Astaxanthin Inhibits Mitochondrial Dysfunction and Interleukin-8 Expression in Helicobacter pylori-Infected Gastric Epithelial Cells



Abstract
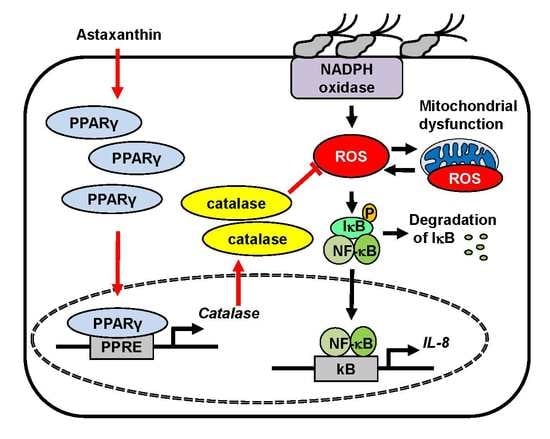
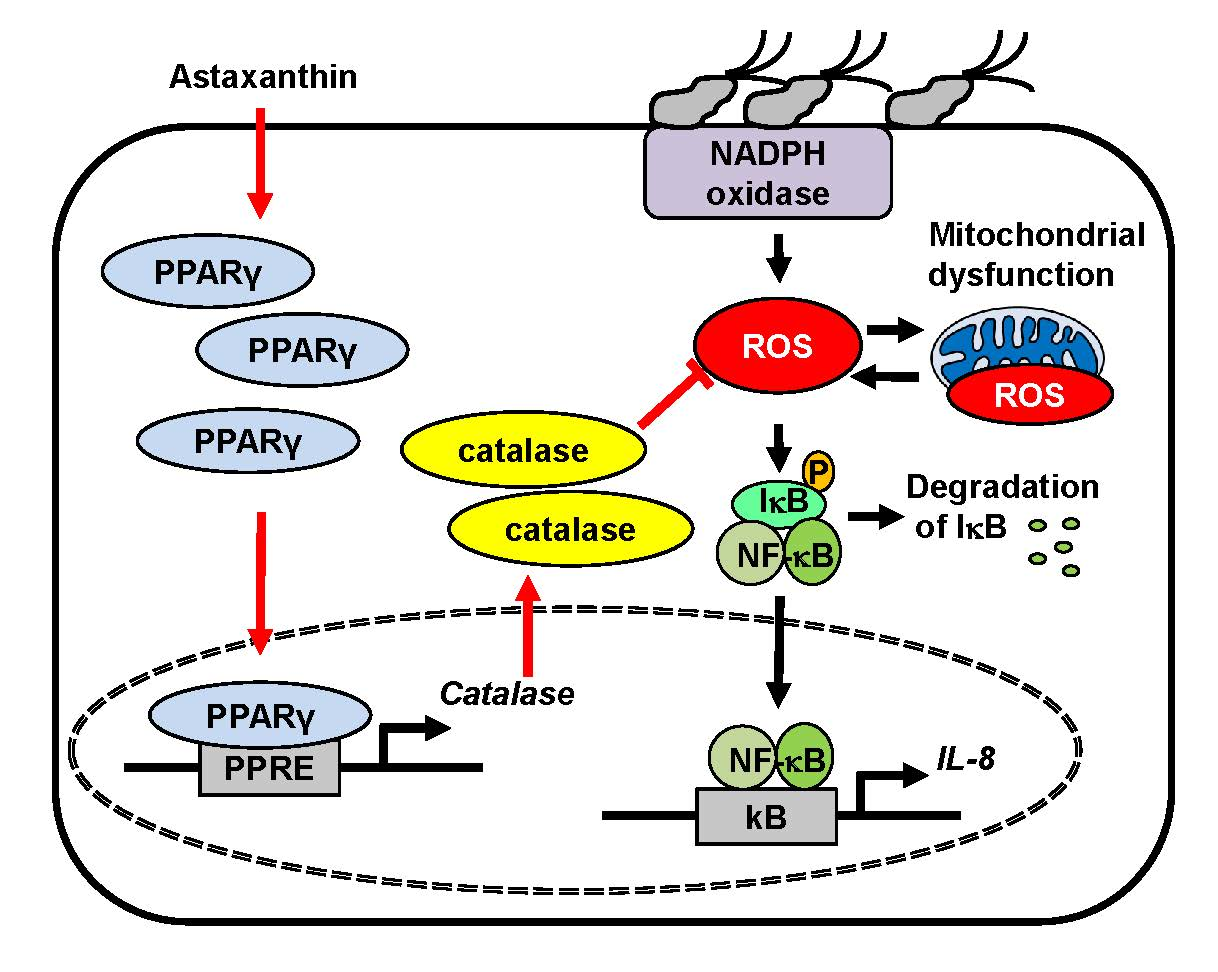{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Line and Culture Condition
2.3. Bacterial Strain and H. pylori Infection
2.4. Experimental Protocol
2.5. Preparation of Cell Extracts
2.6. Real-Time PCR Analysis for IL-8
2.7. Enzyme-Linked Immunosorbent Assay (ELISA) for IL-8
2.8. Measurement of Intracellular and Mitochondrial ROS Levels
2.9. Measurement of Mitochondrial Membrane Potential (MMP) and ATP Level
2.10. Electrophoretic Mobility Shift Assay (EMSA)
2.11. Western Blot Analysis for IκBα, PPAR-γ, and Catalase
2.12. Determination of NADPH Oxidase and Catalase Activities
2.13. Statistical Analysis
3. Results
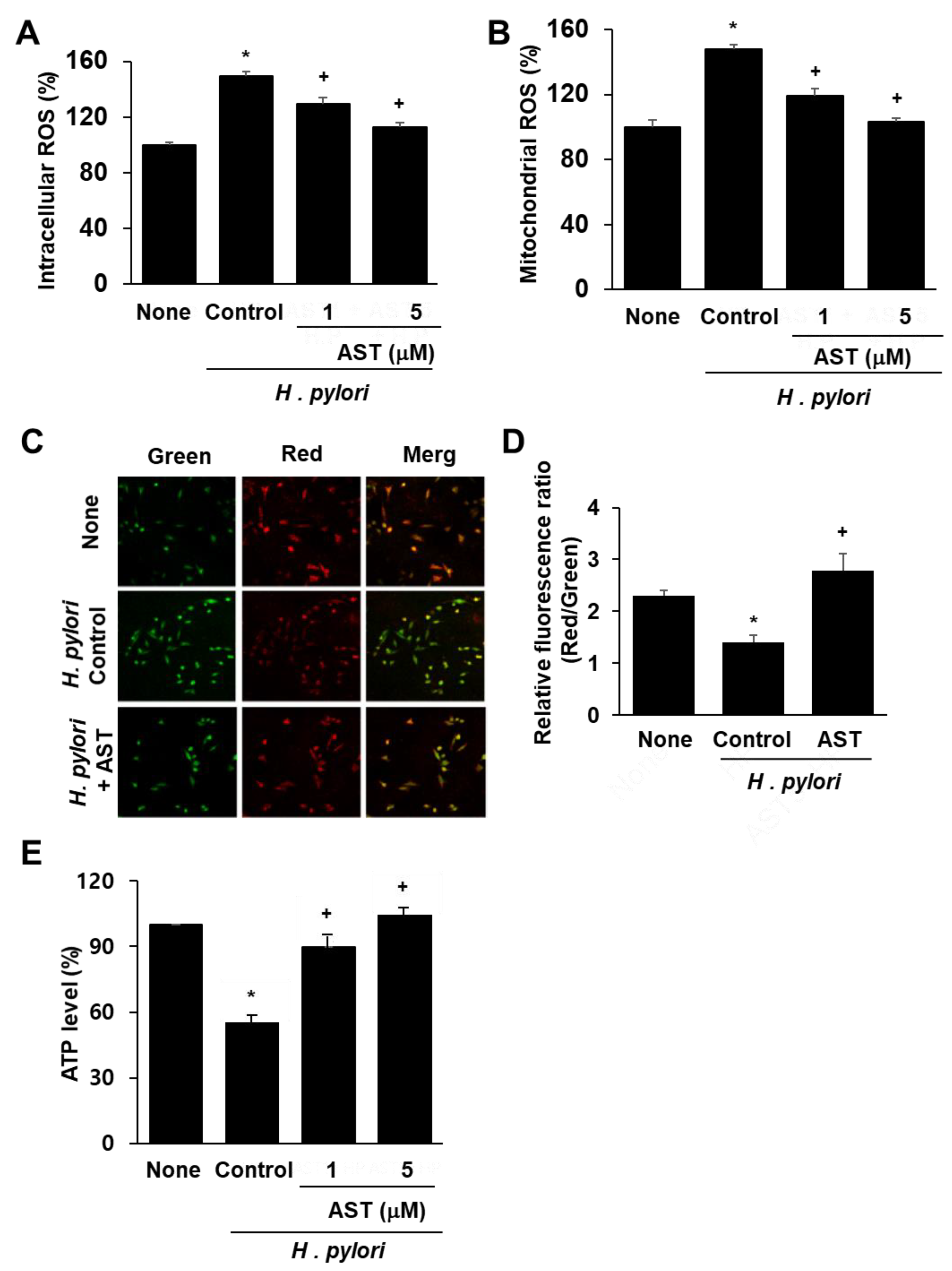
3.1. Astaxanthin Reduces ROS Levels and Inhibits Mitochondrial Dysfunction in H. pylori-Infected AGS Cells
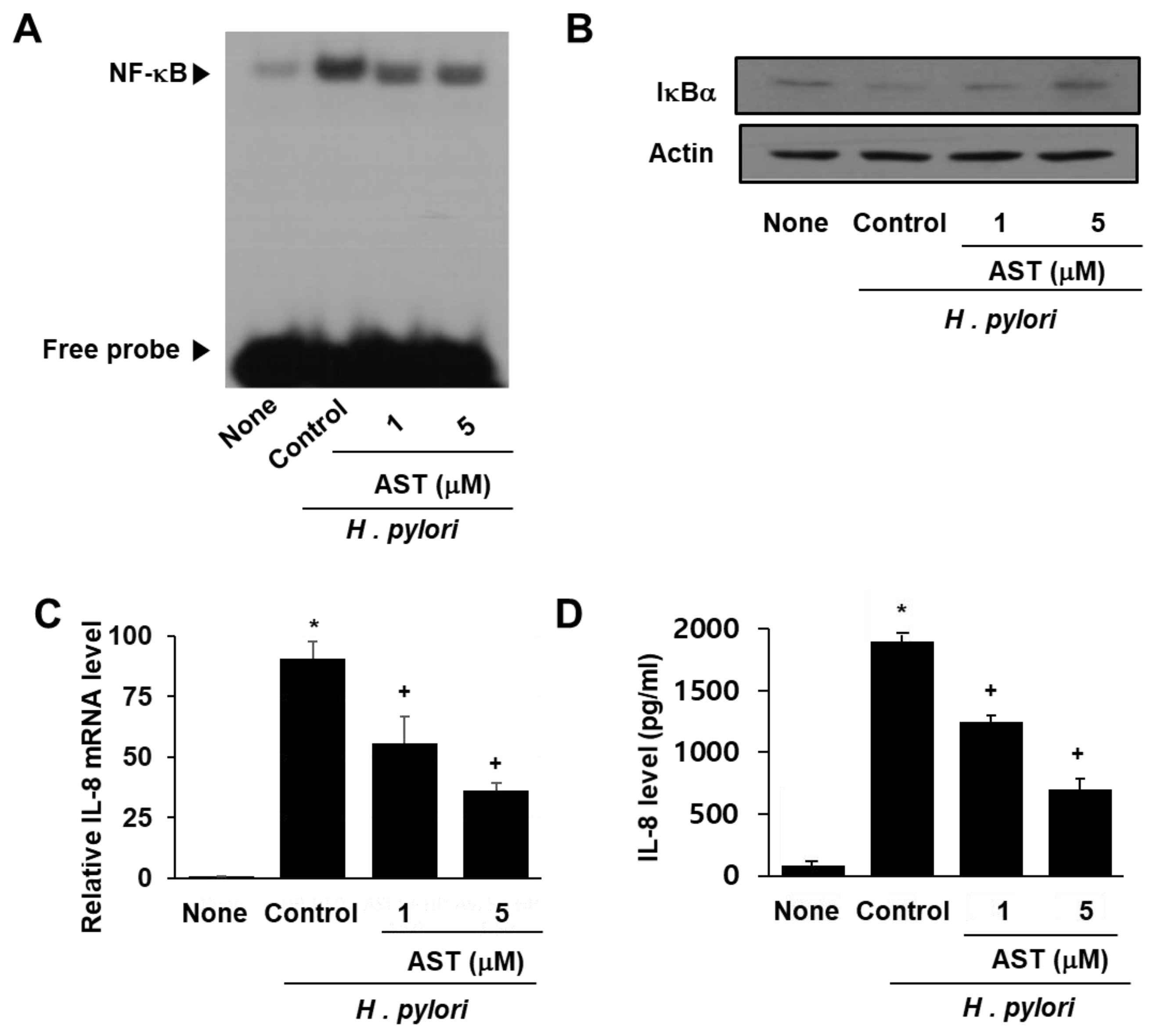
3.2. Astaxanthin Inhibits NF-κB Activation and IL-8 Expression in H. pylori-Infected AGS Cells
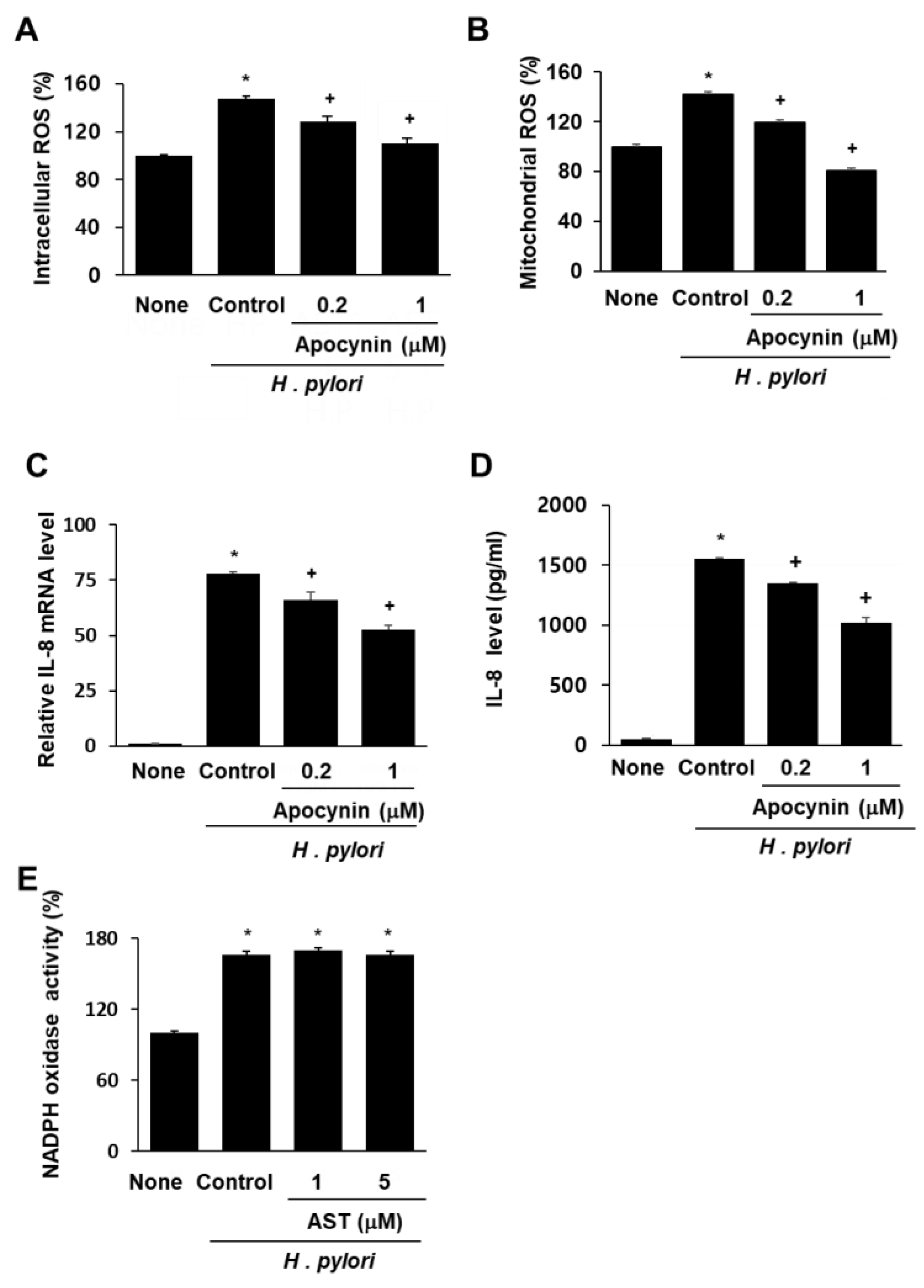
3.3. NADPH Oxidase Inhibitor Apocynin Reduces ROS Levels and Inhibits IL-8 Gene Expression, but Astaxanthin Does Not Affect NADPH Oxidase in H. pylori-Infected AGS Cells
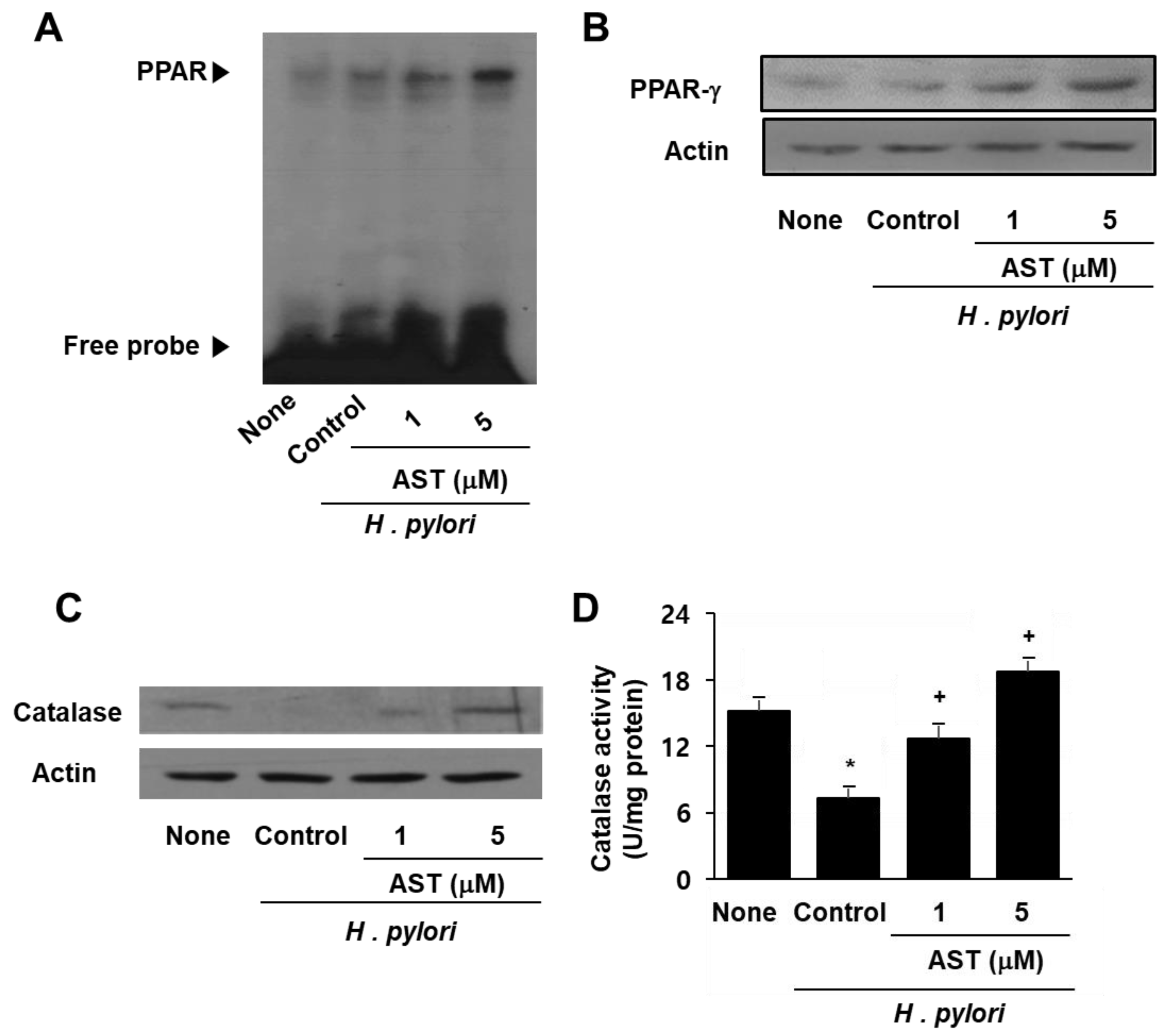
3.4. Astaxanthin Induces Expression and Activation of PPAR-γ and Catalase in H. pylori—Infected AGS Cells
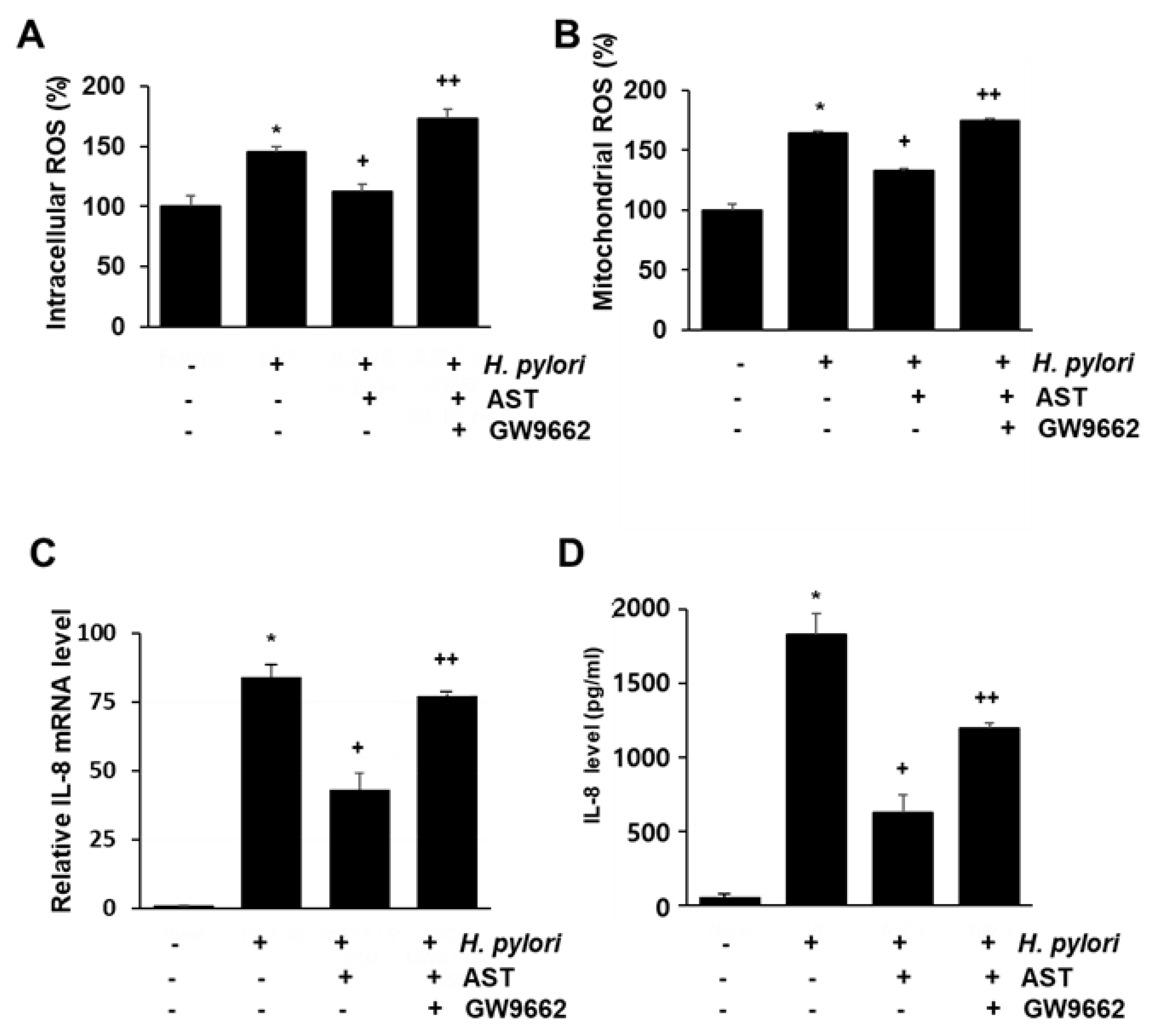
3.5. PPAR-γ Antagonist Abolishes the Inhibitory Effect of Astaxanthin on Elevated ROS Levels and IL-8 Gene Expression in H. pylori—Infected AGS Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Atherton, J.C.; Blaser, M.J. Coadaptation of Helicobacter pylori and humans: Ancient history, modern implications. J. Clin. Investig. 2009, 119, 2475–2487. [Google Scholar] [CrossRef] [PubMed]
- Marshall, B.J.; Warren, J.R. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. J. Lancet 1984, 323, 1311–1315. [Google Scholar] [CrossRef]
- Peek, R.M.; Miller, G.G.; Tham, K.T.; Perez-Perez, G.I.; Zhao, X.; Atherton, J.C.; Blaser, M.J. Heightened inflammatory response and cytokine expression in vivo to cagA+ Helicobacter pylori strains. Lab. Investig. 1995, 73, 760–770. [Google Scholar] [PubMed]
- Fan, X.G.; Chua, A.; Fan, X.J.; Keeling, P.W. Increased gastric production of interleukin-8 and tumour necrosis factor in patients with Helicobacter pylori infection. J. Clin. Pathol. 1995, 48, 133–136. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.E.; Khoi, P.N.; Xia, Y.; Park, J.S.; Joo, Y.E.; Kim, K.K.; Choi, S.Y.; Jung, Y.D. Helicobacter pylori and interleukin-8 in gastric cancer. World J. Gastroenterol. 2013, 45, 8192–8202. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, J.E. Helicobacter pylori and gastric mucosal cytokines: Evidence that CagA-positive strains are more virulent. Lab. Investig. 1995, 73, 742–745. [Google Scholar] [PubMed]
- Keates, S.; Hitti, Y.S.; Upton, M.; Kelly, C.P. Helicobacter pylori infection activates NF-kappa B in gastric epithelial cells. Gastroenterology 1997, 113, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.H.; Lim, J.W.; Kim, H.; Kim, K.H. Helicobacter pylori in a Korean isolate activates mitogen-activated protein kinases, AP-1, and NF-κB and induces chemokine expression in gastric epithelial AGS cells. Lab. Investig. 2004, 84, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Roebuck, K.A. Oxidant stress regulation of IL-8 and ICAM-1 gene expression: Differential activation and binding of the transcription factors AP-1 and NF-kappaB. Int. J. Mol. Med. 1999, 4, 223–253. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.Y.; Kim, K.H.; Kim, H. Role of oxidant-sensitive transcription factors on Helicobacter pylori-induced IL-8 expression in gastric epithelial AGS cells. Korean J. Helicobacter Upper Gastrointest. Res. 2005, 5, 124–133. [Google Scholar]
- Choi, J.H.; Cho, S.O.; Kim, H. α-Lipoic Acid Inhibits Expression of IL-8 by Suppressing Activation of MAPK, Jak/Stat, and NF-κB in H. pylori-Infected Gastric Epithelial AGS Cells. Yonsei Med. J. 2016, 57, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, K.; Kawahara, T.; Sano, T.; Toida, K.; Kuwano, Y.; Sasaki, H.; Kawai, T.; Teshima-Kondo, S.; Rokutan, K. Evidence for cancer-associated expression of NADPH oxidase 1 (Nox1)-based oxidase system in the human stomach. Free Radical Biol. Med. 2007, 43, 1627–1638. [Google Scholar] [CrossRef] [PubMed]
- Cha, B.; Lim, J.W.; Kim, K.H.; Kim, H. 15-deoxy-D12, 14-prostaglandin J2 suppresses RANTES expression by inhibiting NADPH oxidase activation in Helicobacter pylori-infected gastric epithelial cells. J. Physiol. Pharmacol. 2011, 62, 167–174. [Google Scholar] [PubMed]
- Cha, B.; Lim, J.W.; Kim, K.H.; Kim, H. HSP90β interacts with Rac1 to activate NADPH oxidase in Helicobacter pylori-infected gastric epithelial cells. J. Physiol. Pharmacol. 2010, 42, 1455–1461. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef] [PubMed]
- Madamanchi, N.R.; Runge, M.S. Mitochondrial dysfunction in atherosclerosis. Circ. Res. 2007, 100, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.J. Cross talk between mitochondria and NADPH oxidases. Free Radical Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Aguilera, A.; Rull, A.; Rodríguez-Gallego, E.; Riera-Borrull, M.; Luciano-Mateo, F.; Camps, J.; Menéndez, J.A.; Joven, J. Mitochondrial dysfunction: A basic mechanism in inflammation-related non-communicable diseases and therapeutic opportunities. Mediators Inflamm. 2013. [Google Scholar] [CrossRef] [PubMed]
- López-Armada, M.J.; Riveiro-Naveira, R.R.; Vaamonde-García, C.; Valcárcel-Ares, M.N. Mitochondrial dysfunction and the inflammatory response. Mitochondrion 2013, 13, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Ojala, J.; Kaarniranta, K.; Kauppinen, A. Mitochondrial dysfunction and oxidative stress activate inflammasomes: Impact on the aging process and age-related diseases. Cell. Mol. Life Sci. 2012, 69, 2999–3013. [Google Scholar] [CrossRef] [PubMed]
- Ricote, M.; Li, A.C.; Willson, T.M.; Kelly, C.J.; Glass, C.K. The peroxisome proliferator-activated receptor-γ is a negative regulator of macrophage activation. Nature 1998, 391, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Ting, A.T.; Seed, B. PPAR-γ agonists inhibit production of monocyte inflammatory cytokines. Nature 1998, 391, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhang, Y.; Strong, R.; Grotta, J.C.; Aronowski, J. 15d-Prostaglandin J2 activates peroxisome proliferator-activated receptor-γ, promotes expression of catalase, and reduces inflammation, behavioral dysfunction, and neuronal loss after intracerebral hemorrhage in rats. J. Cereb. Blood Flow Metab. 2006, 26, 811–920. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Yang, Q. Peroxisome-proliferator-activated receptors regulate redox signaling in the cardiovascular system. World J. Cardiol. 2013, 5, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Kojima, K.; Shimada, T.; Mitobe, Y.; Yoshiura, K.; Hiraishi, H.; Terano, A. Effect of PPARγ ligands on the viability of gastric epithelial cells. Aliment. Pharmacol. Ther. 2001, 16, 67–73. [Google Scholar] [CrossRef]
- Gupta, R.A.; Polk, D.B.; Krishna, U.; Israel, D.A.; Yan, F.; DuBois, R.N.; Peek, R.M. Activation of peroxisome proliferator-activated receptor γ suppresses nuclear factor κB-mediated apoptosis induced by Helicobacter pylori in gastric epithelial cells. J. Biol. Chem. 2001, 276, 31059–31066. [Google Scholar] [CrossRef] [PubMed]
- Auwerx, J.I. PPARγ in the gastrointestinal tract: Gain or pain? Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 284, G581–G585. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Xu, X. Research Advances in the Correlation between Peroxisome Proliferator-Activated Receptor-γ and Digestive Cancers. PPAR Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kidd, P. Astaxanthin, cell membrane nutrient with diverse clinical benefits and anti-aging potential. Altern. Med. Rev. 2011, 16, 355–364. [Google Scholar] [PubMed]
- Ambati, R.R.; Phang, S.M.; Ravi, S.; Aswathanarayana, R.G. Astaxanthin: Sources, extraction, stability, biological activities and its commercial applications—A review. Mar. Drugs 2014, 12, 128–152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, W.E.; Hu, L.; Zhao, L.; Huang, J. Carotenoids inhibit proliferation and regulate expression of peroxisome proliferators-activated receptor gamma (PPARγ) in K562 cancer cells. Arch. Biochem. Biophys. 2011, 512, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Tanabe, H.; Matsumoto, A.; Takagi, M.; Umegaki, K.; Amagaya, S.; Takahashi, J. Astaxanthin functions differently as a selective peroxisome proliferator-activated receptor γ modulator in adipocytes and macrophages. Biochem. Pharmacol. 2012, 84, 692–700. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; O’toole, P.W.; Doig, P. Stimulation of interleukin-8 production in epithelial cell lines by Helicobacter pylori. Infect. Immun. 1995, 63, 1732–1738. [Google Scholar] [PubMed]
- Eftang, L.L.; Esbensen, Y.; Tannæs, T.M.; Bukholm, I.R.; Bukholm, G. Interleukin-8 is the single most up-regulated gene in whole genome profiling of H. pylori exposed gastric epithelial cells. BMC Microbiol. 2012, 12, 9. [Google Scholar] [CrossRef] [PubMed]
- Kim, H. Oxidative stress in Helicobacter pylori-induced gastric cell injury. Inflammopharmacology 2005, 13, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Kong, Y.; Zhang, H. Oxidative stress, mitochondrial dysfunction, and aging. J. Signal Transduct. 2012. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.H.; Dar, K.B.; Anees, S.; Zargar, M.A.; Masood, A.; Sofi, M.A.; Ganie, S.A. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 2015, 74, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.D.; Sun, J.Y.; Fu, X.T.; Hou, Y.J.; Li, Y.; Yang, M.F.; Fu, X.Y.; Sun, B.L. Astaxanthin Attenuates Homocysteine-Induced Cardiotoxicity In Vitro and In Vivo by Inhibiting Mitochondrial Dysfunction and Oxidative Damage. Front. Physiol. 2017, 8, 1041. [Google Scholar] [CrossRef] [PubMed]
- Pongkan, W.; Takatori, O.; Ni, Y.; Xu, L.; Nagata, N.; Chattipakorn, S.C.; Usui, S.; Kaneko, S.; Takamura, M.; Suigura, M.; et al. β-Cryptoxanthin exerts greater cardioprotective effects on cardiac ischemia-reperfusion injury than astaxanthin by attenuating mitochondrial dysfunction in mice. Mol. Nutr. Food Res. 2017, 61, 1601077. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Shibata, T.; Hisaka, S.; Osawa, T. Astaxanthin inhibits reactive oxygen species-mediated cellular toxicity in dopaminergic SH-SY5Y cells via mitochondria-targeted protective mechanism. Brain Res. 2009, 1254, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Kim, C.S.; Lee, Y.J. Astaxanthin protects against MPTP/MPP+-induced mitochondrial dysfunction and ROS production in vivo and in vitro. Food Chem. Toxicol. 2011, 49, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Son, S.H.; Kim, H.K.; Ji, J.S.; Cho, Y.S.; Kim, S.S.; Chae, H.S.; Choi, H.S.; Han, S.W.; Choi, K.Y.; Chung, I.S.; et al. Expression of peroxisome proliferator-activated receptor (PPAR) gamma in Helicobacter pylori-infected gastric epithelium. Korean J. Gastroenterol. 2007, 49, 72–78. [Google Scholar] [PubMed]
- Konturek, P.C.; Kania, J.; Kukharsky, V.; Raithel, M.; Ocker, M.; Rembiasz, K.; Hahn, E.G.; Konturek, S.J. Implication of peroxisome proliferator-activated receptor γ and proinflammatory cytokines in gastric carcinogenesis: Link to Helicobacter pylori-infection. J. Pharmacol. Sci. 2004, 96, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Slomiany, B.L.; Slomiany, A. Suppression of Gastric Mucosal Inflammatory Responses to Helicobacter pylori Lipopolysaccharide by Peroxisome Proliferator-Activated Receptor γ Activation. IUBMB Life 2002, 53, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Solomonov, Y.; Hadad, N.; Levy, R. The combined anti-inflammatory effect of astaxanthin, Lyc-O-mato and carnosic acid in vitro and in vivo in a mouse model of peritonitis. J. Nutr. Food Sci. 2018, 8, 1000653. [Google Scholar] [CrossRef]
- Kang, H.; Lim, J.W.; Kim, H. Inhibitory effect of Korean red ginseng extract on DNA damage response and apoptosis in Helicobacter pylori-infected gastric epithelial cells. J. Ginseng Res. 2018, in press. [Google Scholar] [CrossRef]
- Jang, S.H.; Cho, S.; Lee, E.S.; Kim, J.M.; Kim, H. The phenyl-thiophenyl propenone RK-I-123 reduces the levels of reactive oxygen species and suppresses the activation of NF-κB and AP-1 and IL-8 expression in Helicobacter pylori-infected gastric epithelial AGS cells. Inflamm. Res. 2013, 62, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Cha, B.; Lim, J.W.; Kim, H. Jak1/Stat3 is an upstream signaling of NF-κB activation in Helicobacter pylori-induced IL-8 production in gastric epithelial AGS cells. Yonsei Med. J. 2015, 56, 862–866. [Google Scholar] [CrossRef] [PubMed]
- Nakachi, N.; Klein, T.W.; Friedman, H.; Yamamoto, Y. Helicobacter pylori infection of human gastric epithelial cells induces IL-8 and TNFK, but not TGFL1 mRNA. FEMS Immunol. Med. Microbiol. 2000, 29, 23–26. [Google Scholar] [PubMed]
- Park, S.A.; Ko, A.; Lee, N.G. Stimulation of growth of the human gastric pathogen Helicobacter pylori by atmospheric level of oxygen under high carbon dioxide tension. BMC Microbiol. 2011, 11, 96. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Xie, T.; He, X.X.; Mao, Z.F.; Jia, L.J.; Wang, W.P.; Zhen, J.L.; Liu, L.M. Astaxanthin rescues neuron loss and attenuates oxidative stress induced by amygdala kindling in adult rat hippocampus. Neurosci. Lett. 2015, 597, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Mosaad, Y.O.; Gobba, N.; Huseein, M.A. Astaxanthin; a promising protector against gentamicin-induced nephrotoxicity in rats. Curr. Pharm. Biotechnol. 2016, 17, 13. [Google Scholar] [CrossRef]
- Bennedsen, M.; Wang, X.; Willén, R.; Wadström, T.; Andersen, L.P. Treatment of H. pylori infected mice with antioxidant astaxanthin reduces gastric inflammation, bacterial load and modulates cytokine release by splenocytes. Immunol. Lett. 1999, 70, 185–189. [Google Scholar] [CrossRef]
- Wang, X.; Willén, R.; Wadström, T. Astaxanthin-rich algal meal and vitamin C inhibit Helicobacter pylori infection in BALB/cA mice. Antimicrob. Agents Chemother. 2000, 44, 2452–2457. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.A. Absorption, transport, and metabolism of carotenoids in humans. Pure Appl. Chem. 1994, 66, 1011–1116. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, H.; Liu, R.; Zhu, H.; Zhan, L.; Tsao, R. Bioaccessibility, cellular uputake, and transport of astaxanthin isomers and their antioxidative effects in human intestinal epithelial Caco-2 cells. J. Agric. Food Chem. 2017, 65, 10223–10232. [Google Scholar] [CrossRef] [PubMed]
- Mimoun-Benarroch, M.; Hogot, C.; Rhazi, L.; Niamba, C.N.; Depeint, F. The bioavailability of astaxanthin is dependent on both the source and the isomeric variants of the molecule. Bull. UASVM Food Sci. Technol. 2016, 73, 61–69. [Google Scholar] [CrossRef]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.H.; Lim, J.W.; Kim, H. Astaxanthin Inhibits Mitochondrial Dysfunction and Interleukin-8 Expression in Helicobacter pylori-Infected Gastric Epithelial Cells. Nutrients 2018, 10, 1320. https://doi.org/10.3390/nu10091320
Kim SH, Lim JW, Kim H. Astaxanthin Inhibits Mitochondrial Dysfunction and Interleukin-8 Expression in Helicobacter pylori-Infected Gastric Epithelial Cells. Nutrients. 2018; 10(9):1320. https://doi.org/10.3390/nu10091320
Chicago/Turabian StyleKim, Suhn Hyung, Joo Weon Lim, and Hyeyoung Kim. 2018. "Astaxanthin Inhibits Mitochondrial Dysfunction and Interleukin-8 Expression in Helicobacter pylori-Infected Gastric Epithelial Cells" Nutrients 10, no. 9: 1320. https://doi.org/10.3390/nu10091320
APA StyleKim, S. H., Lim, J. W., & Kim, H. (2018). Astaxanthin Inhibits Mitochondrial Dysfunction and Interleukin-8 Expression in Helicobacter pylori-Infected Gastric Epithelial Cells. Nutrients, 10(9), 1320. https://doi.org/10.3390/nu10091320