Glycyrrhetinic Acid Improves Insulin-Response Pathway by Regulating the Balance between the Ras/MAPK and PI3K/Akt Pathways


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Reagents, Cell Culture, SDS-PAGE, and Western Blotting
2.3. Enrichment of Target Proteins in Cells
2.4. Colocalization of Target Proteins and GA
2.5. Evaluation of RAS Activation
2.6. PKC Kinase Activity Assay Kit
2.7. Statistical Analysis
3. Results
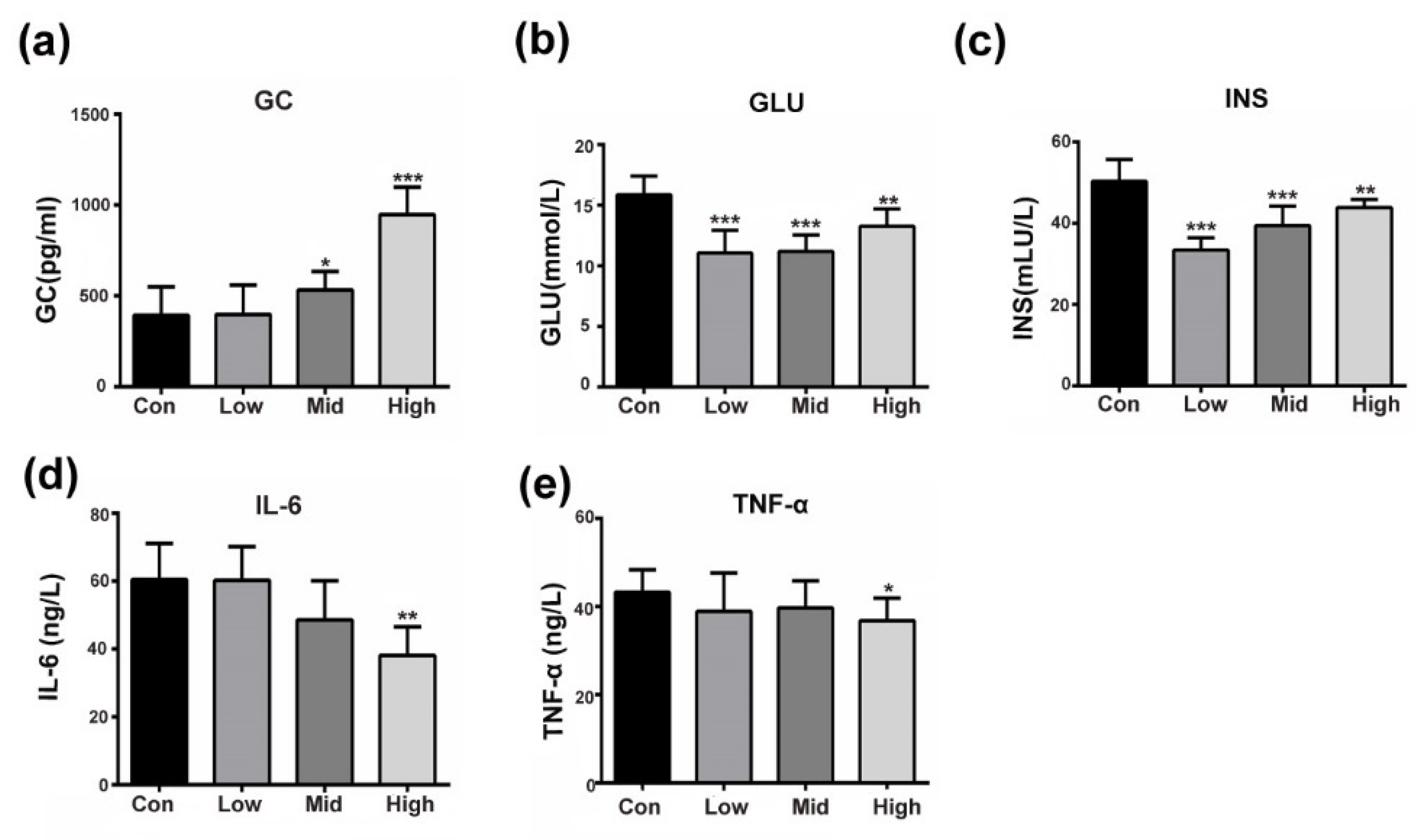
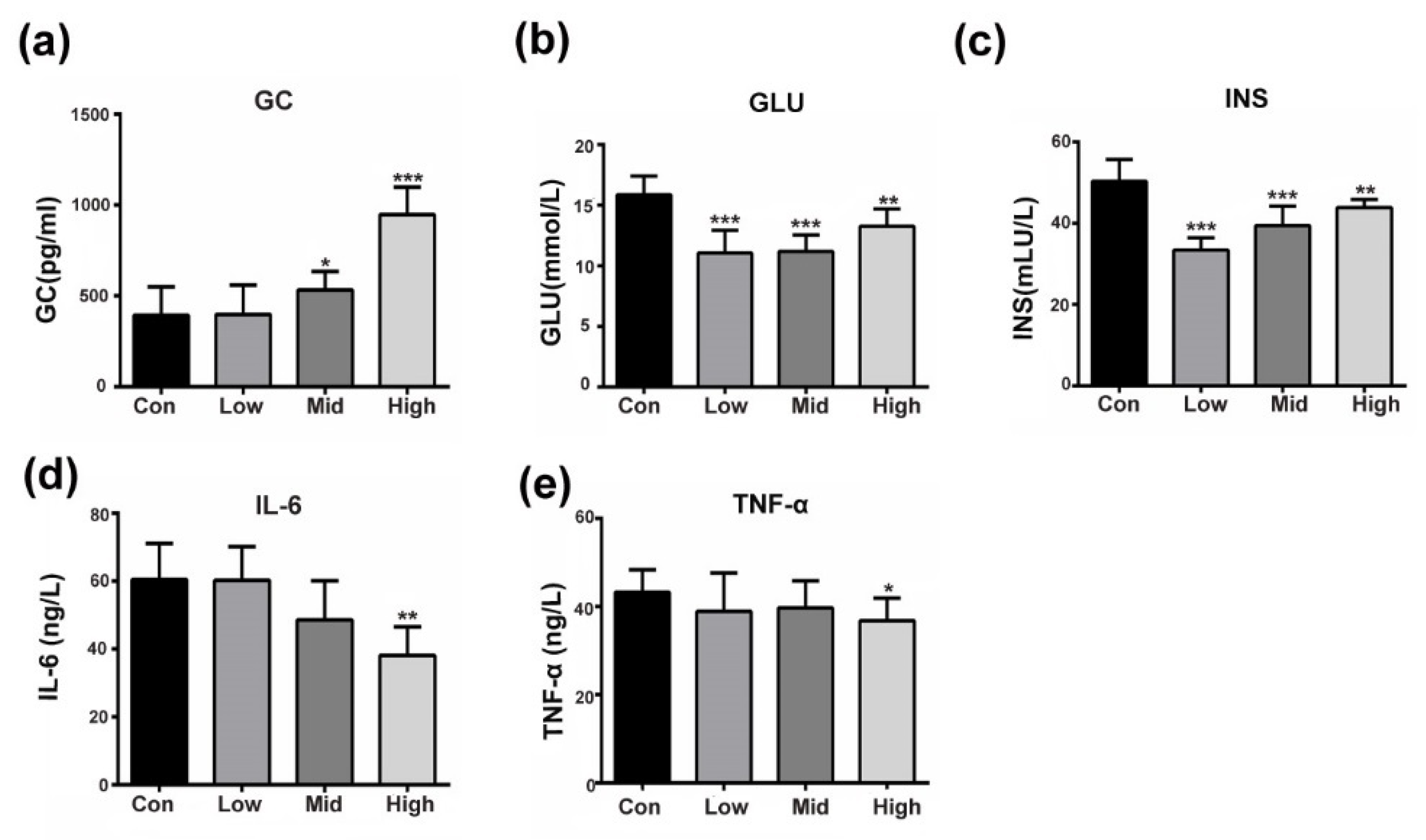
3.1. GA Affects Insulin-Responsive Pathwayin the Whole Body
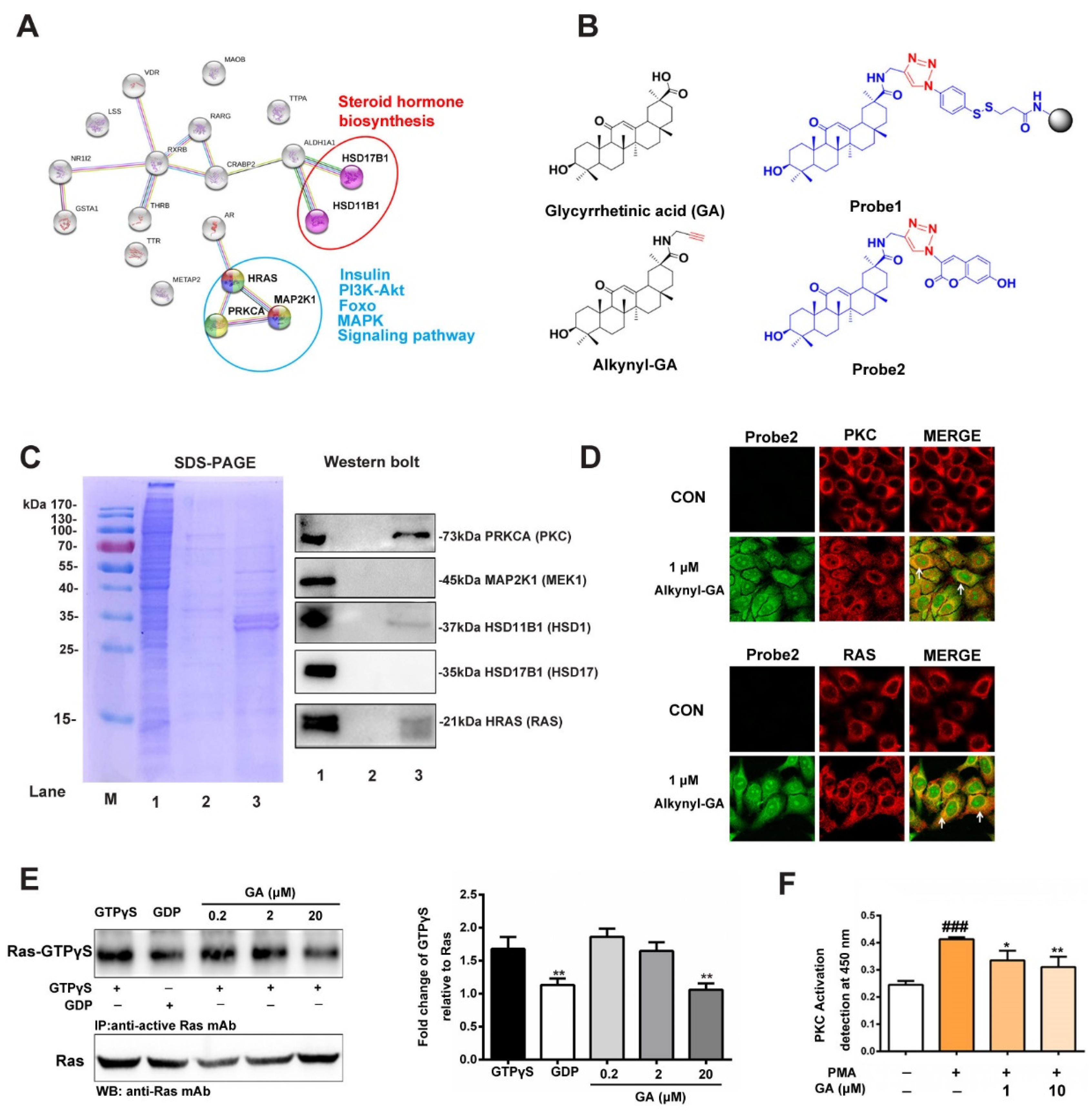
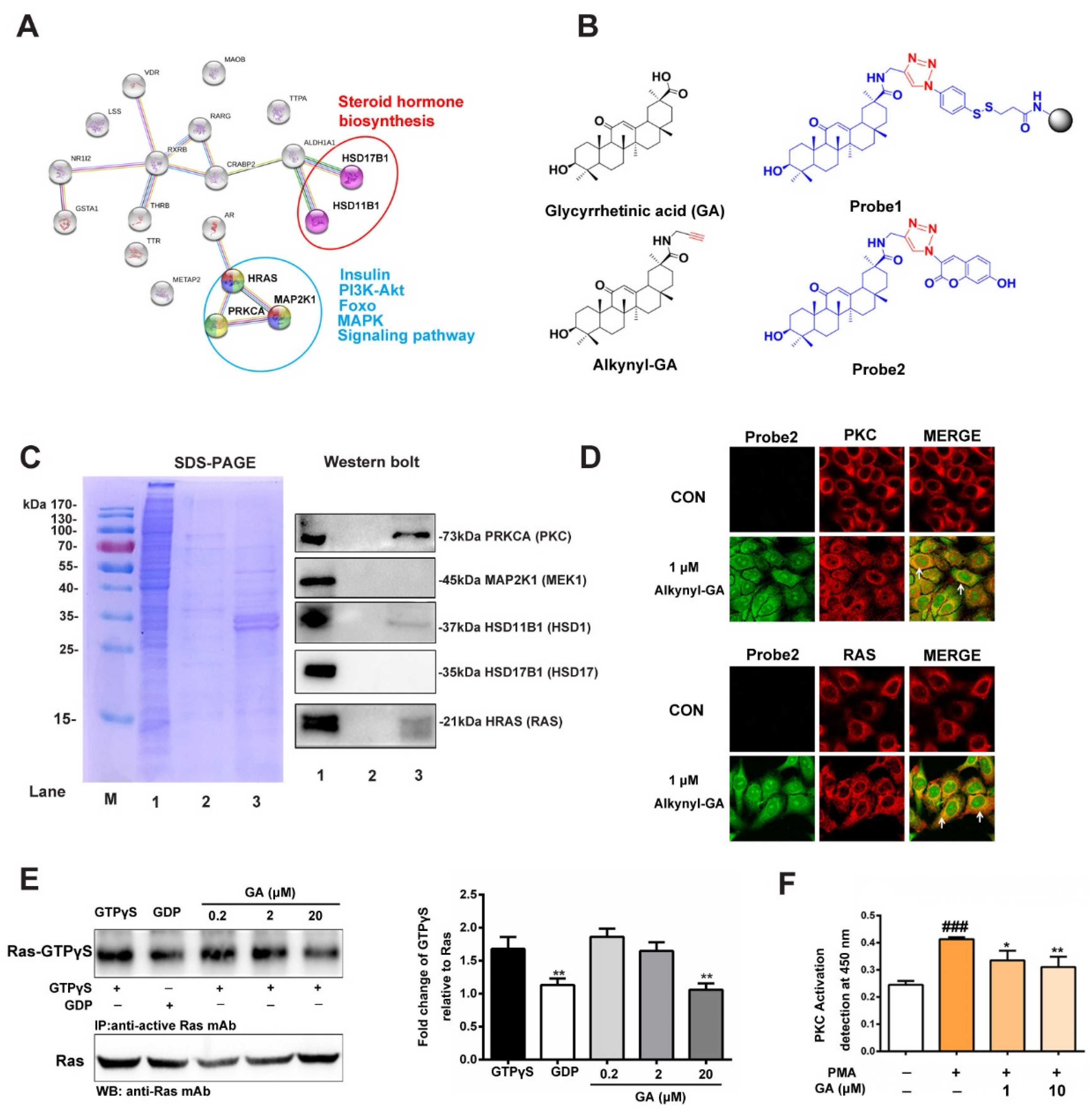
3.2. Prediction and Verification of the Targets of GA
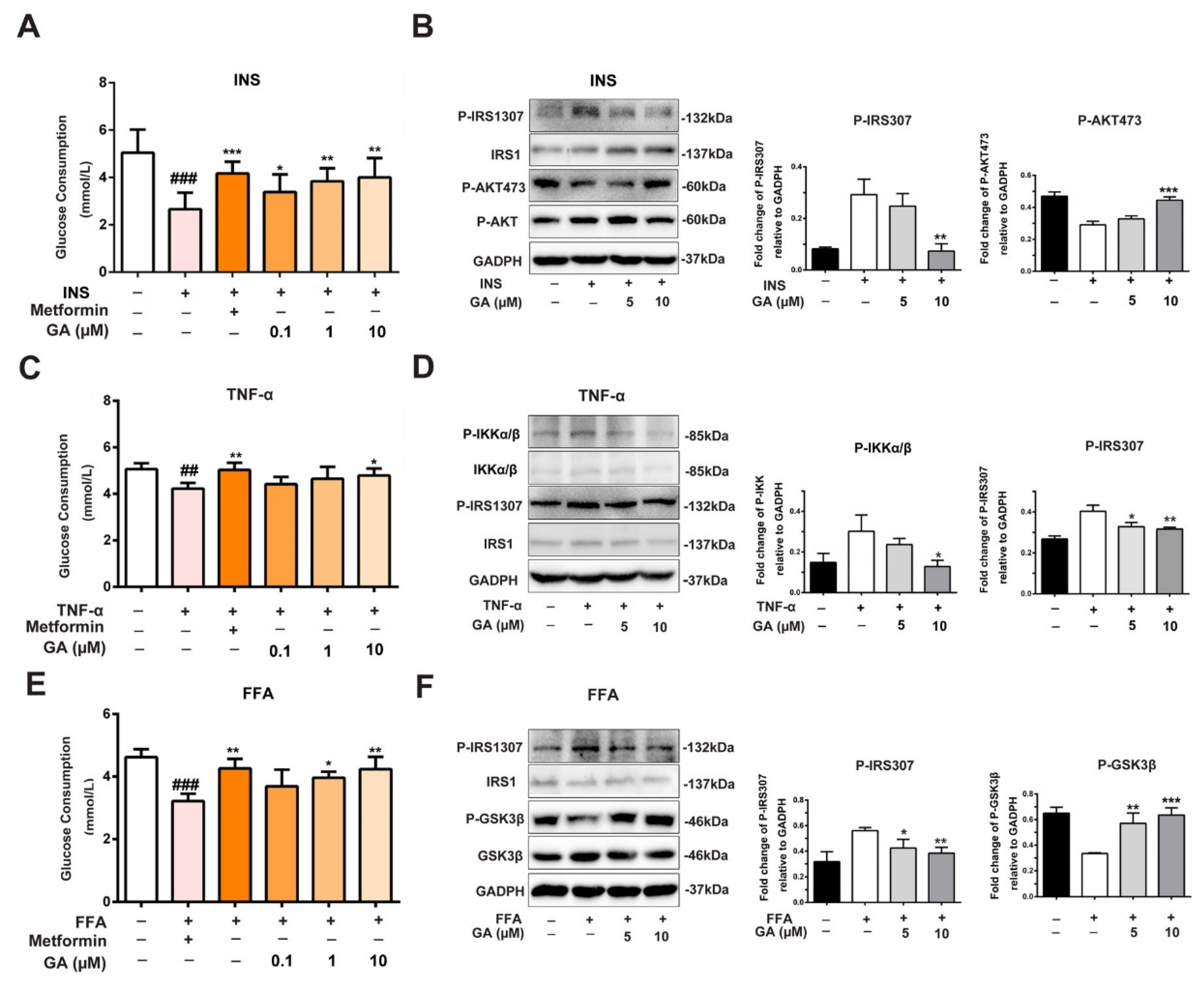
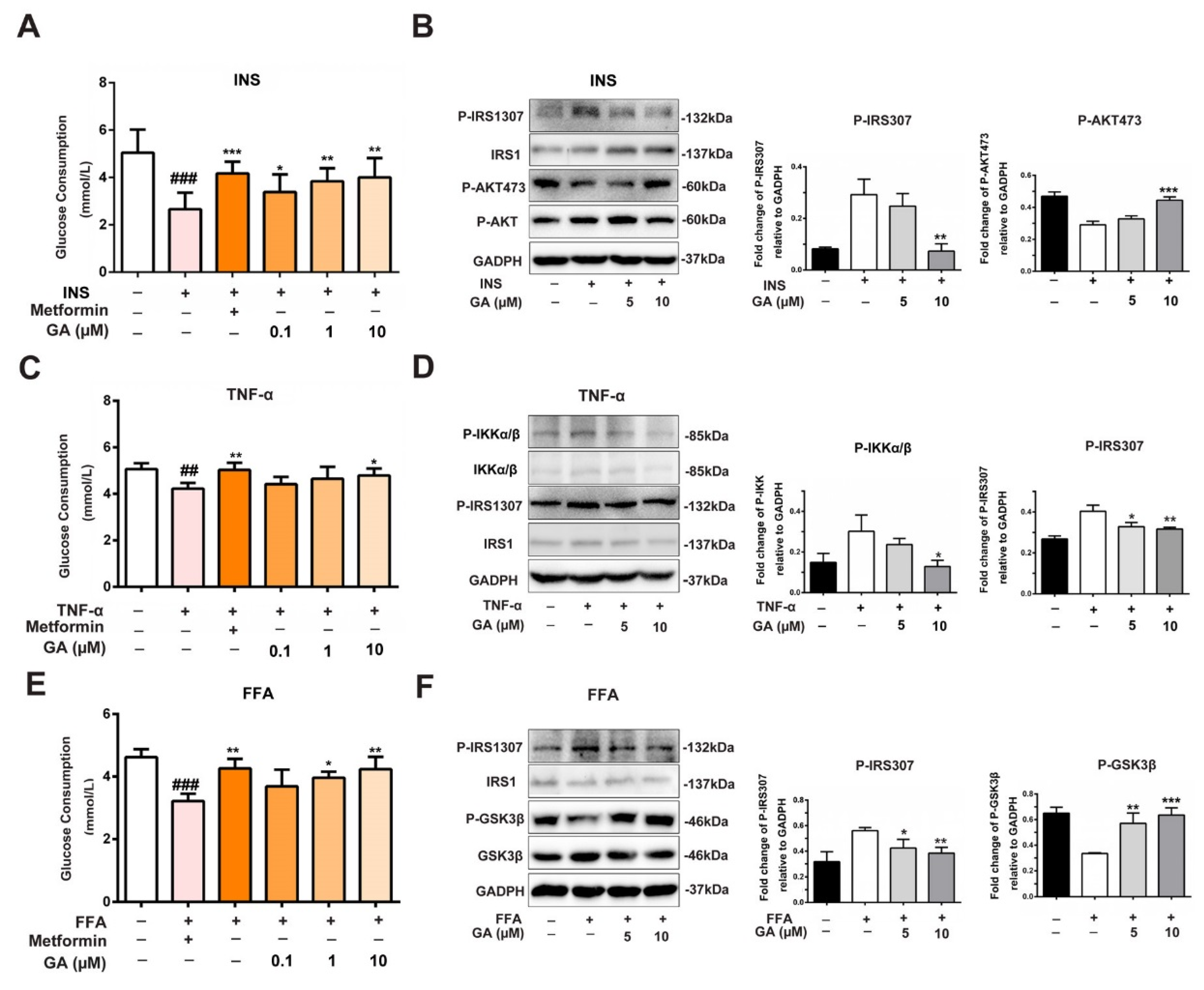
3.3. Effect of GA on Glucose Consumption and Insulin-Responsive Pathway
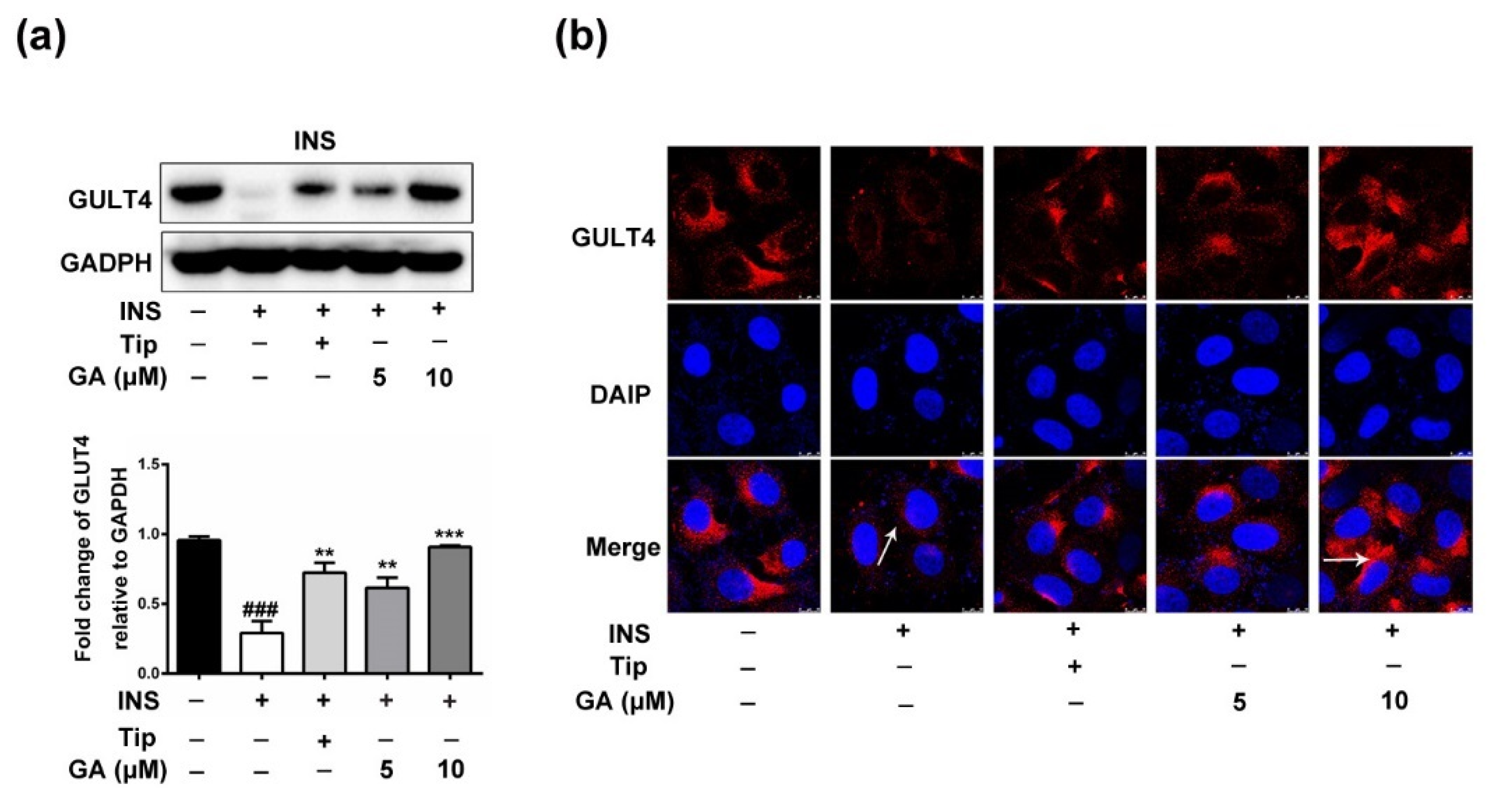
3.4. Effect of GA on GLUT4 Expression
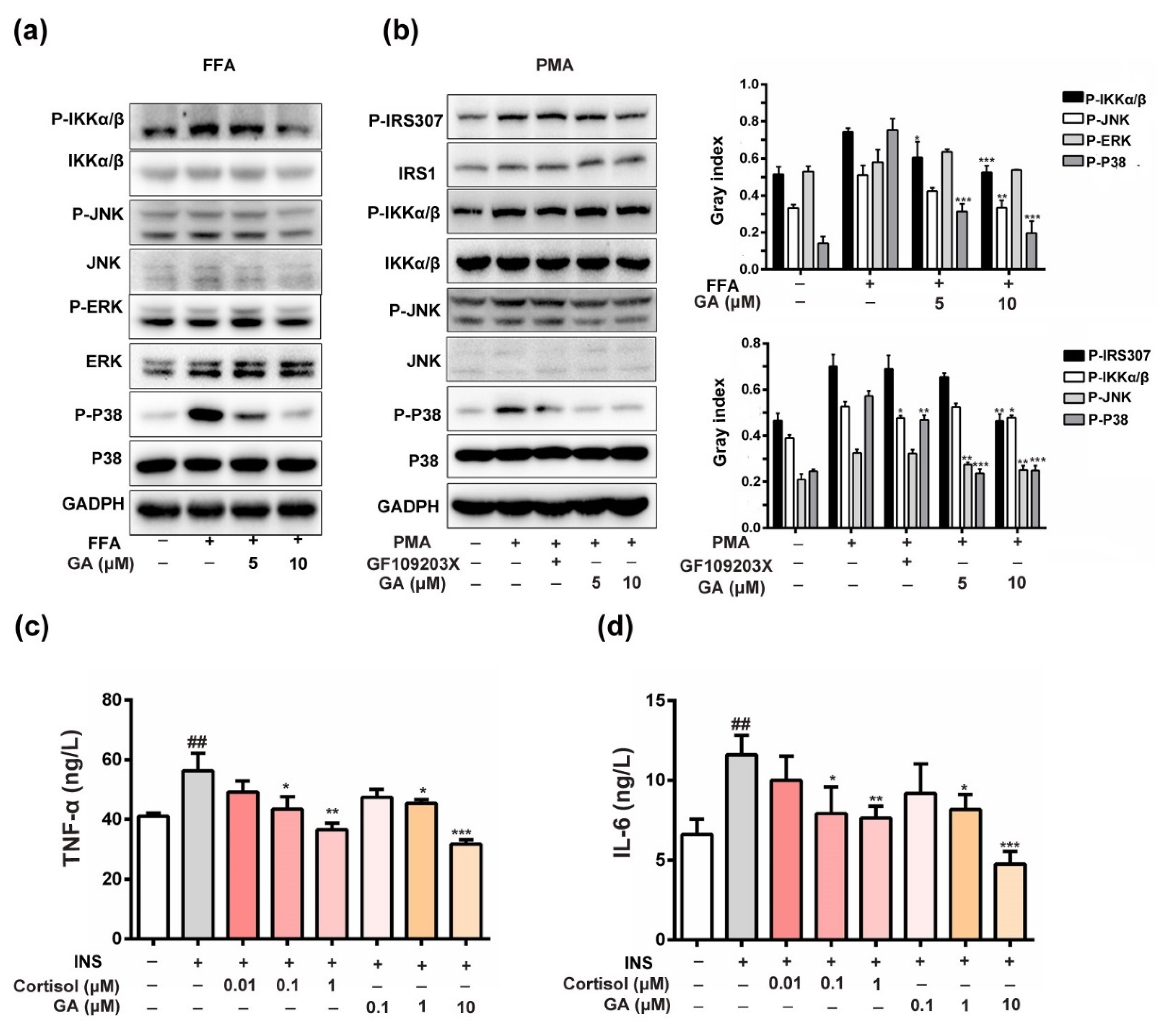
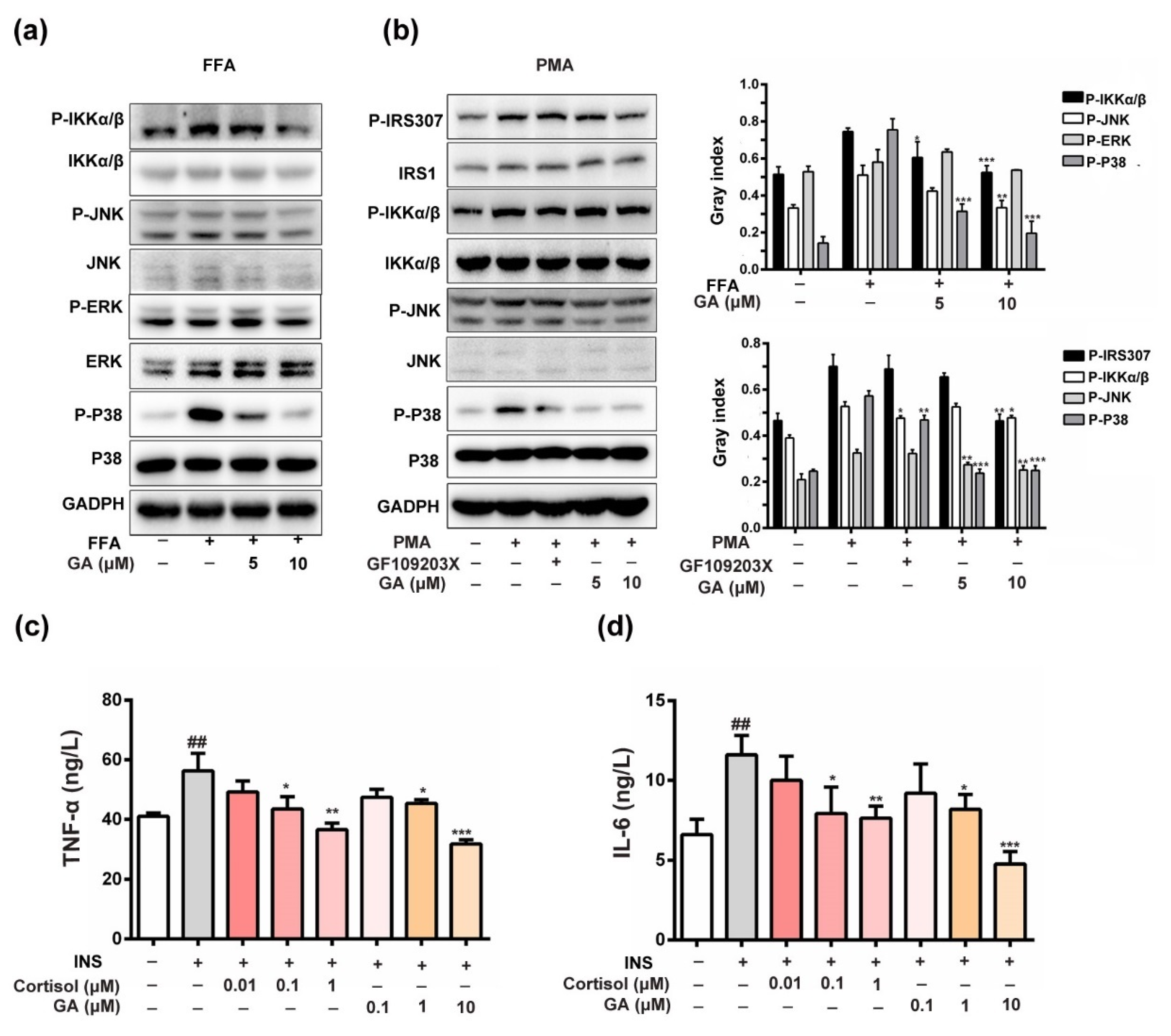
3.5. Effect of GA on Anti-Inflammation and Insulin-Responsive Pathway
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- DeFronzo, R.A.; Hompesch, M.; Kasichayanula, S.; Liu, X.; Hong, Y.; Pfister, M.; Morrow, L.A.; Leslie, B.R.; Boulton, D.W.; Ching, A.; et al. Characterization of renal glucose reabsorption in response to dapagliflozin in healthy subjects and subjects with type 2 diabetes. Diabetes Care 2013, 36, 3169–3176. [Google Scholar] [CrossRef]
- Schmitz-Peiffer, C.; Whitehead, J.P. IRS-1 regulation in health and disease. IUBMB Life 2003, 55, 367–374. [Google Scholar] [CrossRef]
- Perseghin, G.; Ghosh, S.; Gerow, K.; Shulman, G.I. Metabolic defects in lean nondiabetic offspring of NIDDM parents. A cross-sectional study. Diabetes 1997, 46, 1001–1009. [Google Scholar] [CrossRef]
- Aguirre, V.; Uchida, T.; Yenush, L.; Davis, R.; White, M.F. The c-Jun NH2-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser307. J. Biol. Chem. 2000, 275, 9047–9054. [Google Scholar] [CrossRef]
- Gual, P.; Gremeaux, T.; Gonzalez, T.; Le Marchand-Brustel, Y.; Tanti, J.F. Map kinases and mtor mediate insulin-induced phosphorylation of insulin receptor substrate-1 on serine residues 307, 612 and 632. Diabetologia 2003, 46, 1532–1542. [Google Scholar] [CrossRef]
- Rui, L.; Aguirre, V.; Kim, J.K.; Shulman, G.I.; Lee, A.; Corbould, A.; Dunaif, A.; White, M.F. Insulin/igf-1 and tnf-alpha stimulate phosphorylation of irs-1 at inhibitory ser307 via distinct pathways. J. Clin. Investig. 2001, 107, 181–189. [Google Scholar] [CrossRef]
- Yu, C.; Chen, Y.; Cline, G.W.; Zhang, D.; Zong, H.; Wang, Y.; Bergeron, R.; Kim, J.K.; Cushman, S.W.; Cooney, G.J.; et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J. Biol. Chem. 2002, 277, 50230–50236. [Google Scholar] [CrossRef]
- Harrington, L.S.; Findlay, G.M.; Gray, A.; Tolkacheva, T.; Wigfield, S.; Rebholz, H.; Barnett, J.; Leslie, N.R.; Cheng, S.; Shepherd, P.R.; et al. The TSC1-2 tumor suppressor controls insulin-PI3K Signaling via regulation of IRS proteins. J. Cell Biol. 2004, 166, 213–223. [Google Scholar] [CrossRef]
- Sano, H.; Eguez, L.; Teruel, M.N.; Fukuda, M.; Chuang, T.D.; Chavez, J.A.; Lienhard, G.E.; McGraw, T.E. RAB10, a target of the as160 RAB gap, is required for insulin-stimulated translocation of glut4 to the adipocyte plasma membrane. Cell Metab. 2007, 5, 293–303. [Google Scholar] [CrossRef]
- Aksamitiene, E.; Kiyatkin, A.; Kholodenko, B.N. Cross-talk between mitogenic RAS/MAPK and survival PI3K/AKT pathways: A fine balance. Biochem. Soc. Trans. 2012, 40, 139–146. [Google Scholar] [CrossRef]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Csuk, R.; Schwarz, S.; Siewert, B.; Kluge, R.; Strohl, D. Conversions at c-30 of glycyrrhetinic acid and their impact on antitumor activity. Arch. Pharm. 2012, 345, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.Y.; Chia, Y.Y.; Liong, S.Y.; Ton, S.H.; Kadir, K.A.; Husain, S.N. Lipoprotein lipase expression, serum lipid and tissue lipid deposition in orally-administered glycyrrhizic acid-treated rats. Lipids Health Disease 2009, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Chia, Y.Y.; Liong, S.Y.; Ton, S.H.; Kadir, K.B. Amelioration of glucose homeostasis by glycyrrhizic acid through gluconeogenesis rate-limiting enzymes. Eur. J. Pharm. 2012, 677, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Eu, C.H.; Lim, W.Y.; Ton, S.H.; bin Abdul Kadir, K. Glycyrrhizic acid improved lipoprotein lipase expression, insulin sensitivity, serum lipid and lipid deposition in high-fat diet-induced obese rats. Lipids Health Disease 2010, 9, 81. [Google Scholar] [CrossRef] [PubMed]
- Moon, M.H.; Jeong, J.K.; Lee, Y.J.; Seol, J.W.; Ahn, D.C.; Kim, I.S.; Park, S.Y. 18β-Glycyrrhetinic acid inhibits adipogenic differentiation and stimulates lipolysis. Biochem. Biophys. Res. Commun. 2012, 420, 805–810. [Google Scholar] [CrossRef]
- Fang, R.; Cui, Q.; Sun, J.; Duan, X.; Ma, X.; Wang, W.; Cheng, B.; Liu, Y.; Hou, Y.; Bai, G. PDK1/AKT/PDE4D axis identified as a target for asthma remedy synergistic withβ2 AR agonists by a natural agent arctigenin. Allergy 2015, 70, 1622–1632. [Google Scholar] [CrossRef]
- Palacios-Ortega, S.; Varela-Guruceaga, M.; Algarabel, M.; Ignacio Milagro, F.; Alfredo Martinez, J.; de Miguel, C. Effect of TNF-α on caveolin-1 expression and insulin signaling during adipocyte differentiation and in mature adipocytes. Cell. Physiol. Biochem. 2015, 36, 1499–1516. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Peraldi, P.; Budavari, A.; Ellis, R.; White, M.F.; Spiegelman, B.M. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-α- and obesity-induced insulin resistance. Science 1996, 271, 665–668. [Google Scholar] [CrossRef]
- Roden, M.; Price, T.B.; Perseghin, G.; Petersen, K.F.; Rothman, D.L.; Cline, G.W.; Shulman, G.I. Mechanism of free fatty acid-induced insulin resistance in humans. J. Clin. Investig. 1996, 97, 2859–2865. [Google Scholar] [CrossRef]
- Mor, A.; Aizman, E.; George, J.; Kloog, Y. Ras inhibition induces insulin sensitivity and glucose uptake. PLoS ONE 2011, 6, e21712. [Google Scholar] [CrossRef]
- Jiang, G.; Dallas-Yang, Q.; Liu, F.; Moller, D.E.; Zhang, B.B. Salicylic acid reverses phorbol 12-myristate-13-acetate (PMA)- and tumor necrosis factor alpha (TNFα)-induced insulin receptor substrate 1 (IRS1) serine 307 phosphorylation and insulin resistance in human embryonic kidney 293 (HEK293) cells. J. Biol. Chem. 2003, 278, 180–186. [Google Scholar] [CrossRef]
- Boden, G.; She, P.; Mozzoli, M.; Cheung, P.; Gumireddy, K.; Reddy, P.; Xiang, X.; Luo, Z.; Ruderman, N. Free fatty acids produce insulin resistance and activate the proinflammatory nuclear Factor-κB pathway in rat liver. Diabetes 2005, 54, 3458–3465. [Google Scholar] [CrossRef]
- Gao, Z.; Zhang, X.; Zuberi, A.; Hwang, D.; Quon, M.J.; Lefevre, M.; Ye, J. Inhibition of insulin sensitivity by free fatty acids requires activation of multiple serine kinases in 3T3-L1 adipocytes. Mol. Endocrinol. 2004, 18, 2024–2034. [Google Scholar] [CrossRef]
- Lyon, C.J.; Law, R.E.; Hsueh, W.A. Minireview. Adiposity, inflammation, and atherogenesis. Endocrinology 2003, 144, 2195–2200. [Google Scholar] [CrossRef]
- Akash, M.S.; Shen, Q.; Rehman, K.; Chen, S. Interleukin-1 receptor antagonist: A new therapy for type 2 diabetes mellitus. J. Pharm. Sci. 2012, 101, 1647–1658. [Google Scholar] [CrossRef]
- Copps, K.D.; White, M.F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef]
- Hubbard, S.R. The insulin receptor. Both a prototypical and atypical receptor tyrosine kinase. Cold Spring Harb. Perspect. Biol. 2013, 5, a008946. [Google Scholar] [CrossRef]
- Combettes-Souverain, M.; Issad, T. Molecular basis of insulin action. Diabetes Metab. 1998, 24, 477–489. [Google Scholar]
- Steelman, L.S.; Abrams, S.L.; Whelan, J.; Bertrand, F.E.; Ludwig, D.E.; Basecke, J.; Libra, M.; Stivala, F.; Milella, M.; Tafuri, A.; et al. Contributions of the Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways to leukemia. Leukemia 2008, 22, 686–707. [Google Scholar] [CrossRef]
- Zhu, D.; Wang, Y.; Du, Q.; Liu, Z.; Liu, X. Cichoric acid reverses insulin resistance and suppresses inflammatory responses in the glucosamine-induced HepG2 cells. J. Agric. Food Chem. 2015, 63, 10903–10913. [Google Scholar] [CrossRef]
- Hou, Y.; Nie, Y.; Cheng, B.; Tao, J.; Ma, X.; Jiang, M.; Gao, J.; Bai, G. Qingfei Xiaoyan Wan, a traditional Chinese medicine formula, ameliorates Pseudomonas aeruginosa–induced acute lung inflammation by regulation of PI3K/AKT and Ras/MAPK pathways. Acta Pharm. Sin. B 2016, 6, 212–221. [Google Scholar] [CrossRef]
- Satoh, T. Molecular mechanisms for the regulation of insulin-stimulated glucose uptake by small guanosine triphosphatases in skeletal muscle and adipocytes. Int. J. Mol. Sci. 2014, 15, 18677–18692. [Google Scholar] [CrossRef]
- Naruse, K.; Rask-Madsen, C.; Takahara, N.; Ha, S.W.; Suzuma, K.; Way, K.J.; Jacobs, J.R.; Clermont, A.C.; Ueki, K.; Ohshiro, Y.; et al. Activation of vascular protein kinase C-β inhibits Akt-dependent endothelial nitric oxide synthase function in obesity-associated insulin resistance. Diabetes 2006, 55, 691–698. [Google Scholar] [CrossRef]
- Li, J.; DeFea, K.; Roth, R.A. Modulation of insulin receptor substrate-1 tyrosine phosphorylation by an Akt/phosphatidylinositol 3-kinase pathway. J. Biol. Chem. 1999, 274, 9351–9356. [Google Scholar] [CrossRef]
- De Fea, K.; Roth, R.A. Modulation of insulin receptor substrate-1 tyrosine phosphorylation and function by mitogen-activated protein kinase. J. Biol. Chem. 1997, 272, 31400–31406. [Google Scholar] [CrossRef]
- Tsukumo, D.M.; Carvalho-Filho, M.A.; Carvalheira, J.B.; Prada, P.O.; Hirabara, S.M.; Schenka, A.A.; Araujo, E.P.; Vassallo, J.; Curi, R.; Velloso, L.A.; et al. Loss-of-function mutation in toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes 2007, 56, 1986–1998. [Google Scholar] [CrossRef]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef]
- Nandipati, K.C.; Subramanian, S.; Agrawal, D.K. Protein kinases. Mechanisms and downstream targets in inflammation-mediated obesity and insulin resistance. Mol. Cell. Biochem. 2017, 426, 27–45. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Yang, S.; Zhang, M.; Wang, Z.; He, X.; Hou, Y.; Bai, G. Glycyrrhetinic Acid Improves Insulin-Response Pathway by Regulating the Balance between the Ras/MAPK and PI3K/Akt Pathways. Nutrients 2019, 11, 604. https://doi.org/10.3390/nu11030604
Zhang Y, Yang S, Zhang M, Wang Z, He X, Hou Y, Bai G. Glycyrrhetinic Acid Improves Insulin-Response Pathway by Regulating the Balance between the Ras/MAPK and PI3K/Akt Pathways. Nutrients. 2019; 11(3):604. https://doi.org/10.3390/nu11030604
Chicago/Turabian StyleZhang, Yuan, Shengnan Yang, Man Zhang, Zhihua Wang, Xin He, Yuanyuan Hou, and Gang Bai. 2019. "Glycyrrhetinic Acid Improves Insulin-Response Pathway by Regulating the Balance between the Ras/MAPK and PI3K/Akt Pathways" Nutrients 11, no. 3: 604. https://doi.org/10.3390/nu11030604