1. Introduction
Probiotics are defined as: “live microorganisms which when administered in adequate amounts confer a health benefit on the host” [
1,
2]. While they have a number of beneficial health effects in common, it is acknowledged that some functional activities are strain-specific. Consequently, there is a need for rigorous strain-to-strain comparison of the same species in regard to therapeutic activity and efficacy [
1,
2]. Since the functional activity of taxonomically similar or even identical strains manufactured by different producers varies, the health-promoting properties of a specific formulation cannot be extrapolated from one formulation to another without in vitro and in vivo comparative testing [
3,
4]. Fittingly, it is well established that the immune-modulatory effects of two widely investigated Lactobacilli,
L. plantarum and
L. gasseri, i.e., their ability to reciprocally modulate IL-10/IL-12 production in cell cultures, changes widely according to culture conditions and media composition [
5,
6].
Recently, a cluster of studies have reported that different lots of a commercially available multistrain probiotic preparation, available in Europe and North America under the brand name of VSL#3
®, show different biochemical and immunological properties in vitro and exert variable activity in rodent models of intestinal inflammation, depending on the manufacturing site [
7,
8,
9,
10], which suggests that the health-promoting properties of these preparations are affected by the manufacturing processes [
8,
11,
12].
Taken together, these data support the notion that culture conditions, in addition to strain taxonomy, impact on the probiotic properties of genetically similar bacterial strains and that different manufacturing sources might result in probiotic preparations with profound functional variability despite identical taxonomy [
7,
10]. Together, these studies raise the need for a more appropriate definition of the functional properties of the bacterial strains commonly commercialized as probiotics either alone or blended.
In recent years, two different probiotic blends that contain the same mixture of seven taxonomically identical bacterial species have been made commercially available (i.e., VSL#3 and Vivomixx). According to the box labeling, the two blends are mixture of four strains of Lactobacilli, three strains of Bifidobacteria, and one strain of Streptococcus thermophilus. However, the relative proportion of each component in the final mixture and manufacturing conditions are not reported.
Because both VSL#3 and Vivomixx are recommended for use at the same daily dose, and both claim similar health promoting activities, we have set up a study to investigate the effectiveness of the two formulations in modulating the structure of the intestinal microbiota and markers of inflammation in two rodent models of colitis. Additionally, since some of the beneficial effects exerted by probiotics are mediated by their metabolites, including lactate and short-chain fatty acids (SCFA) [
13], we have performed an in vitro/in vivo study to test whether the two mixtures generate similar amounts of lactate and SCFAs.
2. Materials and Methods
2.1. Probiotics
Two different formulations of probiotics were used in this study: VSL#3®, indicated as Blend 1, and Vivomixx®) indicated as Blend 2. Both lots were commercially available in pharmacies in Italy. The lot numbers and expiration dates were the following: VSL#3® lot 705006, expiration 05/2019, and Vivomixx® lot 1719104, expiration 31/07/2019. The list of strains as they appear on their respective commercial packaging is as follow: Streptococcus thermophilus BT01, Lactobacillus plantarum BP06, Bifidobacterium breve BB02, Bifidobacterium longum BL03, Bifidobacterioum infantis BI04, Lactobacillus acidophilus BA05, Lactobacillus paracasei BP07, Lactobacillus debrueckii subsp. bulgaricus BD08, for VSL#3® and Streptococcus thermophilus DSM24731®, Lactobacillus plantarum DSM24730®, Bifidobacterium breve DSM24732®, Lactobacillus paracasei DSM24733®, Lactobacillus delbrueckii subsp. bulgaricus DSM24734®, Lactobacillus acidophilus DSM 24735®, Bifidobacterium longum DSM24736®, Bifidobacterium infantis DSM24737®, for Vivomixx®. The different code names indicate that the bacteria have different sources and are registered in different cell repositories. The two batches were maintained according to the manufacturer instructions until used.
2.2. Bacterial Growth Conditions and Viability Test
Microorganisms from lyophilized probiotic products stored at 4 °C were inoculated onto de Man-Rogosa-Sharpe (MRS) broth (Thermo Scientific™, Waltham, MA, USA) containing 0.05% l-cysteine-HCl (Sigma-Aldrich, St. Louis, MO, USA). Viable microorganisms were determined by plating serial 10-fold dilutions onto MRS-agar (Thermo Scientific™) containing 0.05% l-cysteine-HCl. Tests were performed in duplicate. Colonies were enumerated after incubation of plates at 37 °C for 48 h under anaerobic conditions in Anaerogen system (Thermo Scientific™). Individual strains from Blend 2 were provided by Mendes S.A.-Via Giacometti, 1, 6900 Lugano, Switzerland).
2.3. Residual Carbohydrate Determination and Lactic Acid Production
Ability of lactic acid bacteria to ferment carbohydrates was determined using MRS broth (basal MRS) or MRS supplemented with filter-sterilized fructose. Microorganisms from lyophilized probiotic products were suspended in basal MRS at a concentration of 1010 viable bacteria/ml. The stock suspension was then diluted to 108 viable bacteria/ml in basal MRS broth or MRS supplemented with 0.5 and 2% fructose and cultured at 37 °C for 21 h under anaerobic conditions. Cells were removed by centrifugation (7600× g, 15 min) and supernatants stored at −20 °C for subsequent analyses. Residual glucose and fructose from cell-free culture samples were determined using Enzytec™ Liquid d-Glucose/d-Fructose (R-Biopharm AG, Darmstadt, Germany). Lactic acid production was evaluated with a commercial kit for the determination of D- and L-lactic acid (Test-Combination, UV-method; Boehringer Mannheim/R-Biopharm, Darmstadt, Germany). The NADH increase, stoichiometric to the amount of d- and l-lactic acid, was determined by absorbance at 340 nm, according to manufacturer instruction. Fermentations were carried out in triplicate as independent experiments. The reported data correspond to the median of at least three measurements from three independent experiments. Viable counts at the beginning and the end of each experiment were determined as CFU/mL.
2.4. Animals and Colitis Protocols
Balb/c mice were obtained from Charles River (Lecco, Italy). The colonies were maintained in the animal facility of the University of Perugia. The mice were housed under controlled temperatures (22 °C) and photoperiods (12:12-hour light/dark cycle), allowed unrestricted access to standard mouse chow and tap water, and allowed to acclimate to these conditions for at least 5 days before inclusion in an experiment. The study was conducted in agreement with the Italian law and the protocol was approved by an ethical committee of the University of Perugia and by a National Committee of Italian Ministry of Health permit n° 1126/2016-PR. The health and body conditions of the animals were monitored daily by the veterinarian in the animal facility. Only male mice were used in each experiment. The study protocol caused minor suffering; however, animals losing more than 25% of their initial body weight were euthanized. In the first model, colitis was induced in Balb/c mice using 5% DSS (DSS: Dextran Sulfate, Sodium Salt of Affymetrix USA, molecular mass 40–50 kDa) in drinking water for 8 consecutive days. We used 8–12 animals for each experimental group. Animals were monitored daily. In the second model, the model of colitis induced by 2,4,6-trinitrobenzenesulfonic acid (TNBS), Balb/c mice were fasted for 12 h (day
−1). The day after (day 0), mice were anesthetized, and a 3.5 F catheter inserted into the colon such that the tip was 4 cm proximal to the anus. To induce colitis, 1 mg of TNBS (Sigma Chemical Co., St Louis, MO, USA) in 50% ethanol was administered via a catheter into the lumen using a 1 mL syringe (injection volume of 100 μL); control mice received 50% ethanol alone. We used 5–7 animals for each experimental group. Animals were monitored daily for 4 days. At the end of the experiments, the surviving mice were sacrificed, blood samples collected by cardiac puncture, and the colon was excised, weighed, and evaluated for macroscopic damage. In some groups of mice, we also administered Blend 1 or Blend 2 by oral gavage at the concentration of 50 × 10
9 probiotic cfu/kg of body weight dissolved in saline solution [
7,
14,
15]. Both treatments were administered daily from day 0 to the day of sacrifice. In both animal models, the severity of colitis was measured each day for each mouse by assessing body weight, fecal blood, and stool consistency. Each parameter was scored from 0 to 4 as described previously [
7]. At the end of the study, surviving animals were used for different biochemical and functional analyses as detailed in each Figure, assuring that at least 4 animals for each experimental group were available for each determination.
2.5. Histology
Samples of distal colon (2–3 cm from the anus) were fixed in buffered formalin, cut into 5-µm-thick sections (150 µm between each section, four to eight per fragment per colon), and stained with Hematoxylin and eosin (H&E).
2.6. Isolation of Lamina Propria Cells
The cells were isolated from the colon lamina propria using the Lamina Propria Dissociation Kit (Miltenyi Biotec, Bergisch Gladbach, Germania; 130-097-401), according to the instructions.
2.7. Flow Cytometry
Flow cytometry analyses were carried out using a two-laser standard configuration BD FACSVia™ flow cytometry system. We analyzed 6–10 mice for each experimental group. Data were analyzed using FlowJo software (TreeStar, Ashland, OR, USA). The gates were set using fluorescence minus one (FMO,) control strategy. FMO controls are samples that include all conjugated Abs present in the test samples except one. The channel in which the conjugated Ab is missing is the one for which the FMO provides a gating control. The following mAbs were used: CD4 PerCp-Cy5.5 (RM4-5, eBioscience, San Diego, CA, USA); CD11b Pe-Cy7 (M1/70, eBioscience); Gr1 PE (RB6-8C5, BioLegend); IL-10 FITC (JES5-16E3, eBioscience) and FoxP3 APC (FJK-16s, eBioscience).
2.8. Reverse Transcription of mRNA and Real-Time PCR
Colon samples were immediately frozen in liquid nitrogen and stored at −80 °C until used, mechanically homogenated with the aid of a pestle, and the obtained materials re-suspended in 1 mL of Trizol (Thermo Scientific™). The RNA was extracted according to the manufacturer’s protocol. After purification from genomic DNA using DNase-I treatment (Thermo Scientific™), 1 µg of RNA from each sample was reverse-transcribed using random hexamer primers with Superscript-II (Thermo Scientific™) in a 20 μL reaction volume; 10 ng cDNA were amplified in a 20 μL solution containing 200 nM of each primer and 10 μL of SYBR Select Master Mix (Thermo Scientific™). All reactions were performed in triplicate, and the thermal cycling conditions were as follows: 3 min at 95 °C, followed by 40 cycles of 95 °C for 15 s, 56 °C for 20 s, and 72 °C for 30 s, using a Step One Plus machine (Applied Biosystem). The relative mRNA expression was calculated accordingly to the 2
(−ΔCt) method comparing the expression of different genes to that of GAPDH housekeeping. Primers were designed using the software PRIMER3 (
http://frodo.wi.mit.edu/primer3/) using published data obtained from the NCBI database. For each drawn primer we verified the correct alignment in the target gene using UCSC genome browser (
https://genome.ucsc.edu/cgi-bin/hgBlat) and we verified the efficiency in the laboratory using cDNA at known concentration in scalar dilutions. Alternatively, for some genes the TaqMan probes (Thermo Scientific™) were used, with TaqMan GEX Master Mix (Thermo Scientific™). The primer used was conducted as follows (forward and reverse): Gapdh (for CTGAGTATGTCGTGGAGTCTAC; rev GTTGGTGGTGCAGGATGCATTG; Ifn-γ (for GCTTTGCAGCTCTTCCTCAT; rev ATCCTTTTGCCAGT), Tnf-α (for CCACCACGCTCTTCTGTCTA; rev AGGGTCTGGGCCATAGAACT), Il-6 (for CTTCACAAGTCGGAGGCTTA; rev TTCTGCAAGTGCATCATCGT), Il-1β (for GCTGAAAGCTCTCCACCTCA; rev AGGCCACAGGTATTTTGTCG), Tgf-β (for TTGCTTCAGCTCCACAGAGA; rev TGGTTGTAGAGGGCAAGGAC), Il-10 (for CCCAGAAATCAAGGAGCATT; rev CTCTTCACCTGCTCCACTGC), FoxP3 (for TCTTCGAGGAGCCAGAAGAG; rev AGCTCCCAGCTTCTCCTTTT). We also used the following TaqMan probes: Cd38 (Mm01220906_m1 Thermo Scientific™) and c-myc (Mm00487804_m1 Thermo Scientific™).
2.9. LC/ESI/MS Analysis of SCFAs in Feces
SCFAs in fecal samples of mice were measured using liquid chromatography-mass spectrometry as described by [
16] using a LTQ-FT system (Thermo Scientific™) equipped with a Phenomenex Synergi Polar-RP column (Phenomenex, Torrance, CA, USA). The results are calculated in nmol/mg of fecal sample.
2.10. Metagenomics
DNA extraction. The microbial DNA was purified from mouse stool samples (4 mice for each experimental group), using the PureLink Microbiome DNA Purification Kit (Thermo Scientific™), according the manufacturer’s instructions. Briefly, approximately 100 mg of mouse stool were weighed and transferred to the bead tube and mixed thoroughly with 700 µL of S1-lysis buffer and 100 µL of S2-lysis enhancer to create a homogeneous sample and incubated at 65 °C for 10 min. The bead tubes were homogenized for 10 minutes at maximum speed on the horizontal vortex mixer, then centrifuged at 14,000× g for 5 min, and 400 µL of supernatant was transferred to a clean micro-centrifuge tube and vortexed immediately with 250 µL of S3-cleanup buffer. After 2 min of centrifugation, 500 µL of supernatant was transferred in a new Eppendorf and mixed with 900 µL of S4-binding buffer. Then 700 µL of sample mixture were loaded onto a spin column tube and centrifuged at 14,000× g for 1 min (2×). The spin column was then washed with 500 µL of S5-wash buffer and the flow-through was discarded. Finally, the spin column was placed in a clean tube, and the purified DNA was eluted with 100 µL of S6-elution buffer. The isolated DNA was quantified with a Qubit dsDNA HS Assay Kit on Qubit 3.0 fluorometer (Thermo Scientific™), according the manufacturer’s instructions, and then stored at −20 °C.
16S rRNA sequencing. Sequencing was performed using an Ion 16S Metagenomics Kit (Thermo Scientific™) on the Ion Torrent S5 platform (Thermo Scientific™). Briefly, 3 ng of DNA was subjected to amplification of 16S rRNA libraries using two primer pools to amplify seven hypervariable regions of bacterial 16S rRNA. Primers were partially digested and barcoded adapters (Ion Xpress Barcode Adapters 1-16 Kit) ligated to the amplicons, using the Ion Plus Fragment Library Kit (Thermo Scientific™), purified using Agencourt AMPure XP beads (Beckman Coulter, Brea, CA, USA) according to the manufacturer’s protocol, and stored at −20 °C until further processing. The concentration of each 16S library was determined by qPCR using the Ion Library Quantitation Kit and a Qubit 3.0 fluorometer (Thermo Scientific™). The library was diluted to ~100 pM prior to template preparation. Template preparation of the barcoded libraries was performed using the Ion Chef and the Ion S5 System (Thermo Scientific™). A maximum of 16 barcoded 16S samples were sequenced on a Ion 520 chip (Thermo Scientific™) using the Ion 510 & Ion 520 & Ion 530 Kit-Chef (Thermo Scientific™) according to the manufacturer’s instructions.
Metagenomics analysis. Automated analysis, annotation, and taxonomical assignment were generated using Ion Reporter Software—Metagenomics Workflow (Ion Reporter 5.10.2.0, Thermo Scientific™). The Ion Reporter Software enables the rapid identification (at genus or species level) of microbes present in each sample, using both curated Greengenes and premium curated MicroSEQ ID 16S rRNA reference databases. The Ion Reporter metagenomics workflow also provides primer information, classification information, percent ID, and mapping information.
Data visualization and statistical analyses of taxonomy. Data visualization and statistical analyses were performed using Krona and QIIME™ analysis software, and related packages were used for diversity and correlation analyses. Principal coordinates analysis (PCoA) was conducted with identified reads/OTUs using classical multidimensional scaling (Bray-Curtis) to analyze distribution of dissimilarities and analysis of variance using abundance data.
2.11. Statistical analysis
The ANOVA followed by a non-parametric Mann-Whitney U test or a two-tailed unpaired Student t test were used for statistical comparisons (* p < 0.05) using the Prism 6.0 software (GraphPad, San Diego, CA, USA).
4. Discussion
The relevance of industrial production processes in modulating the functionality of probiotics in vivo is documented and we and others have recently provided evidence that different lots of the same product manufactured at different industrial sites, exert different in vitro and in vivo properties [
7,
17].
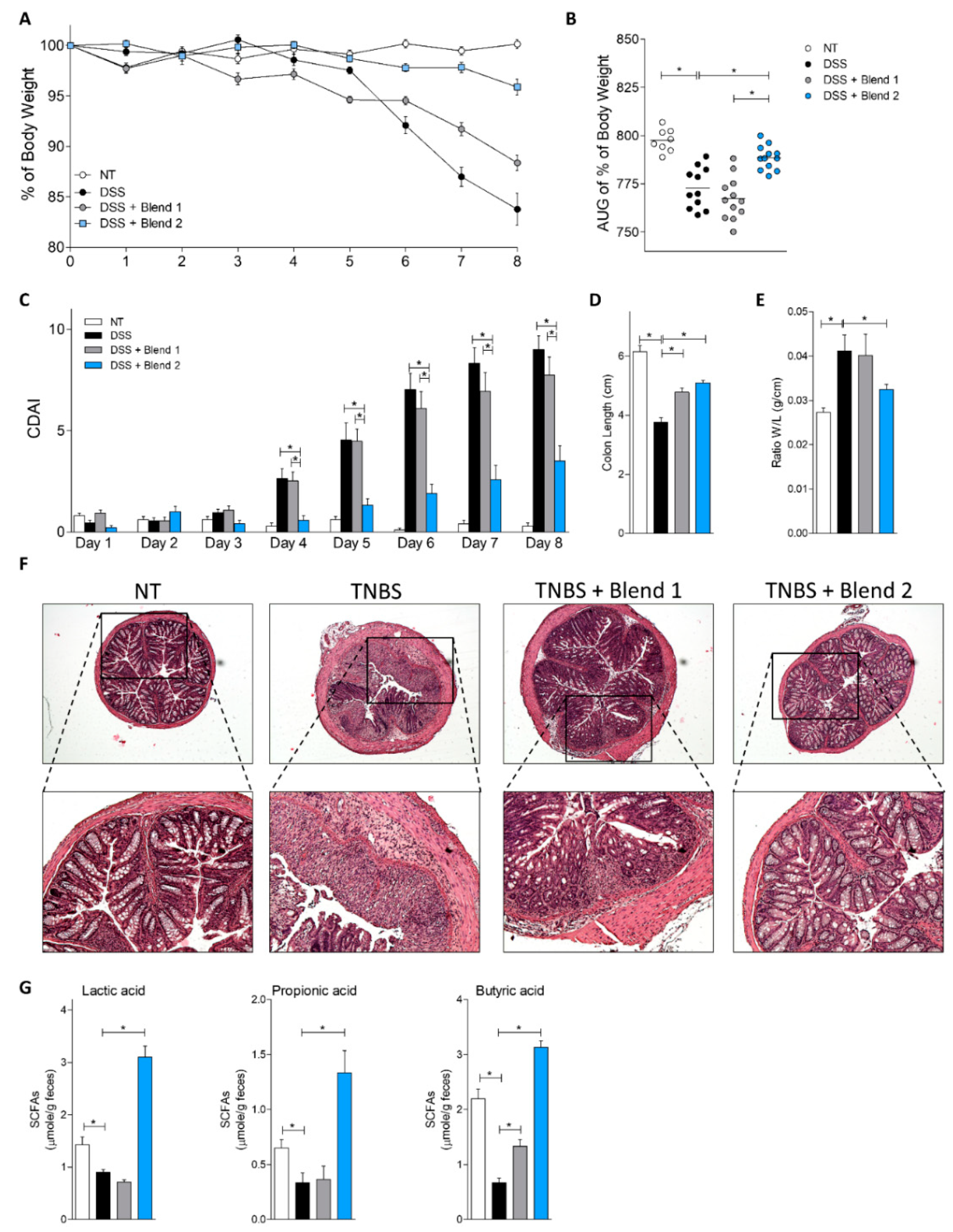
In the present study we have investigated the functionality of two multistrain preparations of probiotics in rodent models of colitis. As stated on their respective packages, the two preparations used in these studies are claimed to contain a blend of seven eight probiotic species. However, it should be noted that the two mixtures differ on several characteristics. First of all, that probiotic strains included in each preparation are labeled in a different manner suggesting a genetic diversity and/origin. Second, while both mixtures are reported to contain eight bacterial strains, the relative composition in terms of amount of each strain in the whole mixture is not known and might be different. Because individual probiotic strains perform different metabolic and immunological functions, variability in the amount of a specific strain in the final blend is likely to impact on mixture functionality. Third, the two preparations originate from different industrial sites and the composition of media used for industrial manufacturing are not known in detail, though one formulation (Blend 1) claims to be dairy-free, and this fact may introduce another robust source of variability.
Beside the above-mentioned difference, the two probiotic mixtures differ in their functionality. In vitro investigations on the two blends demonstrated that the two mixtures differed significantly in their ability to produce d- and l-Lactic acid, and that the Blend 2 preparation generated a significantly higher amount of both d- and l-lactic acids than the counterpart. Because lactic acid is a part of the probiotic functionality and is a negative regulator of pro-inflammatory mediators such matrix metalloproteases, TNFα, CCL2 and CCL7, and NF-KB in monocytes [
18,
19], while inducing the expression of anti-inflammatory markers such as IL-23 [
18,
20] and EGR2 (a marker of M2 phenotype [
20]), we speculated that the enhanced generation of d-and l-lactic acid by the Blend 2 formulation might contribute to its anti-inflammatory activity. Our in vitro analysis using individual probiotic species demonstrated that lactic acid is essentially produced, as expected, by the various Lactobacillus species, with a minor contribution by Bifidobacteria. Lactic acid is actively used by intestinal bacteria to generate propionate and butyrate, i.e., two of the best characterized bacterial metabolites in the gut [
21]. Because the SCFAs are key bacterial metabolites and represent an essential component of probiotic functionality [
22,
23], we have examined whether these in vitro findings translate in vivo. Fittingly, results obtained in the DSS model of colitis demonstrated that while intestinal inflammation in DSS-treated mice resulted in a dramatic reduction of fecal content of lactate and two SCFAs, i.e., propionate and butyrate (
Figure 3G), this pattern was reversed by treating mice with Blend 2 (i.e., the multistrain formulation that effectively released more lactate in vitro). Finally, since in vivo generation of lactate and butyrate in the intestine is also contributed to by Bacteroides species, the fact that Blend 2 administration reversed the Bacteroidetes reduction caused by DSS (
Figure 5A) is further confirmation of the different functionality of the two blends [
24,
25].
Of relevance exposure to DSS resulted in a robust modification of intestinal microbiota with an increase in representation of
Proteobacteriaceae and a concomitant reduction of
Bacteroides without any significant changes in
Firmicutes [
26]. This finding confirms previous reports in various mice models of colitis, showing that progression of the disease is positively correlated to an increase of
Proteobacteria, especially of the
Escherichia genus [
27]. The increase in facultative anaerobes, such as
Enterobacteriaceae, in response to intestinal inflammation has been explained according to the “oxygen hypothesis” [
28]. Thus, under normal conditions, colonic epithelial cells deplete oxygen levels in the lumen through beta-oxidation processes, generating an anaerobic environment. In the setting of local inappropriate and persistent inflammatory response the beta-oxidation capacity of the colonic epithelial cells decreases, leading to an increased availability of oxygen, which, in turn, promotes dysbiosis and is associated with increased
Proteobacteria. Alternatively, it has been suggested that predominance of
Enterobacteriaceae during inflammatory processes might be due the augmented availability of nitrates, produced by the host during inflammation, or the absence of butyrate-producing microbiota leading to a loss of PPAR-γ regulation of iNOS (inducible nitric oxide synthase) expression [
29,
30]. While the treatment with Blend 1 failed to impact on the structure of intestinal microbiota associated with DSS administration, the microbiological profile of the animals treated with Blend 2 was characterized by an appreciable similarity to what was observed in naïve animals assessed using principal coordinate analysis (PCoA) using the Bray–Curtis dissimilarity matrices.
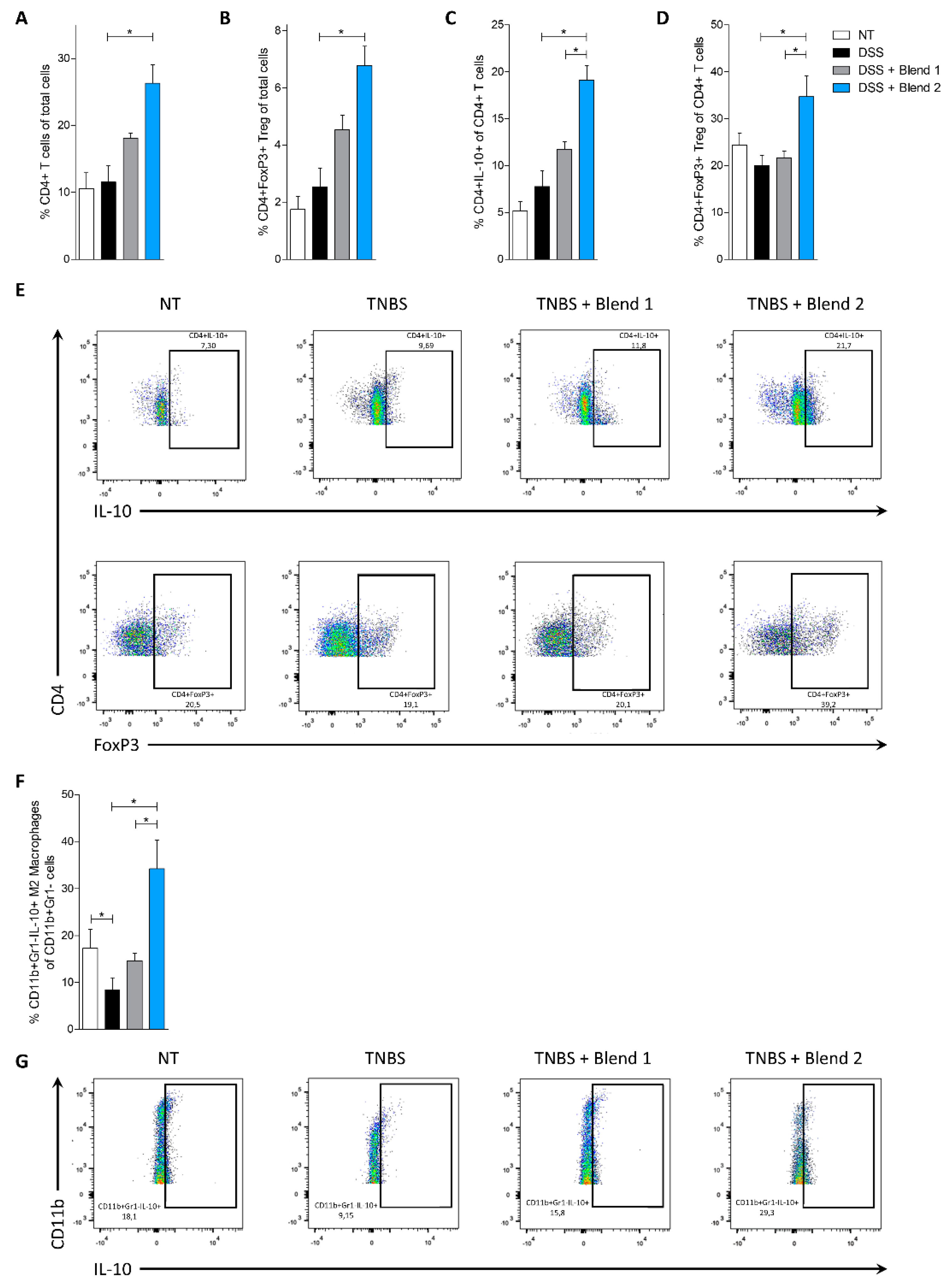
Because both blends claim efficacy in treating ulcerative colitis and ileal pouchitis, the functional effectiveness of the two multistrain preparations was examined in vivo in two rodent models of colitis. In both models, only treating mice with Blend 2 (Vivomixx) effectively reversed intestinal inflammation induced by TNBS and DSS. Results shown in
Figure 1,
Figure 2,
Figure 3 and
Figure 4, demonstrated that this blend significantly attenuated development of signs and symptoms of colitis, and that these beneficial effects associated with increased content of CD4
+Foxp3
+ cells and CD4
+IL-10
+ cells (Treg) in colon lamina propria. Thus, while the proportion of Treg was significantly reduced in mice treated with TNBS and DSS alone, this pattern was reversed by treating mice with Blend 2, while the other mixture failed to reach the same results. Treg cells are critical for maintaining immune homeostasis and establishing tolerance to non-pathogenic antigens, including commensal bacteria and food. These cells release suppressive cytokines, such as IL-10 and TGF-β, to regulate the balance of Th1/Th2 cells and maintain mucosal tolerance in the intestine [
31,
32]. Importantly, Treg formation is disabled in acute inflammation, and non-functional or absent Tregs or genetic mutations in Foxp3 induces hypersensitivity to bacterial antigens, altering the ability of the intestinal mucosal immunity to respond to inflammation [
33]. In the present study, we found that the percentage of CD4
+ Foxp3
+ T cells (
Figure 2D and
Figure 4D) in the
lamina propria was significantly reduced by treating mice with both TNBS and DSS, which also promoted a disturbance of Th1/Th2 cytokines balance and overexpression of pro-inflammatory mediators such as Il-1, Tnf-α, and Ifn-γ. This pattern was completely reversed by treating mice with Blend 2, which promoted a robust expansion of the number of CD4
+ Foxp3 T cells and their signature cytokine, Il-10. In this study, the Blend 2 effectively increased the number of CD4
+Foxp3
+T cells and restored their suppressive function. Since SCFA play a critical role in controlling Treg generation and maturation [
34,
35,
36], we speculated that Blend 2 alleviated inflammation and immune dysfunction by promoting a SCFA-dependent maturation of CD4
+ Foxp3
+T cells.
In addition to SCFA, Blend 2 could modulate the formation of Foxp3+ Treg cells by restructuring the intestinal microbiota. For example, commensal
Bacteroides fragilis polysaccharide promotes TLR2 dependent development of IL-10 producing Tregs and tolerance in experimental colitis [
35]. Fittingly, as shown in
Figure 5A, Blend 2 increased the amount of
Bacteroidetes species in mice administered with DSS.
One limitation of the present study is that we have investigated the functionality of the two probiotics in preclinical models of intestinal inflammation, and therefore the relevance of our results to humans need to be confirmed. Additionally, since the inflamed mice developed a specific microbiota, it is unclear whether effects observed with the two blends will extend to non colitic mice.

 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}