Diabetes Mellitus–Induced Glomerular Hyperfiltration and Proteinuria in Rat. Hypertension 2014, 64, 323–329. [Google Scholar] [CrossRef]
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef]
- Baumgardt, S.L.; Paterson, M.; Leucker, T.M.; Fang, J.; Zhang, D.X.; Bosnjak, Z.J.; Ge, Z.D. Chronic Co-Administration of Sepiapterin and l -Citrulline Ameliorates Diabetic Cardiomyopathy and Myocardial Ischemia/Reperfusion Injury in Obese Type 2 Diabetic Mice. Circ. Heart Fail. 2016, 9, e002424. [Google Scholar] [CrossRef]
- Xuan, C.; Lun, L.-M.; Zhao, J.-X.; Wang, H.W.; Wang, J.; Ning, C.P.; Liu, Z.; Zhang, B.B.; He, G.W. L-citrulline for protection of endothelial function from, ADMA-induced injury in porcine coronary artery. Sci. Rep. 2015, 5, 10987. [Google Scholar] [CrossRef]
- Chien, S.-J.; Lin, K.-M.; Kuo, H.-C.; Huang, C.F.; Lin, Y.J.; Huang, L.T.; Tain, Y.L. Two different approaches to restore renal nitric oxide and prevent hypertension in young spontaneously hypertensive rats: L-citrulline and nitrate. Transl. Res. 2014, 163, 43–52. [Google Scholar] [CrossRef]
- Orozco-Gutiérrez, J.J.; Castillo-Martínez, L.; Orea-Tejeda, A.; Vázquez-Díaz, O.; Valdespino-Trejo, A.; Narváez-David, R.; Keirns-Davis, C.; Carrasco-Ortiz, O.; Navarro-Navarro, A.; Sánchez-Santillán, R. Effect of L-arginine or L-citrulline oral supplementation on blood pressure and right ventricular function in heart failure patients with preserved ejection fraction. Cardiol. J. 2010, 17, 612–618. [Google Scholar]
- Barkhidarian, B.; Khorshidi, M.; Shab-Bidar, S.; Hashemi, B. Effects of L-citrulline supplementation on blood pressure: A systematic review and meta-analysis. Avicenna J. Phytomed. 2019, 9, 10–20. [Google Scholar]
- Kim, I.-Y.; Schutzler, S.E.; Schrader, A.; Spencer, H.J.; Azhar, G.; Deutz, N.E.; Wolfe, R.R. Acute ingestion of citrulline stimulates nitric oxide synthesis but does not increase blood flow in healthy young and older adults with heart failure. Am. J. Physiol. Metab. 2015, 309, E915–E924. [Google Scholar] [CrossRef] [Green Version]
- Churchward-Venne, T.A.; Cotie, L.M.; MacDonald, M.J.; Mitchell, C.J.; Prior, T.; Baker, S.K.; Phillips, S.M. Citrulline does not enhance blood flow, microvascular circulation, or myofibrillar protein synthesis in elderly men at rest or following exercise. Am. J. Physiol. Metab. 2014, 307, E71–E83. [Google Scholar] [CrossRef] [Green Version]
- Voet, D.; Voet, J.G.; Pratt, C.W. Fundamentals of Biochemistry: Life at the Molecular Level; Wiley: Hoboken, NJ, USA, 2006. [Google Scholar]
- Sharif Kashani, B.; Tahmaseb Pour, P.; Malekmohammad, M.; Behzadnia, N.; Sheybani-Afshar, F.; Fakhri, M.; Chaibakhsh, S.; Naghashzadeh, F.; Aidenlou, S. Oral l-citrulline malate in patients with idiopathic pulmonary arterial hypertension and Eisenmenger Syndrome: A clinical trial. J. Cardiol. 2014, 64, 231–235. [Google Scholar] [CrossRef] [Green Version]
- Callis, A.; Magnan de Bornier, B.; Serrano, J.J.; Bellet, H.; Saumade, R. Activity of citrulline malate on acid-base balance and blood ammonia and amino acid levels. Study in the animal and in man. Arzneimittelforschung 1991, 41, 660–663. [Google Scholar]
- Takeda, K.; Machida, M.; Kohara, A.; Omi, N.; Takemasa, T. Effects of citrulline supplementation on fatigue and exercise performance in mice. J. Nutr. Sci. Vitaminol. (Tokyo) 2011, 57, 246–250. [Google Scholar] [CrossRef]
- Hwang, P.; Morales Marroquín, F.E.; Gann, J.; Andre, T.; McKinley-Barnard, S.; Kim, C.; Morita, M.; Willoughby, D.S. Eight weeks of resistance training in conjunction with glutathione and L-Citrulline supplementation increases lean mass and has no adverse effects on blood clinical safety markers in resistance-trained males. J. Int. Soc. Sports Nutr. 2018, 15, 30. [Google Scholar] [CrossRef]
- Glenn, J.M.; Gray, M.; Jensen, A.; Stone, M.S.; Vincenzo, J.L. Acute citrulline-malate supplementation improves maximal strength and anaerobic power in female, masters athletes tennis players. Eur. J. Sport Sci. 2016, 16, 1095–1103. [Google Scholar] [CrossRef]
- Glenn, J.M.; Gray, M.; Wethington, L.N.; Stone, M.S.; Stewart, R.W.; Moyen, N.E. Acute citrulline malate supplementation improves upper- and lower-body submaximal weightlifting exercise performance in resistance-trained females. Eur. J. Nutr. 2017, 56, 775–784. [Google Scholar] [CrossRef]
- Chappell, A.J.; Allwood, D.M.; Simper, T.N. Citrulline Malate Fails to Improve German Volume Training Performance in Healthy Young Men and Women. J. Diet. Suppl. 2018, 1–12. [Google Scholar] [CrossRef]
- Gonzalez, A.M.; Spitz, R.W.; Ghigiarelli, J.J.; Sell, K.M.; Mangine, G.T. Acute effect of citrulline malate supplementation on upper-body resistance exercise performance in recreationally resistance-trained men. J. Strength Cond. Res. 2017, 1. [Google Scholar] [CrossRef]
- Farney, T.M.; Bliss, M.V.; Hearon, C.M.; Salazar, D.A. The Effect of Citrulline Malate Supplementation On Muscle Fatigue Among Healthy Participants. J. Strength Cond. Res. 2017, 1. [Google Scholar] [CrossRef]
- da Silva, D.; Jacinto, J.; de Andrade, W.B.; Roveratti, M.C.; Estoche, J.M.; Balvedi, M.C.W.; de Oliveira, D.B.; da Silva, R.A.; Aguiar, A.F. Citrulline Malate Does Not Improve Muscle Recovery after Resistance Exercise in Untrained Young Adult Men. Nutrients 2017, 9, 1132. [Google Scholar] [CrossRef]
- Brazeau, A.-S.; Rabasa-Lhoret, R.; Strychar, I.; Mircescu, H. Barriers to physical activity among patients with type 1 diabetes. Diabetes Care 2008, 31, 2108–2109. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCarthy, O.; Moser, O.; Eckstein, M.L.; Bain, S.C.; Pitt, J.; Bracken, R. Supplementary Nitric Oxide Donors and Exercise as Potential Means to Improve Vascular Health in People with Type 1 Diabetes: Yes to NO? Nutrients 2019, 11, 1571. https://doi.org/10.3390/nu11071571
McCarthy O, Moser O, Eckstein ML, Bain SC, Pitt J, Bracken R. Supplementary Nitric Oxide Donors and Exercise as Potential Means to Improve Vascular Health in People with Type 1 Diabetes: Yes to NO? Nutrients. 2019; 11(7):1571. https://doi.org/10.3390/nu11071571
Chicago/Turabian StyleMcCarthy, Olivia, Othmar Moser, Max L. Eckstein, Stephen C. Bain, Jason Pitt, and Richard Bracken. 2019. "Supplementary Nitric Oxide Donors and Exercise as Potential Means to Improve Vascular Health in People with Type 1 Diabetes: Yes to NO?" Nutrients 11, no. 7: 1571. https://doi.org/10.3390/nu11071571