Lactational High-Fat Diet Exposure Programs Metabolic Inflammation and Bone Marrow Adiposity in Male Offspring

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Husbandry
2.2. Intraperitoneal Glucose and Insulin Tolerance Tests (IPGTT, ITT)
2.3. Homeostatic Model Assessment of Insulin Resistance (HOMA-IR)
2.4. Immunofluorescence Staining
2.5. RNA Extraction, Reverse Transcription, Quantitative Real-Time PCR
2.6. Adipose Tissue Stromal Vascular Fraction (SVF) Isolation and Flow Cytometry
2.7. Methocellulose Colony Forming Unit Assay
2.8. Bone µCT
2.9. Bone Histology
2.10. Statistical Analysis
3. Results
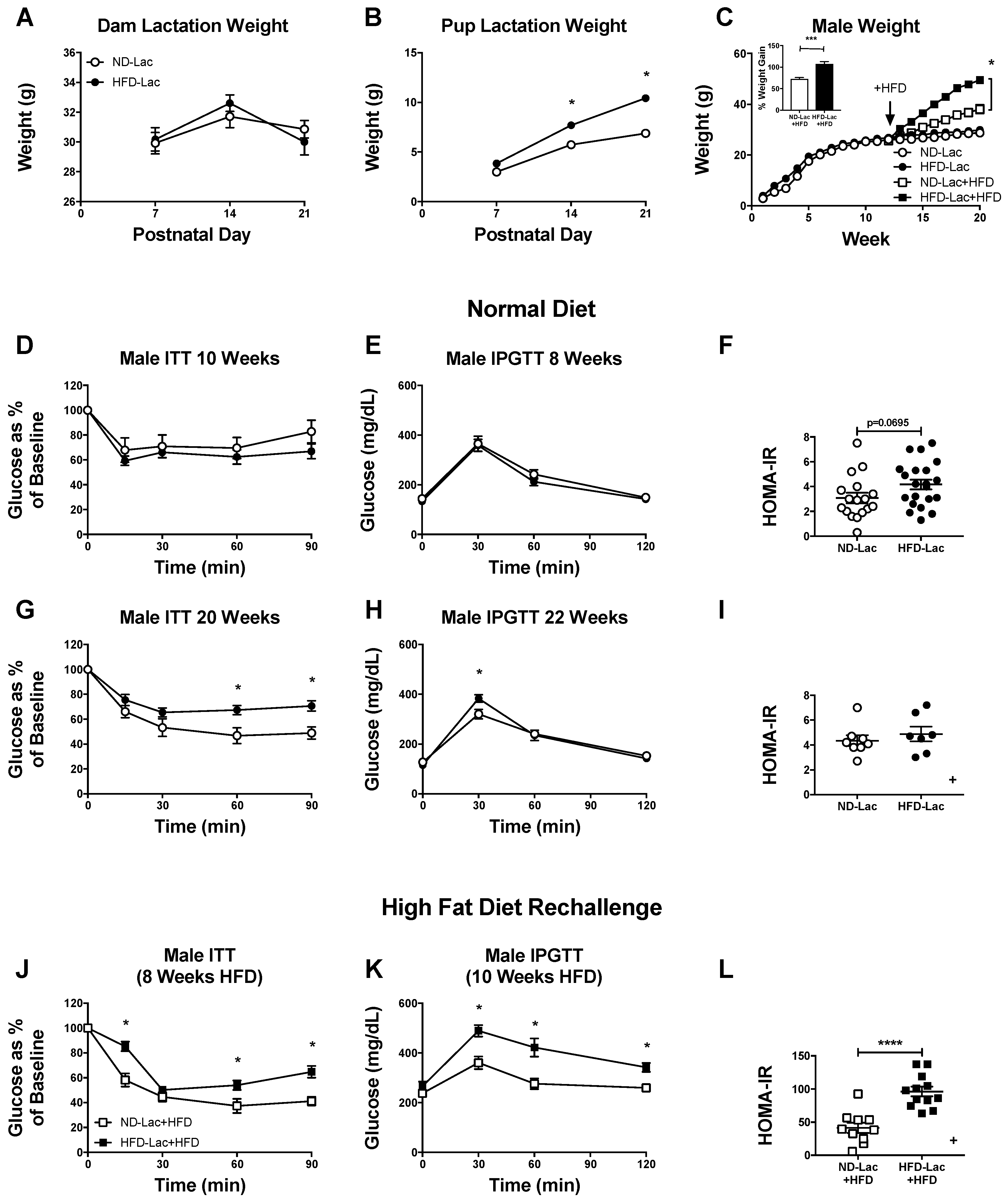
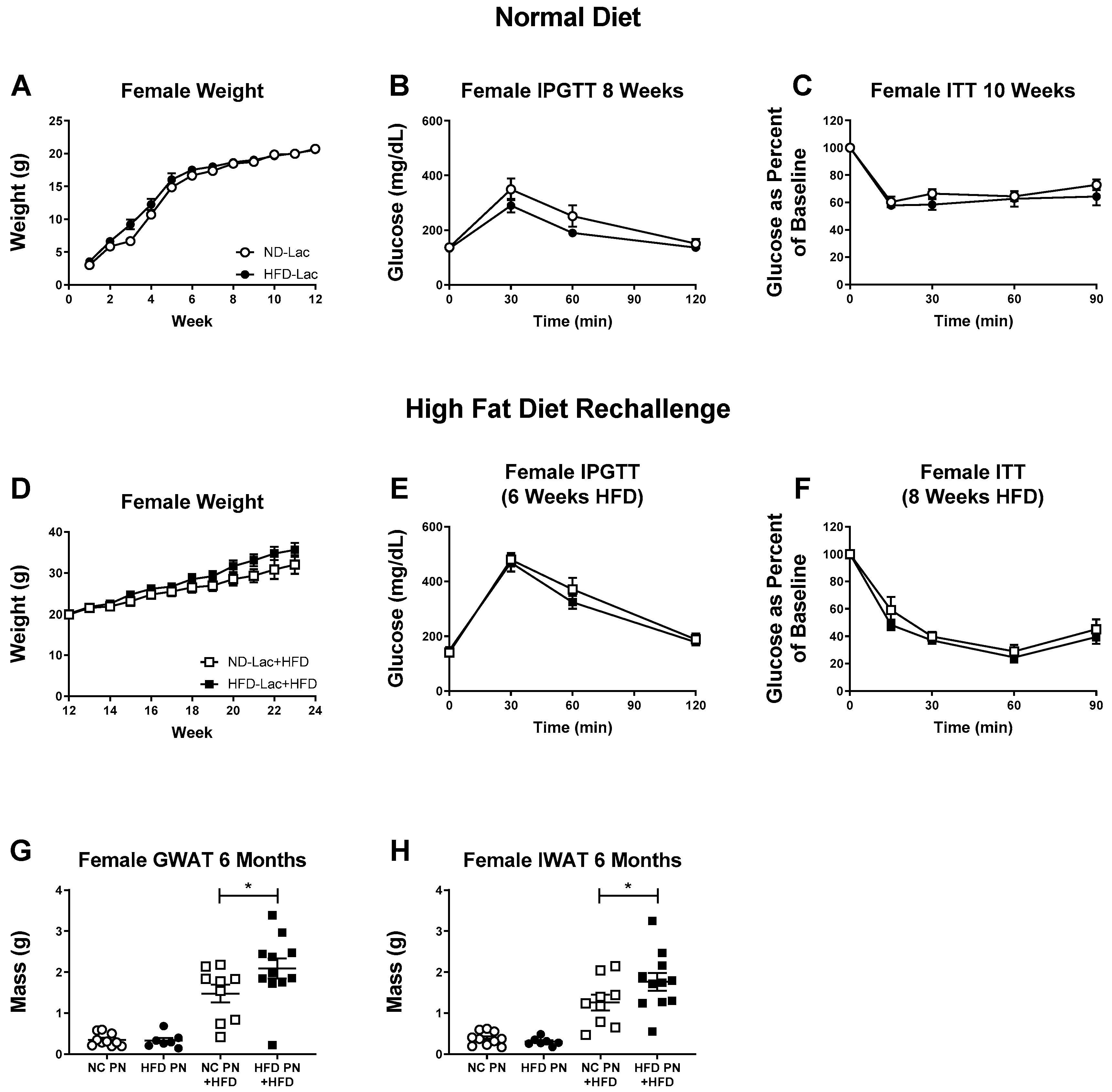
3.1. Male Lactation HFD Offspring Show Insulin Resistance and Mild Glucose Intolerance on Normal Diet
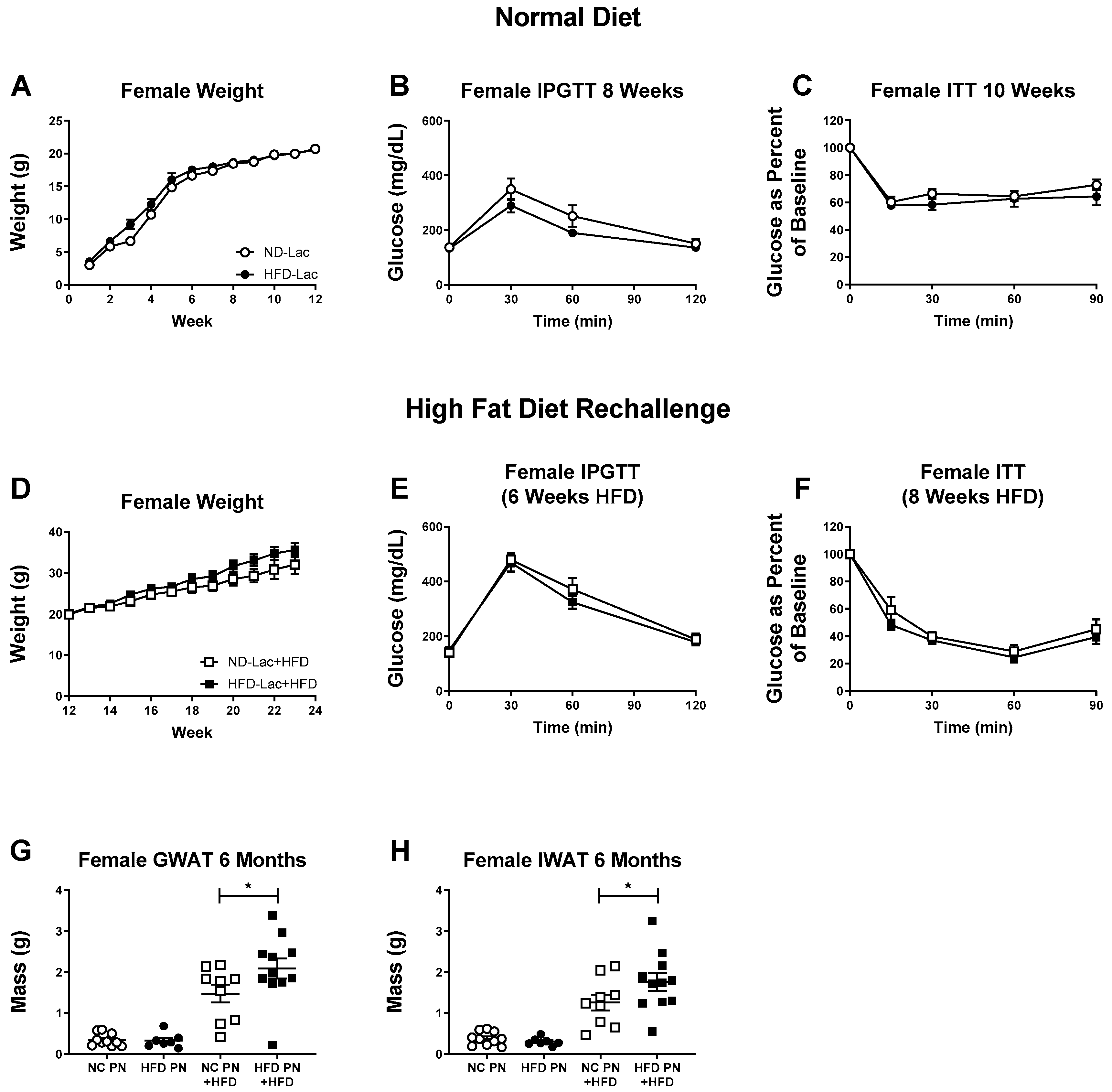
3.2. Female Offspring Had no Evident Metabolic Changes after Lactational HFD Exposure
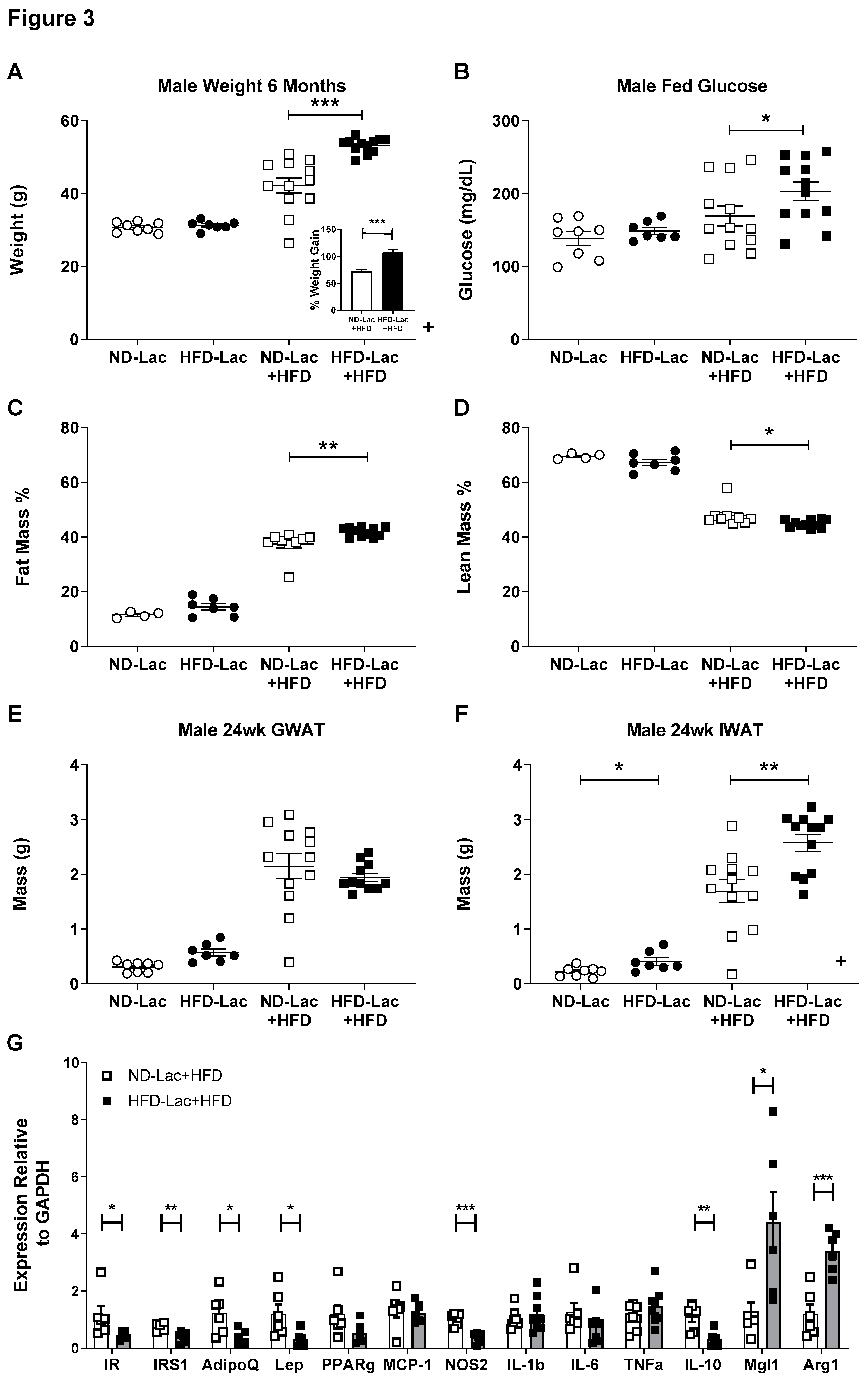
3.3. Re-Challenged Male HFD-Lac Offspring Have Increased Fat Mass
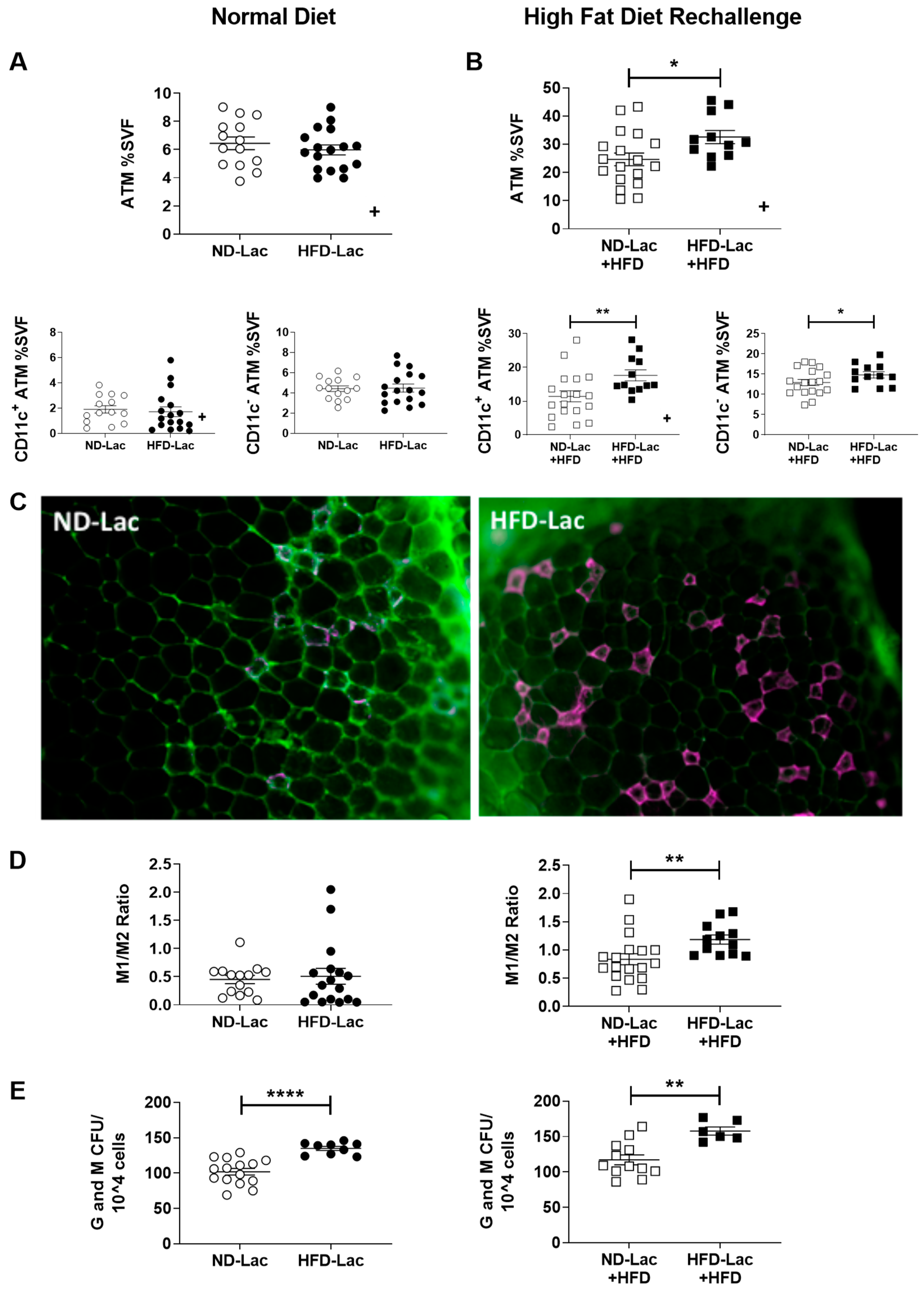
3.4. Enhanced Myeloid Inflammation in HFD-Lac Males Challenged to HFD
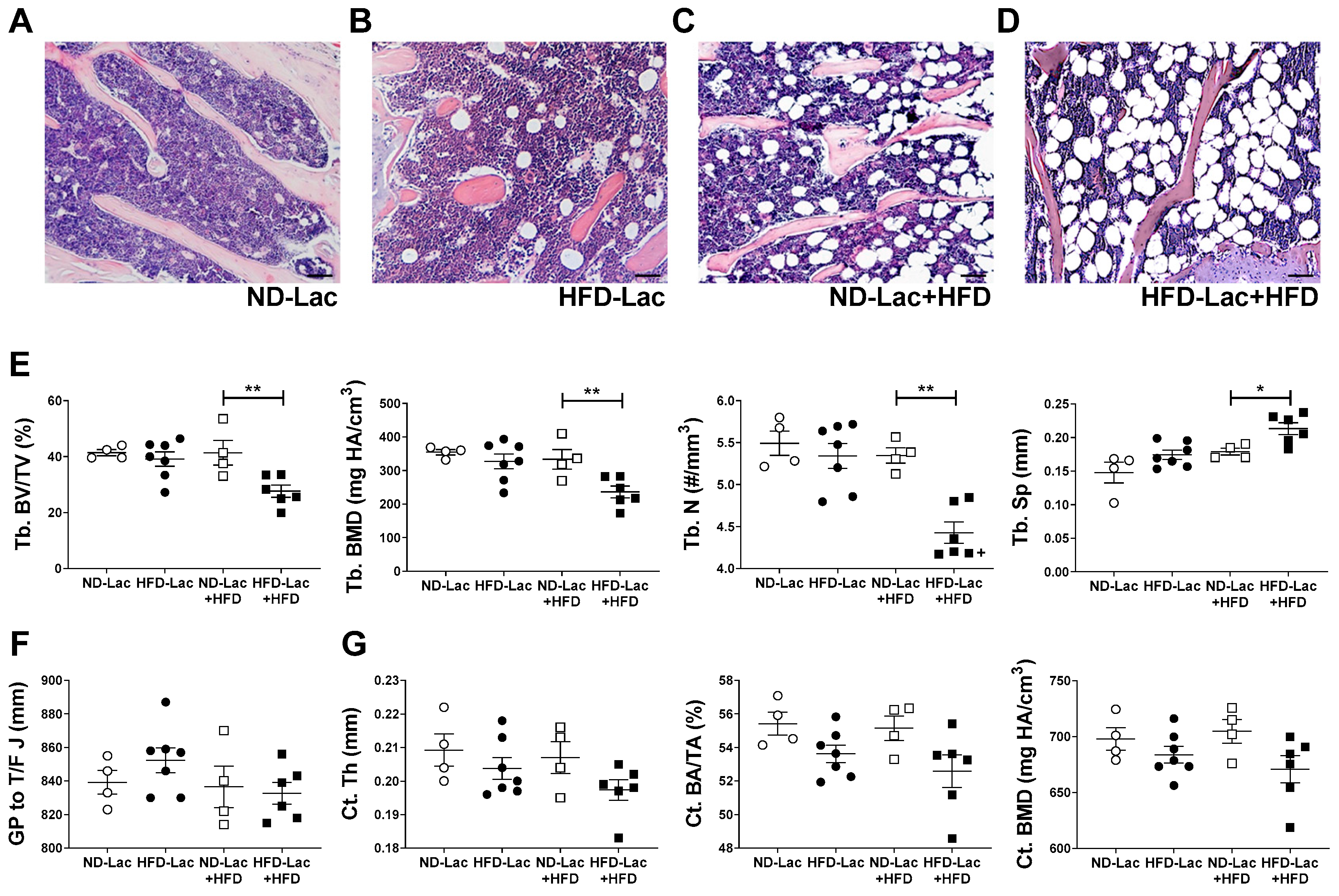
3.5. HFD Re-Challenge after Exposure to HFD during Lactation Causes Long-Term Bone Loss and Expansion of Bone Marrow Adiposity
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ong, T.P.; Ozanne, S.E. Developmental programming of type 2 diabetes: Early nutrition and epigenetic mechanisms. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Scaglia, L.; Cahill, C.J.; Finegood, D.T.; Bonner-Weir, S. Apoptosis participates in the remodeling of the endocrine pancreas in the neonatal rat. Endocrinology 1997, 138, 1736–1741. [Google Scholar] [CrossRef] [PubMed]
- Zorn, A.M. Liver development. StemBook 2008. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Zhang, T.; Xu, H. Human adipose dynamics and metabolic health. Ann. N.Y. Acad. Sci. 2013, 1281, 160–177. [Google Scholar] [CrossRef] [PubMed]
- Ellsworth, L.; Harman, E.; Padmanabhan, V.; Gregg, B. Lactational programming of glucose homeostasis: A window of opportunity. Reproduction 2018, 156, R23–R42. [Google Scholar] [CrossRef] [PubMed]
- Petrik, J.; Reusens, B.; Arany, E.; Remacle, C.; Coelho, C.; Hoet, J.J.; Hill, D.J. A low protein diet alters the balance of islet cell replication and apoptosis in the fetal and neonatal rat and is associated with a reduced pancreatic expression of insulin-like growth factor-II. Endocrinology 1999, 140, 4861–4873. [Google Scholar] [CrossRef]
- Masuyama, H.; Hiramatsu, Y. Additive effects of maternal high fat diet during lactation on mouse offspring. PLoS ONE 2014, 9, e92805. [Google Scholar] [CrossRef]
- Liang, X.; Yang, Q.; Zhang, L.; Maricelli, J.W.; Rodgers, B.D.; Zhu, M.J.; Du, M. Maternal high-fat diet during lactation impairs thermogenic function of brown adipose tissue in offspring mice. Sci. Rep. 2016, 6, 34345. [Google Scholar] [CrossRef] [Green Version]
- Vogt, M.C.; Paeger, L.; Hess, S.; Steculorum, S.M.; Awazawa, M.; Hampel, B.; Neupert, S.; Nicholls, H.T.; Mauer, J.; Hausen, A.C.; et al. Neonatal insulin action impairs hypothalamic neurocircuit formation in response to maternal high-fat feeding. Cell 2014, 156, 495–509. [Google Scholar] [CrossRef]
- Plagemann, A.; Heidrich, I.; Gotz, F.; Rohde, W.; Dorner, G. Obesity and enhanced diabetes and cardiovascular risk in adult rats due to early postnatal overfeeding. Exp. Clin. Endocrinol. 1992, 99, 154–158. [Google Scholar] [CrossRef]
- Berends, L.M.; Fernandez-Twinn, D.S.; Martin-Gronert, M.S.; Cripps, R.L.; Ozanne, S.E. Catch-up growth following intra-uterine growth-restriction programmes an insulin-resistant phenotype in adipose tissue. Int. J. Obes. 2013, 37, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Torow, N.; Hornef, M.W. The Neonatal window of opportunity: Setting the stage for life-long host-microbial interaction and immune homeostasis. J. Immunol. 2017, 198, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Murabayashi, N.; Sugiyama, T.; Zhang, L.; Kamimoto, Y.; Umekawa, T.; Ma, N.; Sagawa, N. Maternal high-fat diets cause insulin resistance through inflammatory changes in fetal adipose tissue. Eur. J. Obstet. Gynecol. Reprod. Biol. 2013, 169, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Yu, Y.; Wang, D.; Chai, S.; Liu, D.; Xiao, X.; Huang, Y. Maternal high-fat diet feeding during pregnancy and lactation augments lung inflammation and remodeling in the offspring. Respir. Physiol. Neurobiol. 2015, 207, 1–6. [Google Scholar] [CrossRef]
- Mennitti, L.V.; Oyama, L.M.; De Oliveira, J.L.; Hachul, A.C.; Santamarina, A.B.; De Santana, A.A.; Okuda, M.H.; Ribeiro, E.B.; Oller do Nascimento, C.M.; Pisani, L.P. Maternal supplementation with oligofructose (10%) during pregnancy and lactation leads to increased pro-inflammatory status of the 21-D-old offspring. PLoS ONE 2015, 10, e0132038. [Google Scholar] [CrossRef] [PubMed]
- Kruse, M.; Keyhani-Nejad, F.; Isken, F.; Nitz, B.; Kretschmer, A.; Reischl, E.; De las Heras Gala, T.; Osterhoff, M.A.; Grallert, H.; Pfeiffer, A.F. High-fat diet during mouse pregnancy and lactation targets GIP-regulated metabolic pathways in adult male offspring. Diabetes 2016, 65, 574–584. [Google Scholar] [CrossRef]
- Monks, J.; Orlicky, D.J.; Stefanski, A.L.; Libby, A.E.; Bales, E.S.; Rudolph, M.C.; Johnson, G.C.; Sherk, V.D.; Jackman, M.R.; Williamson, K.; et al. Maternal obesity during lactation may protect offspring from high fat diet-induced metabolic dysfunction. Nutr. Diabetes 2018, 8, 18. [Google Scholar] [CrossRef] [Green Version]
- Boullu-Ciocca, S.; Achard, V.; Tassistro, V.; Dutour, A.; Grino, M. Postnatal programming of glucocorticoid metabolism in rats modulates high-fat diet-induced regulation of visceral adipose tissue glucocorticoid exposure and sensitivity and adiponectin and proinflammatory adipokines gene expression in adulthood. Diabetes 2008, 57, 669–677. [Google Scholar] [CrossRef]
- Butruille, L.; Marousez, L.; Pourpe, C.; Oger, F.; Lecoutre, S.; Catheline, D.; Gors, S.; Metges, C.C.; Guinez, C.; Laborie, C.; et al. Maternal high-fat diet during suckling programs visceral adiposity and epigenetic regulation of adipose tissue stearoyl-CoA desaturase-1 in offspring. Int. J. Obes. 2019. [Google Scholar] [CrossRef]
- Gregg, B.E.; Botezatu, N.; Brill, J.D.; Hafner, H.; Vadrevu, S.; Satin, L.S.; Alejandro, E.U.; Bernal-Mizrachi, E. Gestational exposure to metformin programs improved glucose tolerance and insulin secretion in adult male mouse offspring. Sci. Rep. 2018, 8, 5745. [Google Scholar] [CrossRef] [Green Version]
- Westcott, D.J.; Delproposto, J.B.; Geletka, L.M.; Wang, T.; Singer, K.; Saltiel, A.R.; Lumeng, C.N. MGL1 promotes adipose tissue inflammation and insulin resistance by regulating 7/4hi monocytes in obesity. J. Exp. Med. 2009, 206, 3143–3156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- RRID: AB_2792981. Available online: https://scicrunch.org/resolver/AB_2792981 (accessed on 10 April 2019).
- Shults, C.W.; Haas, R.; Oakes, D.; Kieburtz, K.; Plumb, S.; Shoulson, I.; Beal, M.F.; Juncos, J.; Nutt, J. Measuring the effects of therapy in Parkinson disease. JAMA 2004, 291, 2430–2431. [Google Scholar] [CrossRef] [PubMed]
- RRID: AB_837133. Available online: https://scicrunch.org/resolver/AB_837133 (accessed on 10 April 2019).
- Singer, K.; Maley, N.; Mergian, T.; DelProposto, J.; Cho, K.W.; Zamarron, B.F.; Martinez-Santibanez, G.; Geletka, L.; Muir, L.; Wachowiak, P.; et al. Differences in hematopoietic stem cells contribute to sexually dimorphic inflammatory responses to high fat diet-induced obesity. J. Biol. Chem. 2015, 290, 13250–13262. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, M.; Song, F.; Aalinkeel, R.; Patel, M.S. Molecular adaptations in islets from neonatal rats reared artificially on a high carbohydrate milk formula. J. Nutr. Biochem. 2001, 12, 575–584. [Google Scholar] [CrossRef]
- Song, F.; Srinivasan, M.; Aalinkeel, R.; Patel, M.S. Use of a cDNA array for the identification of genes induced in islets of suckling rats by a high-carbohydrate nutritional intervention. Diabetes 2001, 50, 2053–2060. [Google Scholar] [CrossRef]
- Aalinkeel, R.; Srinivasan, M.; Song, F.; Patel, M.S. Programming into adulthood of islet adaptations induced by early nutritional intervention in the rat. Am. J. Physiol. Endocrinol. Metab. 2001, 281, E640–648. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Srinivasan, M.; Aalinkeel, R.; Patel, M.S.; Laychock, S.G. Mitochondrial-encoded gene regulation in rat pancreatic islets. Metabolism 2001, 50, 200–206. [Google Scholar] [CrossRef] [PubMed]
- RRID:SCR_017119. Available online: https://scicrunch.org/resolver/SCR_017119 (accessed on 24 April 2019).
- Parlee, S.D.; Lentz, S.I.; Mori, H.; MacDougald, O.A. Quantifying size and number of adipocytes in adipose tissue. Methods Enzymol. 2014, 537, 93–122. [Google Scholar] [CrossRef] [PubMed]
- March of Dimes Peristats. Available online: https://www.marchofdimes.org/Peristats/ViewSubtopic.aspx?reg=99&top=17&stop=350&lev=1&slev=1&obj=1 (accessed on 10 April 2019).
- Zamarron, B.F.; Mergian, T.A.; Cho, K.W.; Martinez-Santibanez, G.; Luan, D.; Singer, K.; DelProposto, J.L.; Geletka, L.M.; Muir, L.A.; Lumeng, C.N. Macrophage proliferation sustains adipose tissue inflammation in formerly obese mice. Diabetes 2017, 66, 392–406. [Google Scholar] [CrossRef] [PubMed]
- Masuyama, H.; Mitsui, T.; Nobumoto, E.; Hiramatsu, Y. The effects of high-fat diet exposure in utero on the obesogenic and diabetogenic traits through epigenetic changes in adiponectin and leptin gene expression for multiple generations in female mice. Endocrinology 2015, 156, 2482–2491. [Google Scholar] [CrossRef] [PubMed]
- Varghese, M.; Griffin, C.; McKernan, K.; Eter, L.; Lanzetta, N.; Agarwal, D.; Abrishami, S.; Singer, K. Sex differences in inflammatory responses to adipose tissue lipolysis in diet-induced obesity. Endocrinology 2019, 160, 293–312. [Google Scholar] [CrossRef] [PubMed]
- Singer, K.; DelProposto, J.; Morris, D.L.; Zamarron, B.; Mergian, T.; Maley, N.; Cho, K.W.; Geletka, L.; Subbaiah, P.; Muir, L.; et al. Diet-induced obesity promotes myelopoiesis in hematopoietic stem cells. Mol. Metab. 2014, 3, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Kamimae-Lanning, A.N.; Krasnow, S.M.; Goloviznina, N.A.; Zhu, X.; Roth-Carter, Q.R.; Levasseur, P.R.; Jeng, S.; McWeeney, S.K.; Kurre, P.; Marks, D.L. Maternal high-fat diet and obesity compromise fetal hematopoiesis. Mol. Metab. 2015, 4, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Sharkey, D.; Symonds, M.E.; Budge, H. Adipose tissue inflammation: Developmental ontogeny and consequences of gestational nutrient restriction in offspring. Endocrinology 2009, 150, 3913–3920. [Google Scholar] [CrossRef] [PubMed]
- Morris, D.L.; Singer, K.; Lumeng, C.N. Adipose tissue macrophages: Phenotypic plasticity and diversity in lean and obese states. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 341–346. [Google Scholar] [CrossRef]
- Li, Z.; Hardij, J.; Bagchi, D.P.; Scheller, E.L.; MacDougald, O.A. Development, regulation, metabolism and function of bone marrow adipose tissues. Bone 2018, 110, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Scheller, E.L.; Cawthorn, W.P.; Burr, A.A.; Horowitz, M.C.; MacDougald, O.A. Marrow Adipose tissue: trimming the fat. Trends Endocrinol. Metab. 2016, 27, 392–403. [Google Scholar] [CrossRef]
- Liang, C.; Oest, M.E.; Jones, J.C.; Prater, M.R. Gestational high saturated fat diet alters C57BL/6 mouse perinatal skeletal formation. Birth Defects Res. B Devel. Reprod. Toxicol. 2009, 86, 362–369. [Google Scholar] [CrossRef]
- Chen, J.-R.; Lazarenko, O.P.; Zhao, H.; Alund, A.W.; Shankar, K. Maternal obesity impairs skeletal development in adult offspring. J. Endocrinol. 2018, 239, 33–47. [Google Scholar] [CrossRef] [Green Version]
- Parlee, S.D.; MacDougald, O.A. Maternal nutrition and risk of obesity in offspring: The Trojan horse of developmental plasticity. Biochim. Biophys. Acta 2014, 1842, 495–506. [Google Scholar] [CrossRef]
- Sun, B.; Purcell, R.H.; Terrillion, C.E.; Yan, J.; Moran, T.H.; Tamashiro, K.L. Maternal high-fat diet during gestation or suckling differentially affects offspring leptin sensitivity and obesity. Diabetes 2012, 61, 2833–2841. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, A.M.; Matthews, P.A.; Argenton, M.; Christie, M.R.; McConnell, J.M.; Jansen, E.H.; Piersma, A.H.; Ozanne, S.E.; Twinn, D.F.; Remacle, C.; et al. Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: A novel murine model of developmental programming. Hypertension 2008, 51, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Howie, G.J.; Sloboda, D.M.; Kamal, T.; Vickers, M.H. Maternal nutritional history predicts obesity in adult offspring independent of postnatal diet. J. Physiol. 2009, 587, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, J.; Farr, O.M.; Mantzoros, C.S. The role of leptin in regulating bone metabolism. Metabolism 2015, 64, 105–113. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward 5′-3′ | Reverse 5′-3′ |
|---|---|---|
| IR | TTTGTCATGGATGGAGGCTA | CCTCATCTTGGGGTTGAACT |
| IRS1 | CGATGGCTTCTCAGACGTG | CAGCCCGCTTGTTGATGTTG |
| AdipoQ | GCAGGCATCCCAGGACATC | GCGATACATATAAGCGGCTTCT |
| Lep | GAGACCCCTGTGTCGGTTC | CTGCGTGTGTGAAATGTCATTG |
| PPARg | GGAAGACCACTCGCATTCCTT | TCGCACTTTGGTATTCTTGGAG |
| MCP-1 | TTAAAAACCTGGATCGGAACCAA | GCATTAGCTTCAGATTTACGGGT |
| NOS-2 | CCAAGCCCTCACCTACTTCC | CTCTGAGGGCTGACACAAGG |
| IL-1b | AAATACCTGTGGCCTTGGGC | CTTGGGATCCACACTCTCCAG |
| IL-6 | TAGTCCTTCCTACCCCAATTTCC | TTGGTCCTTAGCCACTCCTTC |
| TNFa | TAGCCCACGTCGTAGCAAAC | ACAAGGTACACCCCATCGGC |
| Mgl1 | TGAGAAAGGCTTTAAGAACTGGG | GACCACCTGTAGTGATGTGGG |
| Arg1 | CTCCAAGCCAAAGTCCTTAGAG | AGGAGCTGTCATTAGGGACATC |
| IL-10 | GCTCTTACTGACTGGCATGAG | CGCAGC TCT AGG AGC ATG TG |
| GAPDH | TGAAGCAGGCATCTGAGGG | CGAAGGTGGAAGAGTGGGAG |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hafner, H.; Chang, E.; Carlson, Z.; Zhu, A.; Varghese, M.; Clemente, J.; Abrishami, S.; Bagchi, D.P.; MacDougald, O.A.; Singer, K.; et al. Lactational High-Fat Diet Exposure Programs Metabolic Inflammation and Bone Marrow Adiposity in Male Offspring. Nutrients 2019, 11, 1393. https://doi.org/10.3390/nu11061393
Hafner H, Chang E, Carlson Z, Zhu A, Varghese M, Clemente J, Abrishami S, Bagchi DP, MacDougald OA, Singer K, et al. Lactational High-Fat Diet Exposure Programs Metabolic Inflammation and Bone Marrow Adiposity in Male Offspring. Nutrients. 2019; 11(6):1393. https://doi.org/10.3390/nu11061393
Chicago/Turabian StyleHafner, Hannah, Eric Chang, Zach Carlson, Allen Zhu, Mita Varghese, Jeremy Clemente, Simin Abrishami, Devika P. Bagchi, Ormond A. MacDougald, Kanakadurga Singer, and et al. 2019. "Lactational High-Fat Diet Exposure Programs Metabolic Inflammation and Bone Marrow Adiposity in Male Offspring" Nutrients 11, no. 6: 1393. https://doi.org/10.3390/nu11061393