Sustainable Zero-Valent Metal (ZVM) Water Treatment Associated with Diffusion, Infiltration, Abstraction, and Recirculation

Abstract
:
1. Introduction
- specific design storms for storm runoff,
- abstraction/infiltration/runoff flow rate,
- the amount of ZVM placed in an aquifer for static diffusion treatment (and time), and
- the abstraction and infiltration/injection rate when aquifer/plume/GWM treatment strategy requires the entire Eh and pH of the water body to be changed to new defined limits.
- treated drinking water (including removal of leached metals, micro-organisms, and organic chemicals),
- remediated GWMs and plumes,
- treated GWMs and plumes to support specific agriculture activities and remove the adverse effects of leaching/infiltration of pollutants,
- improved water quality in aquifers, and
- low cost, rapid supply, high volume sources of treated water in the aftermath of natural/ anthropogenic disasters.
1.1. ZVM Treatment


1.2. Sustainable ZVM Treatment Design Issues Addressed
2. Methodology and Permeability
2.1. Basic Principals
2.1.1. ZVM Remediation
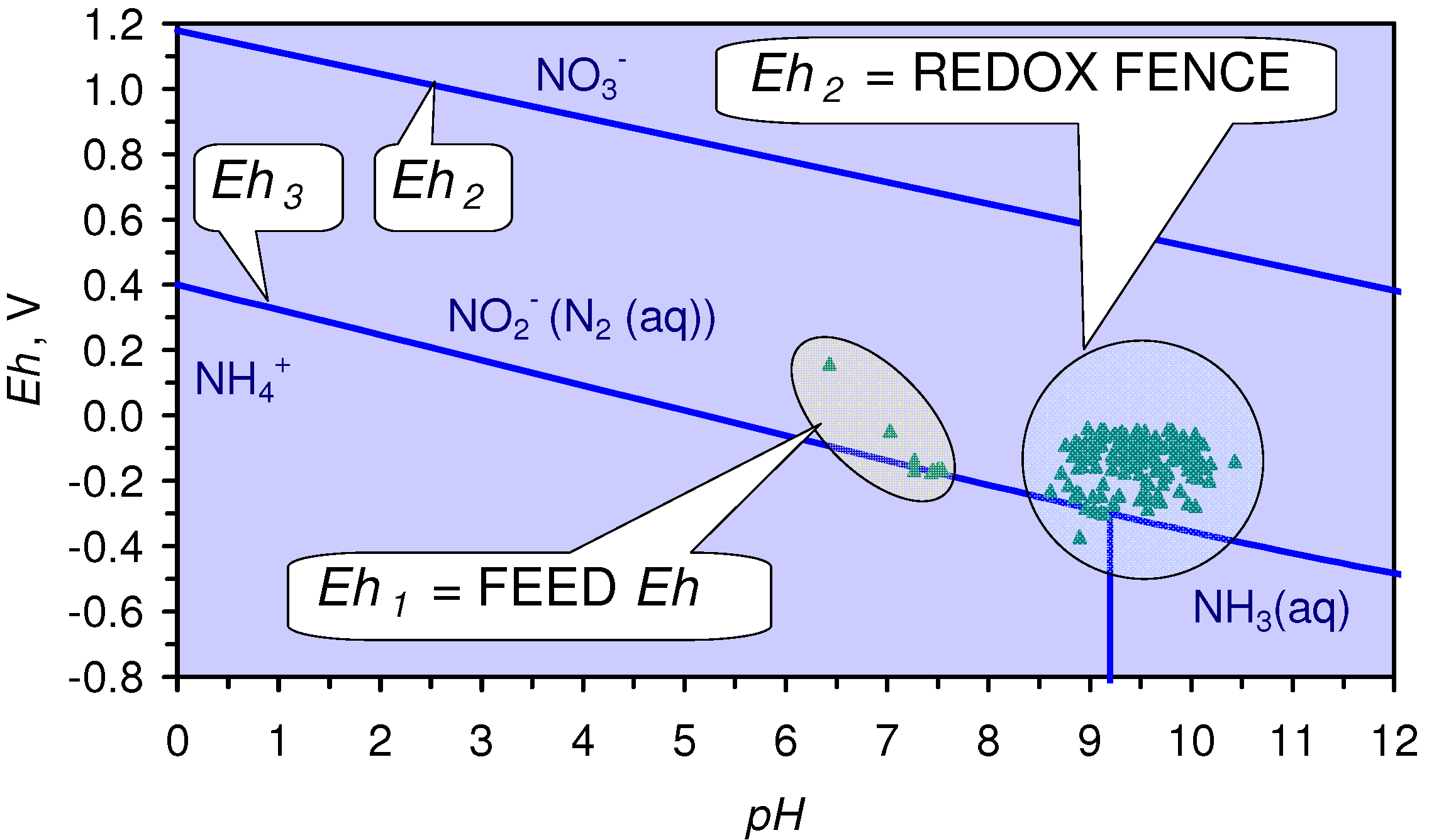
NO3− + H3O+[H2O]2 = NH4+ + 3OH− + 1.5O2(g)
Eh = Eh(cathode) – Eh(anode)
2HO2− = O2(g) + 2OH− [associated with Eh decrease and no change in pH]
6H2O = O2(g) + 4H3O+[associated with no change in Eh and pH decrease]
2.1.2. Static Diffusion
2.1.3. Permeability
k = Q/ΔP
2.1.4. Space Velocity
2.1.5. Materials and Methods
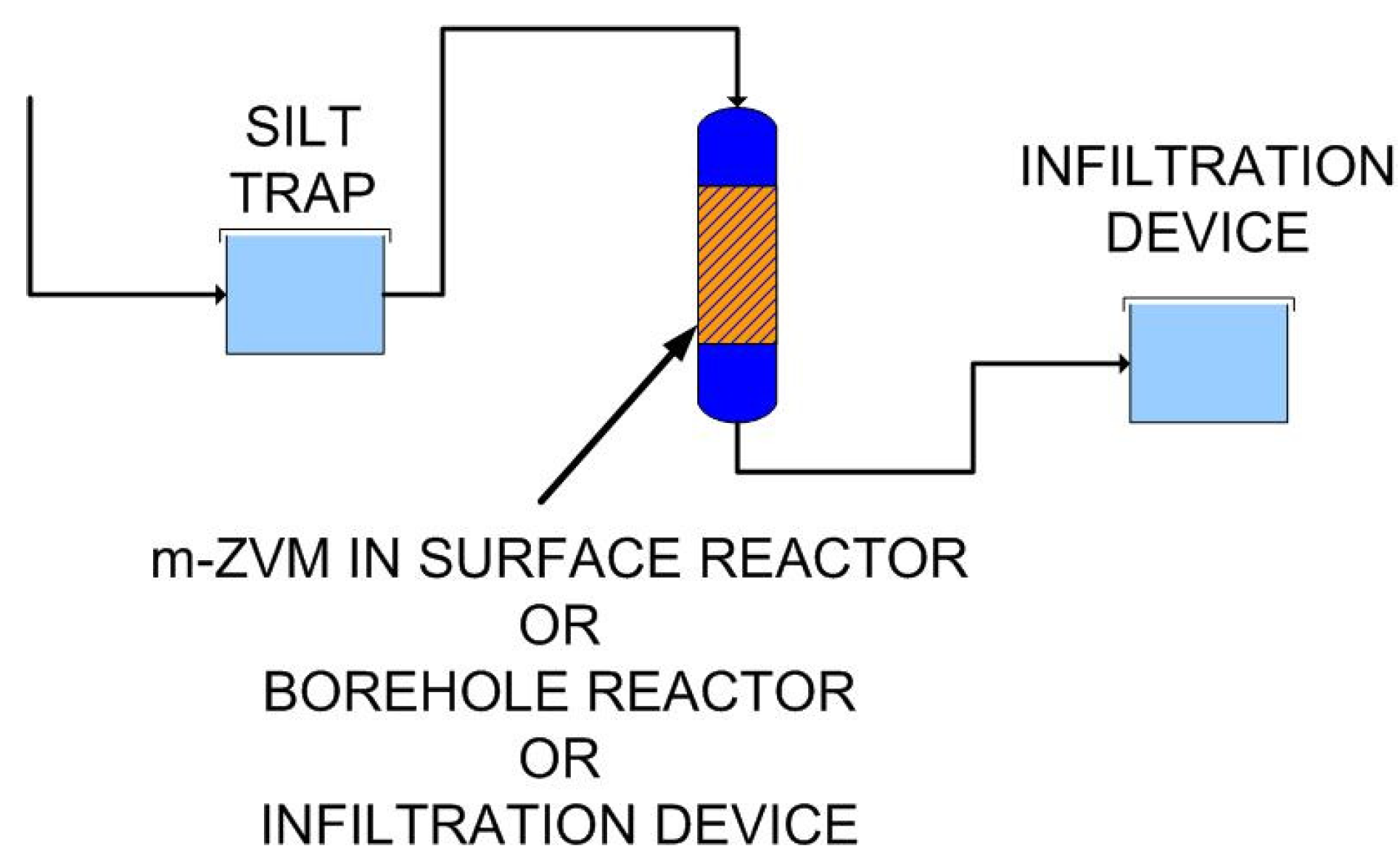
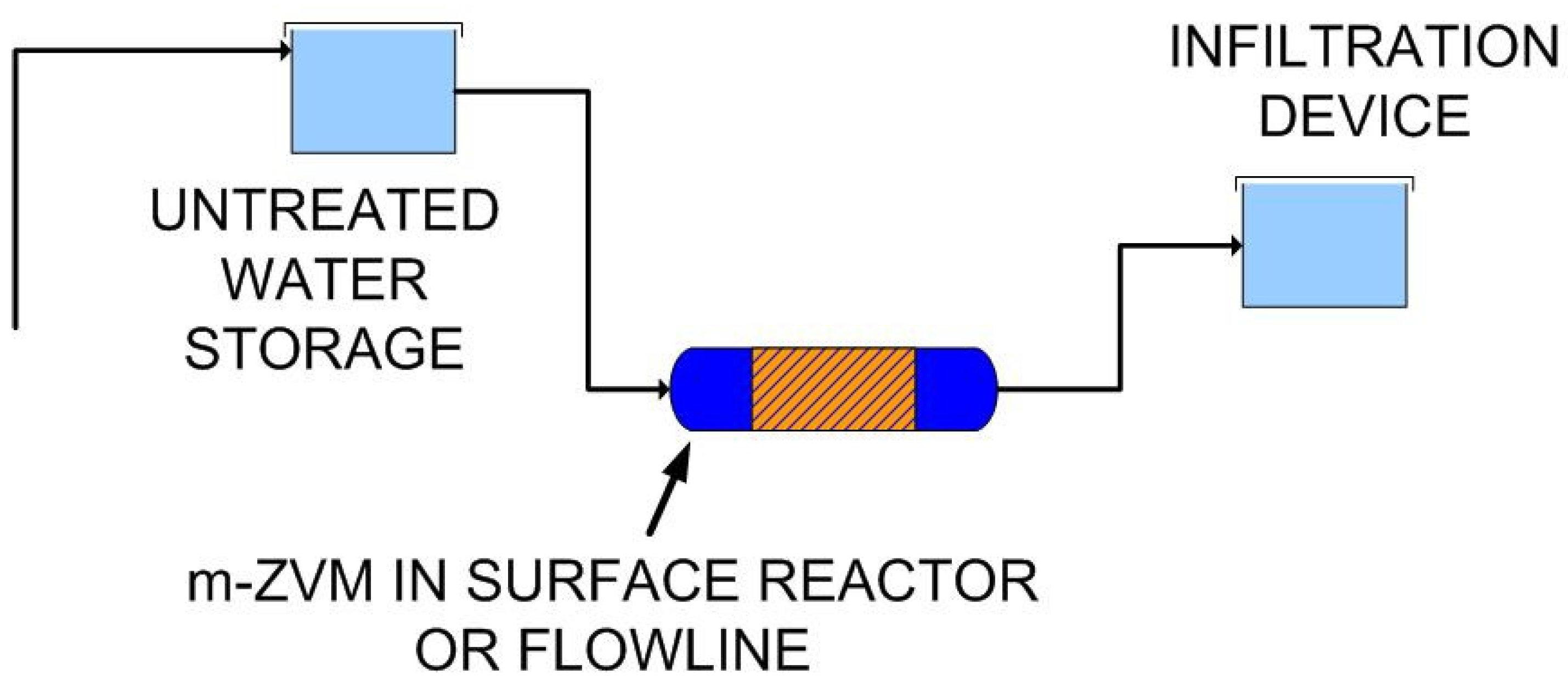
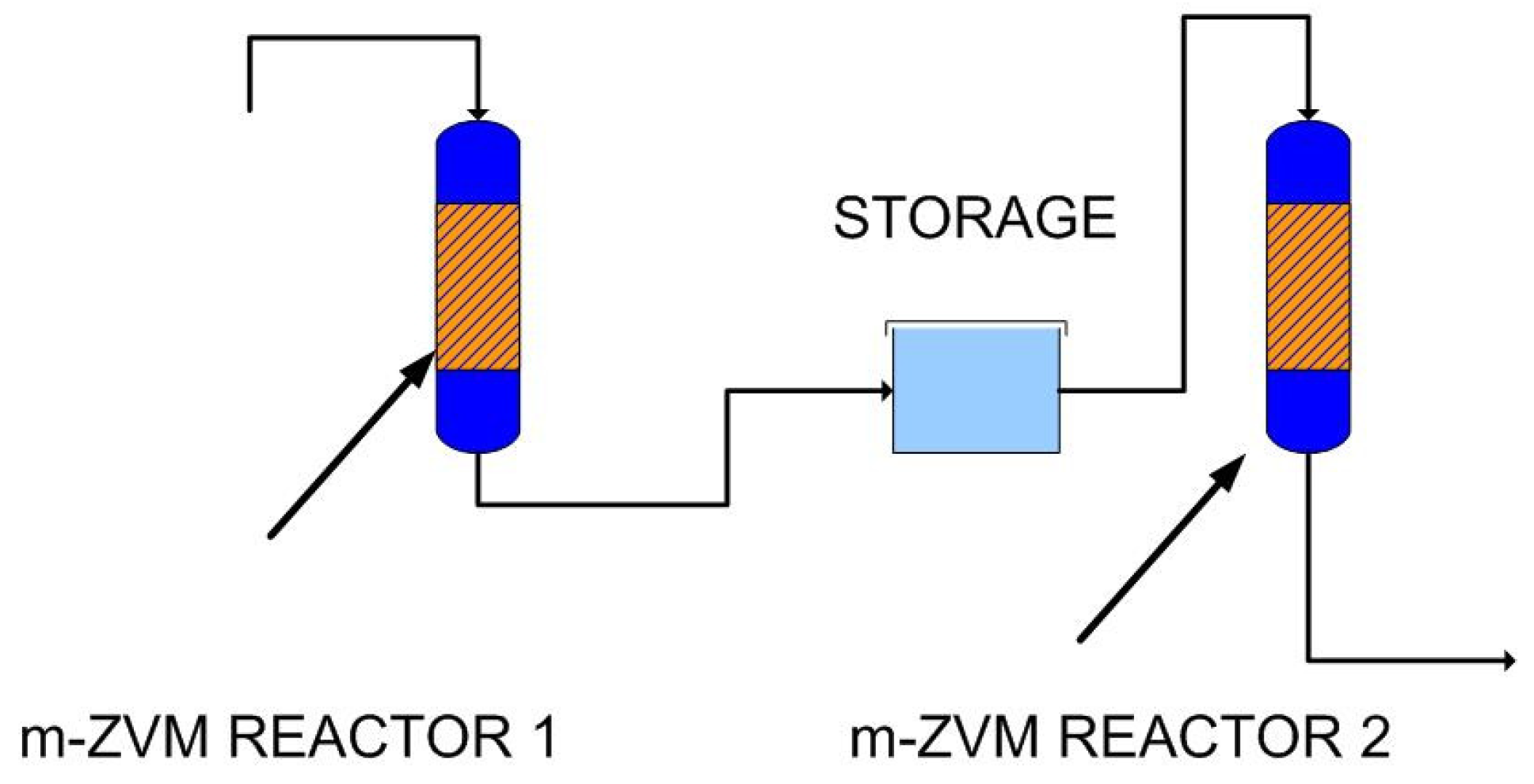
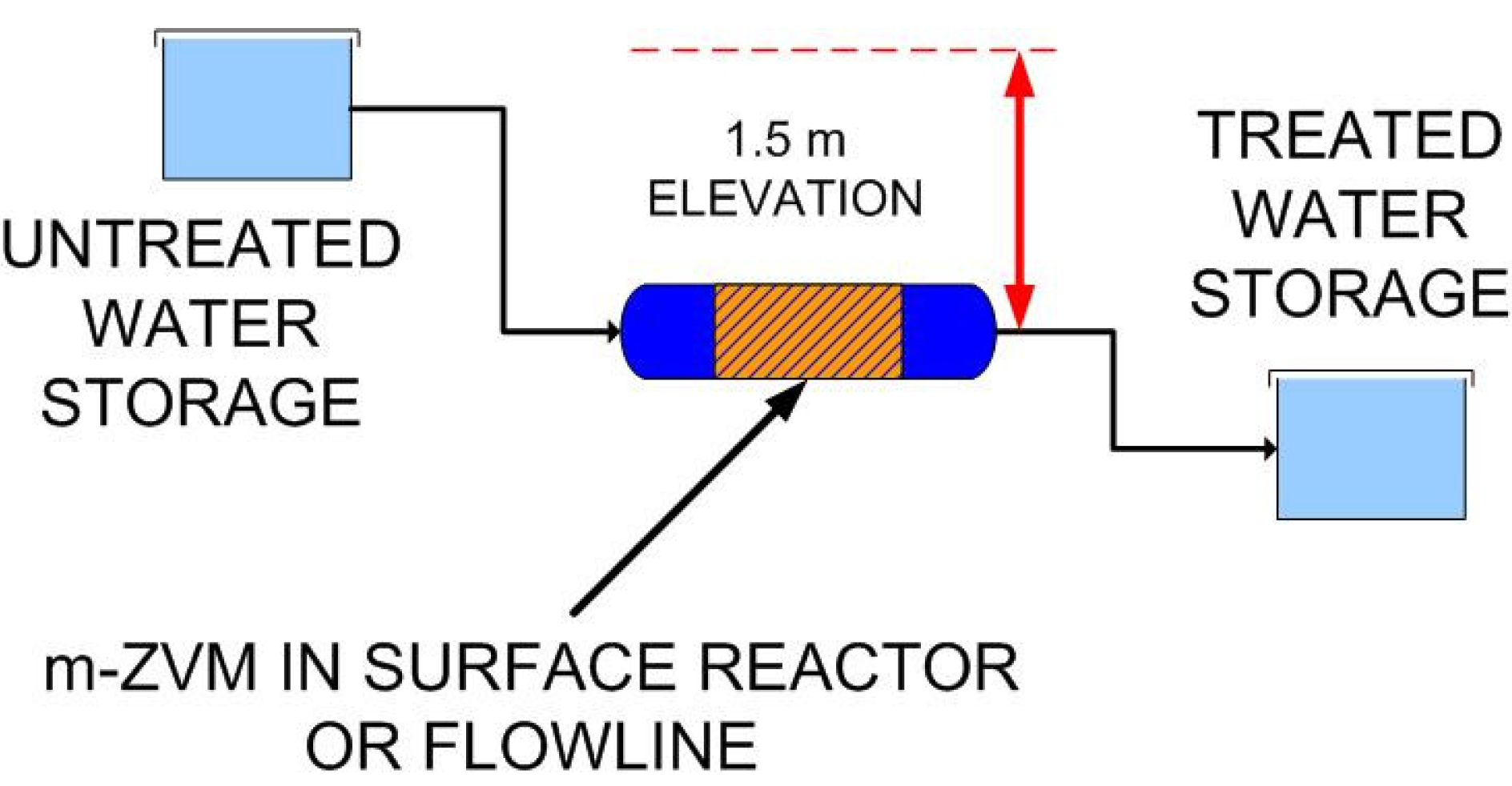
2.1.6. Borehole Reactor Structure: m-ZVM
2.1.7. Static Diffusion Reactor Structure: m-ZVM
2.1.8. Continuous Flow Reactor Structure: n-ZVM
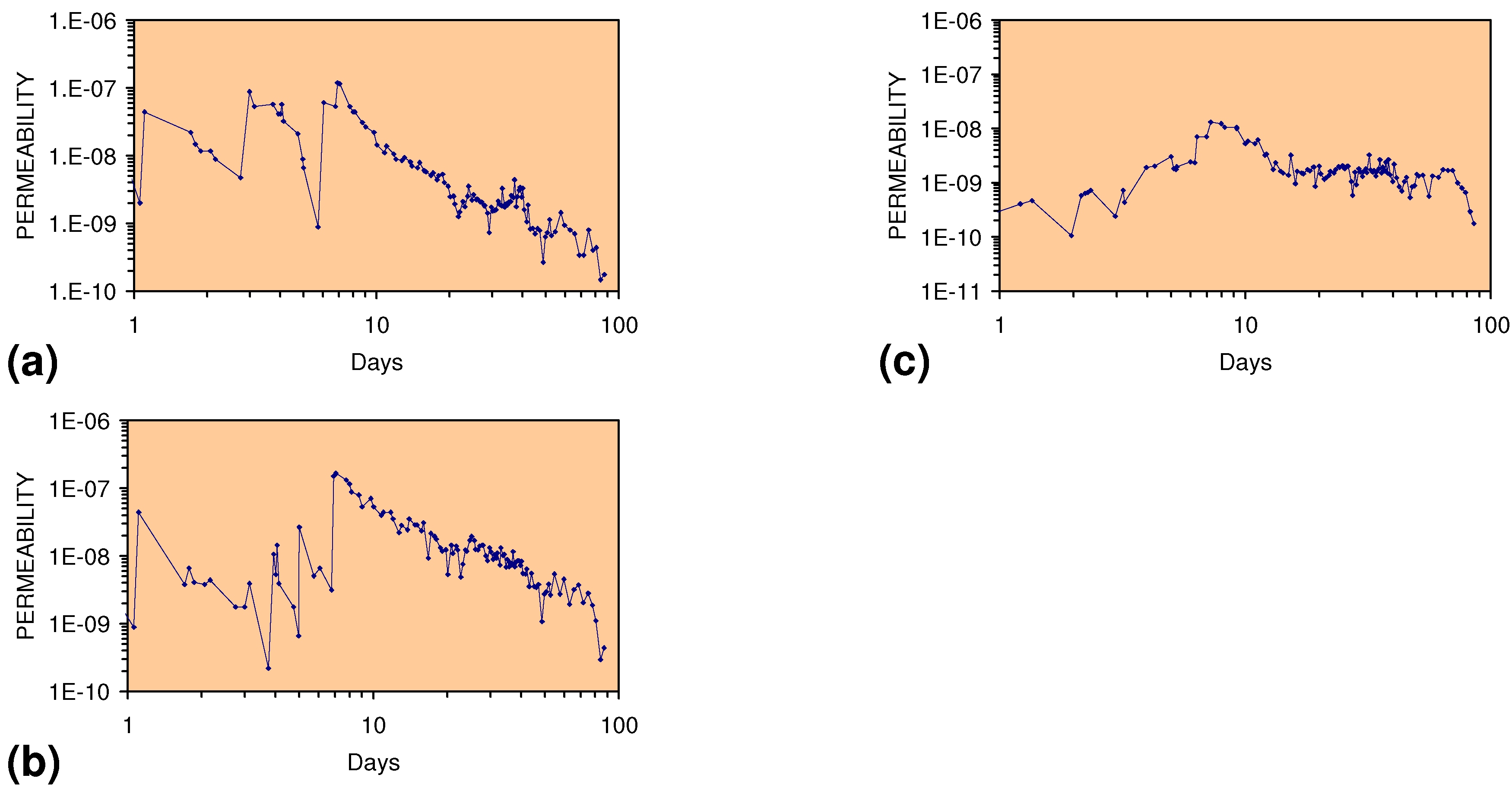
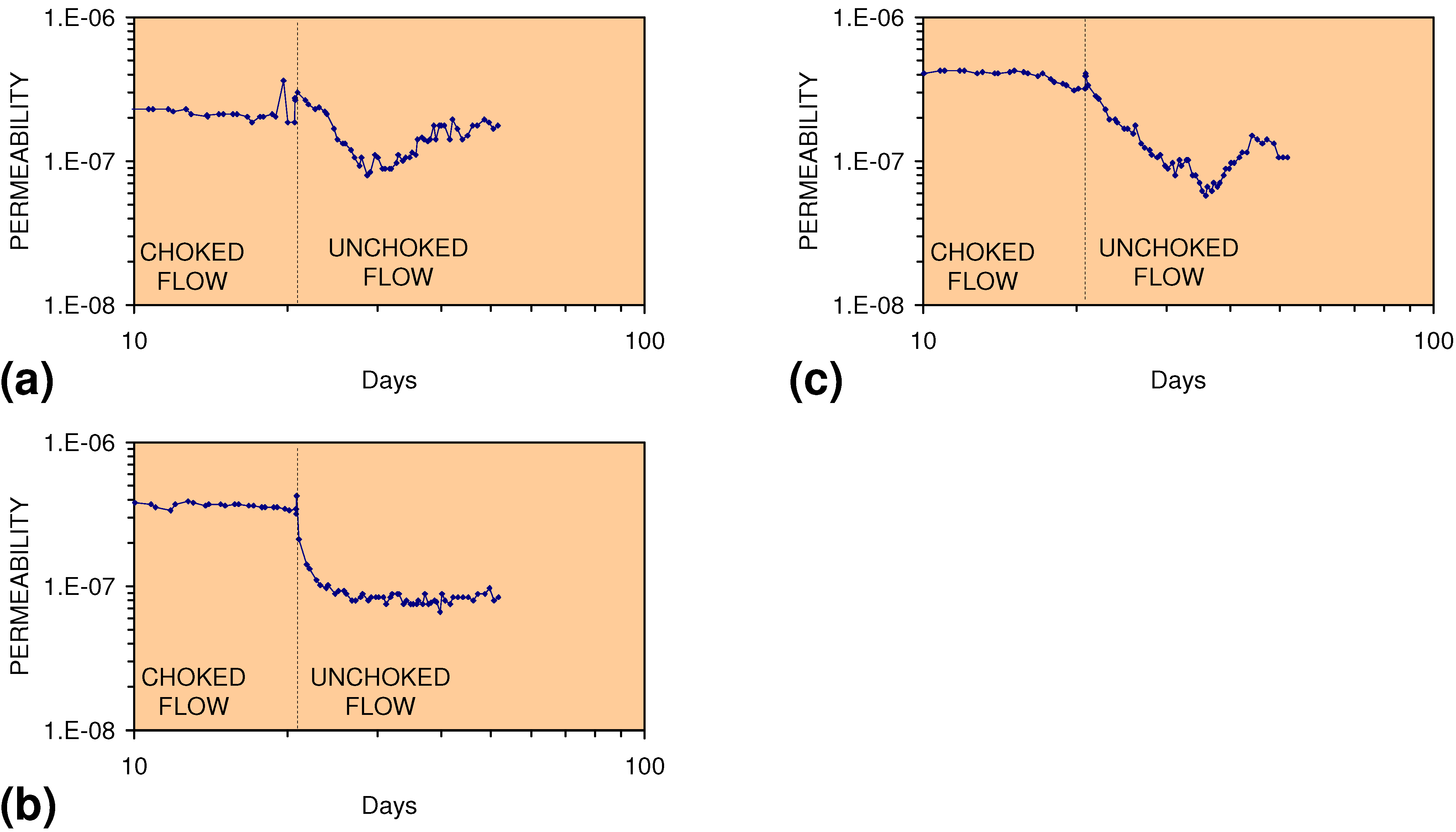
2.2. Permeability of ZVM
2.2.1. Permeability of n-ZVM
- The n-ZVM becomes increasingly consolidated due to Fe-oxide formation and Fe particle growth
- O2 gas bubbles start to discharge from the n-ZVM bed as the permeability reduces. This results in pore throat occlusion with an associated decrease in permeability [1].
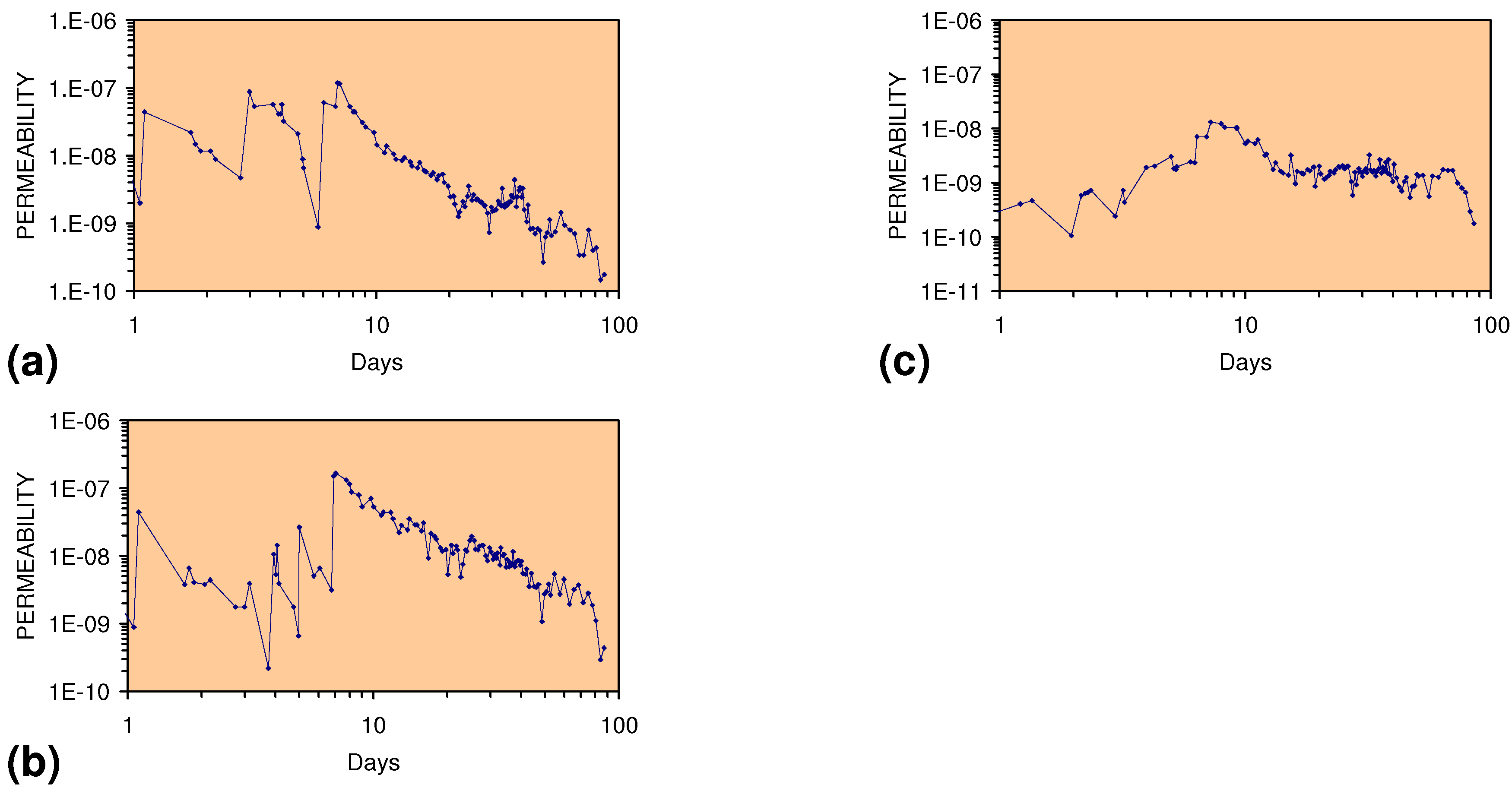
2.2.2. Permeability of m-ZVM

2.2.3. Significance of Permeability Observations
- The size of the facility required to process a specific water flow rate is reduced by a factor of 100
- The weight of ZVM required to process a specific water flow rate is reduced by a factor of 100
- The volume of water which can be processed/unit time by a specific weight of ZVM is increased by a factor of 100.
2.3. Impact of O2(g) Generation by ZVM on GWM Permeability
2.3.1. Creation of Macropores Associated with O2 generation




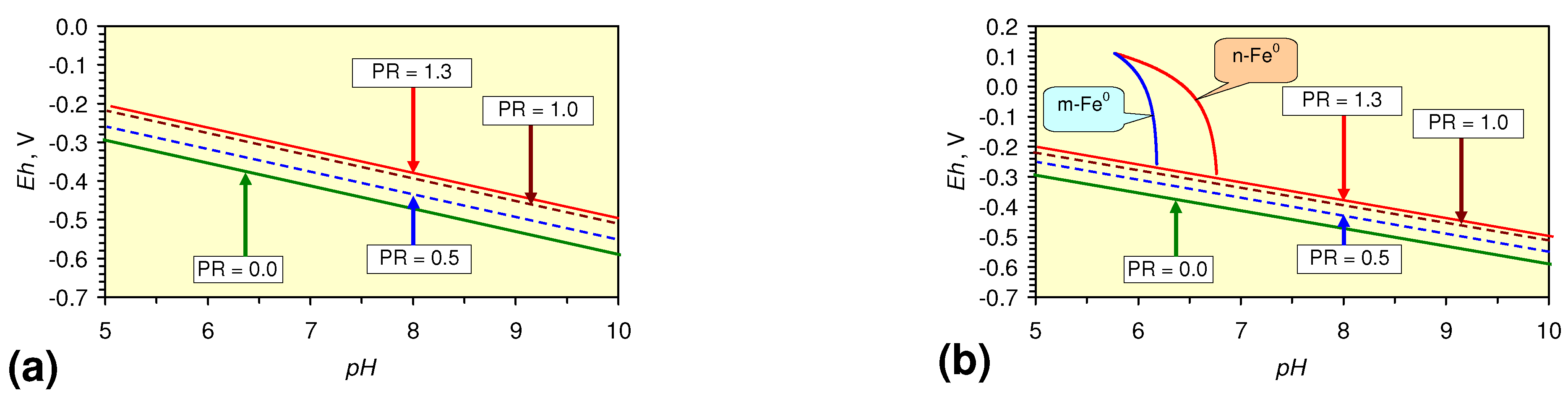
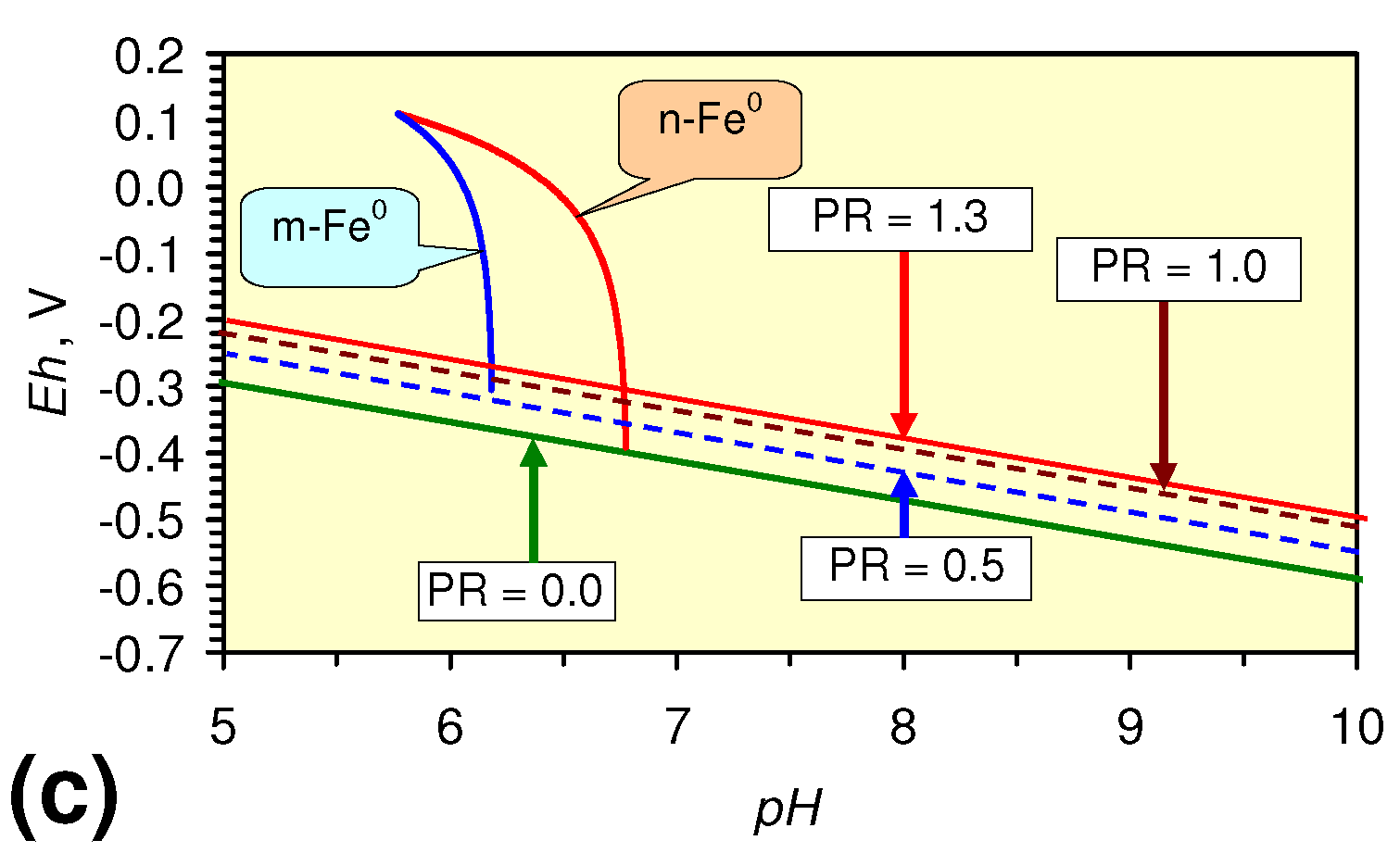
2.3.2. n-Fe0 and n-Fe0 + n-Cu0
2.3.3. n-Fe0 + n-Al0 + n-Cu0
2.3.4. Significance of O2

3. Results
3.1. Static Diffusion Reactor Results
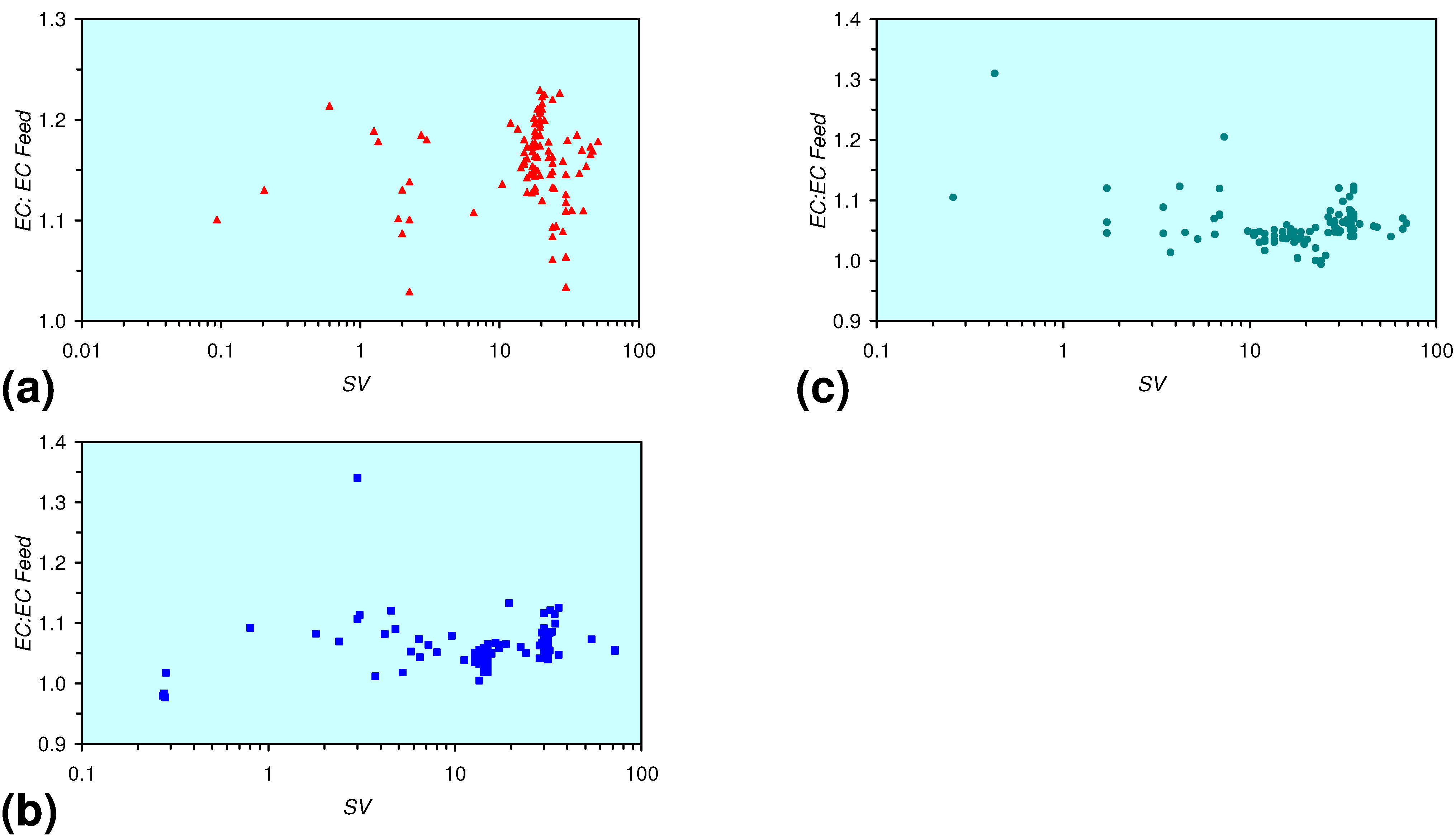
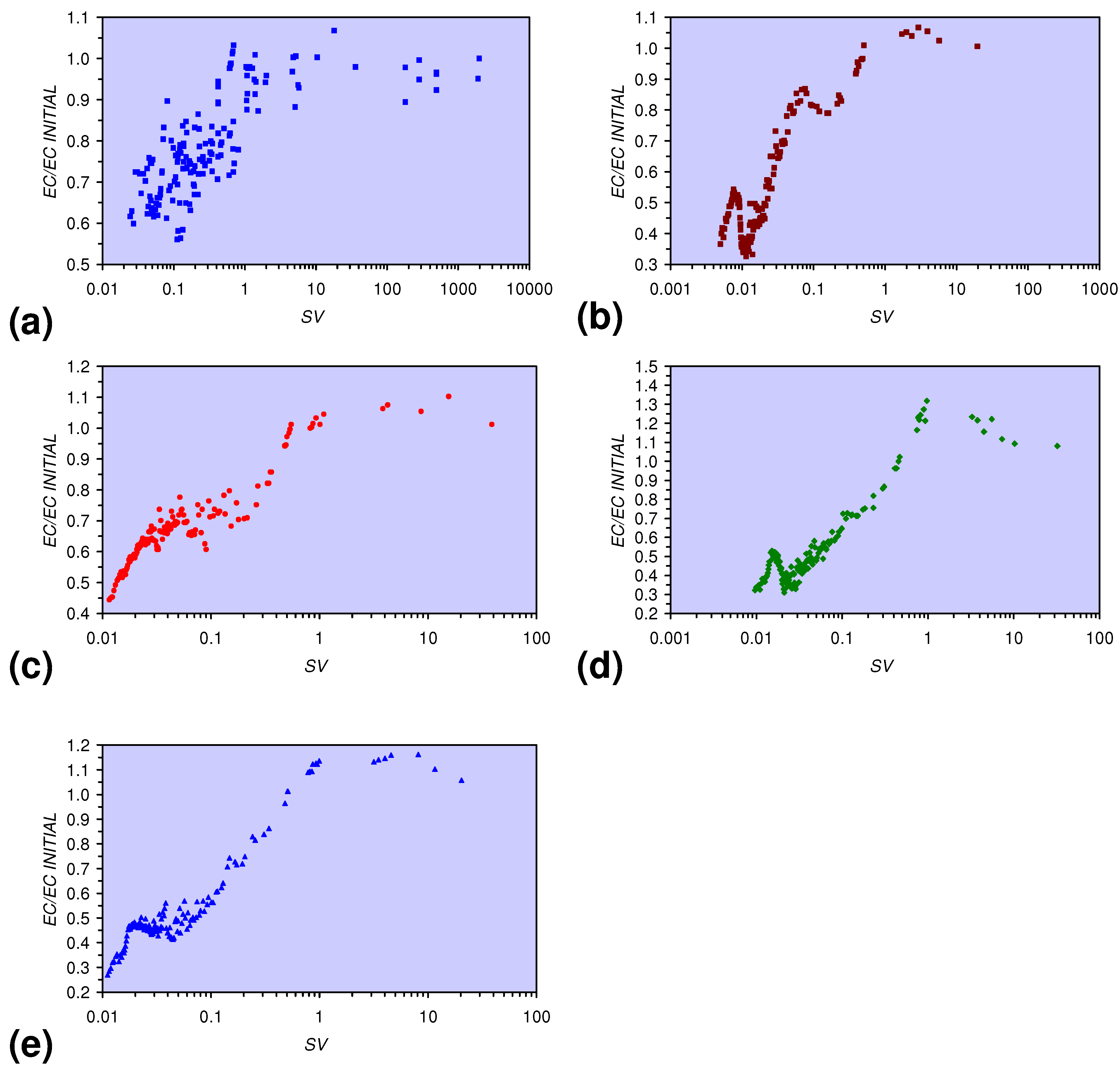
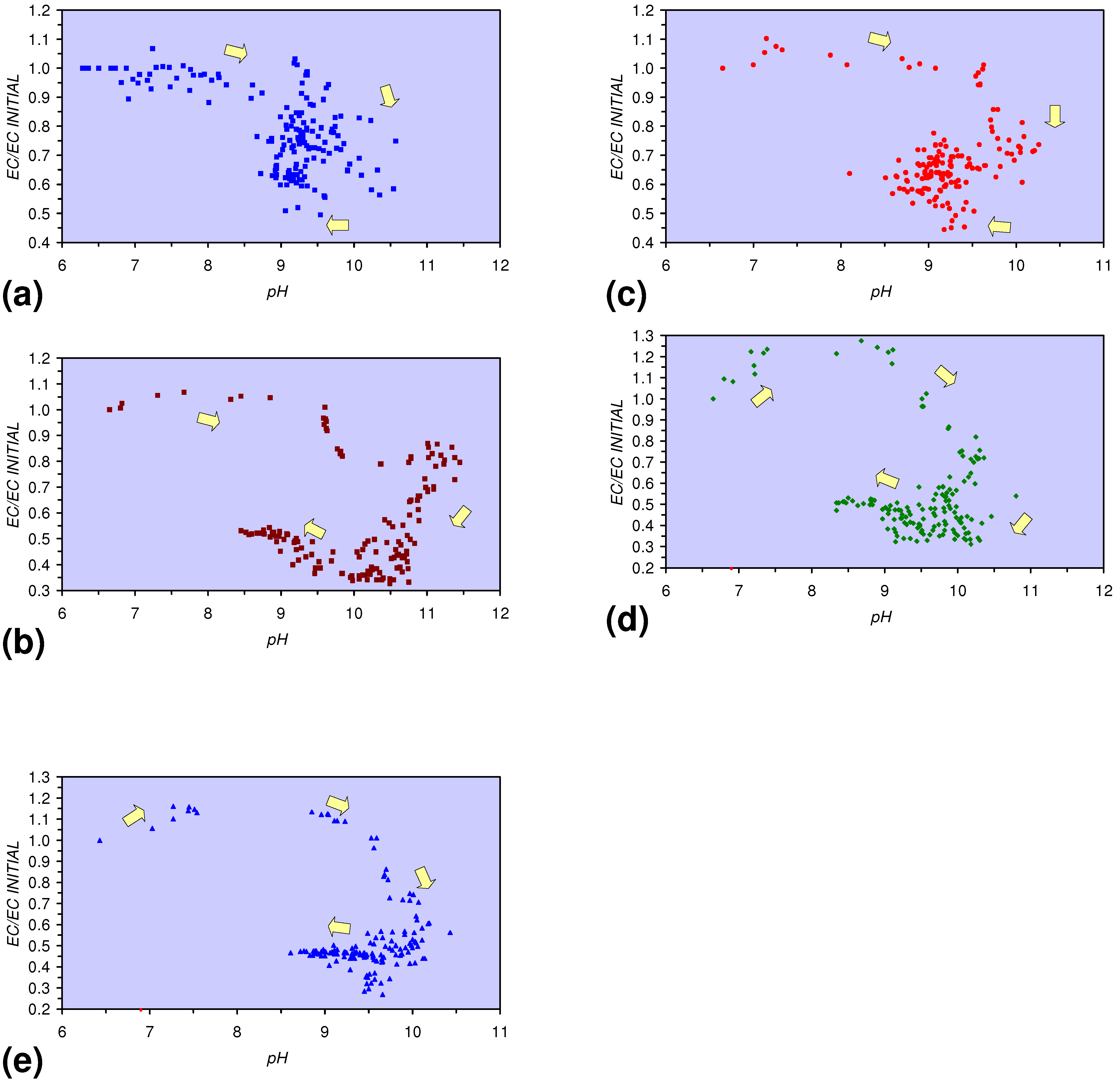
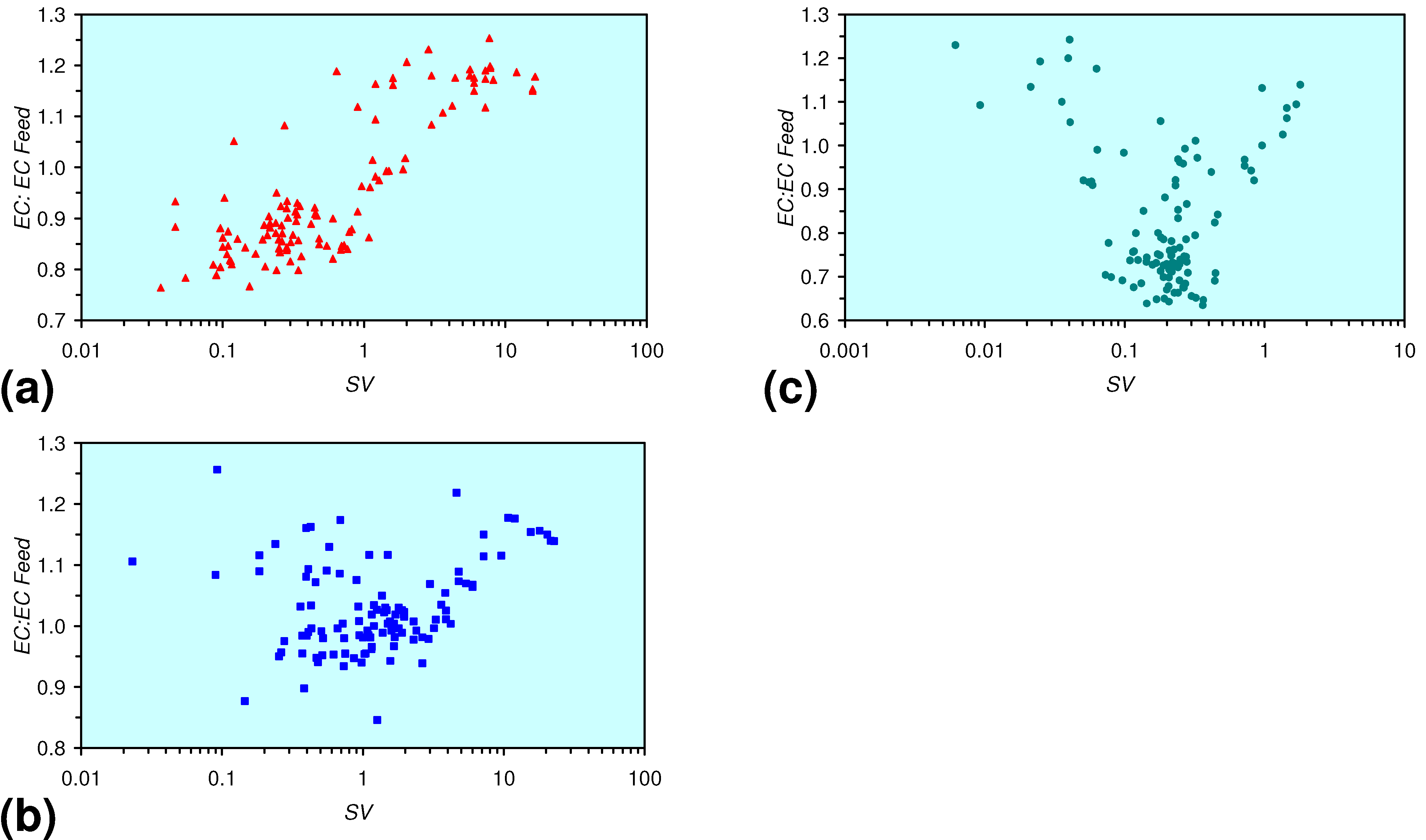
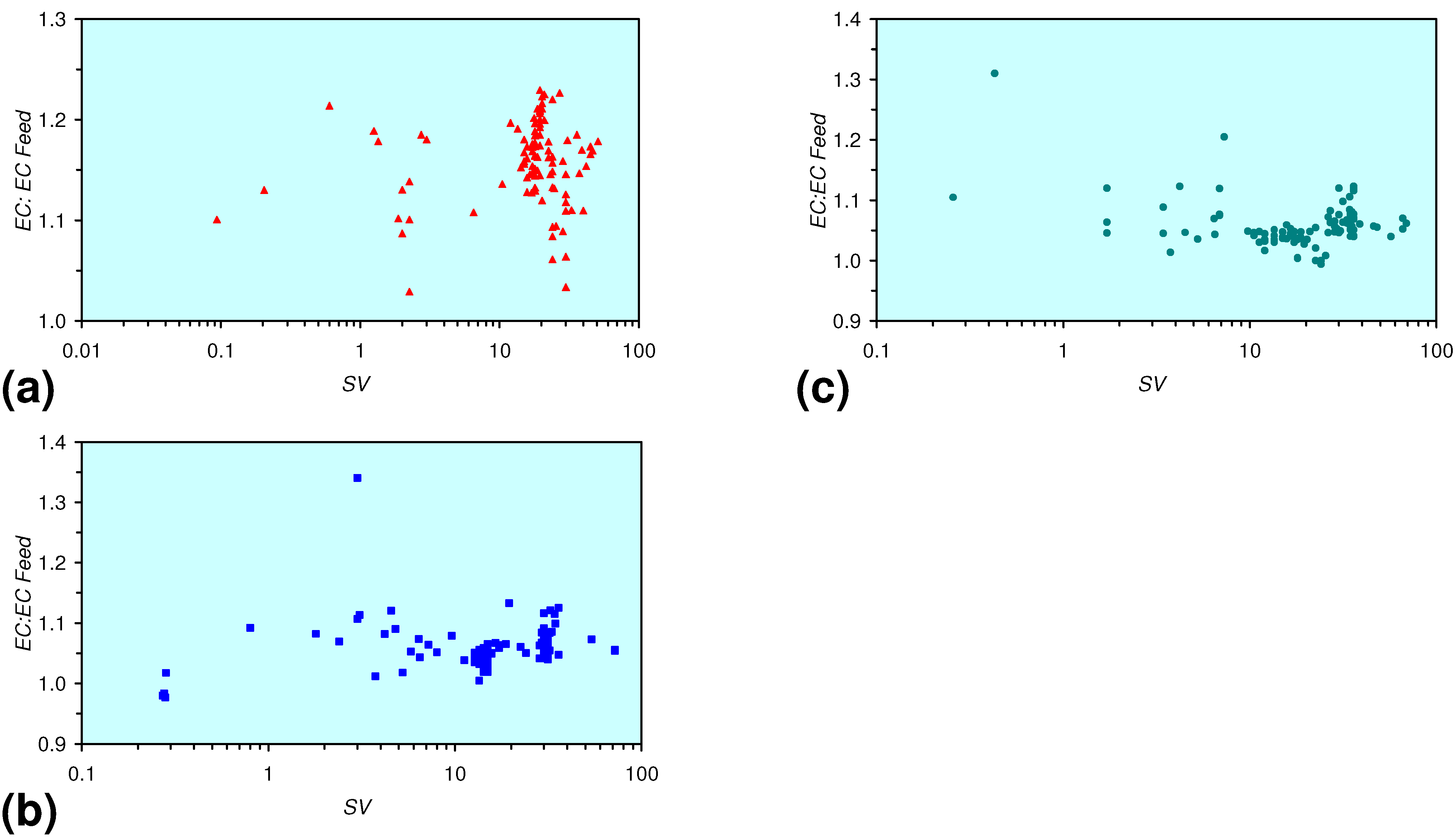
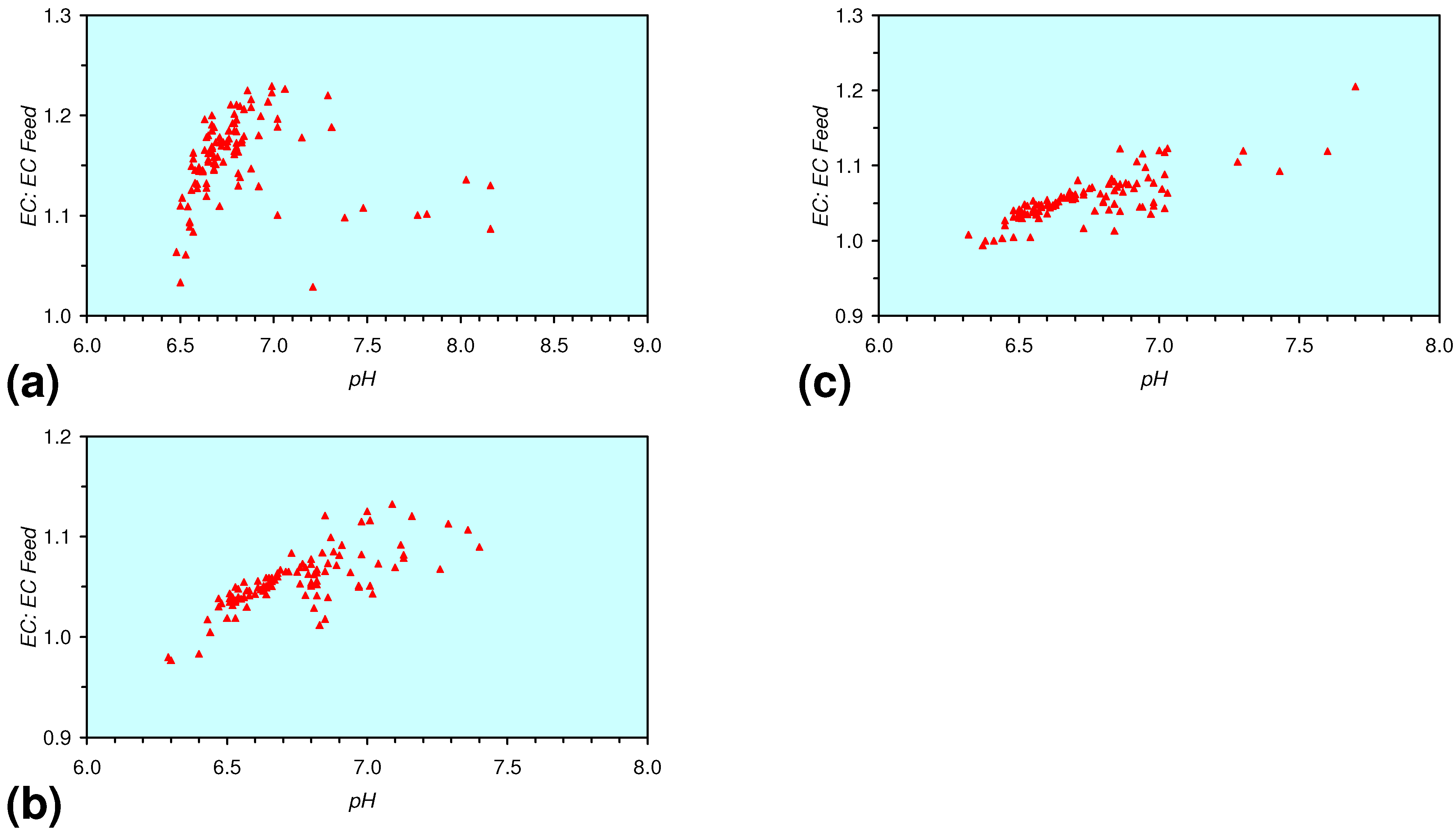
- The EC:EC Initial ratio initially rises with decreasing SV, before subsequently reducing. The ratio may reduce to <30% (Figure 13), indicating that m-ZVM can be an effective remover of dissolved ions within water.
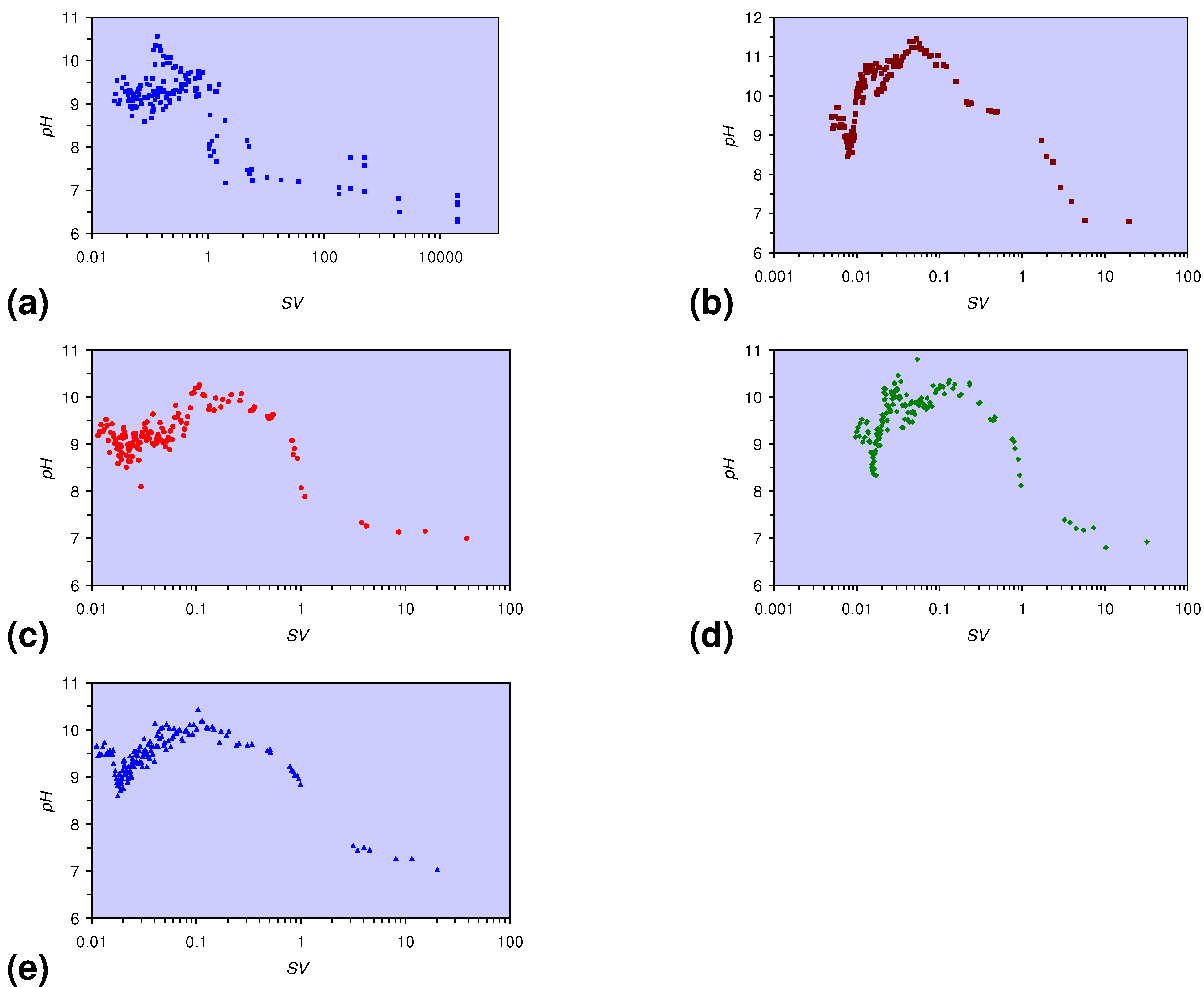
- The EC:EC Initial ratio initially rises with increasing pH but decreases as the pH declines with decreasing SV (Figure 15).
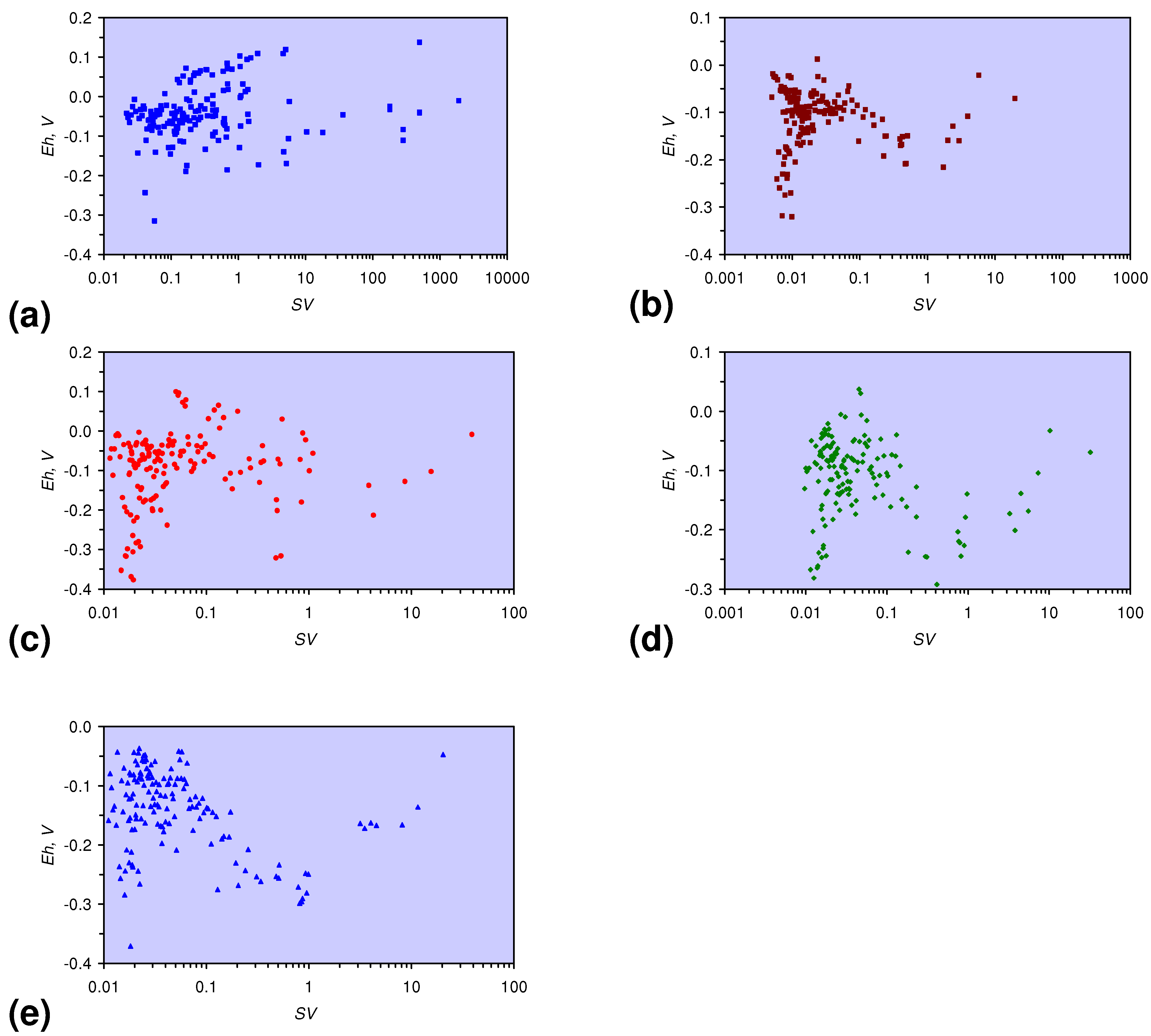
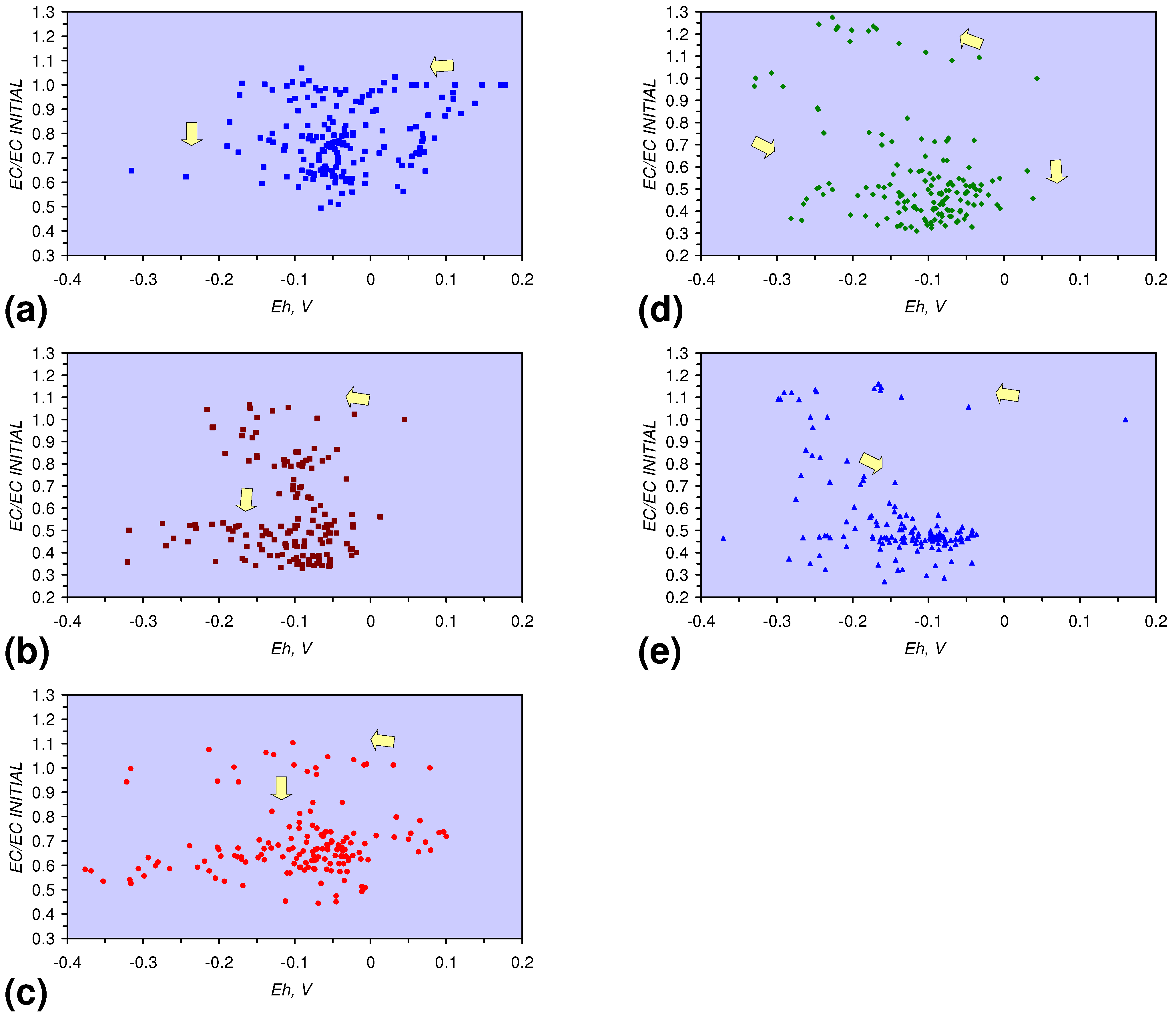
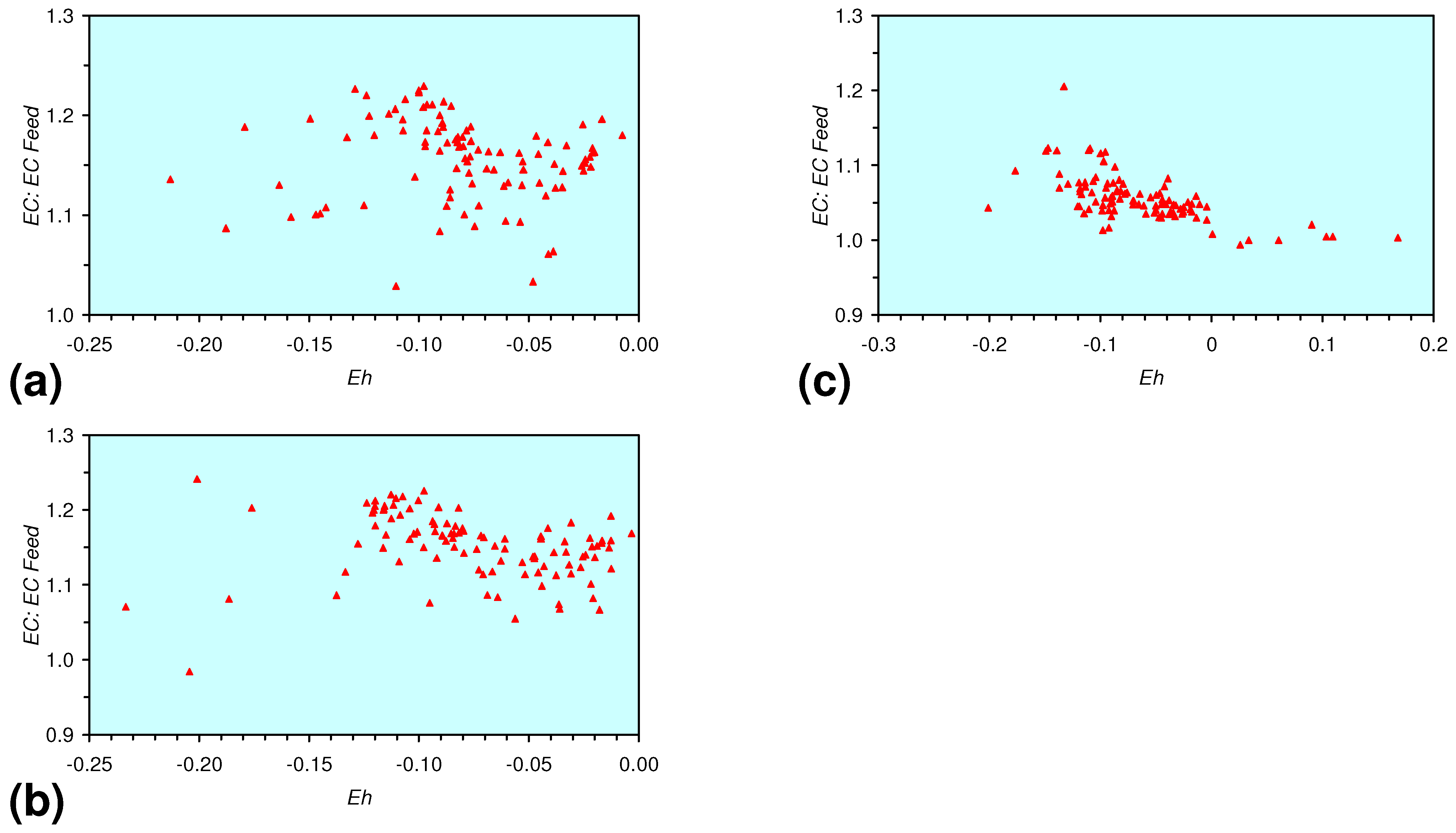
- The EC:EC Initial ratio varies with both Eh and SV. The exact relationship varies with m-ZVM type (Figure 16).
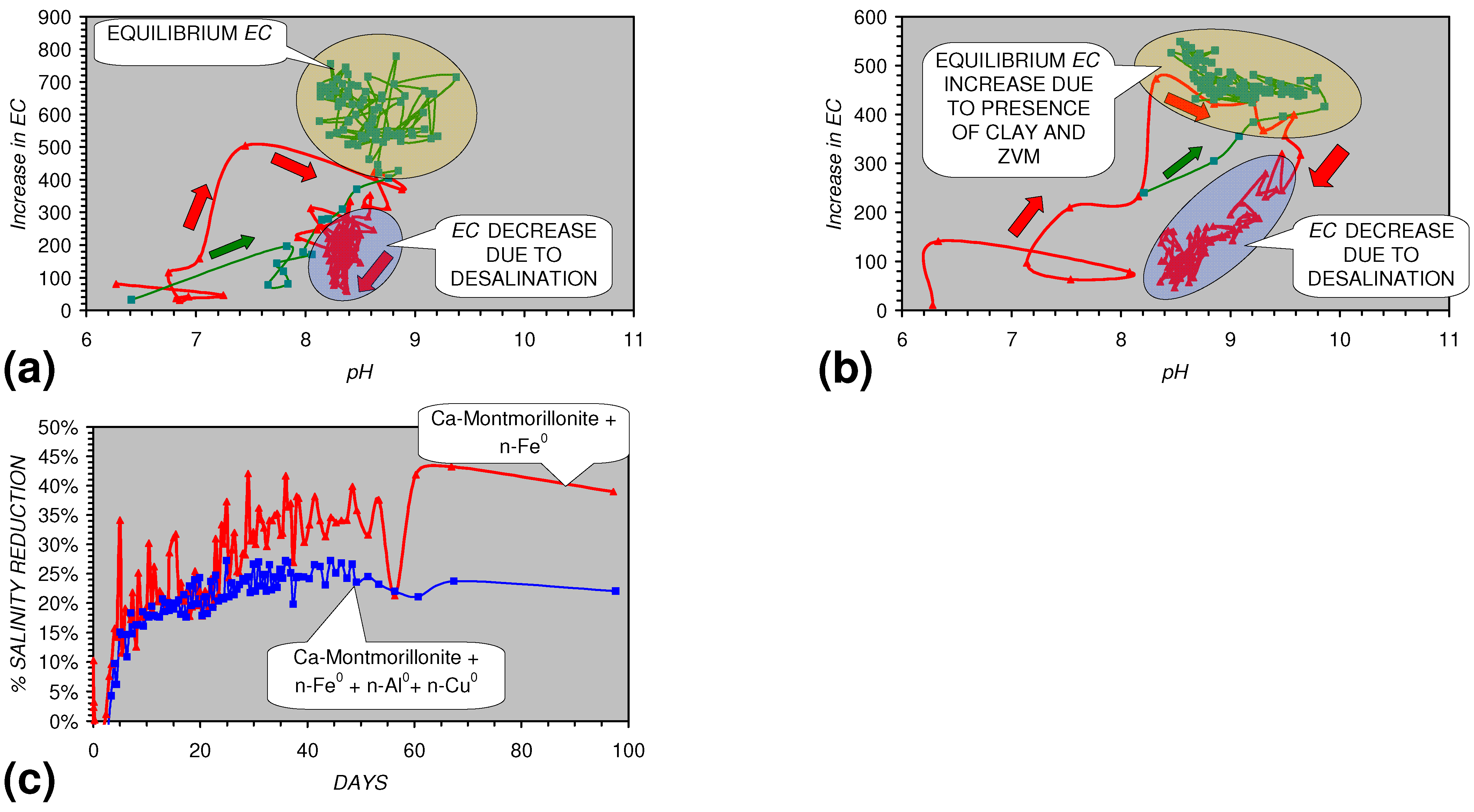
- The presence of more than 1 metal type (or a combination of n-ZVM and m-ZVM of the same metal type) reduces the variance associated with a specific parameter (e.g., Eh, pH, EC).
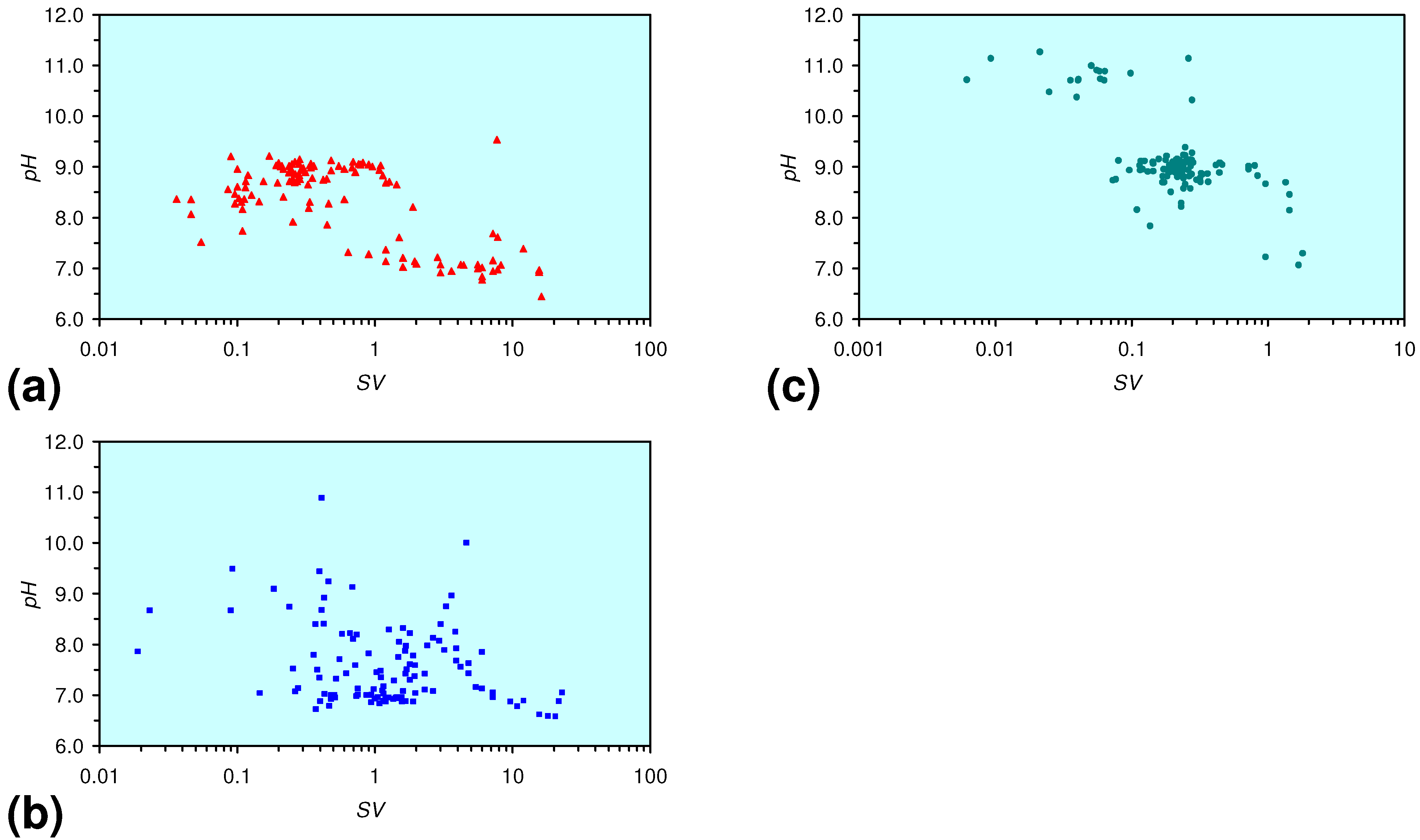
3.1.1. MWR1
pH = −0.5243 × Log(0.13 < SV < 6) + 8.796; R2 = 0.508;
pH = 0.1887 × Log(SV < 0.13) + 9.743; R2 = 0.061; pHInitial = 6.43;
EC:ECInitial = 0.0693 × Log(SV < 1) + 0.8791; R2 = 0.424; ECInitial = 0.339 mS cm−1;
3.1.2. MWR2
Eh = −0.0335 × Log(0.02 < SV < 1) − 0.1932; R2 = 0.629;
Eh = 0.0185 × Log(SV < 0.02) − 0.374; R2 = 0.005; EhInitial = 0.045 V
pH = −0.8708 × Log(0.05 < SV < 5) + 8.787; R2 = 0.972;
pH = 0.5784 × Log(0.01 < SV < 0.05) + 12.89; R2 = 0.559;
pH = 4.9227 × Log(0.0077 < SV < 0.01) + 32.364; R2 = 0.725;
pH = −1.3165 × Log(SV < 0.0077) + 2.5779; R2 = 0.471; pHInitial = 6.65;
EC:ECInitial = 0.1153 × Log(0.1 < SV < 1) + 1.0281; R2 = 0.877;
EC:ECInitial = −0.1431 × Log(0.07 < SV < 0.1) + 0.487; R2 = 0.851;
EC:ECInitial = 0.2862 × Log(0.01 < SV < 0.07) + 1.6361; R2 = 0.948;
EC:ECInitial = −0.6428 × Log(0.0077 < SV < 0.01) – 2.5634; R2 = 0.734;
EC:ECInitial = 0.3692 × Log(SV < 0.0077) + 2.332: R2 = 0.961; ECInitial = 0.328 mS cm−1;
3.1.3. MWR3
Eh = −0.0359 × Log(0.08 < SV < 1) − 0.12; R2 = 0.145;
Eh = 0.0772 × Log(SV < 08) + 0.1764; R2 = 0.144; EhInitial = 0.078 V;
pH = −1.8857 × Log(0.5 < SV < 1) + 8.4663; R2 = 0.879;
pH = −0.2833 × Log(0.1 < SV < 0.5) + 9.4166; R2 = 0.590;
pH = 0.307 × Log(SV < 0.1) + 10.234: R2 = 0.263; pHInitial = 6.65;
EC:ECInitial = 0.2208 × Log(0.2 < SV < 1) + 1.0834; R2 = 0.896;
EC:ECInitial = 0.0468 × Log(0.025 < SV < 0.2) + 0.822; R2 = 0.354;
EC:ECInitial = 0.2477 × Log(SV < 0.025) + 1.563; R2 = 0.968; ECInitial = 0.331 mS cm−1;
3.1.4. MWR4
Eh = −0.0516 × Log(0.03 < SV < 0.4) − 0.2427; R2 = 0.297;
Eh = 0.052 × Log(SV < 0.03) + 0.097; R2 = 0.051; EhInitial = 0.043 V;
pH = −1.1926 × Log(0.2 < SV < 1) + 8.5343; R2 = 0.911;
pH = 0.2224 × Log(0.03 < SV < 0.2) + 10.569; R2 = 0.171;
pH = 2.4307 × Log(0.0155 < SV < 0.03) + 18.846; R2 = 0.744;
pH = −1.4766 × Log(SV < 0.0155) + 2.639; R2 = 0.450; pHInitial = 6.65;
EC:ECInitial = 0.2253 × Log(0.02 < SV < 1) + 1.1981; R2 = 0.964;
EC:ECInitial = −0.4157 × Log(0.015 < SV < 0.02) – 1.209; R2 = 0.757;
EC:ECInitial = 0.4138 × Log(SV < 0.015) + 2.218; R2 = 0.846; ECInitial = 0.332 mS cm−1;
3.1.5. MWR5
Eh = −0.0483 × Log(0.02 > SV < 0.8) − 0.2753; R2 = 0.459;
Eh = −0.0413 × Log(SV < 0.02) − 0.324: R2 = 0.009; EhInitial = 0.160 V;
pH = −0.6104 × Log(0.1 < SV < 3) + 8.8984; R2 = 0.900;
pH = 0.7137 × Log(0.0177 < SV < 0.1) + 11.897; R2 = 0.777;
pH = −1.6348 × Log(SV < 0.0177) + 2.4307: R2 = 0.583; pHInitial = 6.43;
EC:ECInitial = 0.2253 × Log(0.04 < SV < 1) + 1.1281; R2 = 0.979;
EC:ECInitial = 0.0174 × Log(0.02 < SV < 0.04) + 0.5314; R2 = 0.020;
EC:ECInitial = 0.3838 × Log(SV < 0.02) + 1.9909; R2 = 0.930;
ECInitial = 0.334 mS cm−1; EC:ECInitial = Observed EC/ECInitial.



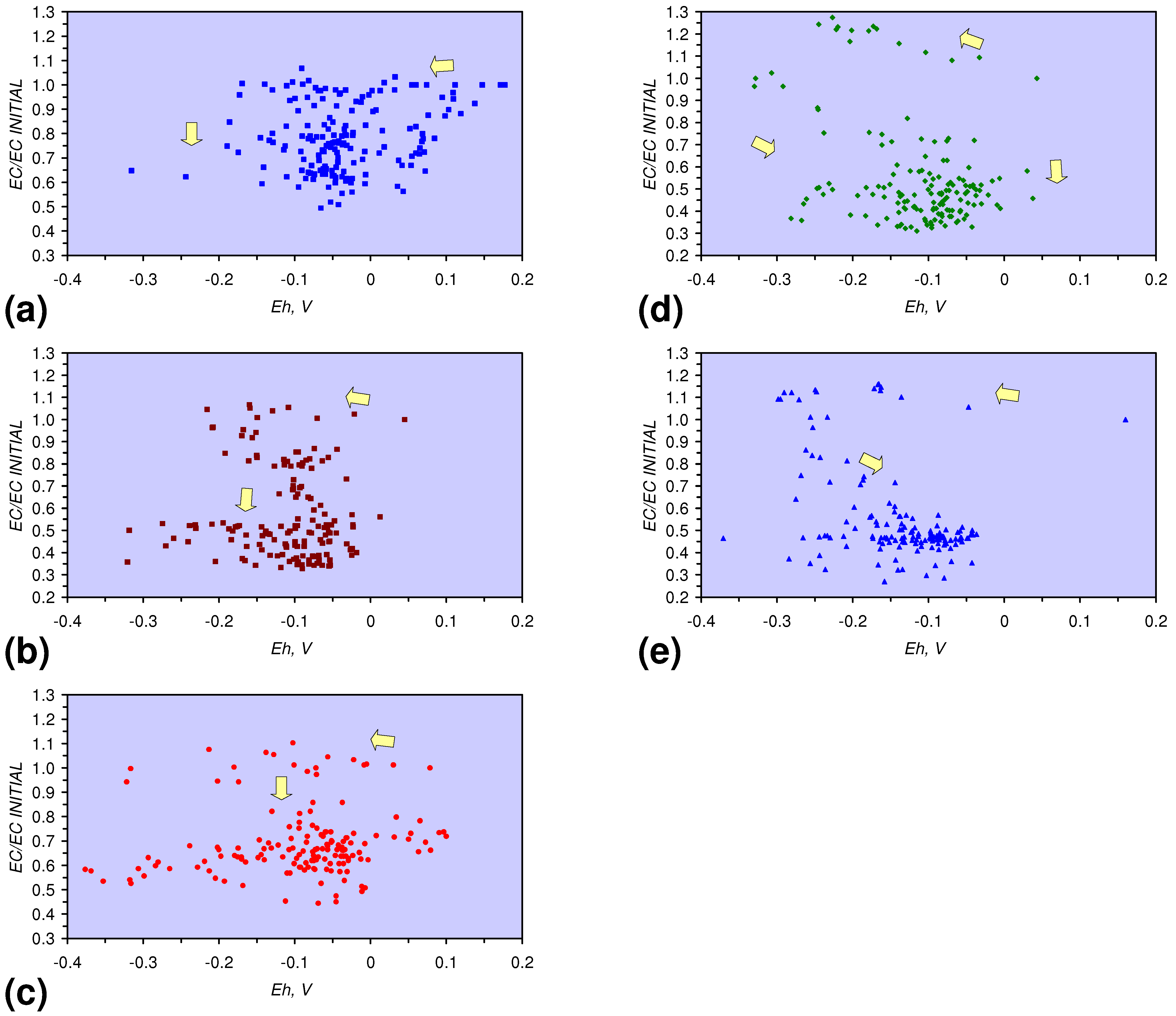
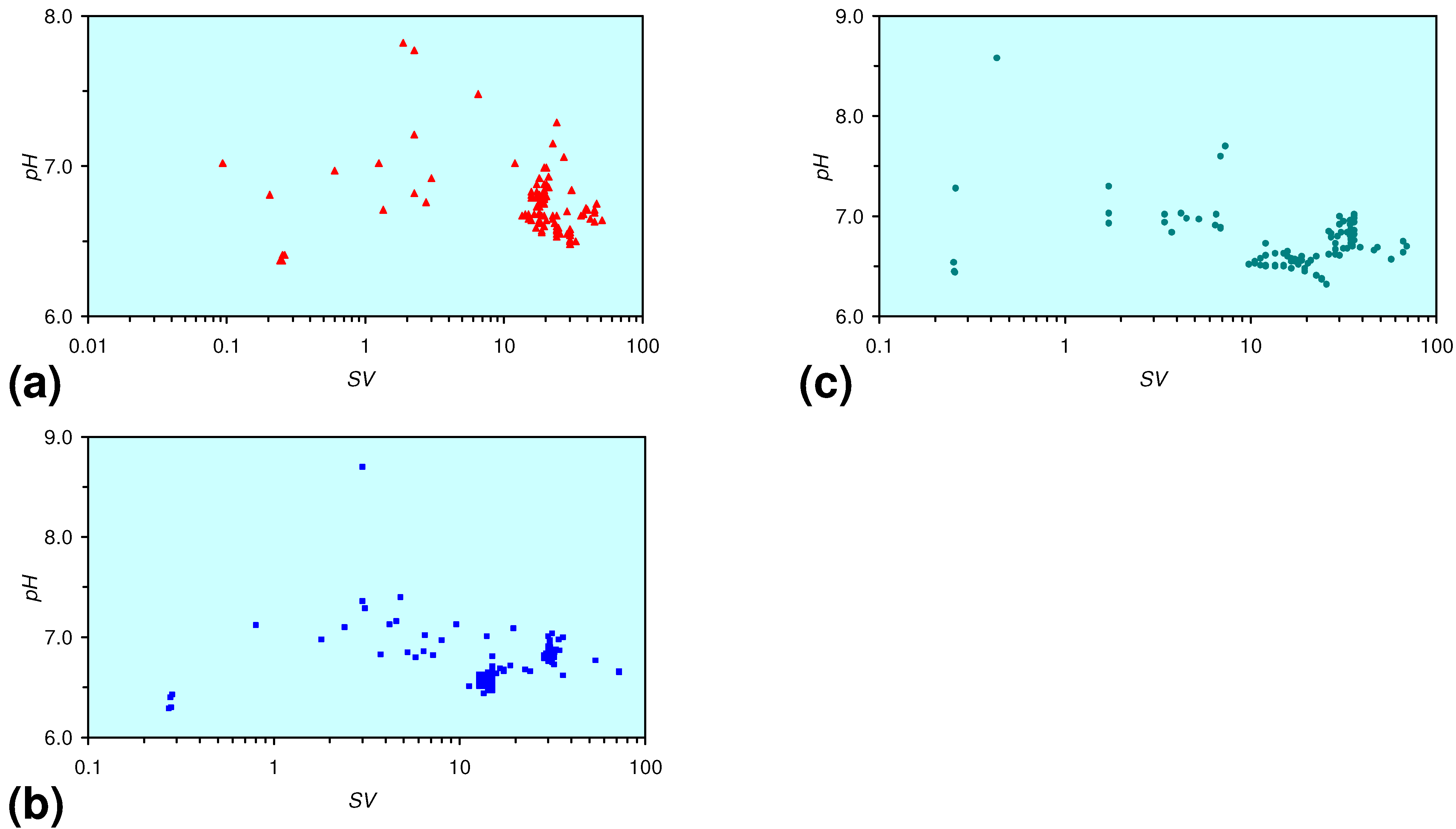
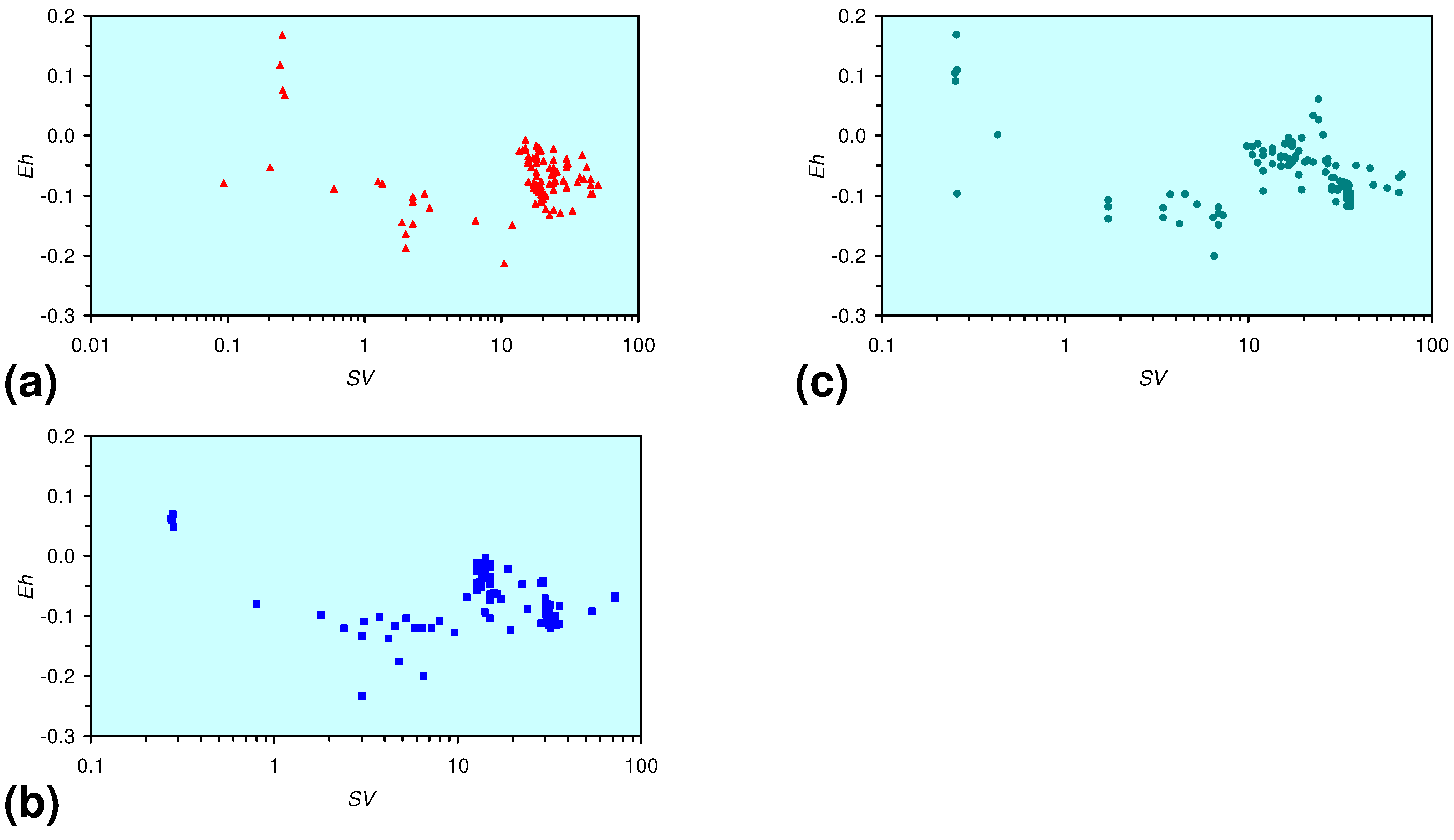
3.2. Continuous Flow Reactor Results: n-ZVM
3.2.1. DR1
3.2.2. DR2
3.2.3. DR3
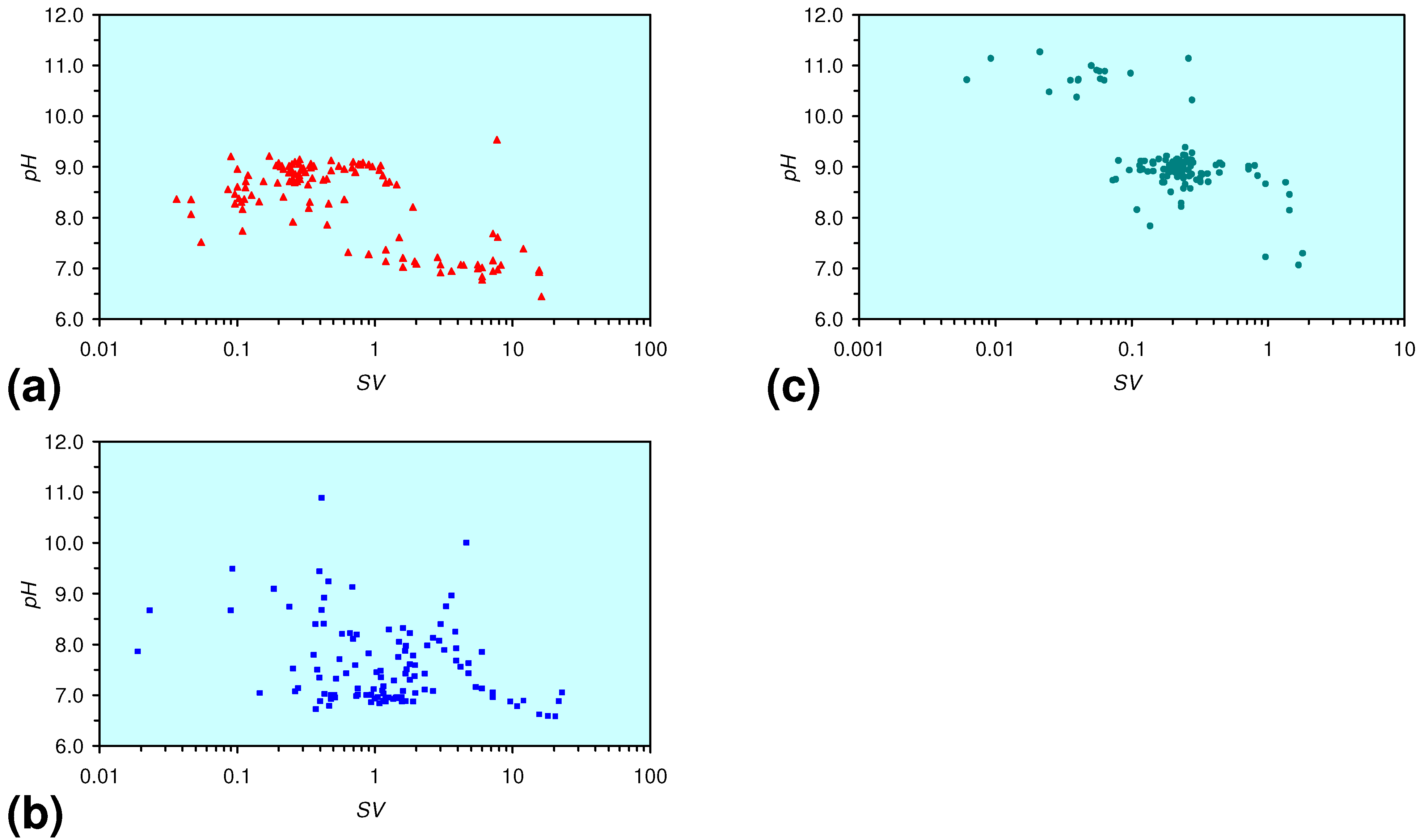
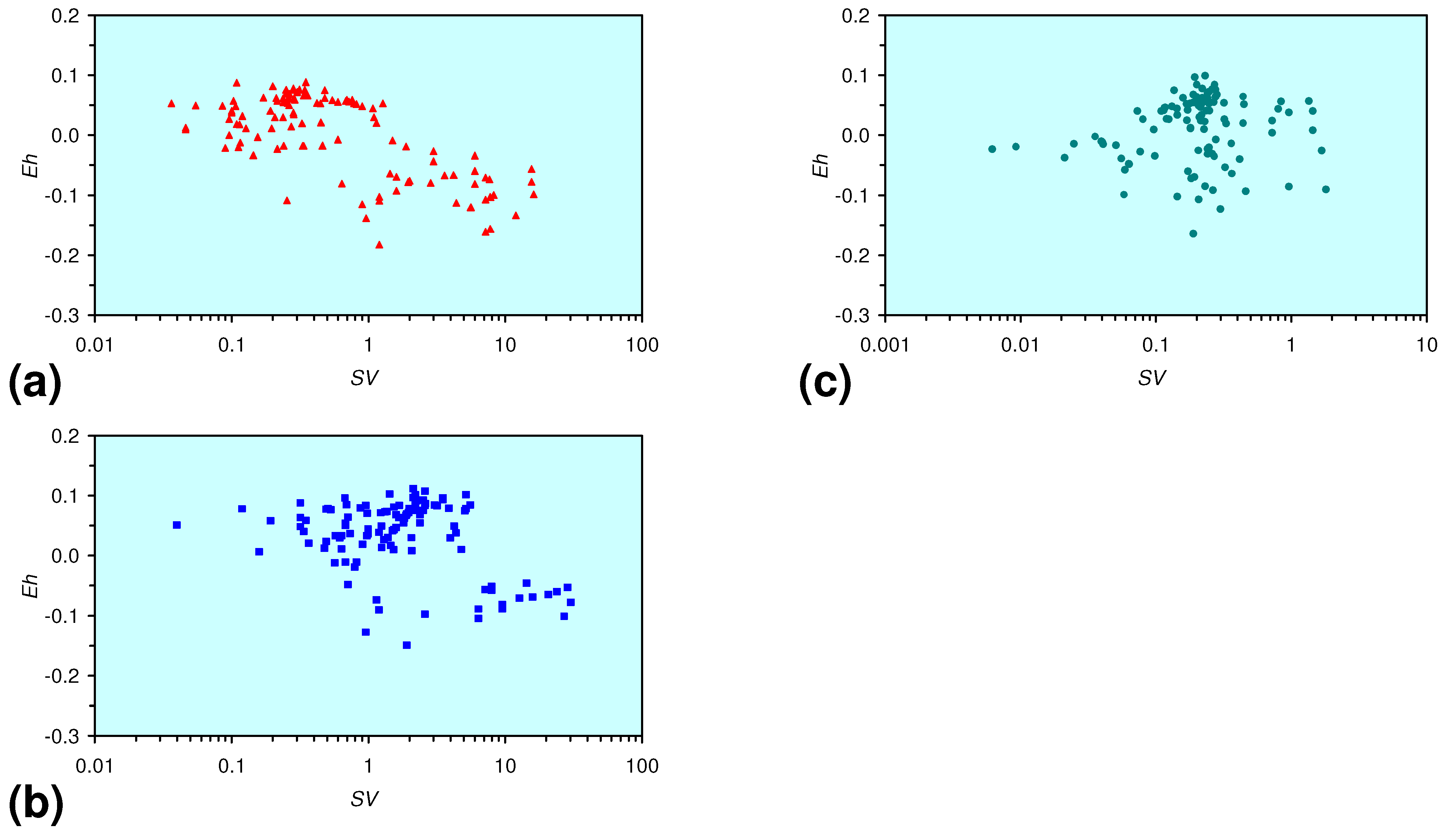
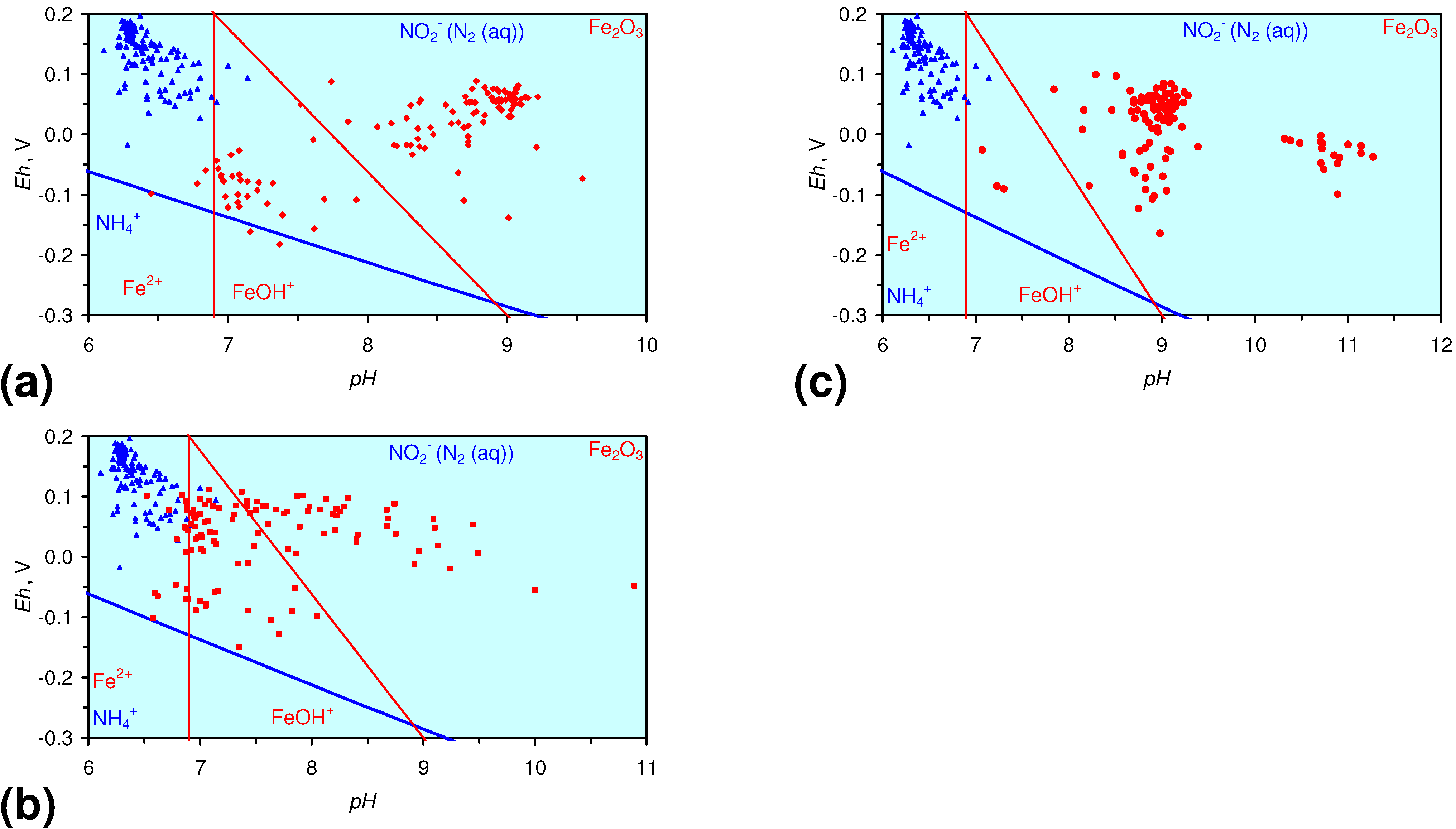
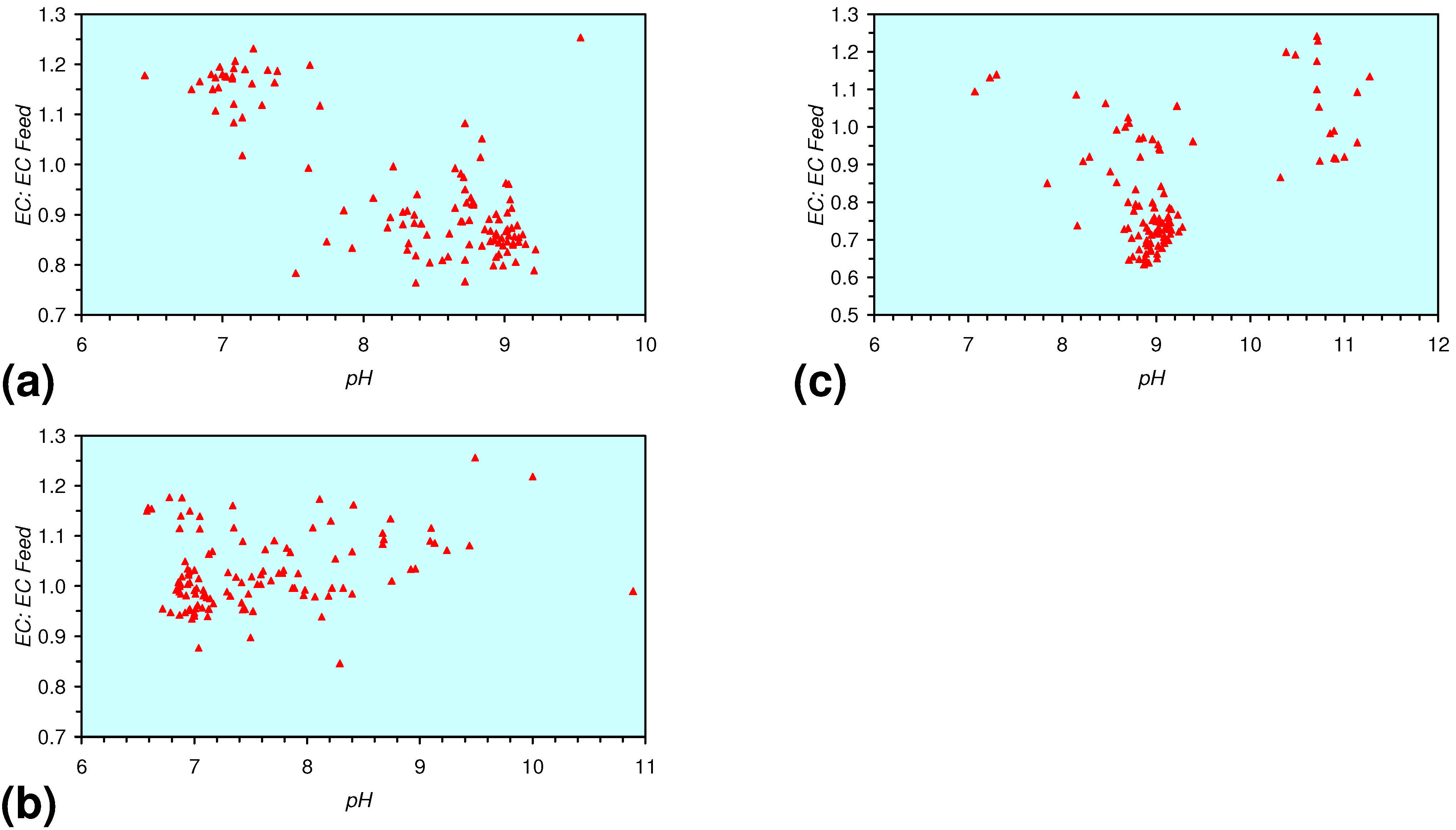
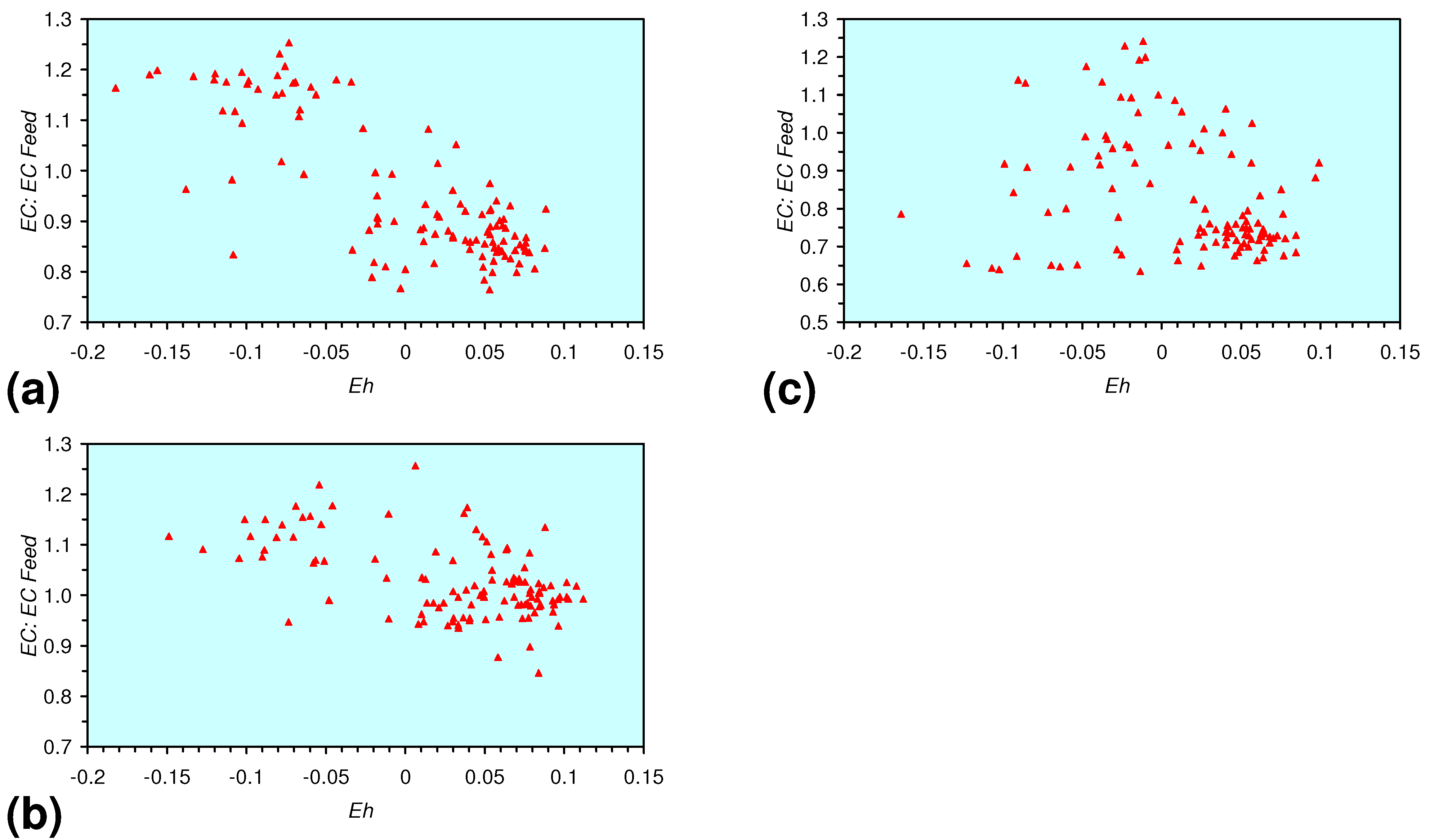
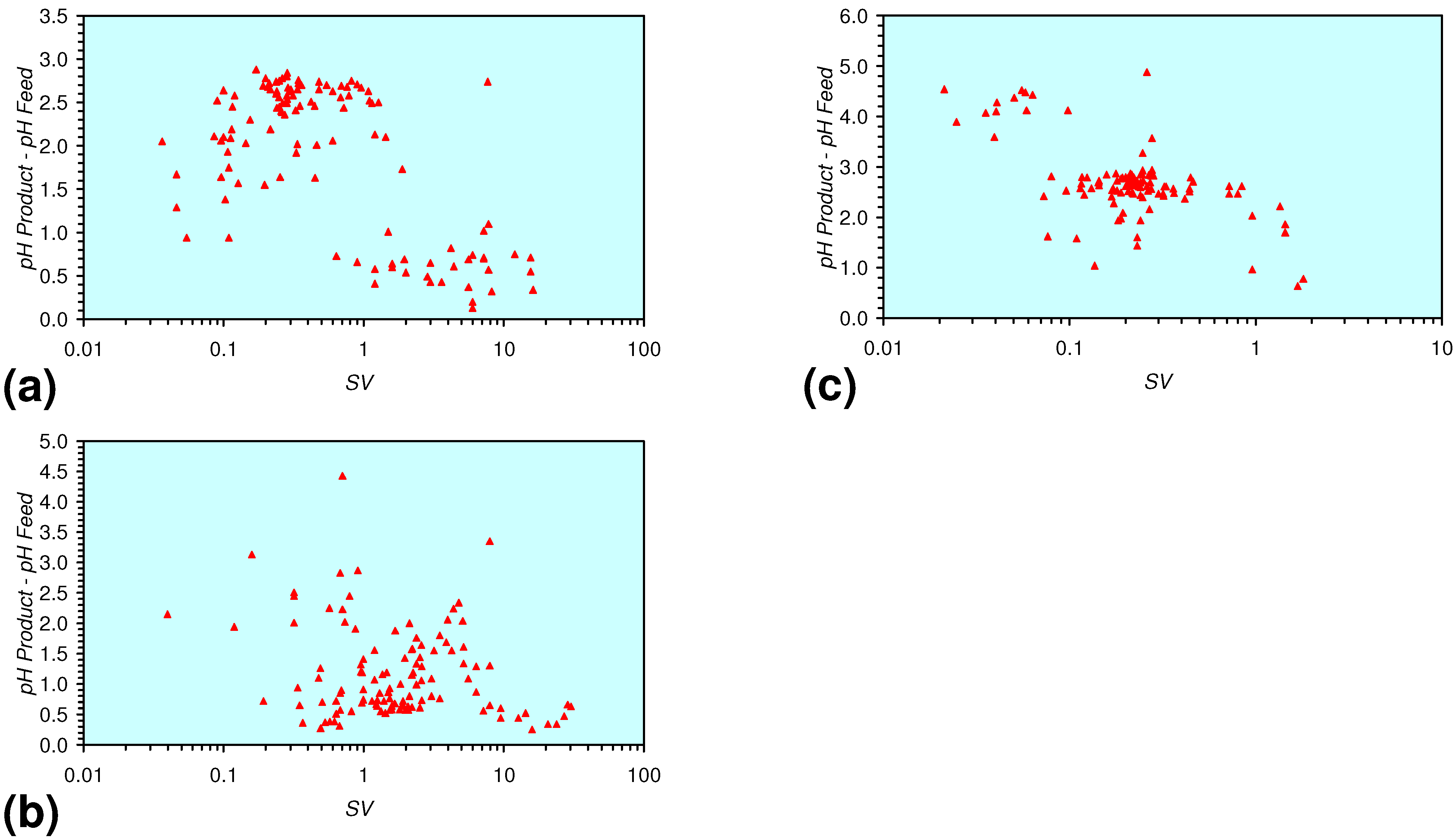
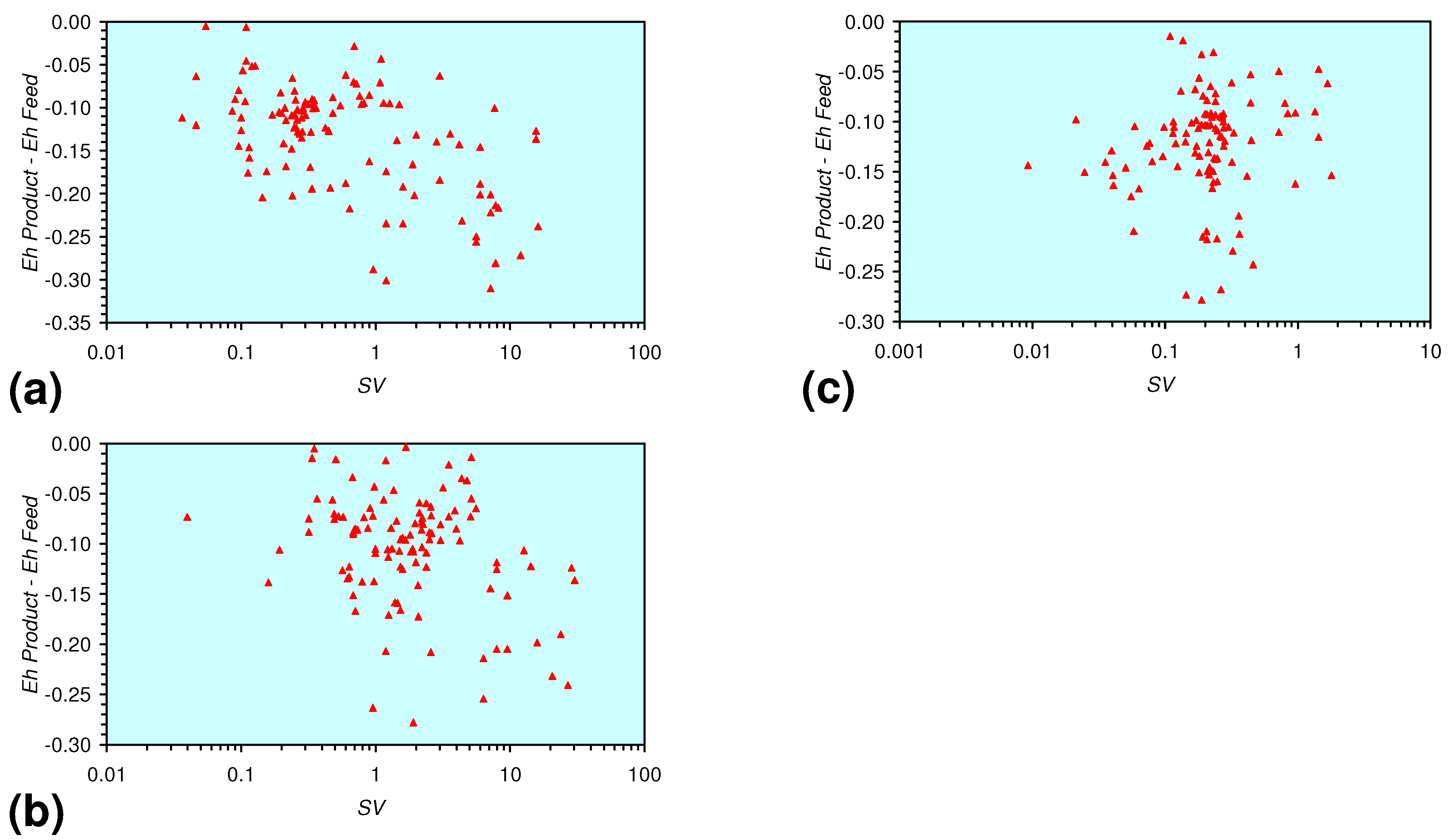
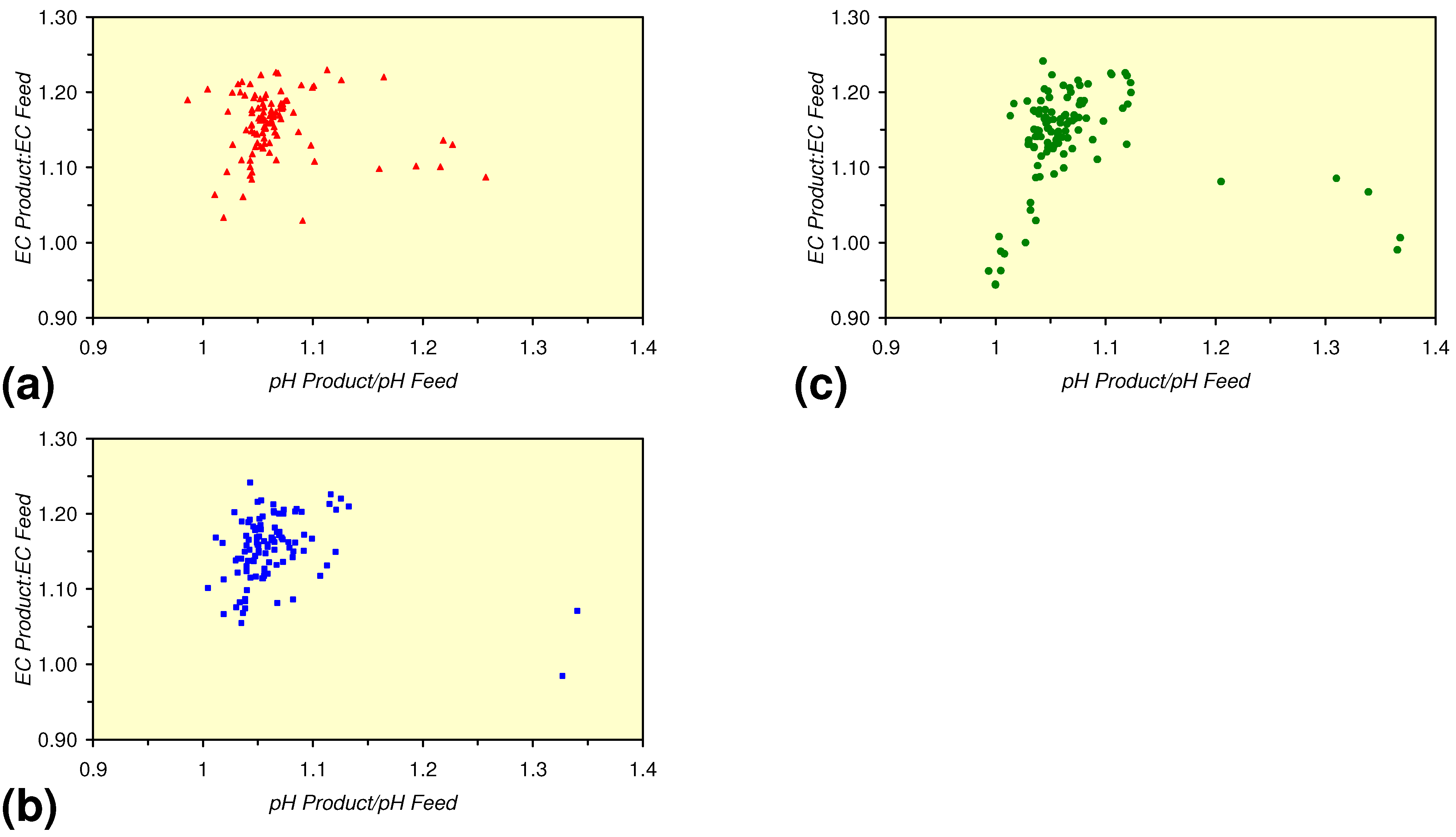
3.3. Continuous Flow Reactor Results: m-ZVM
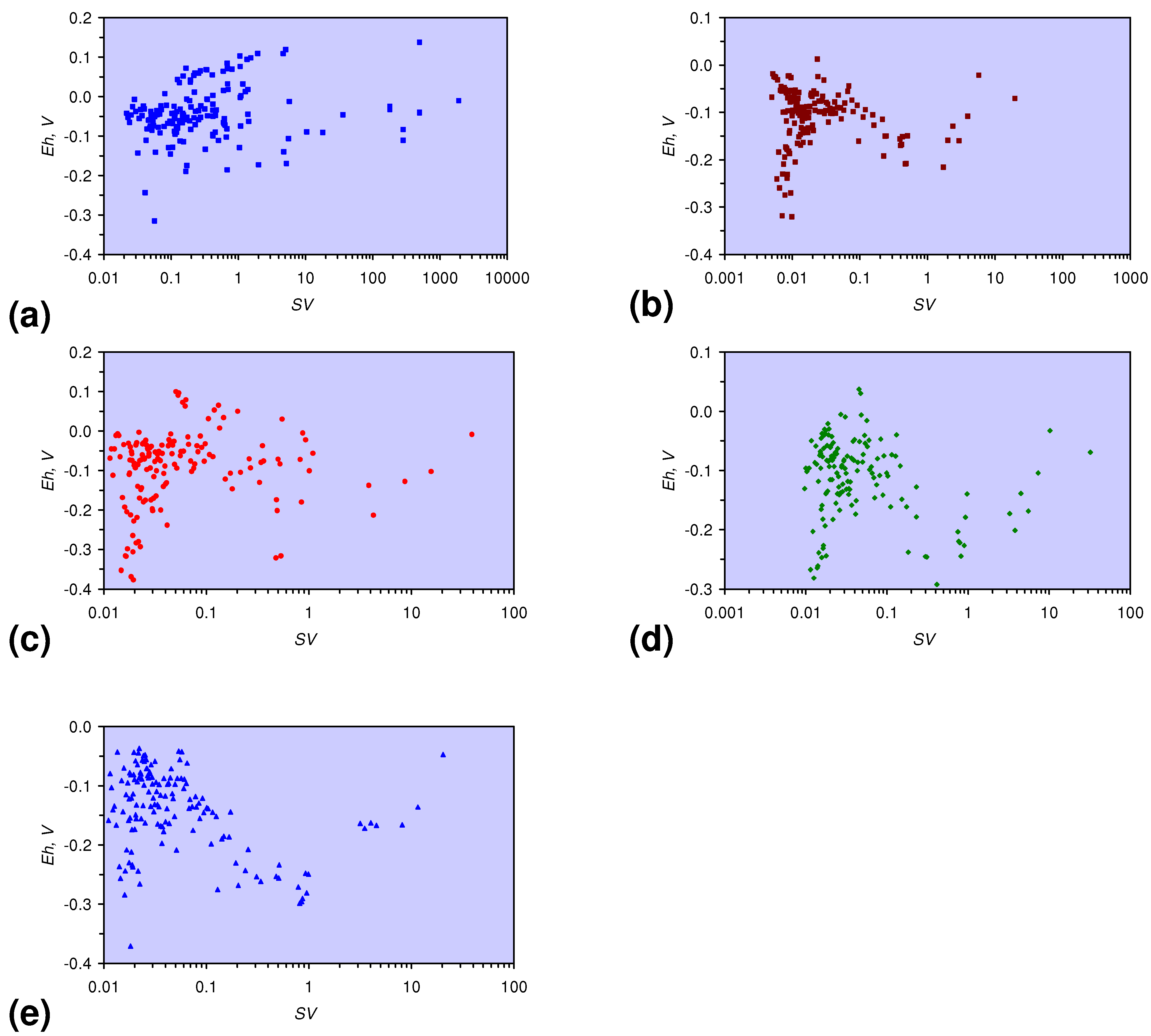
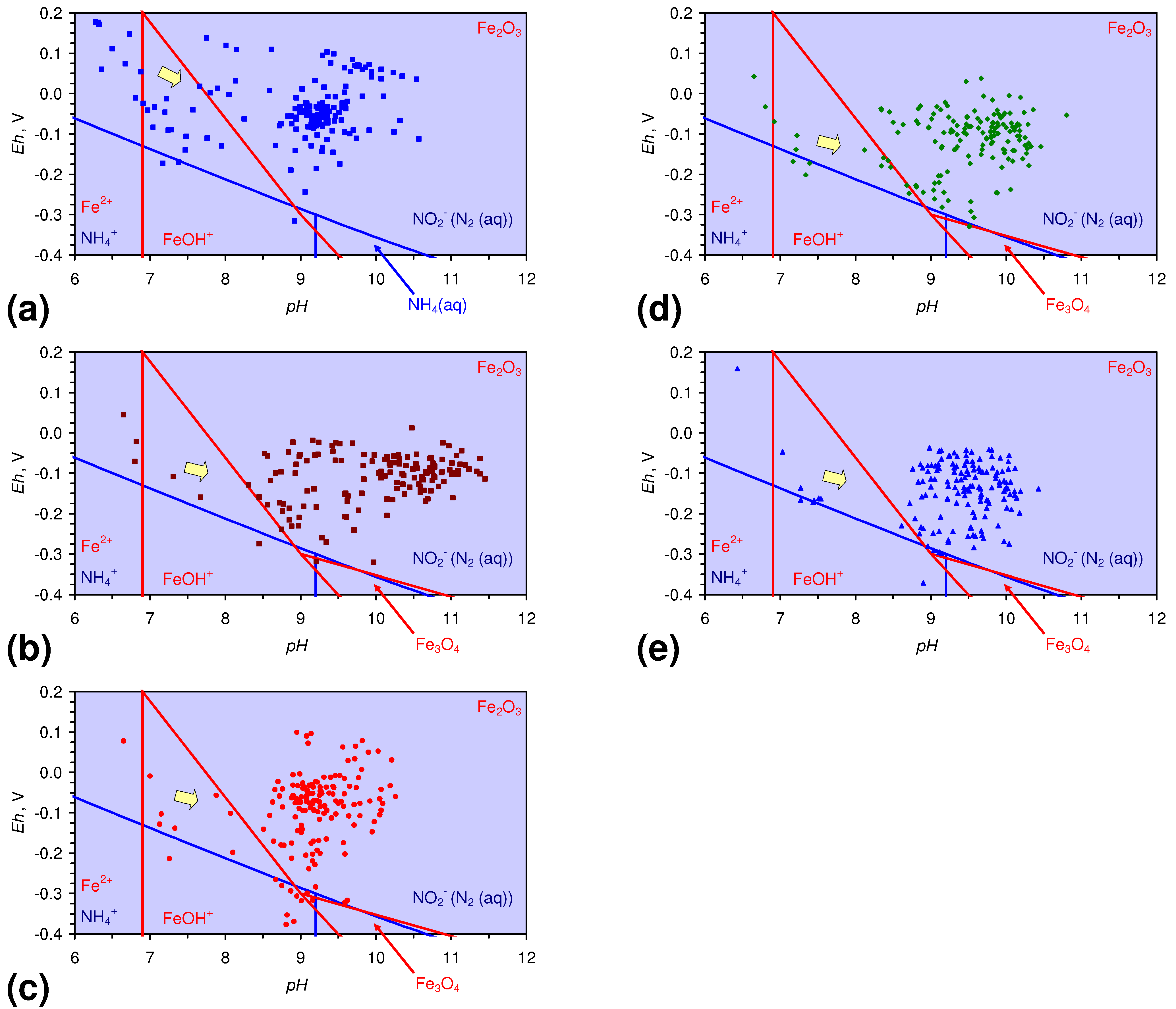
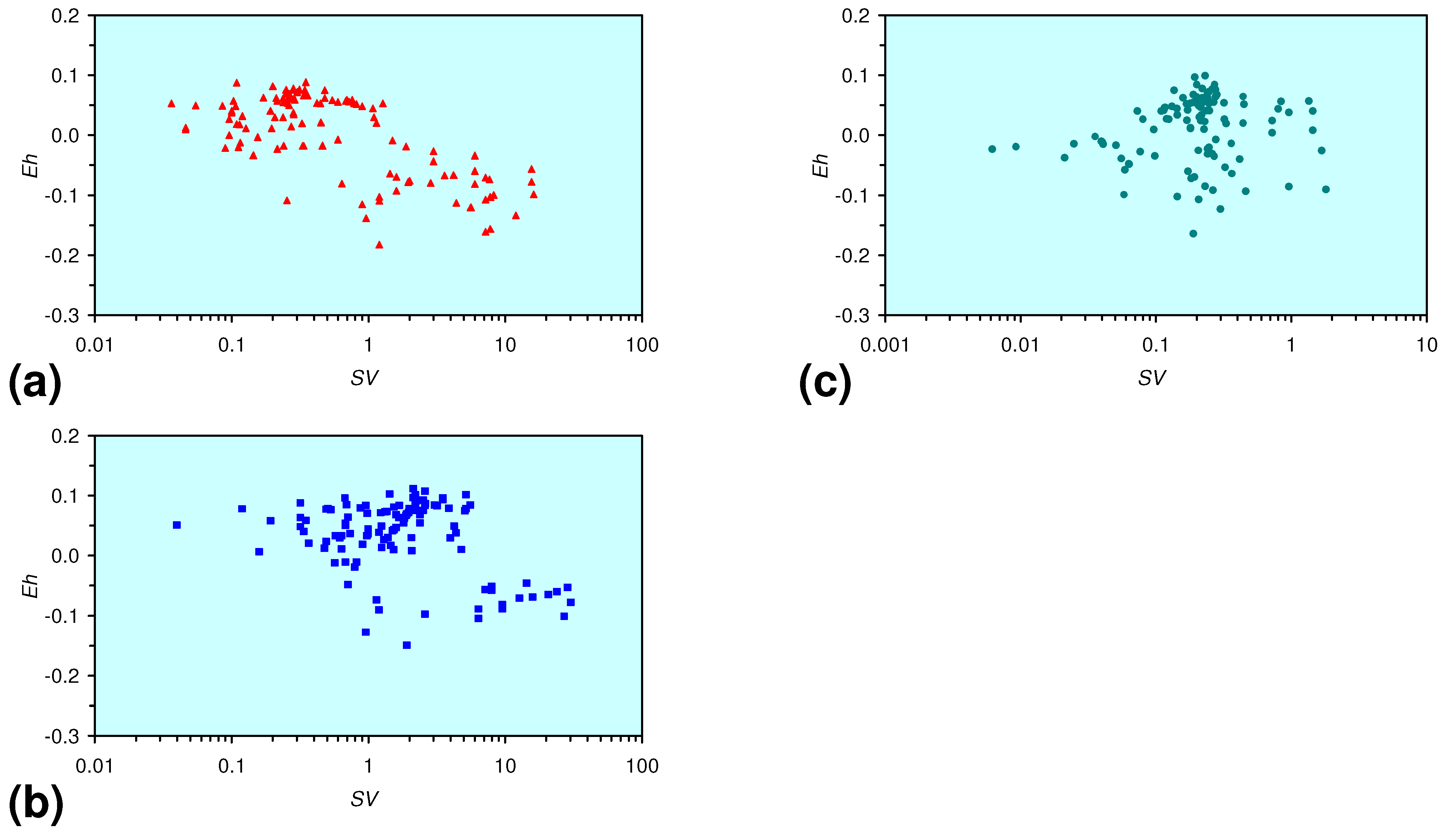
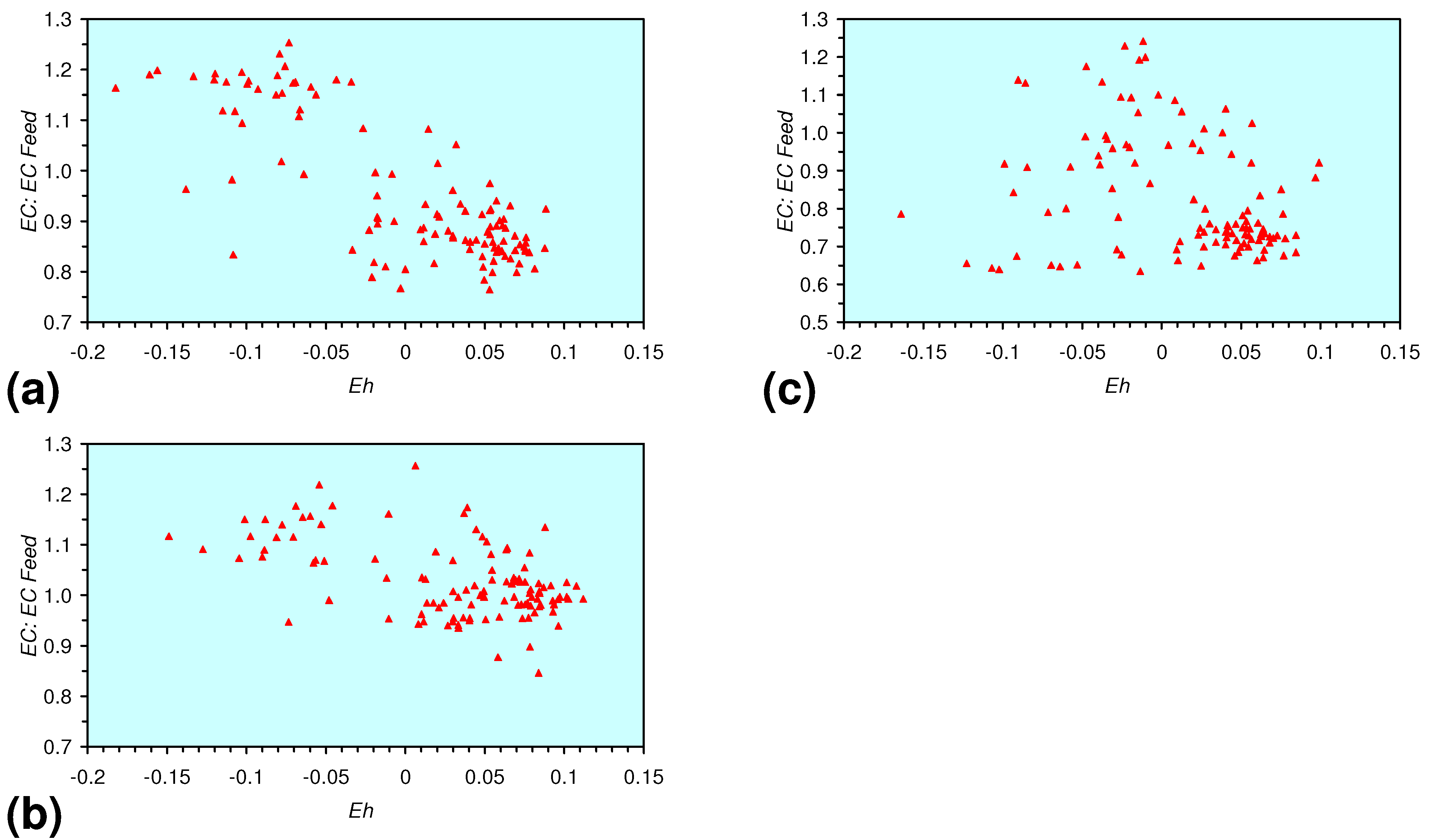
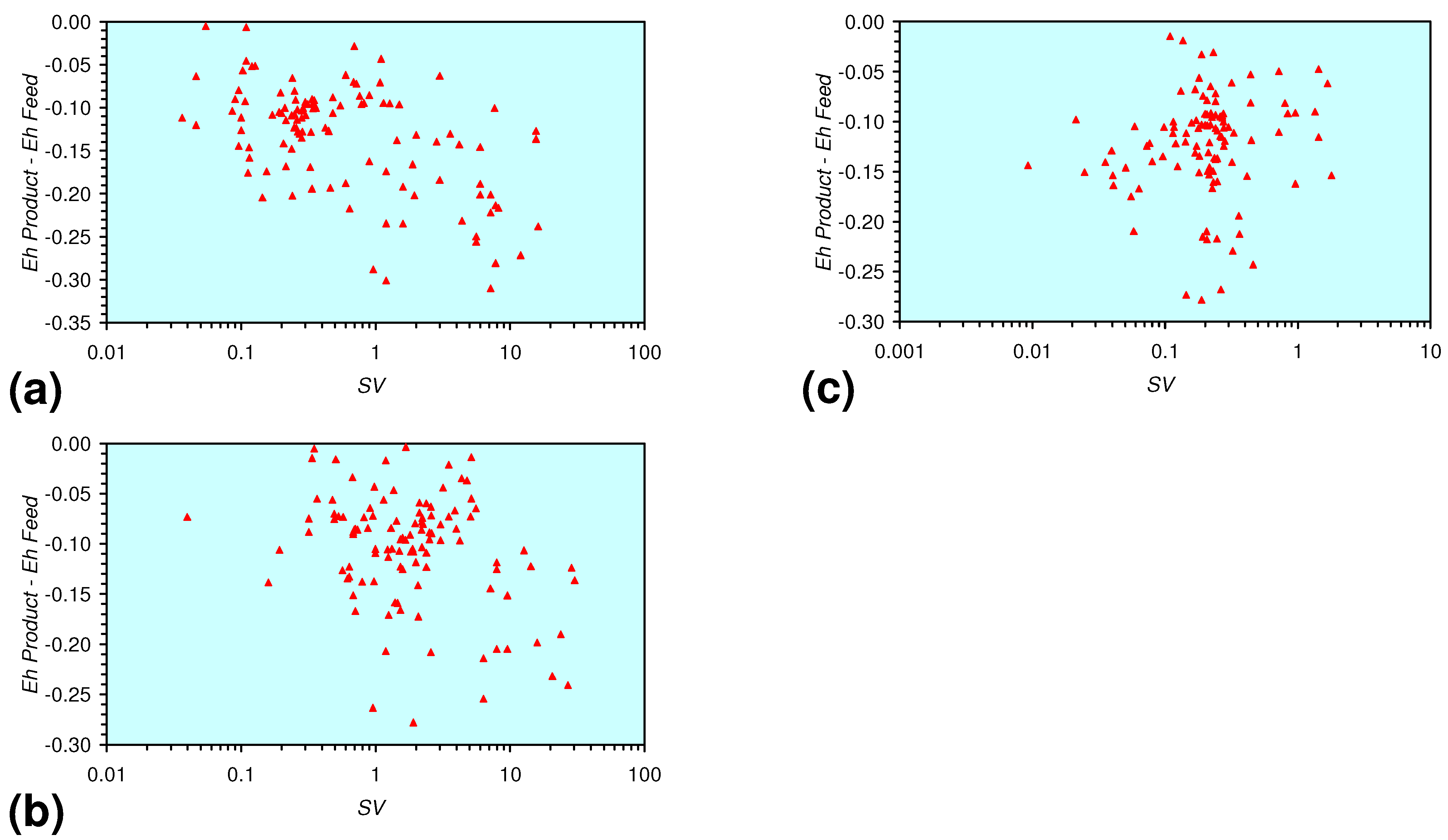
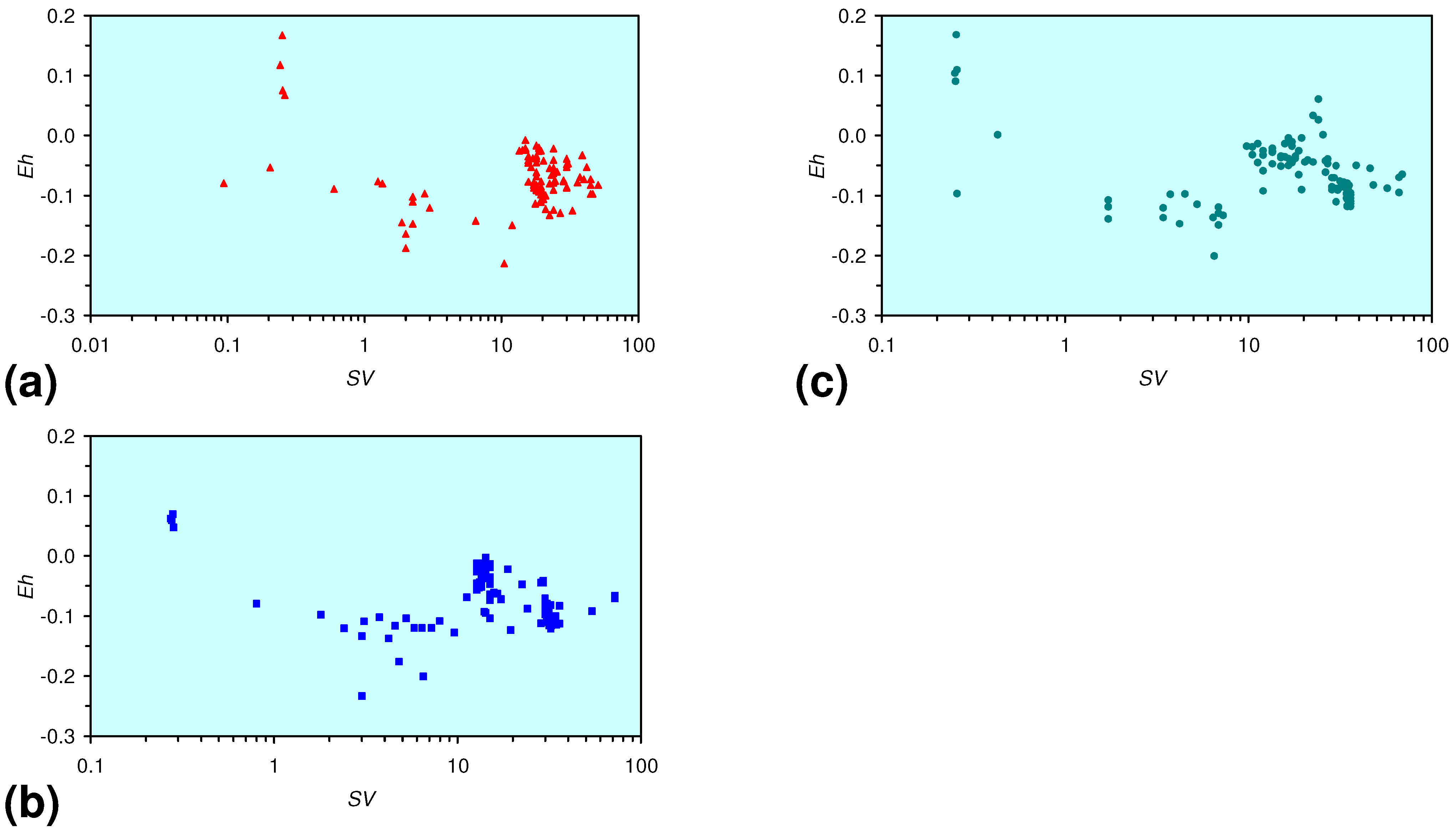
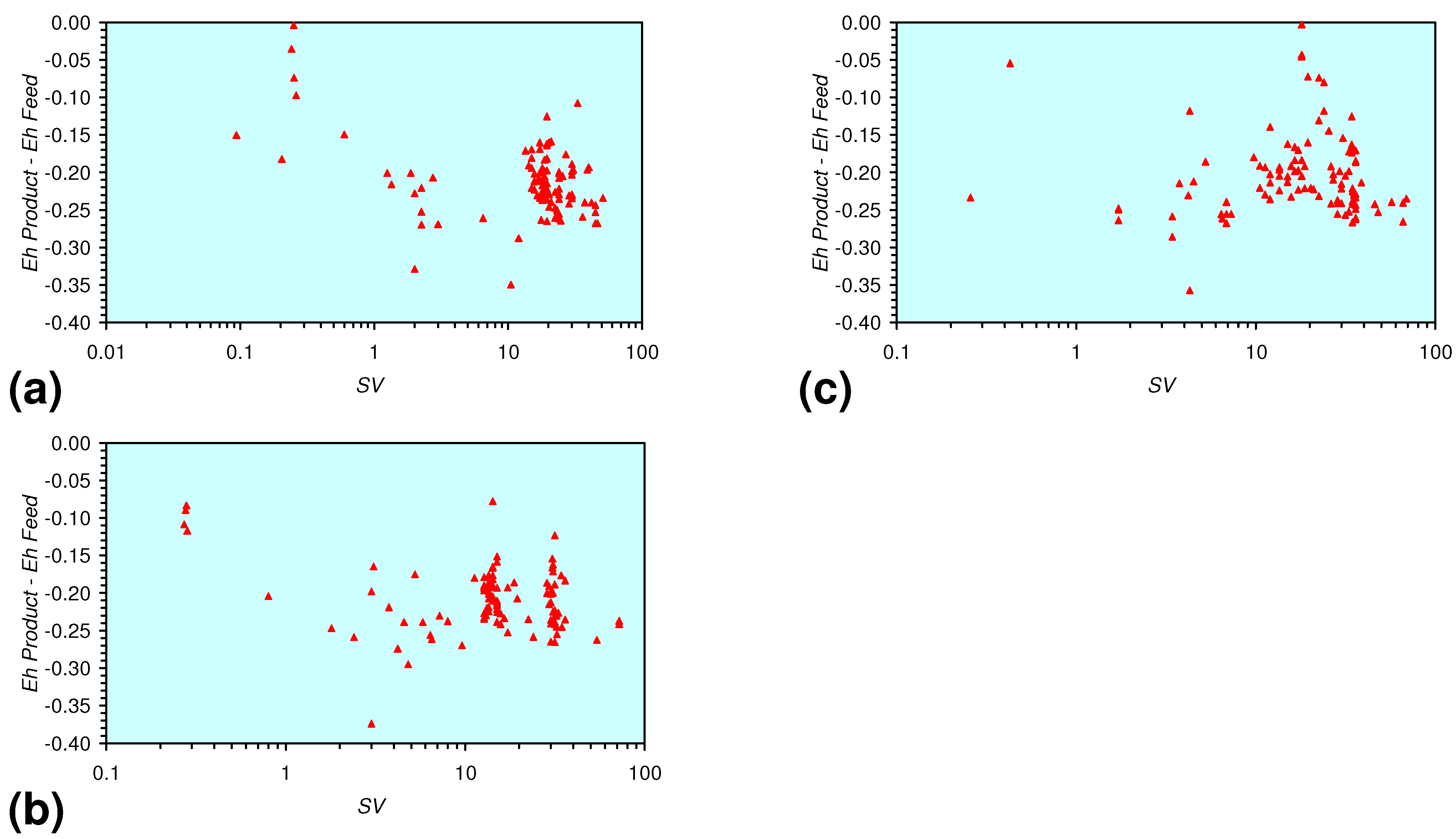
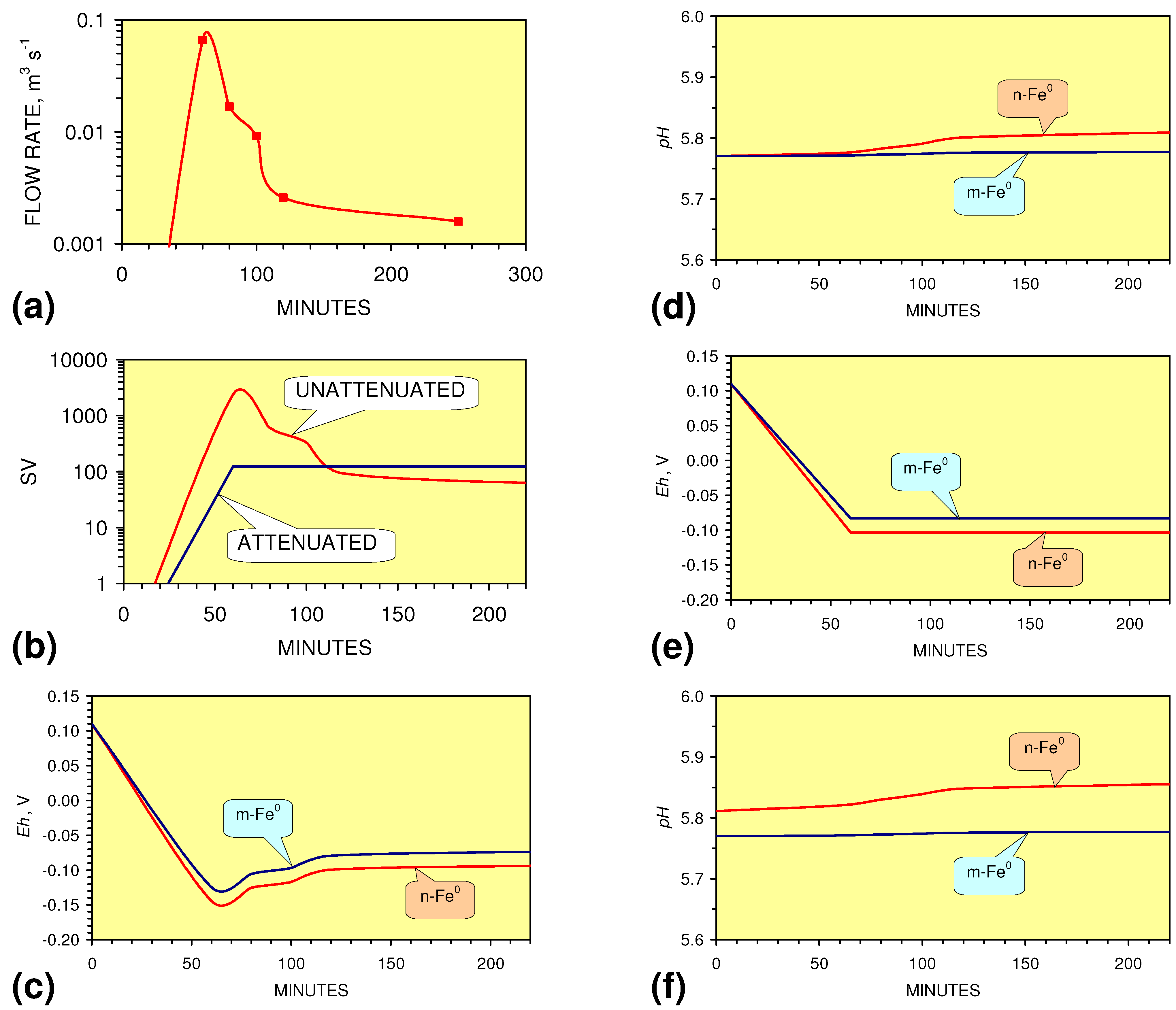
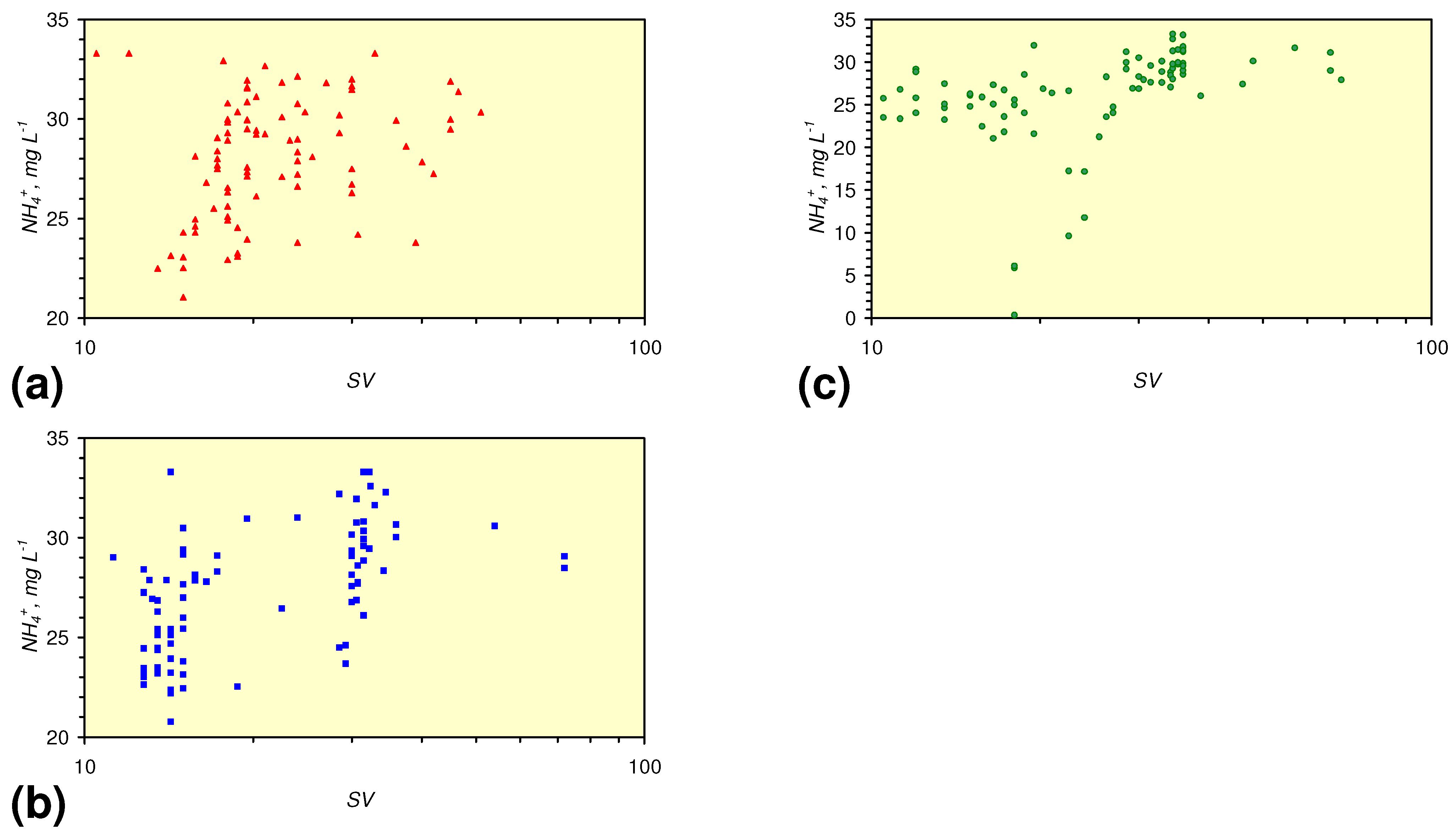
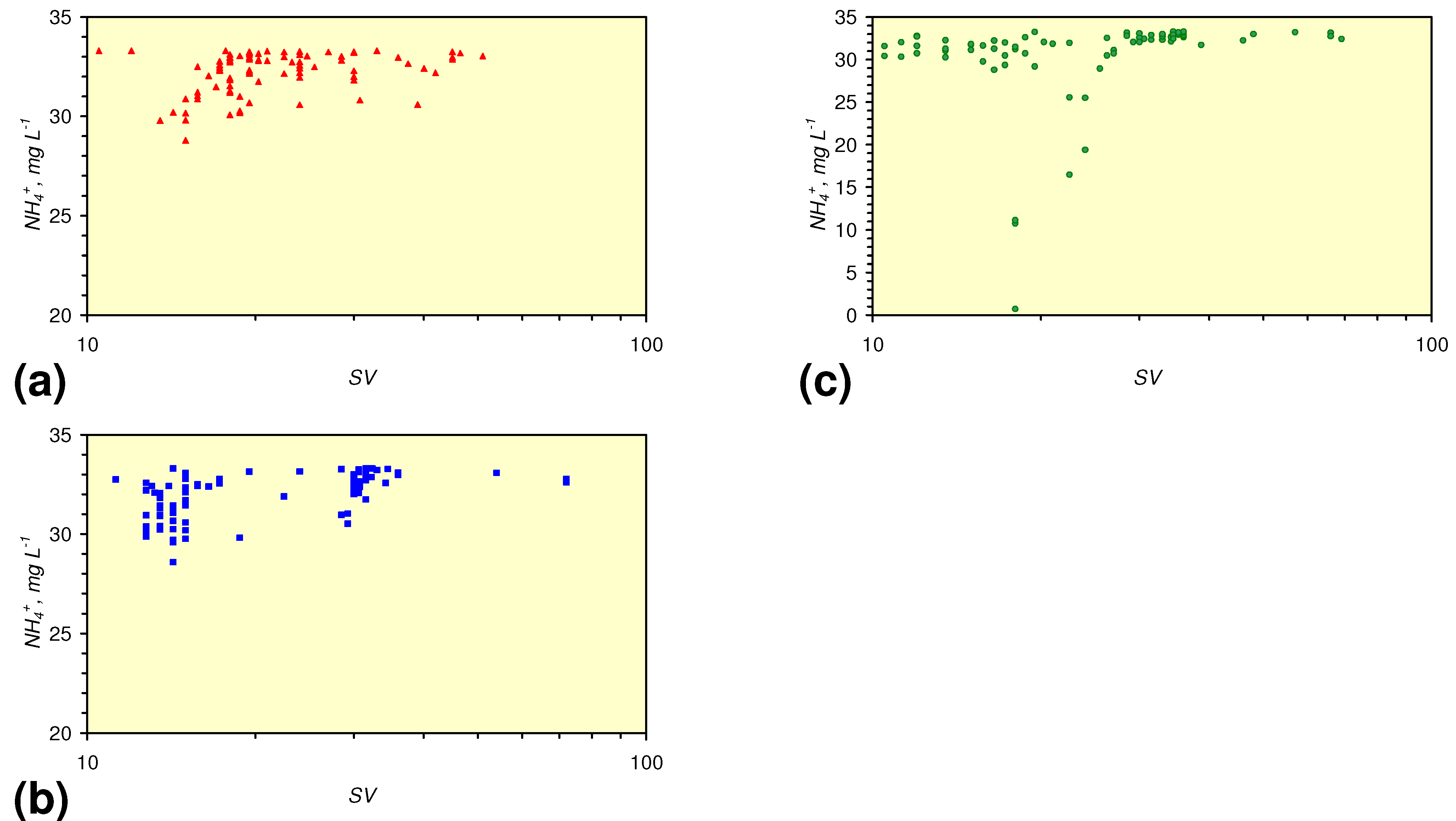
- There is a general trend of increasing Eh with decreasing SV. The lowest Eh values were observed over the range 1 < SV < 10 (Figure 26).
- The m-ZVM consistently moves the product water Eh:pH to the NH4+:NOx− redox fence (Figure 28).
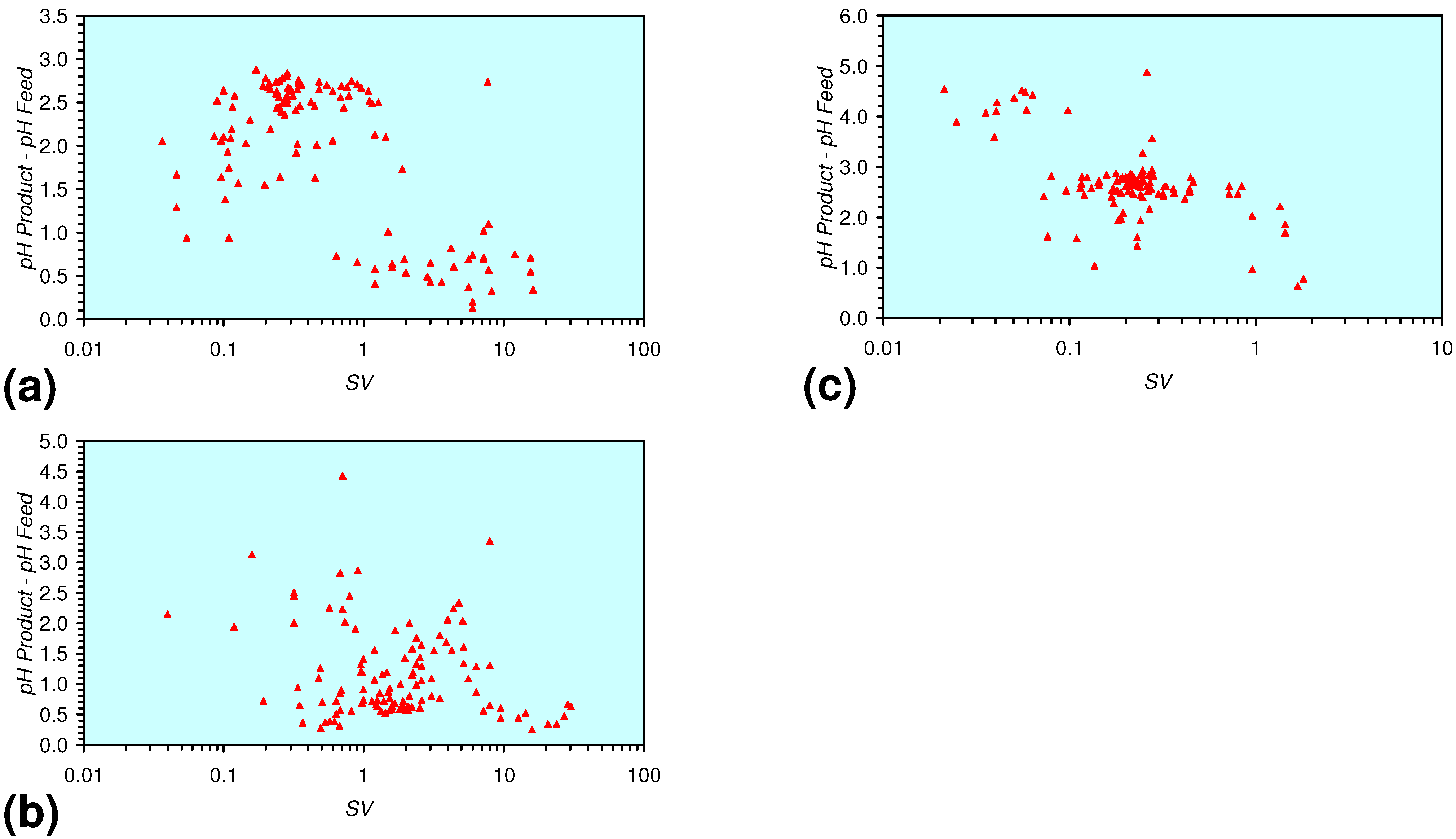
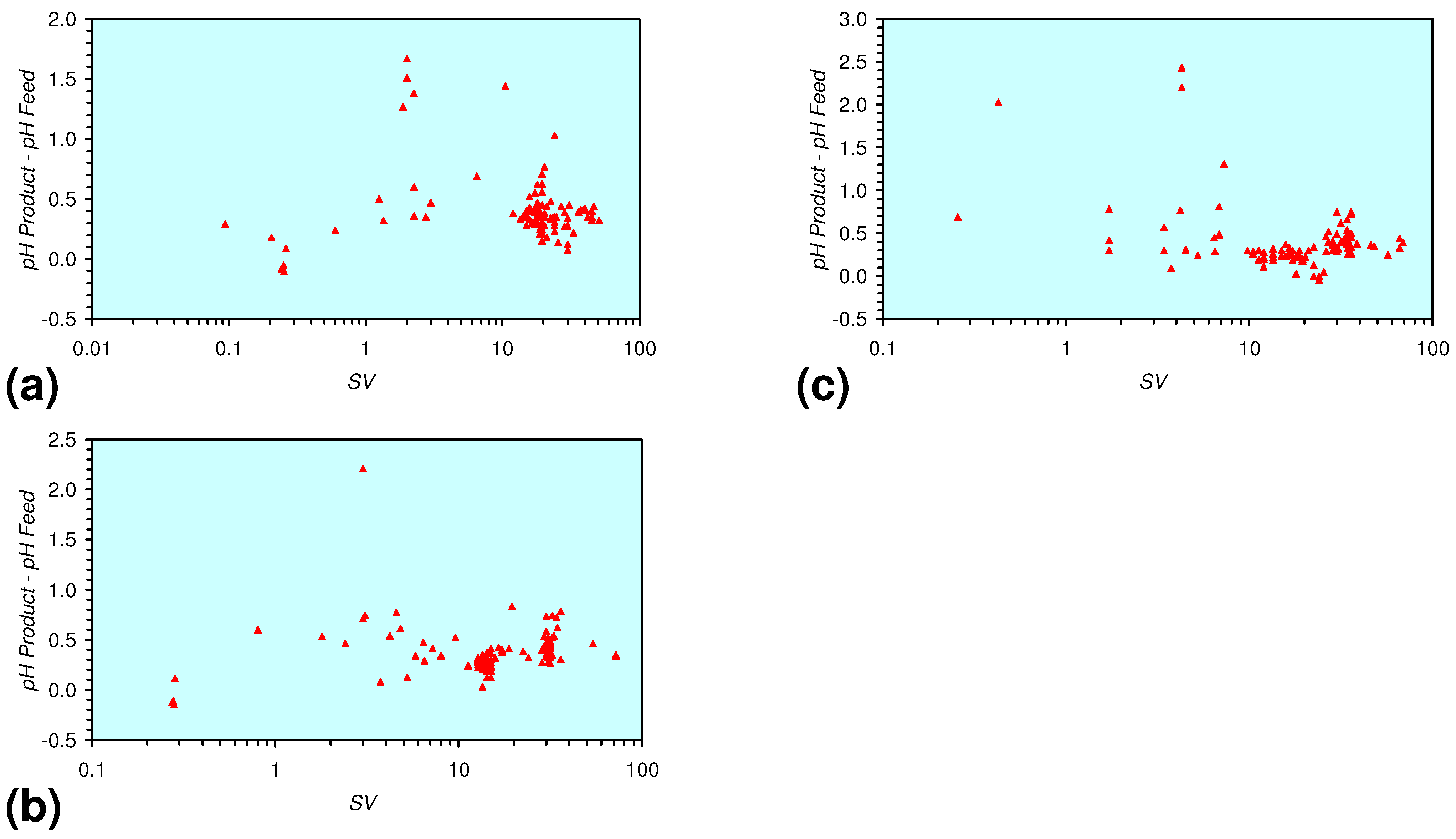
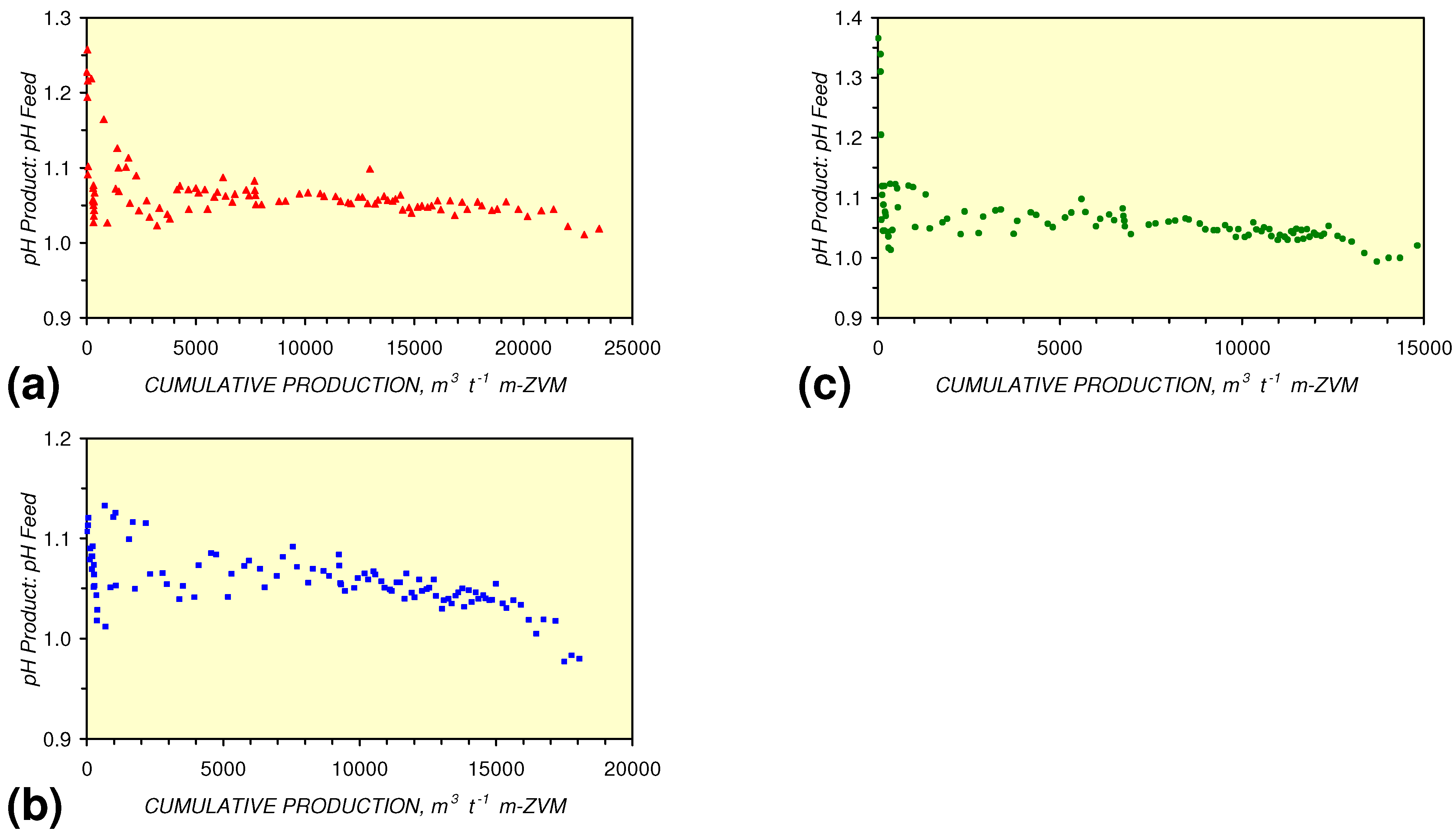
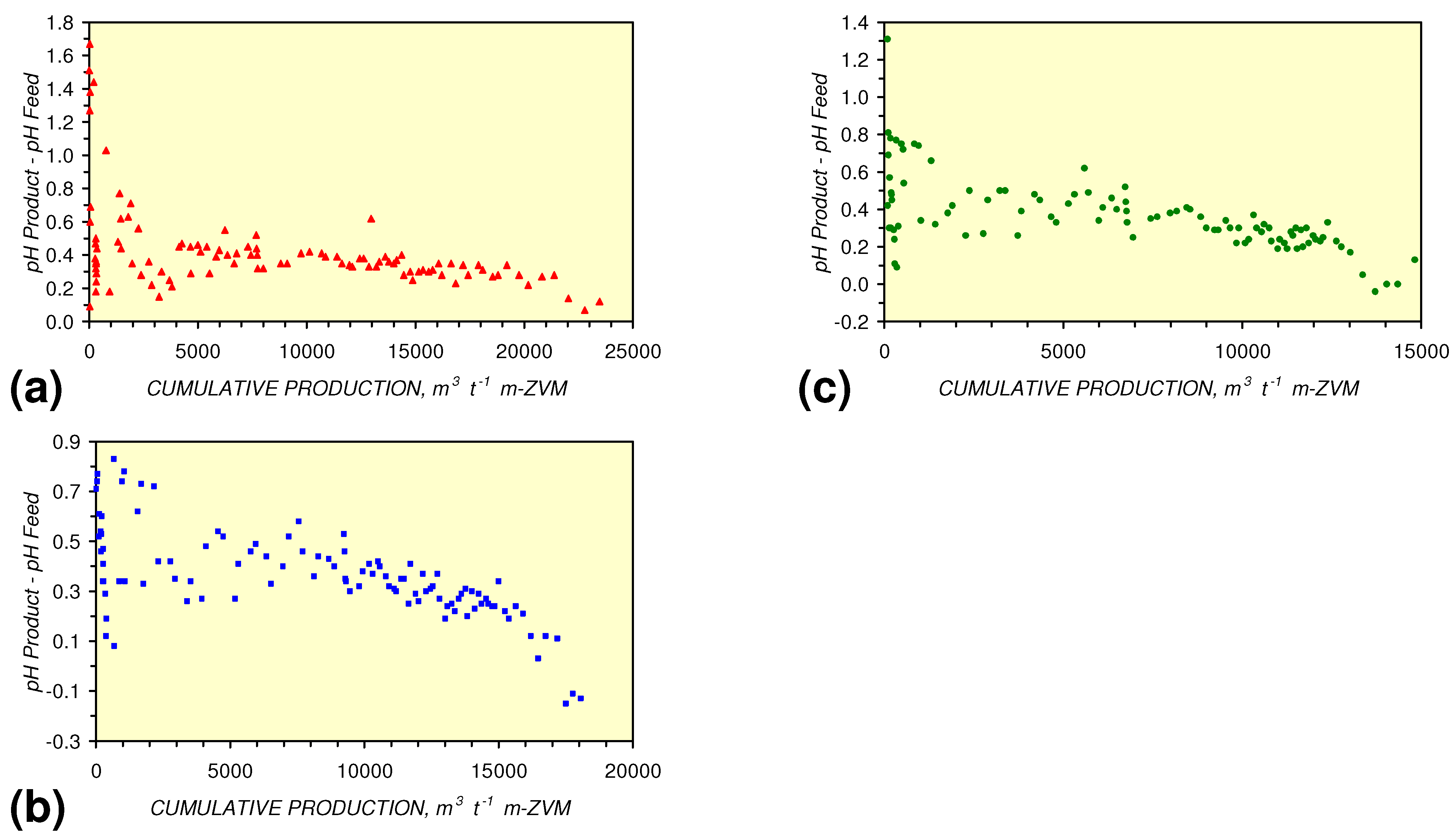
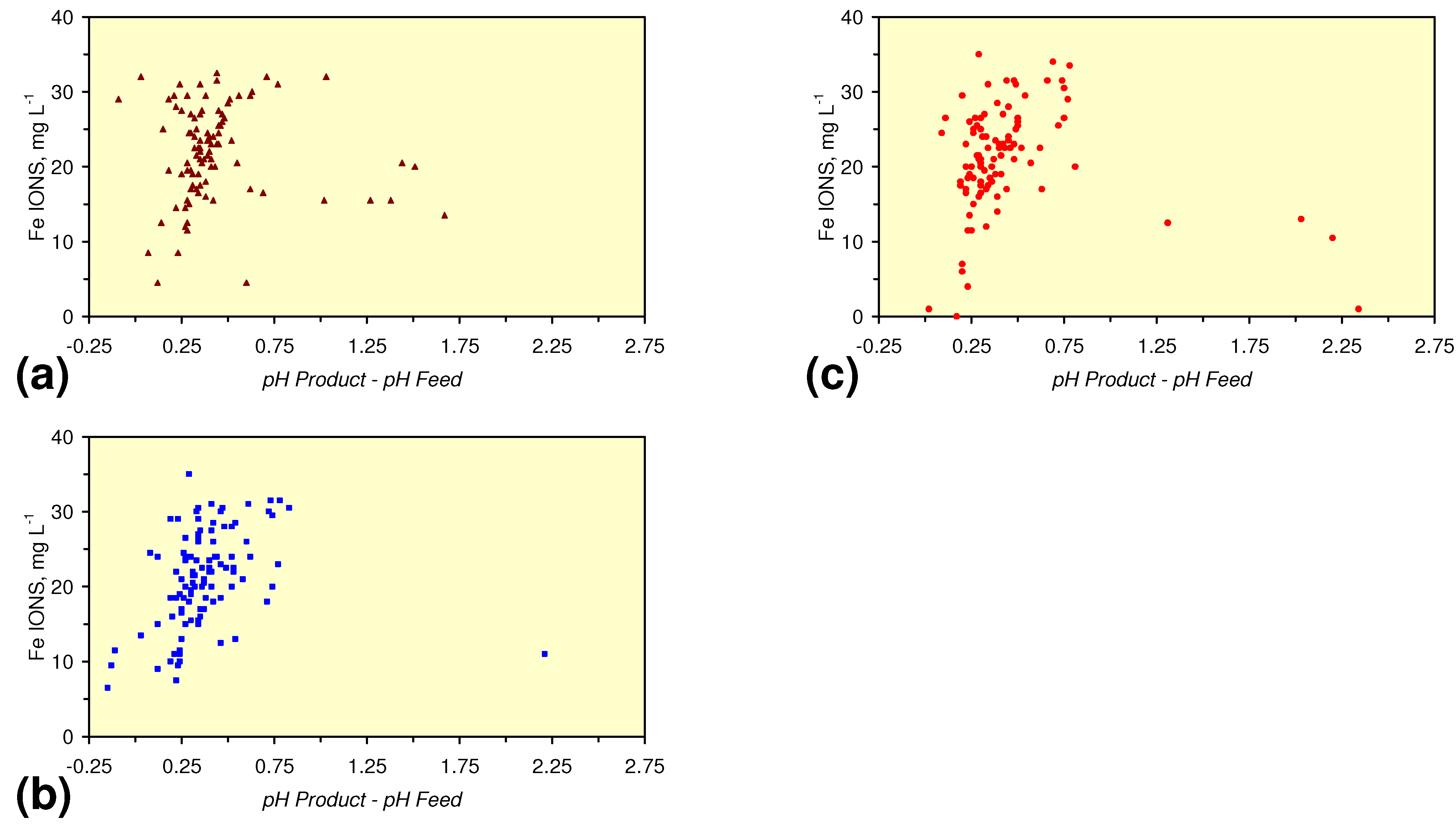
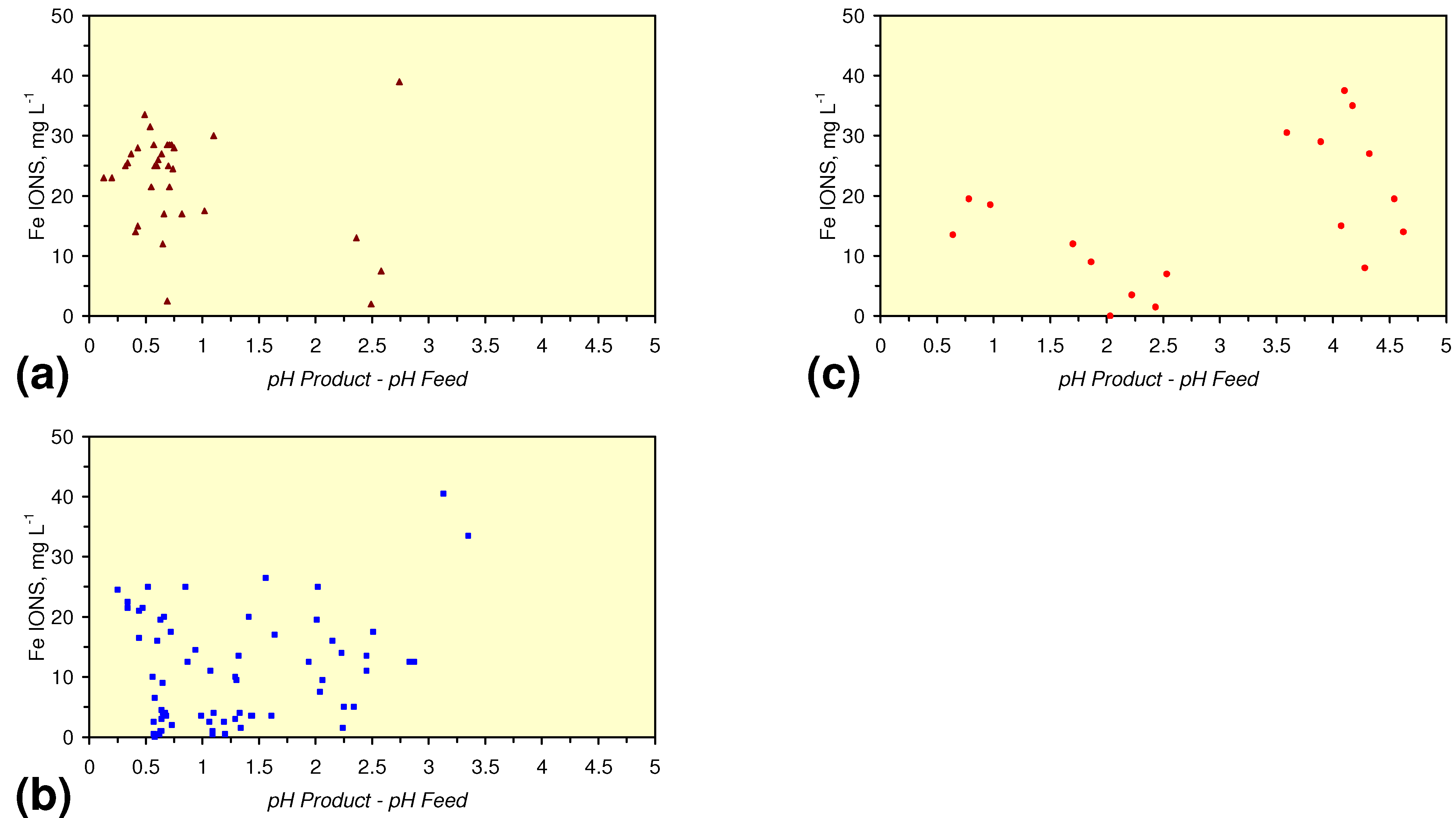
- m-Fe0, or m-Fe0 + m-Cu0 show a general trend where [pHproduct – pHFeed] increases with decreasing SV to a maximum value where 1 < SV < 10 (Figure 31). [pHproduct – pHFeed] then decreases with decreasing SV. The addition of m-Al0 resulted in [pHproduct– pHFeed] increasing with decreasing SV when SV < 1 (Figure 31).
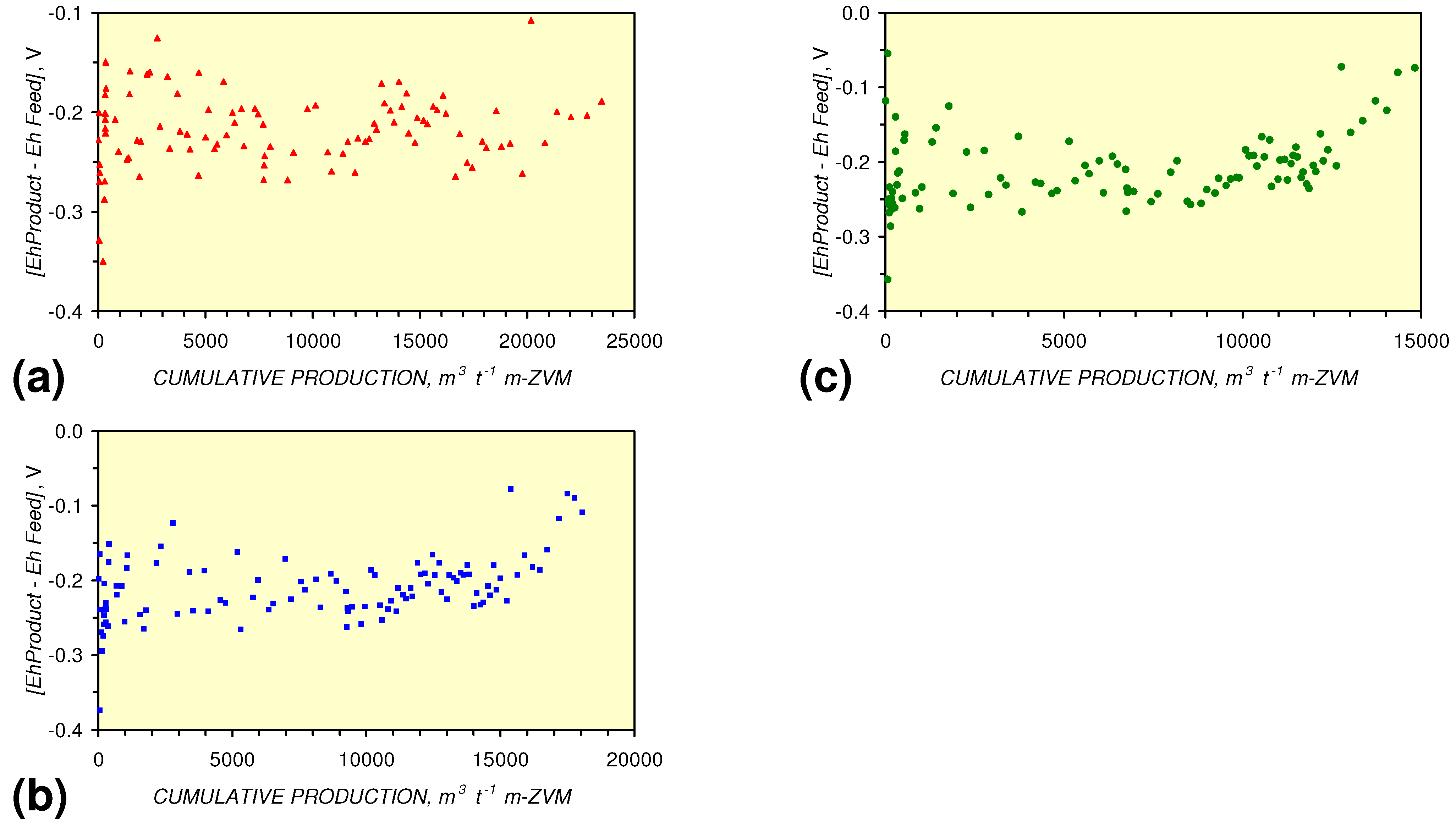
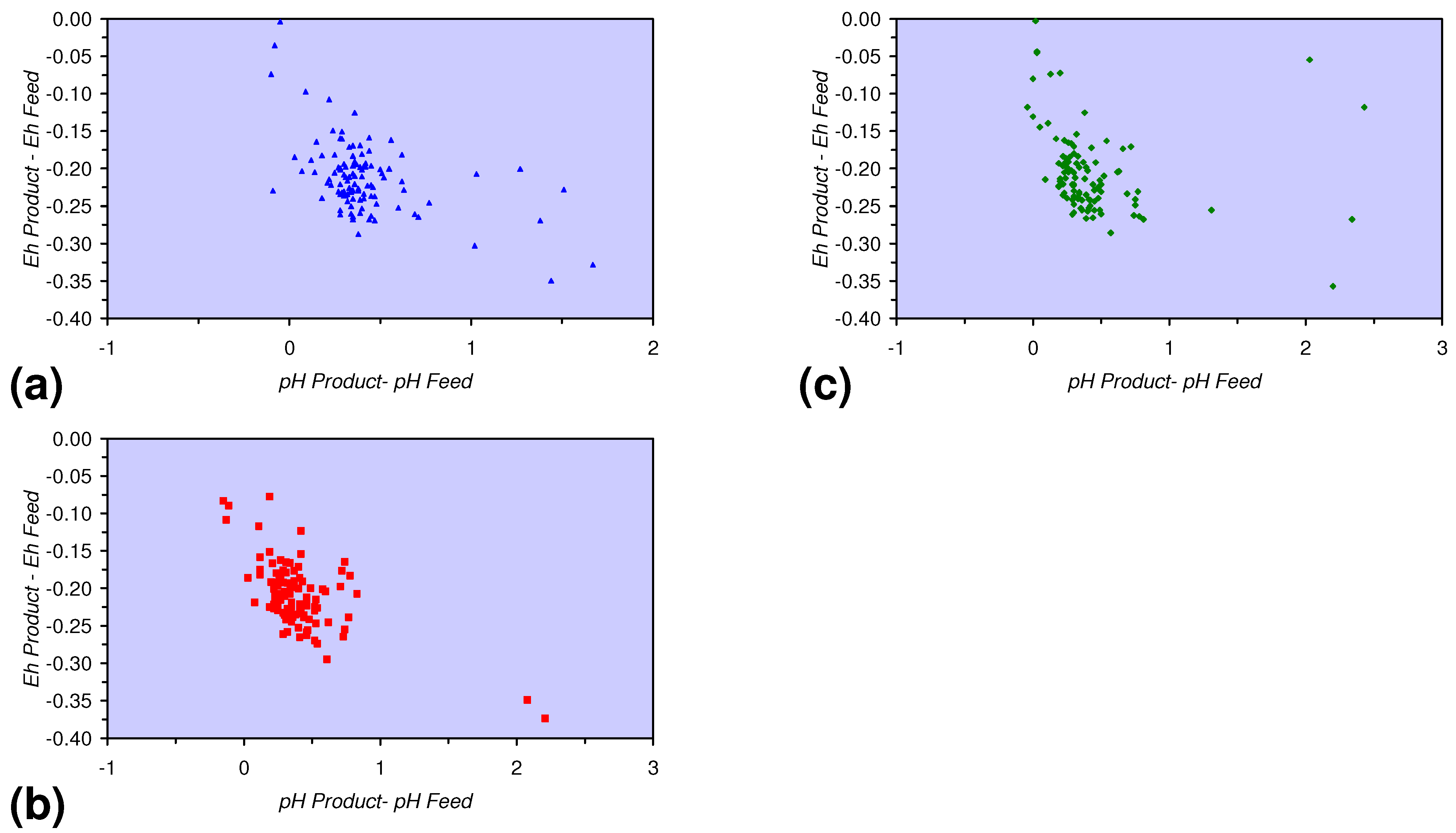
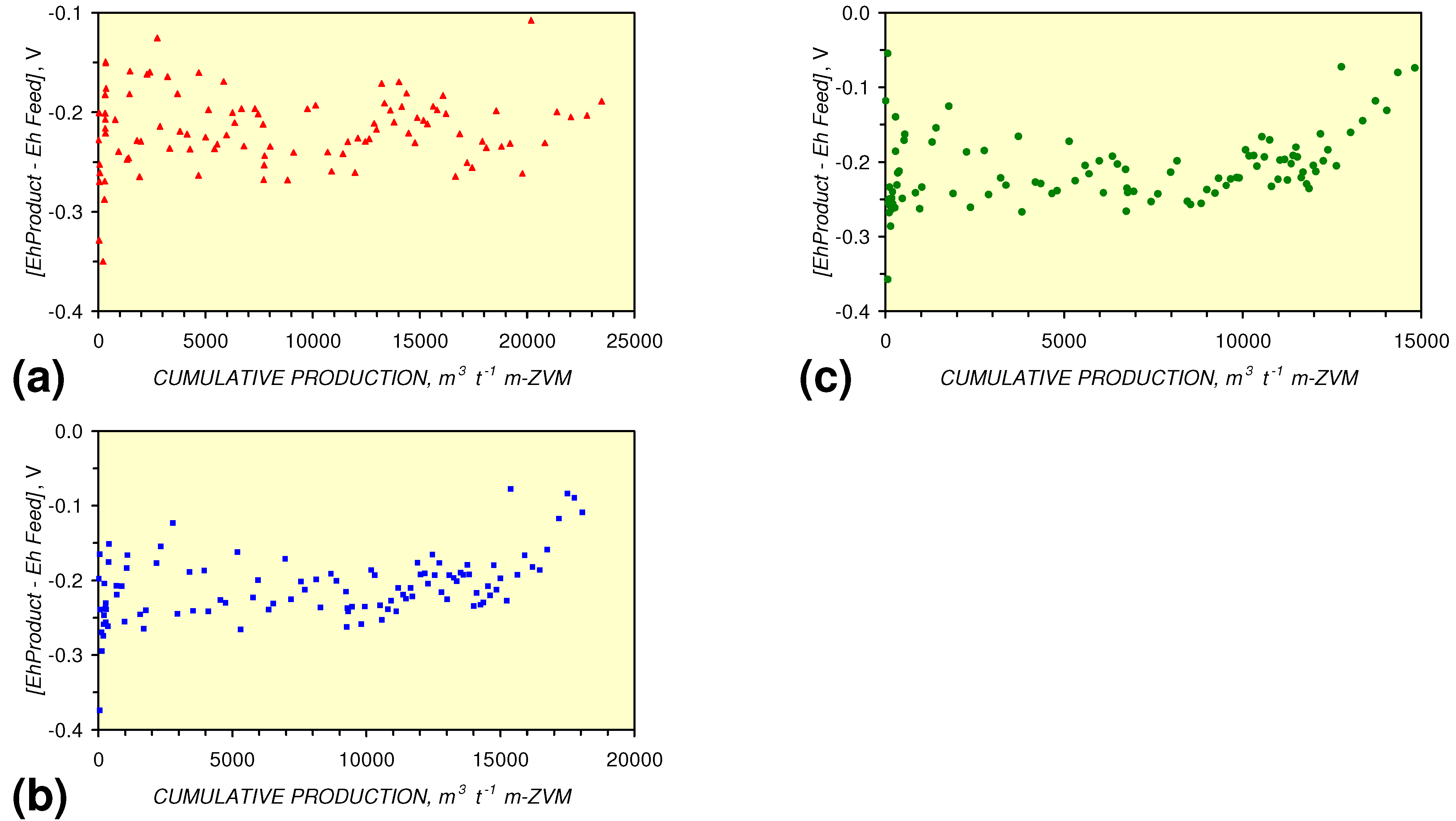
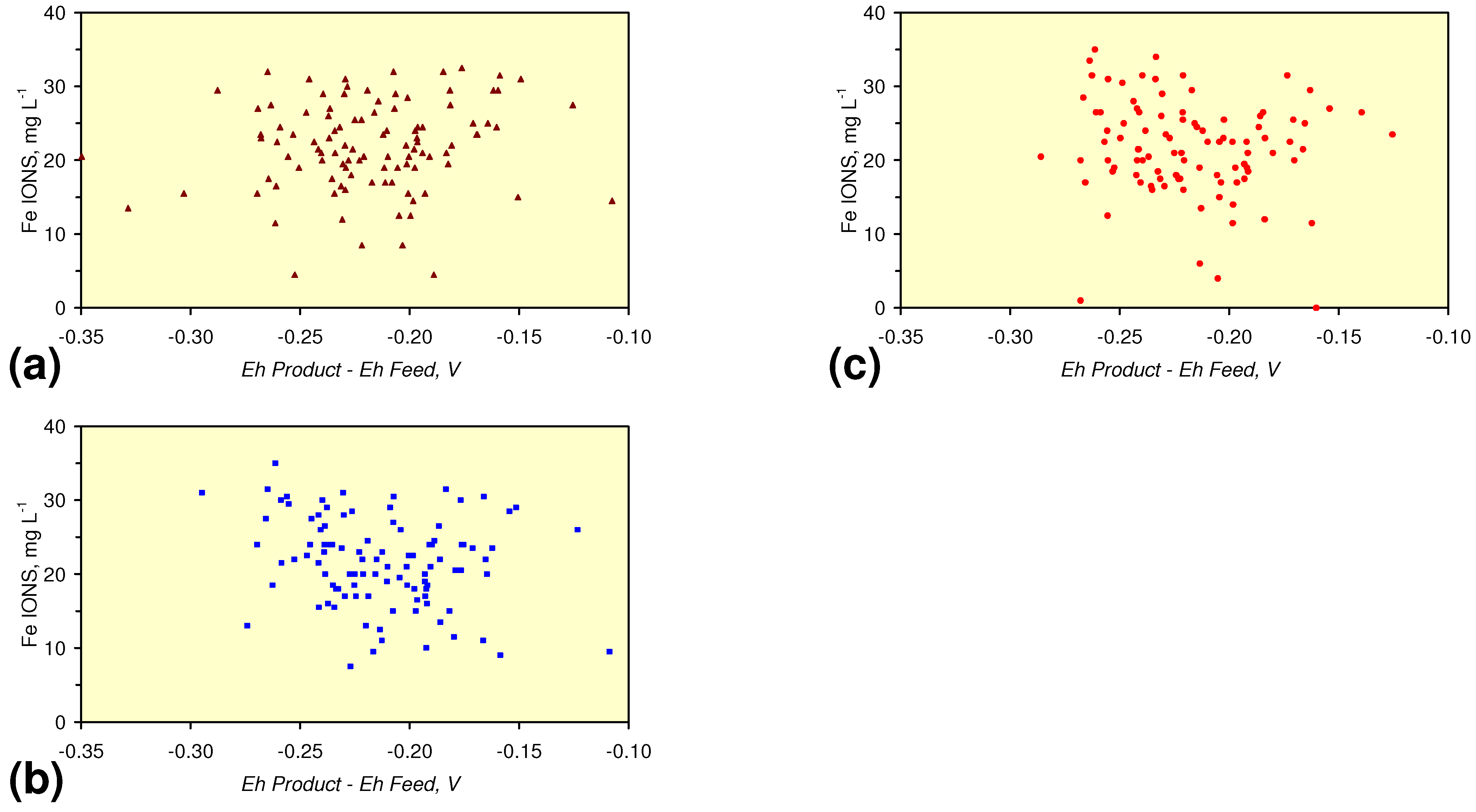
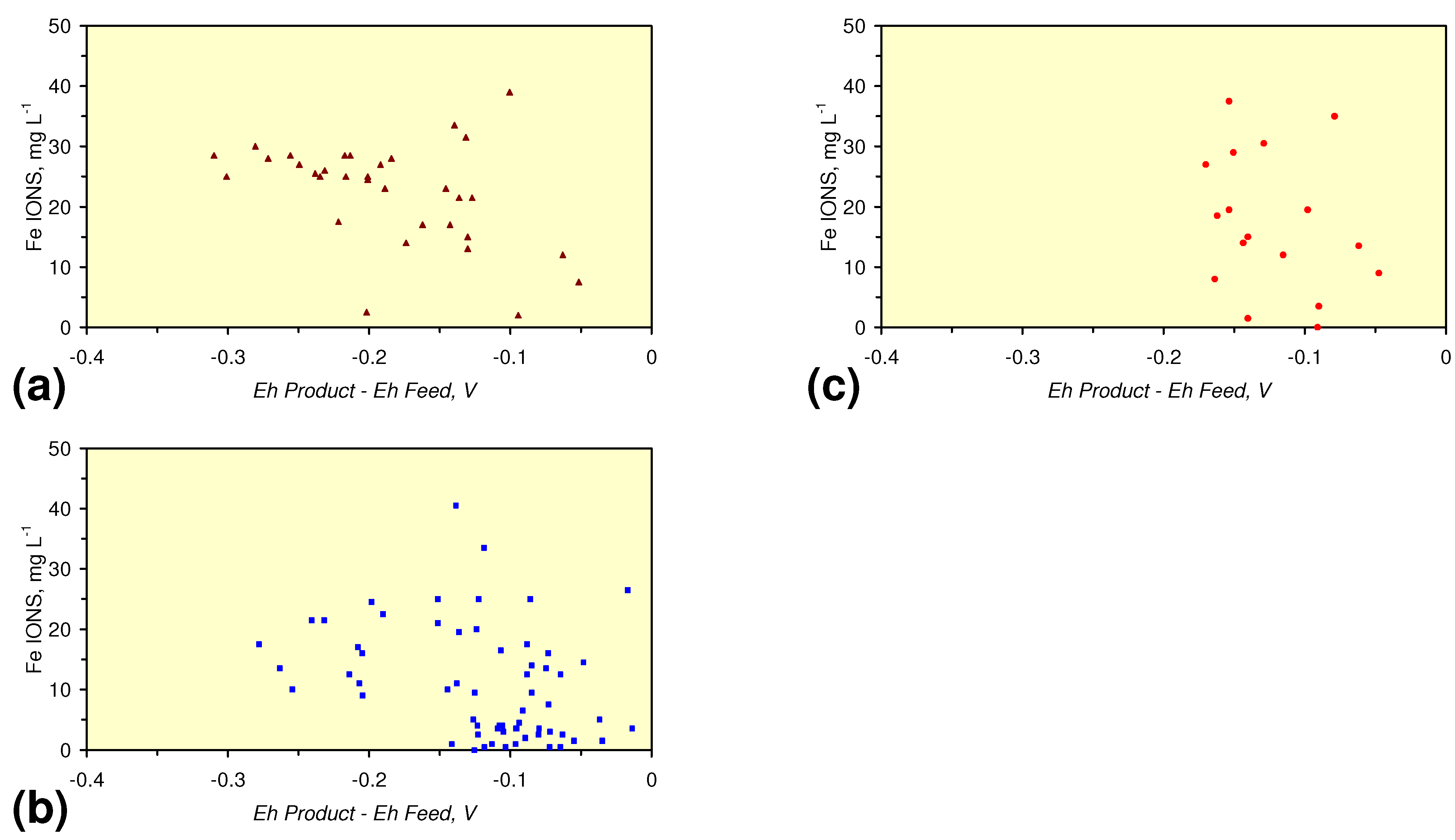
- A general trend of [Ehproduct – EhFeed] becoming increasingly negative with increasing [pHproduct – pHFeed] is present (Figure 33). This indicates that the chemical process of Eh reduction and pH increase is related.
3.3.1. BR1
3.3.2. BR2
3.3.3. BR3
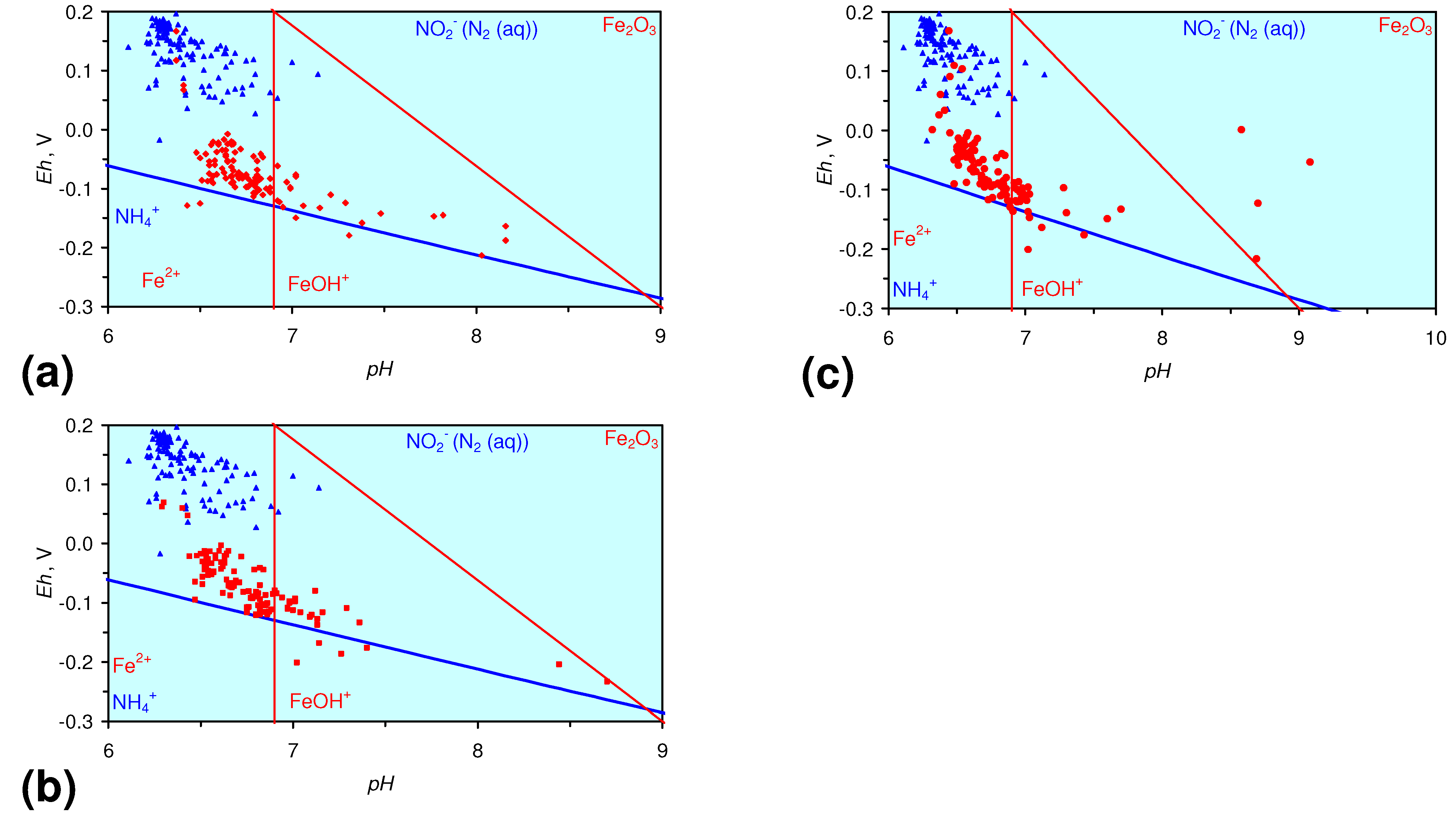
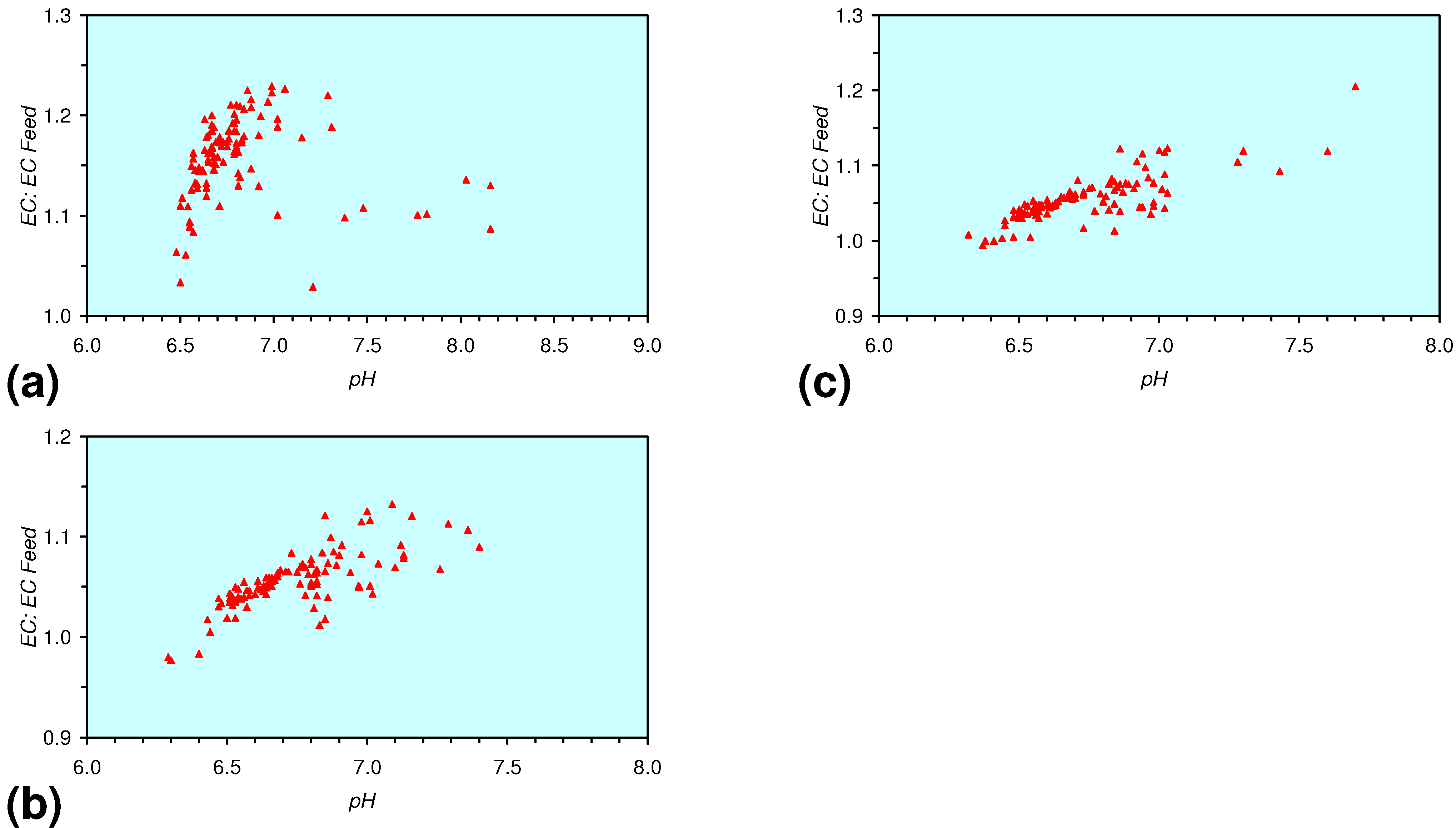
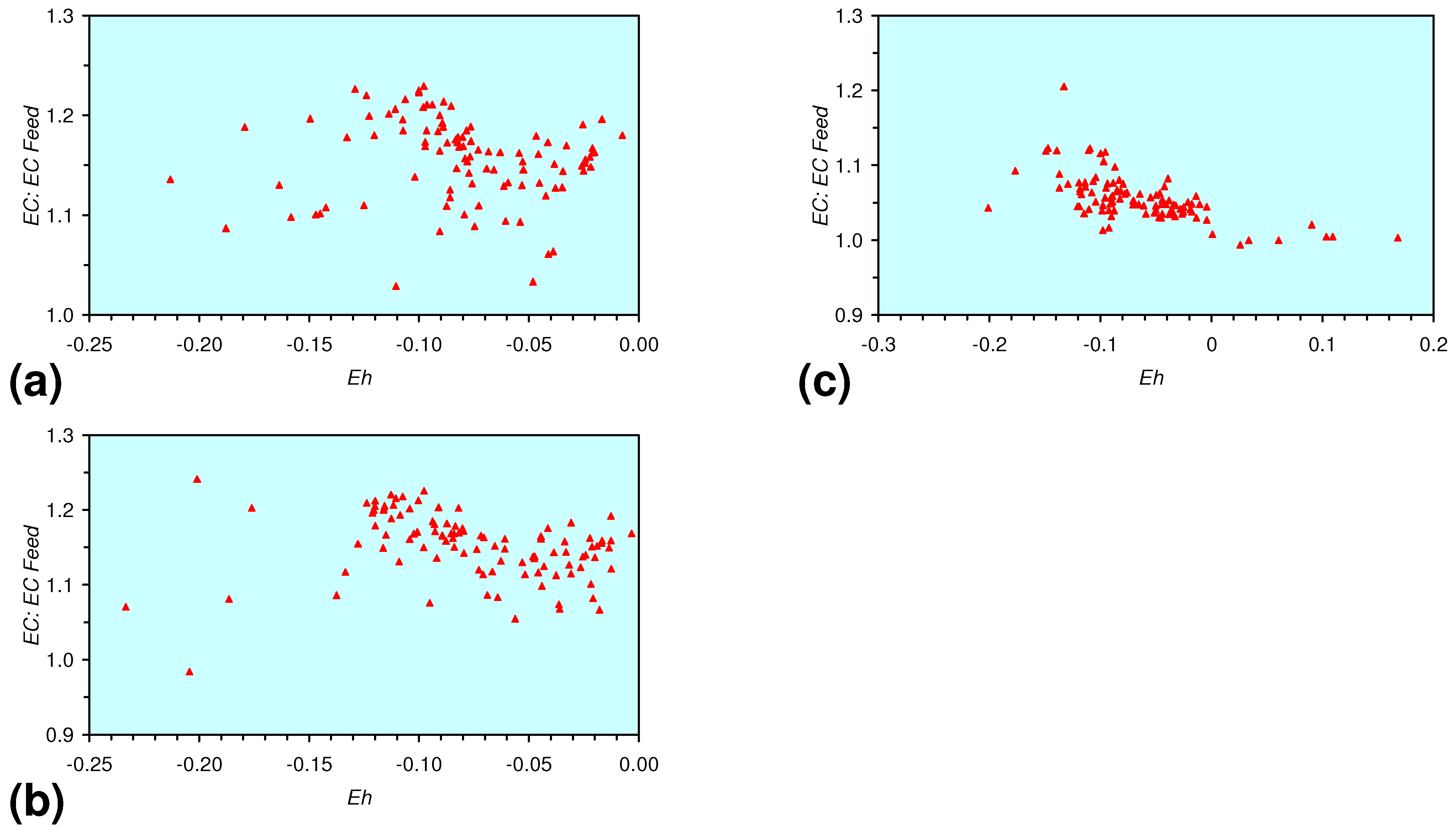
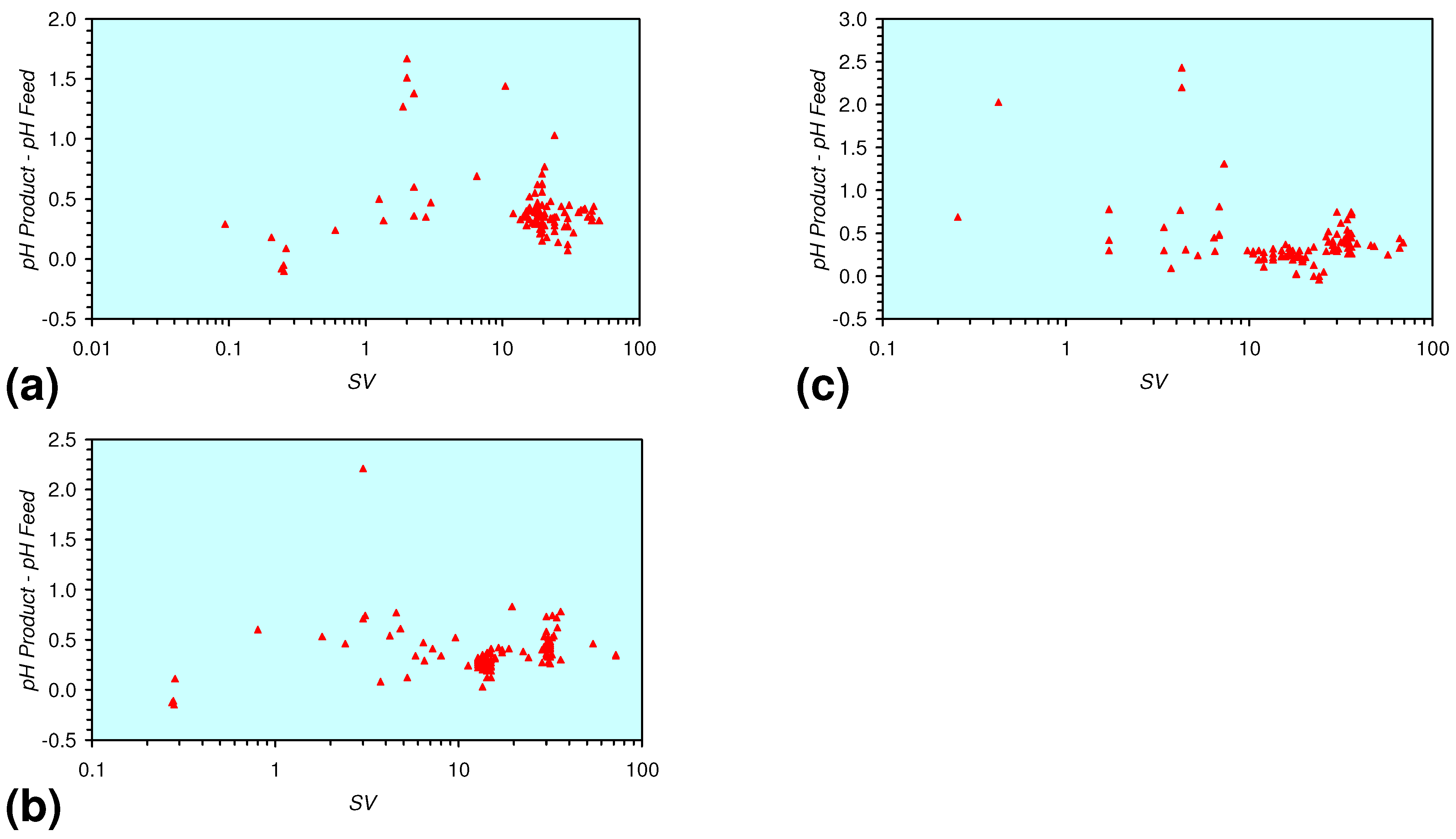

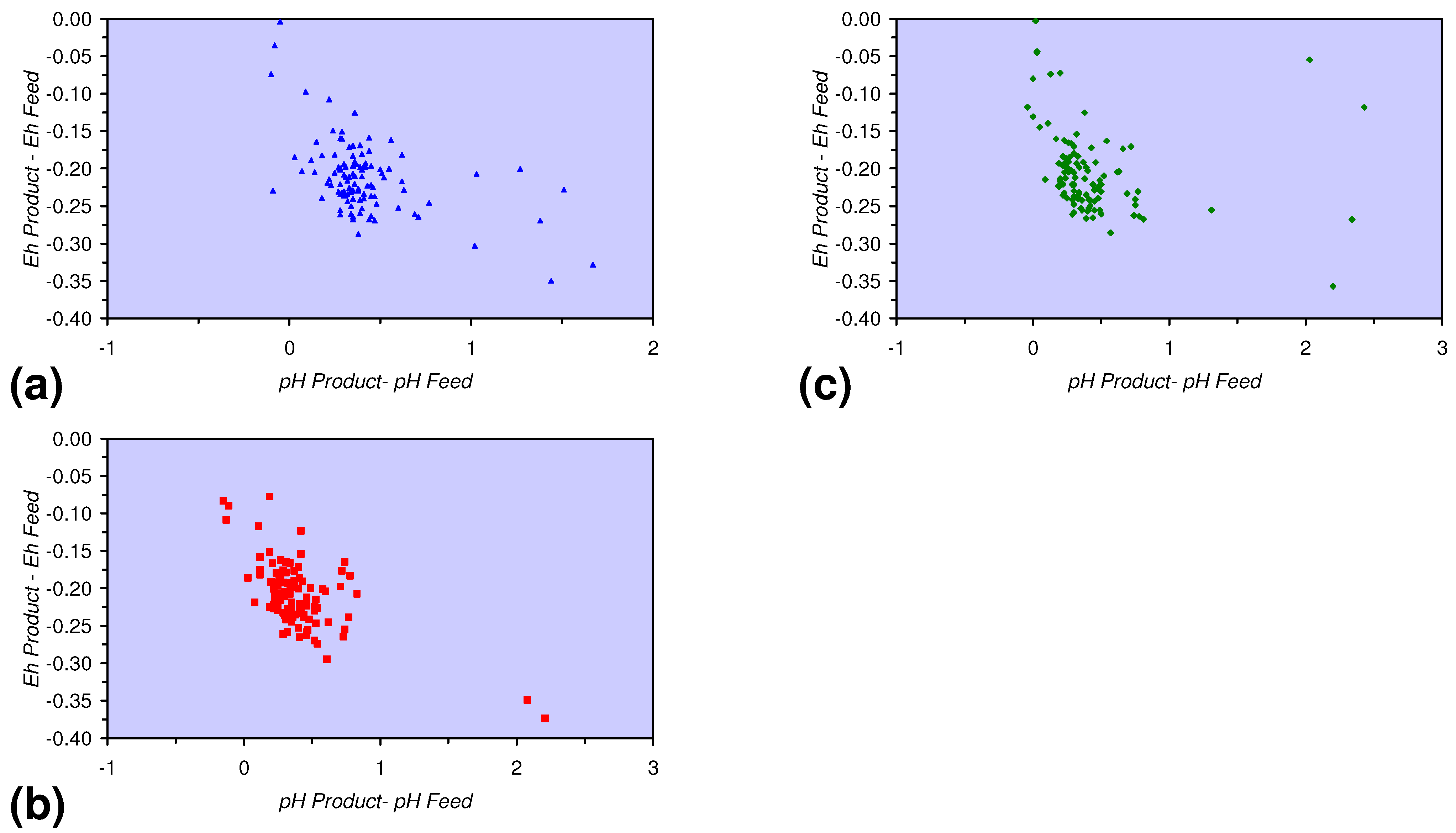
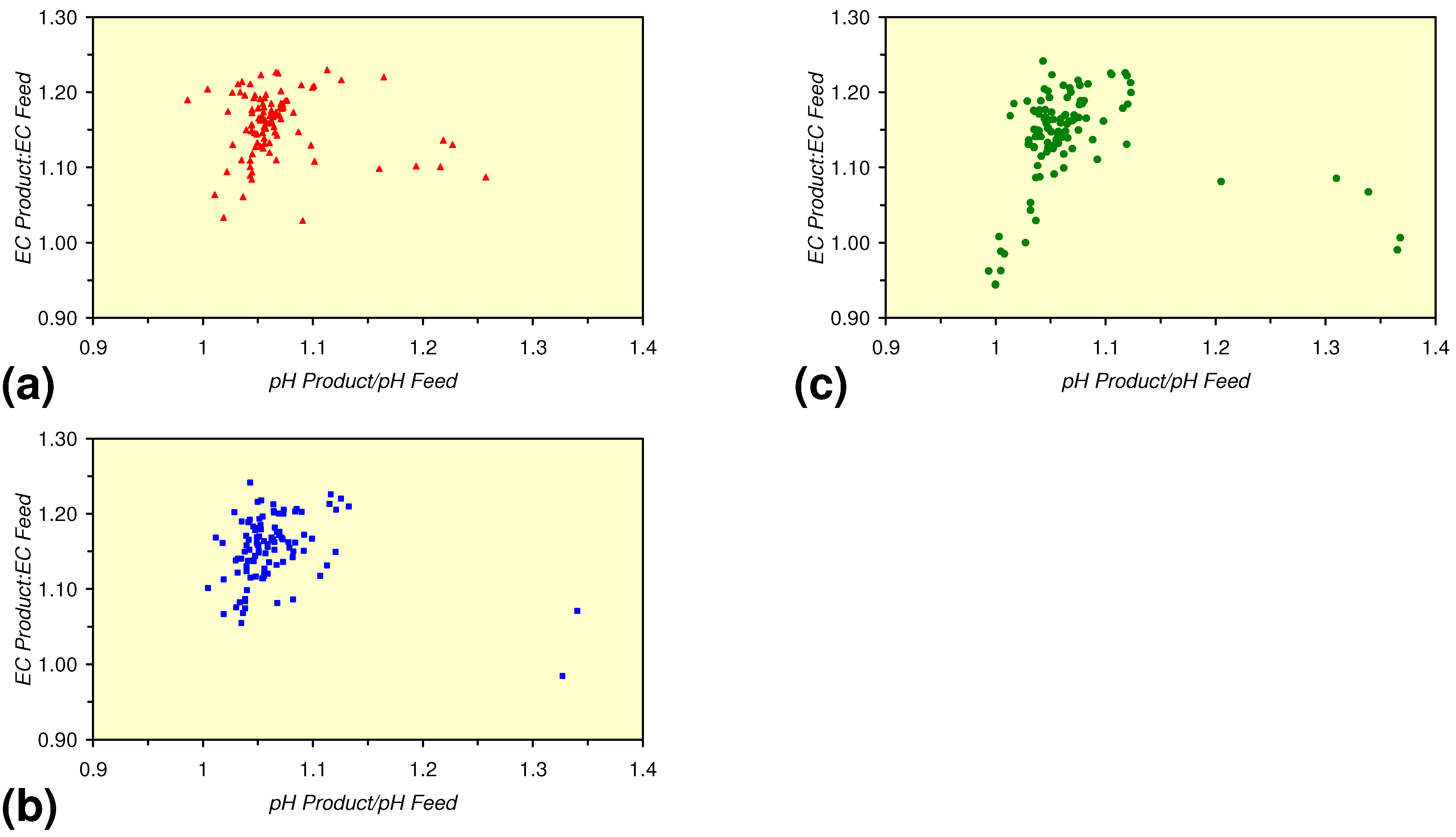
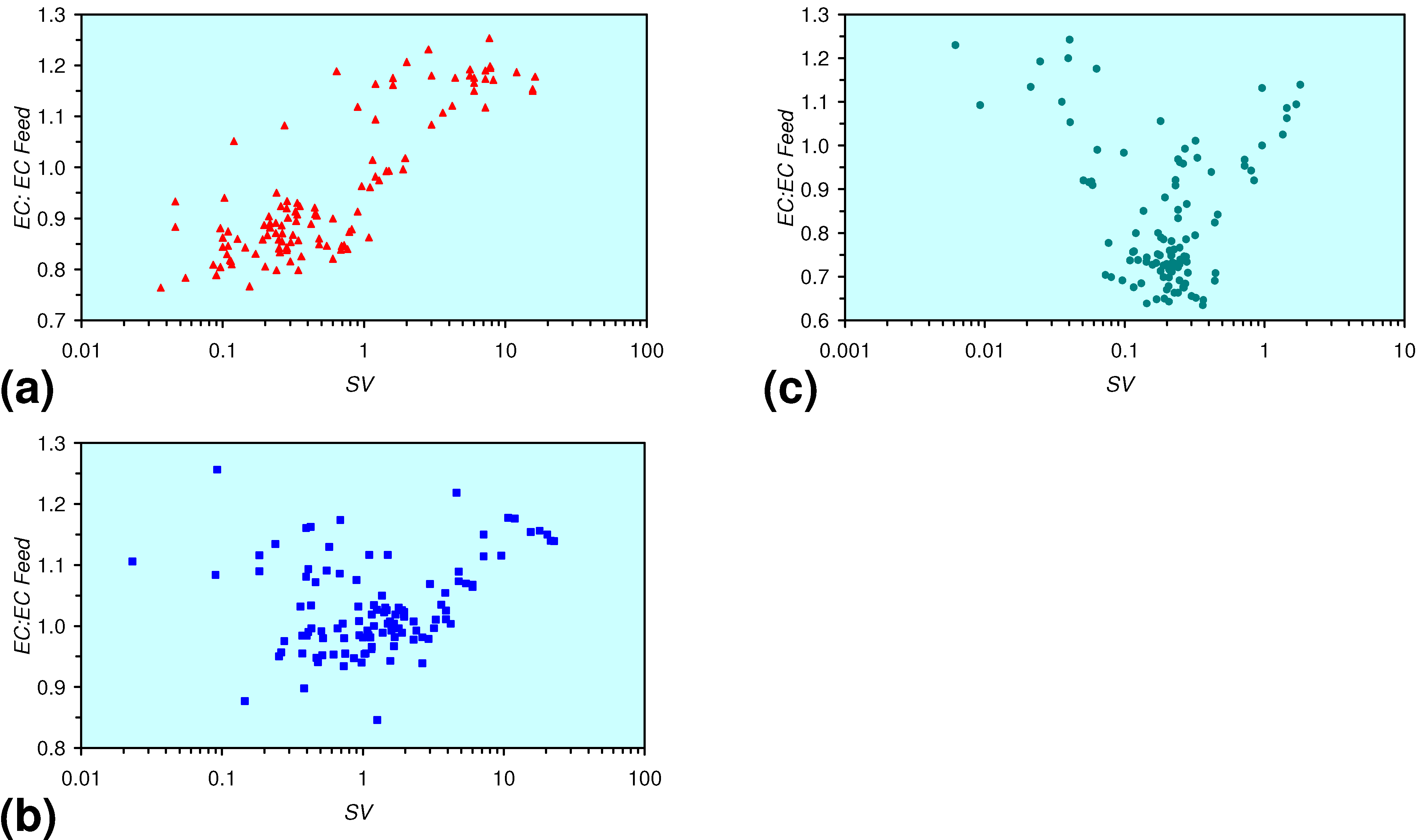
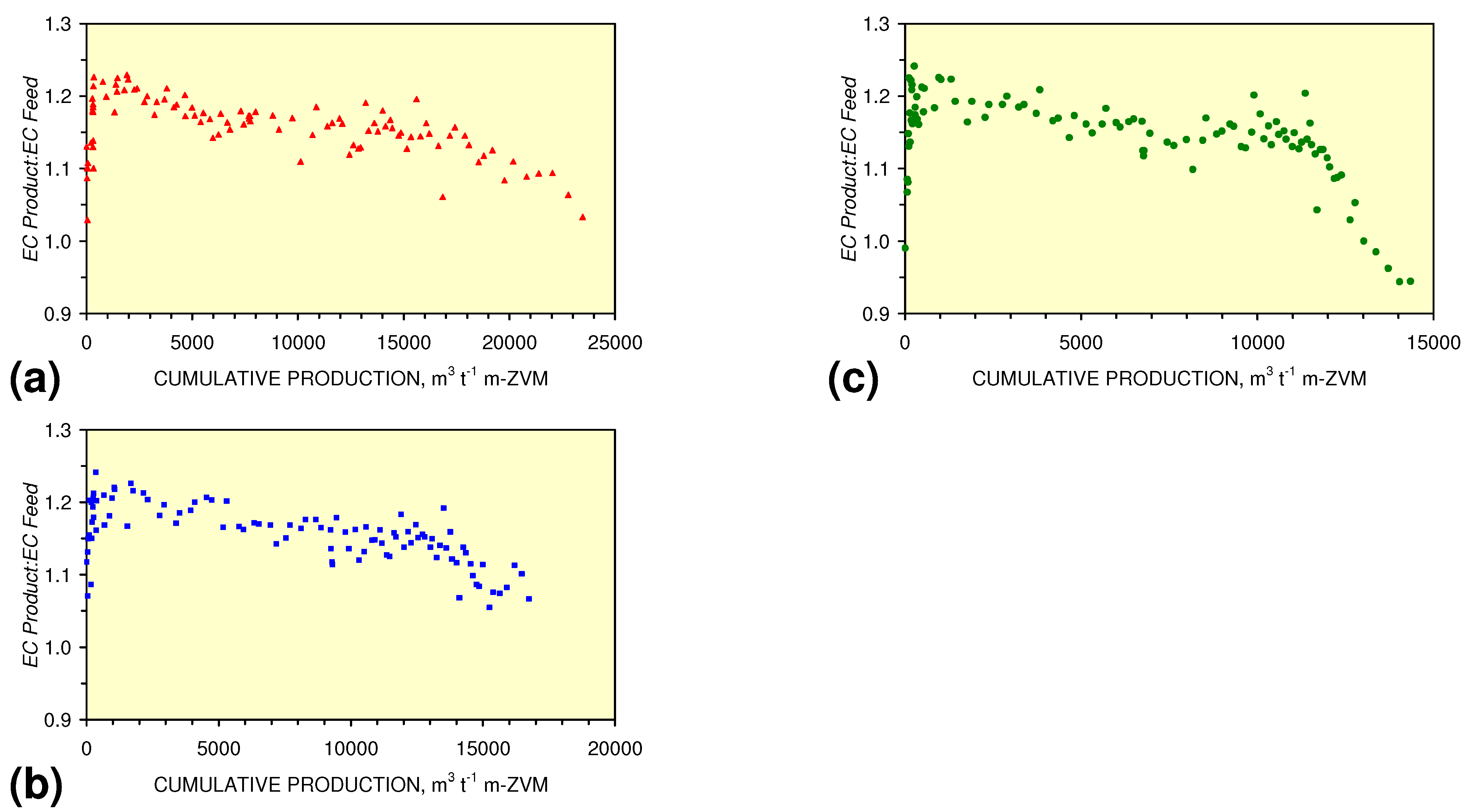
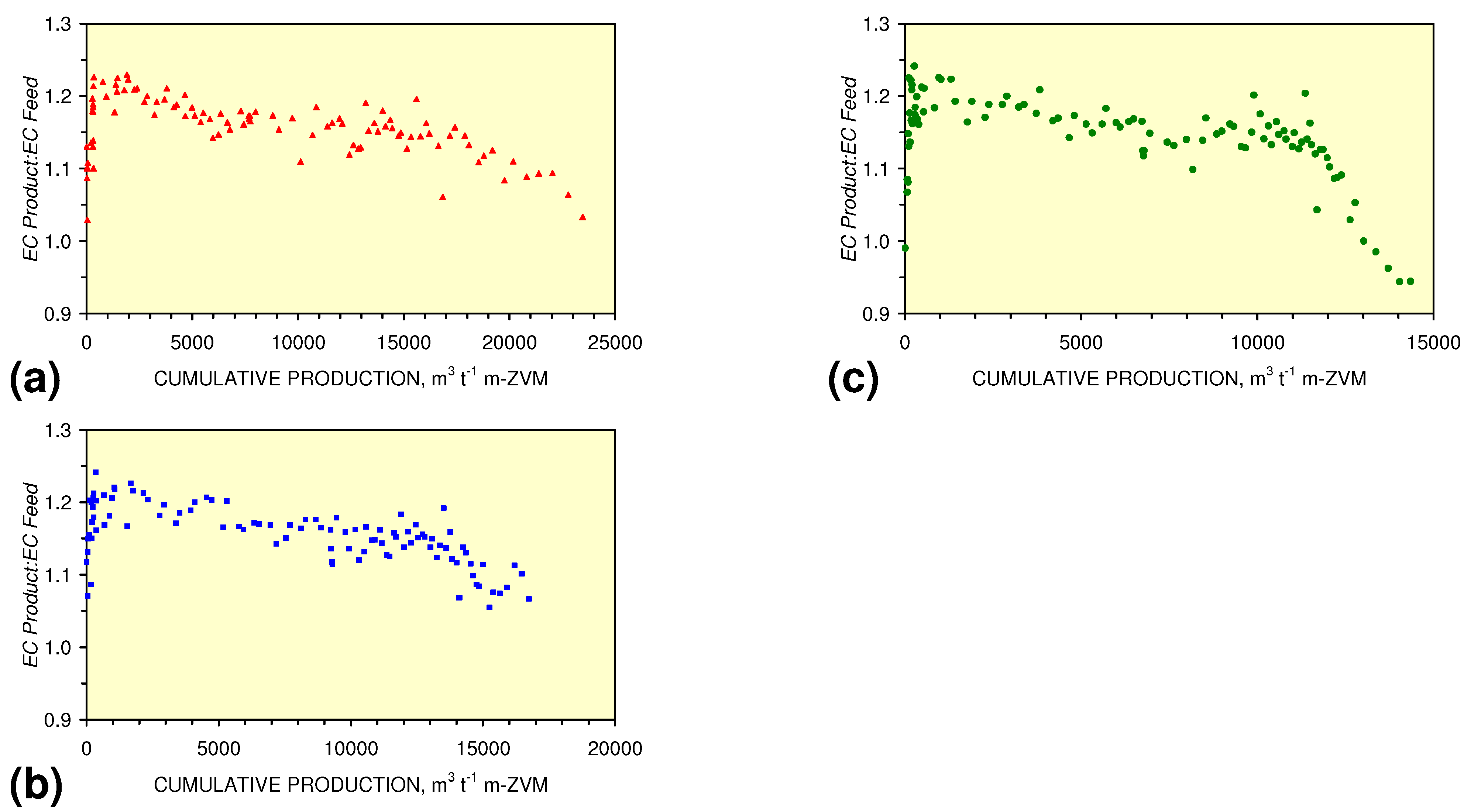
3.3.4. Cumulative Water Volume Processed
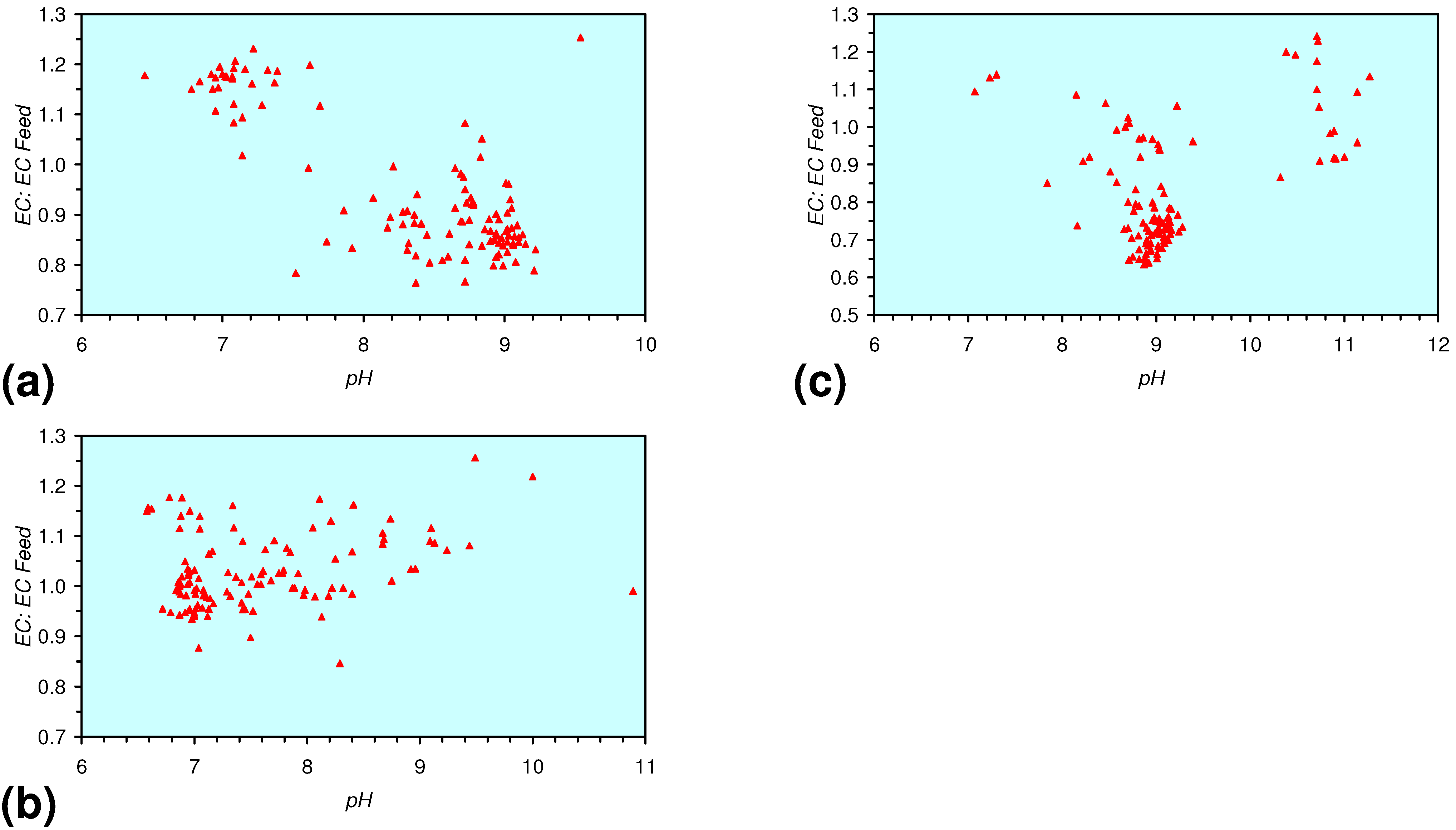
[pHProduct:pHFeed] = −2.26 × 10−5 × (12,000 > Vc > 4,000) + 1.3167; R2 = 0.843;
[ECProduct:ECFeed] = −1.56 × 10−5 × (Vc > 17,000) + 1.4171; R2 = 0.856;
[ECProduct:ECFeed] = −4.4 × 10−6 × (Vc > 13,000) + 1.599; R2 = 0.004;
[ECProduct:ECFeed] = −7.6 × 10−5 × (12,000 > Vc > 4,000) + 2.0096; R2 = 0.974;
[EhProduct:EhFeed] = 4.3 × 10−5 × (Vc > 12,000) − 0.7098; R2 = 0.5;
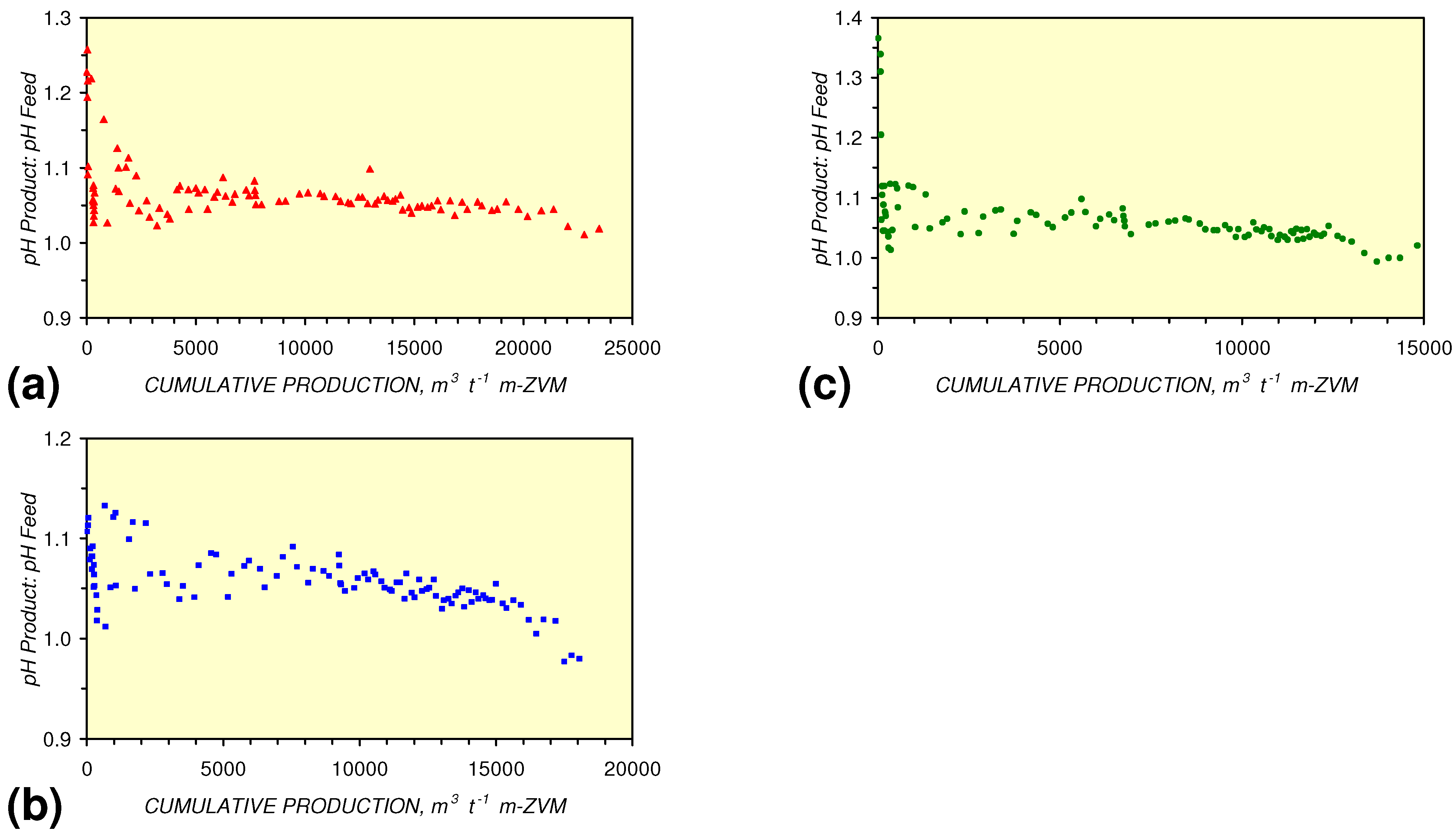
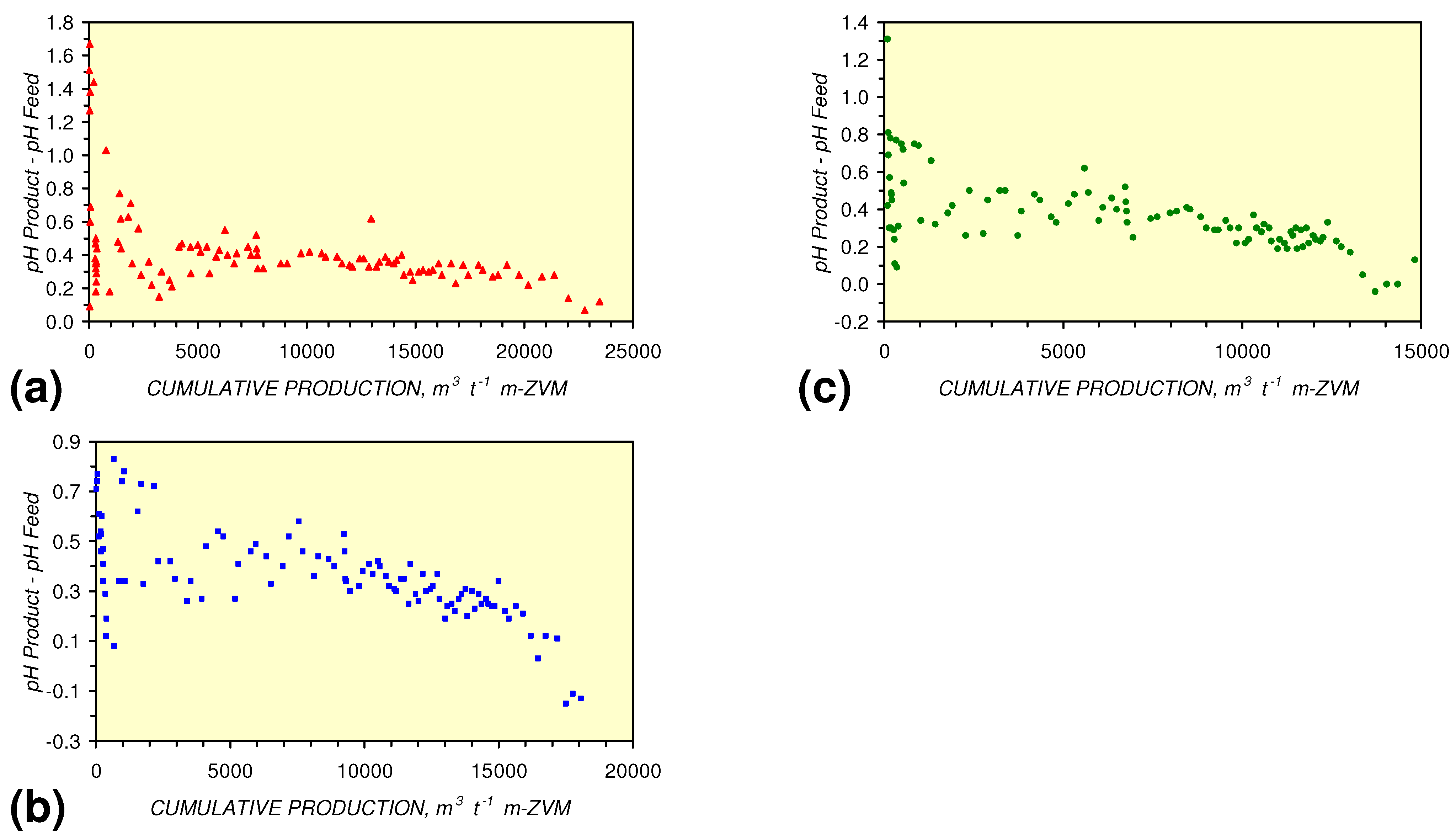
4. Discussion/Implications/Applications
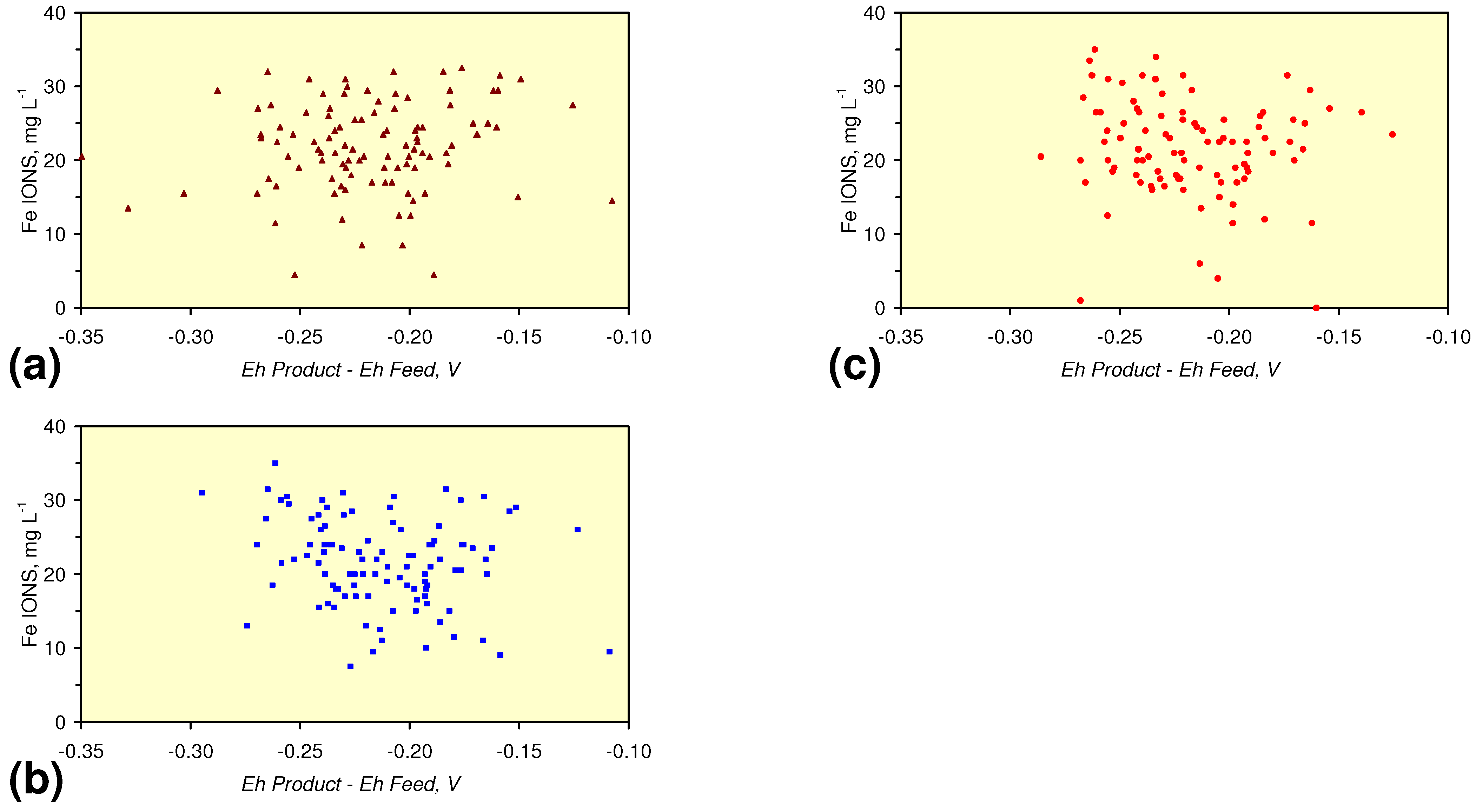
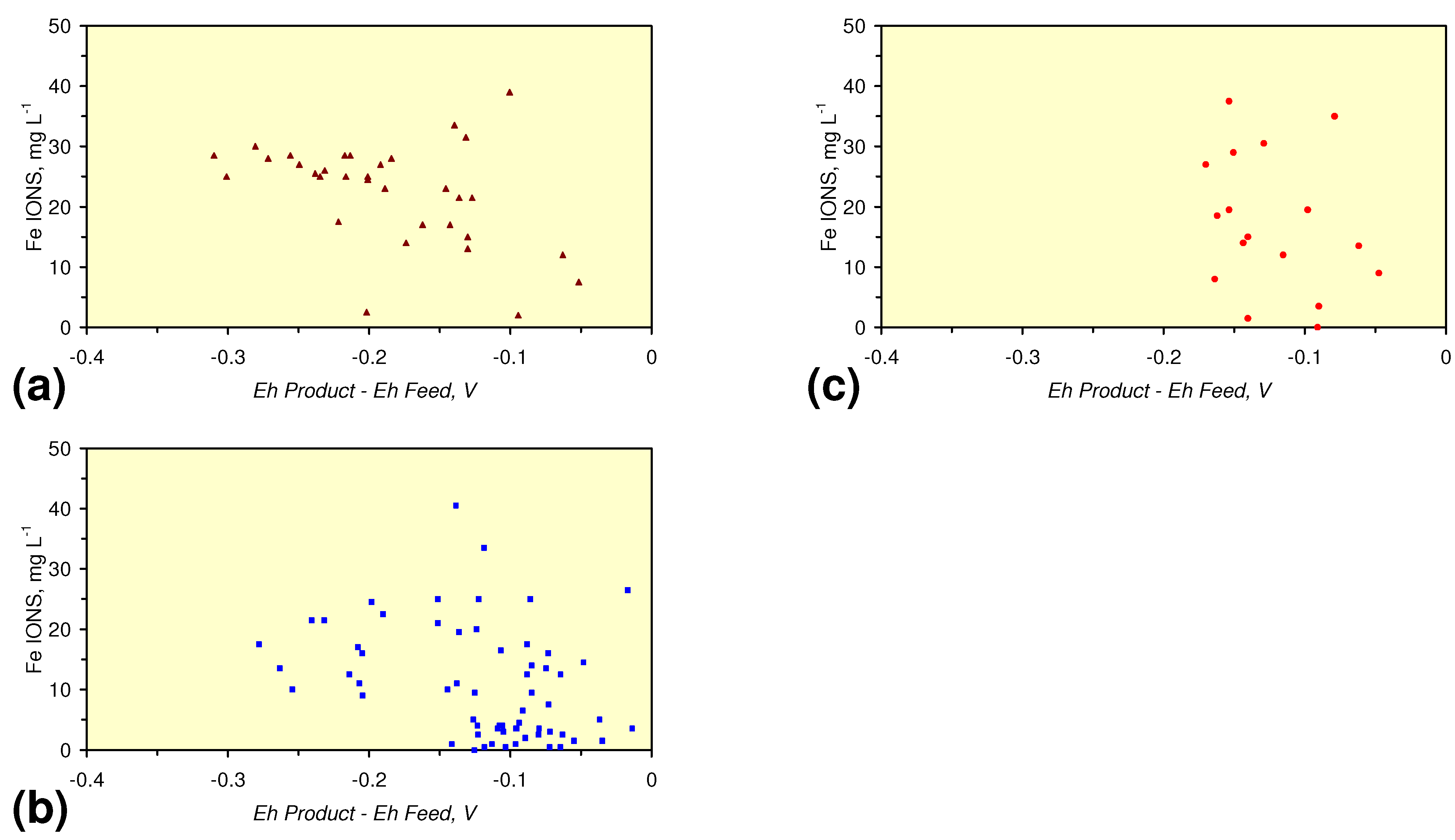
4.1. Fe Ion Type

4.1.1. Permeability Implications


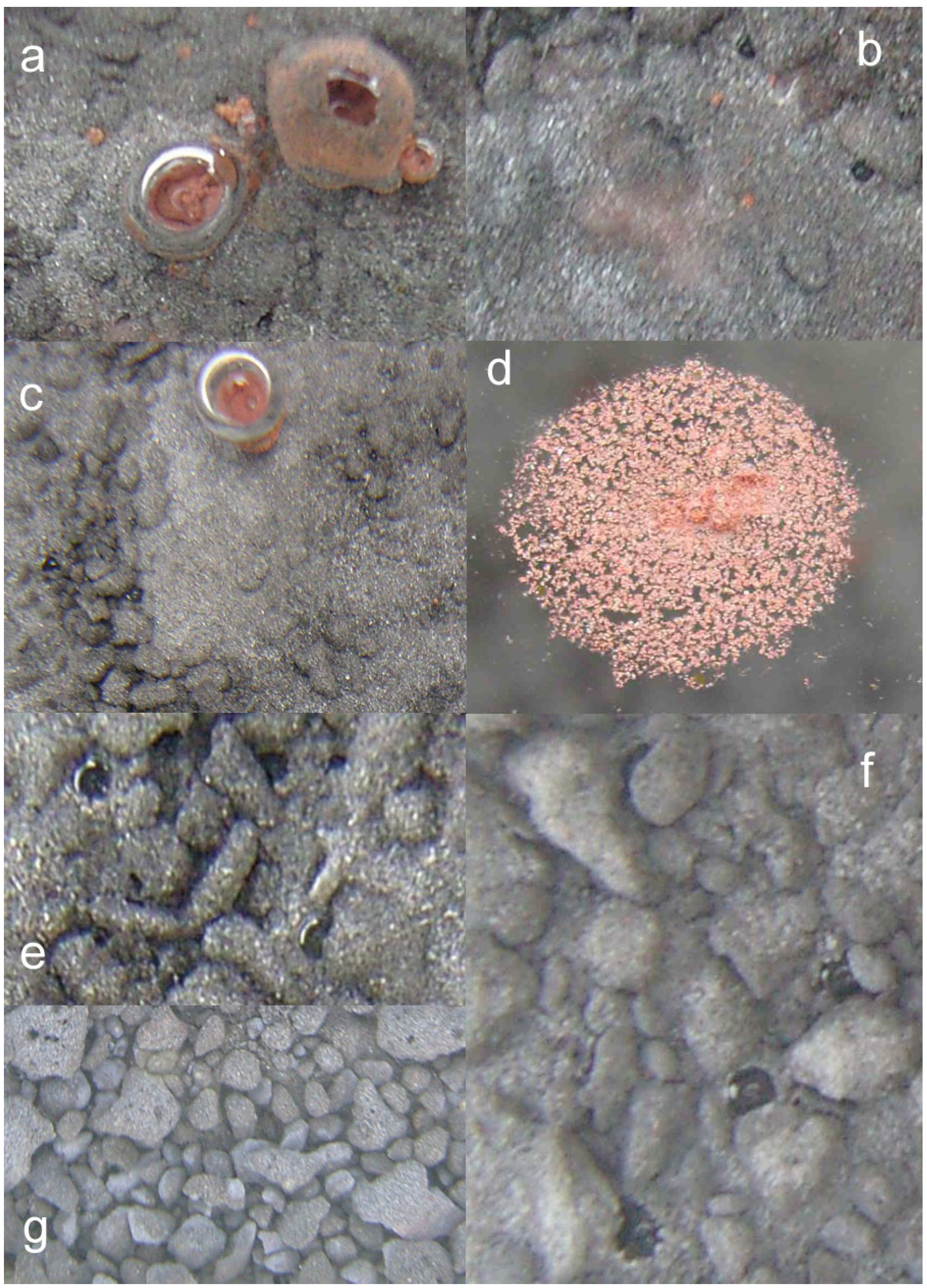
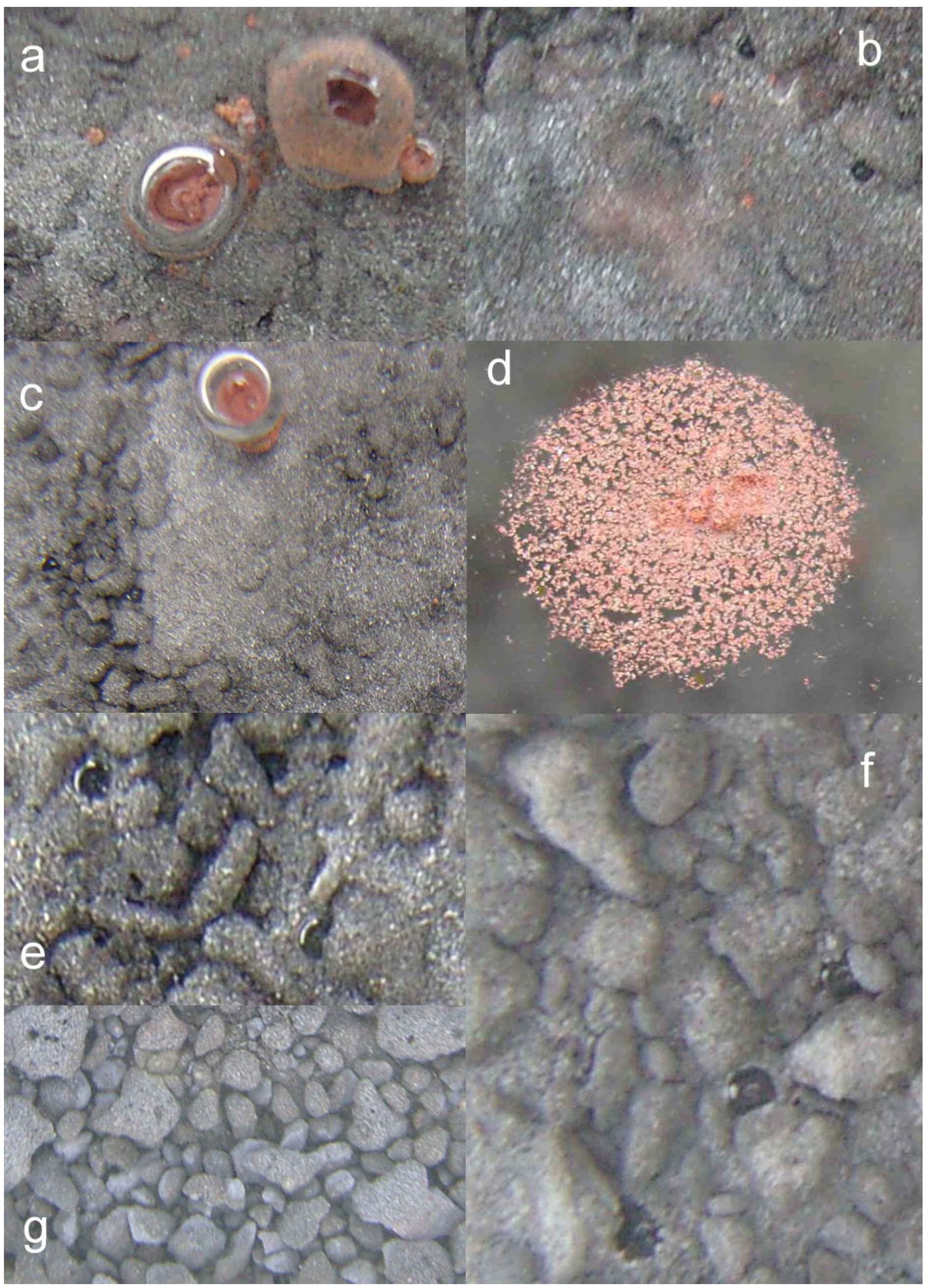
- extensive development of goethite within the ZVM bed and extensive n-Al0 diapirism (Figure 40b).
- raised particulate clod surfaces can be directly related to underlying n-Al0 diapirism (Figure 40c). Cracks within the n-Fe0 bed can be directly linked to the adjacent n-Al0 diapirs (Figure 40a, c). O2 gas bubbles are present within the n-Fe0 layers (Figure 40a, c and Figure 41), while the goethite is restricted to layers where n-Al0 and n-Fe0 are in contact (Figure 40 and Figure 41).
- The active venting associated with O2 discharge results in the ZVM bed becoming density graded with concentrations of n-Al0 confined to diapiric structures (Figure 41a, b). Active O2(g) formation occurs in the vugs and macropores radiating from the n-Al0 diapirs (Figure 41c). The resulting gas bubbles rise through the crystalline n-ZVM bed into the amorphous n-ZVM bed immediately under the n-ZVM-water contact before discharging into the water (Figure 41b).
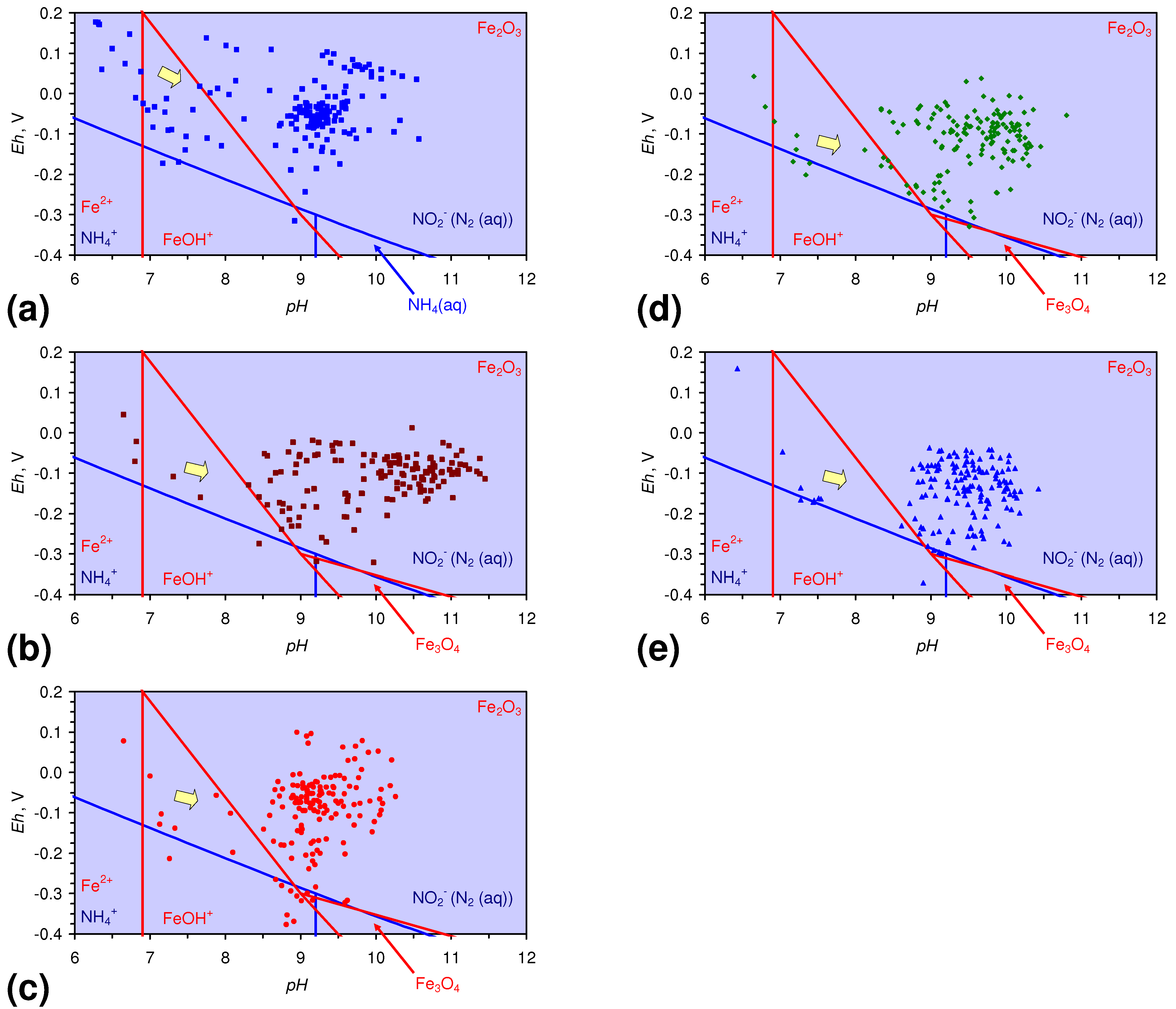
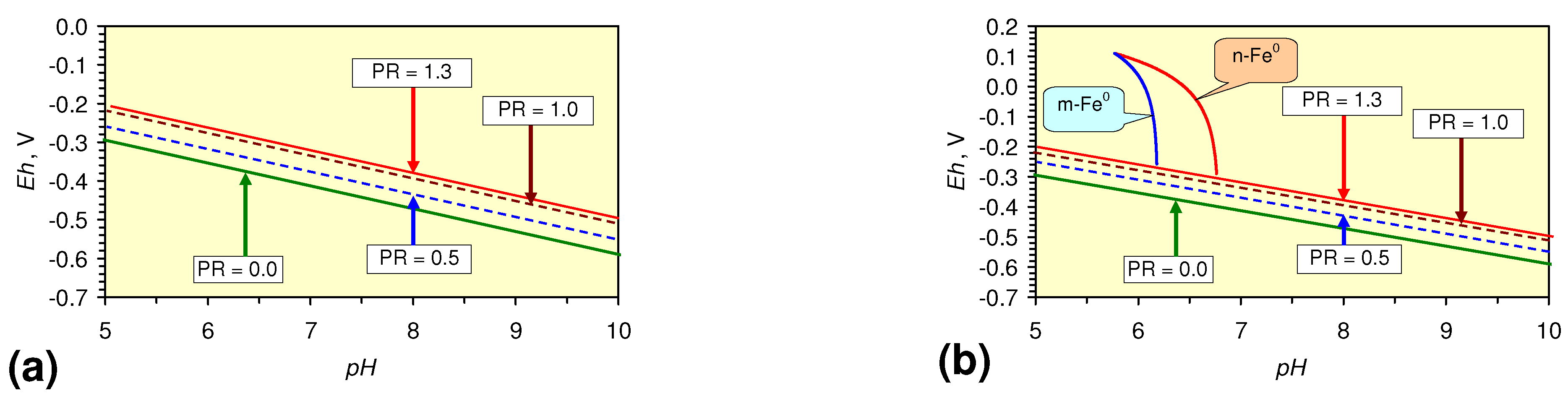
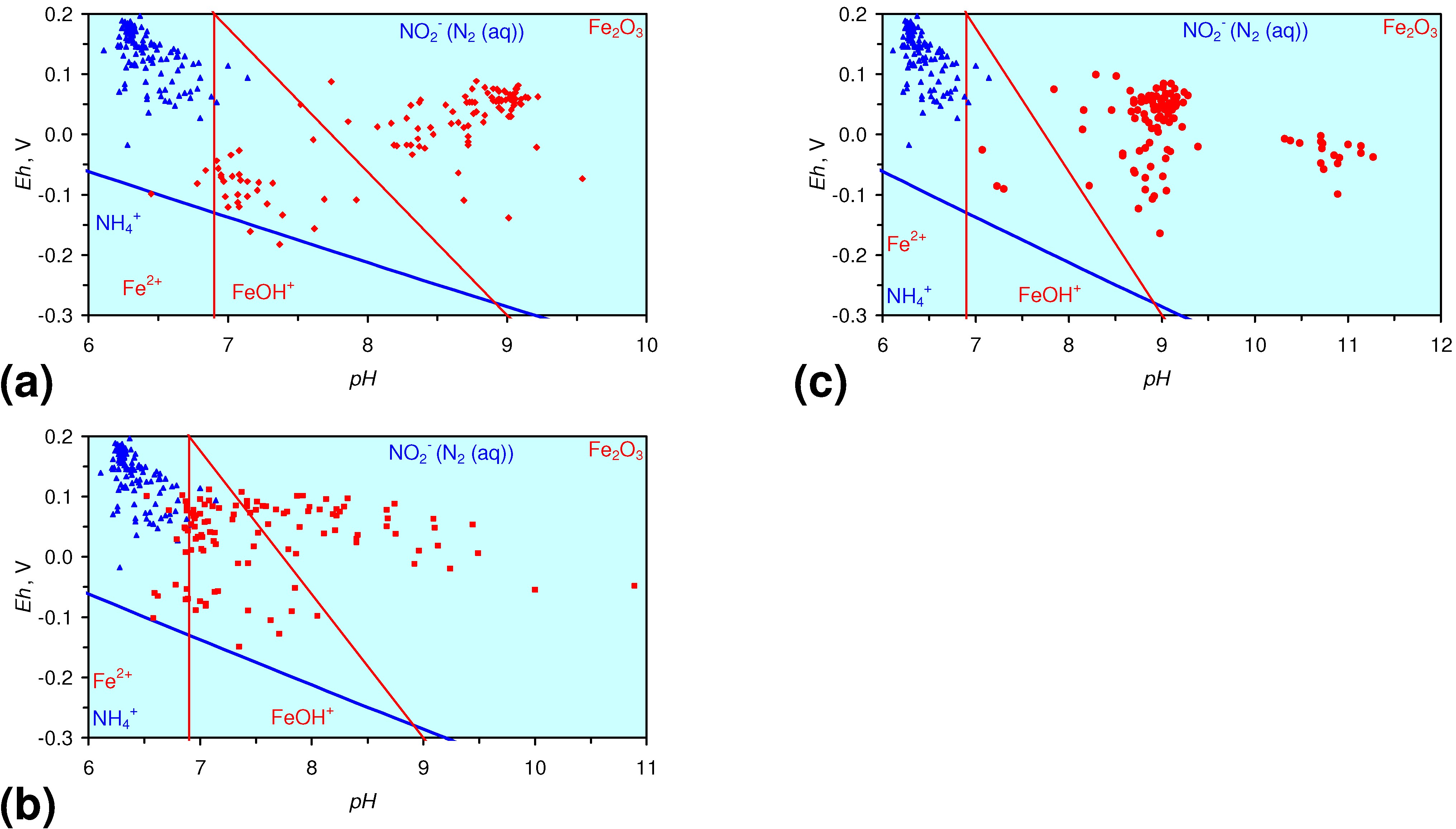
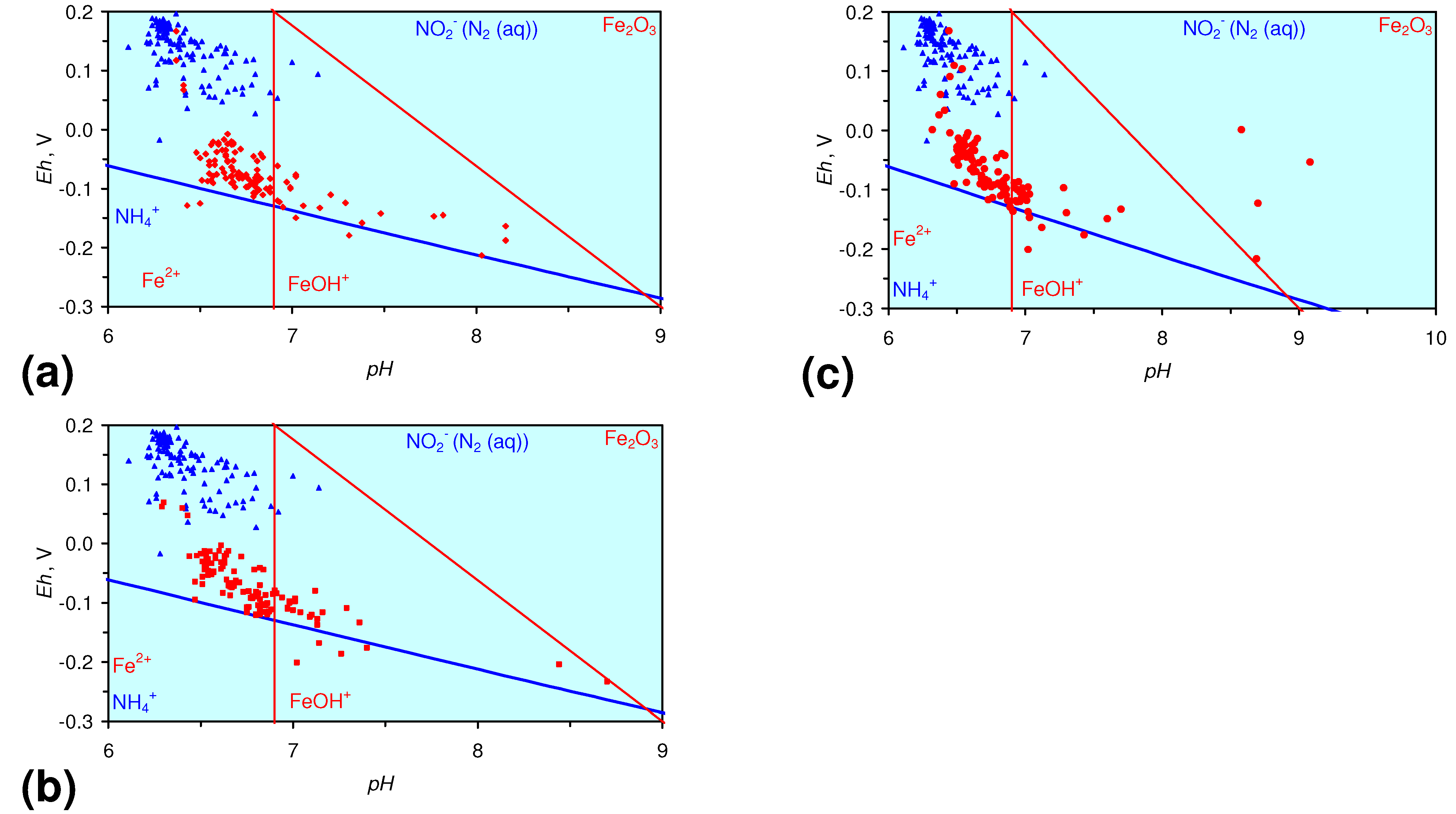
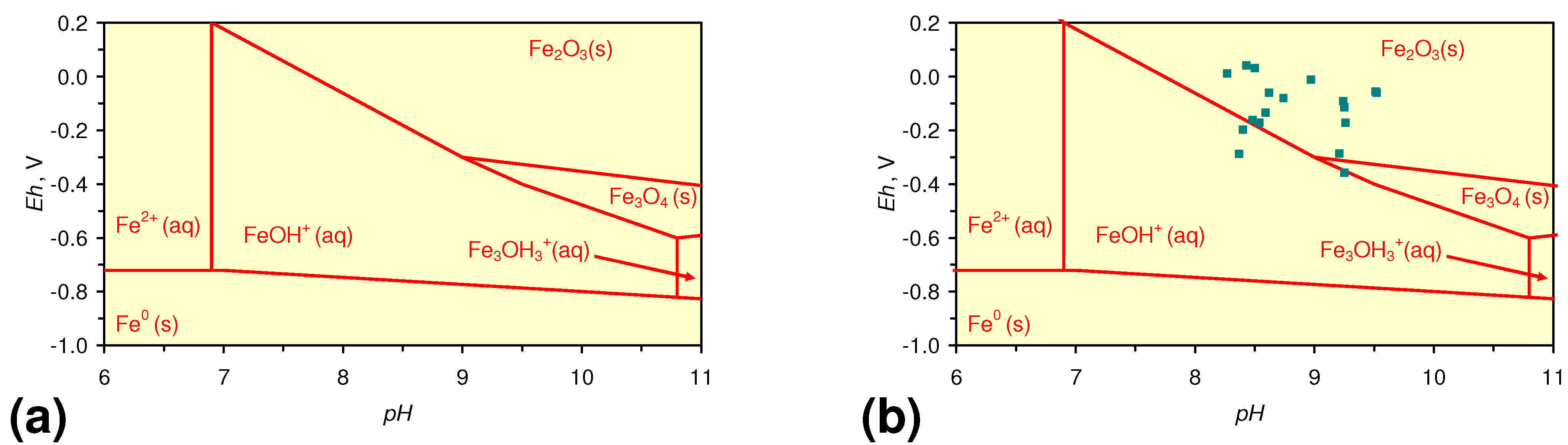
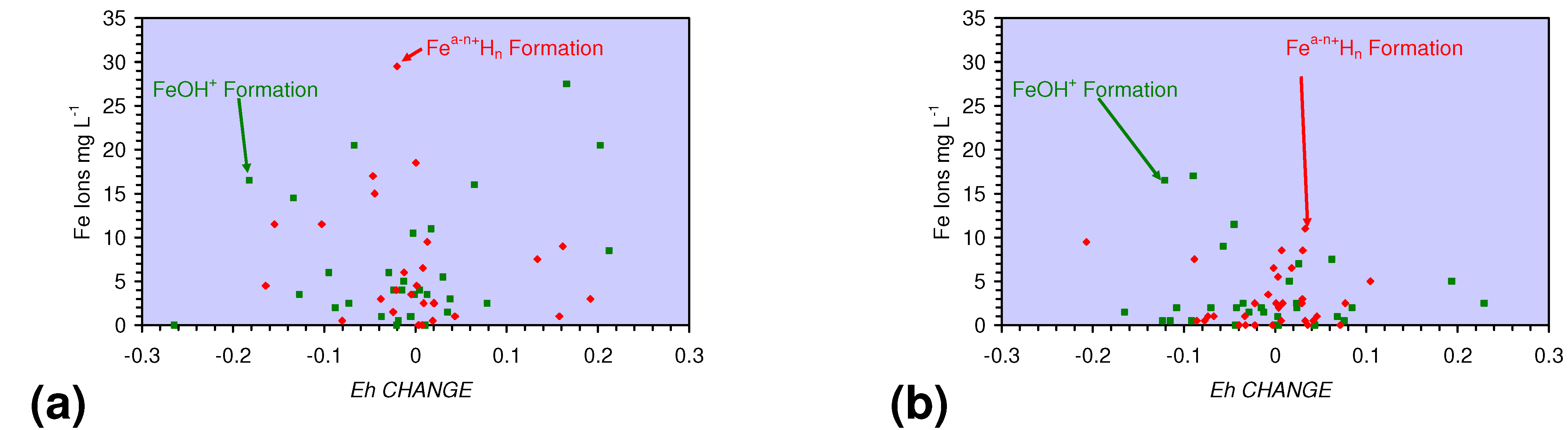
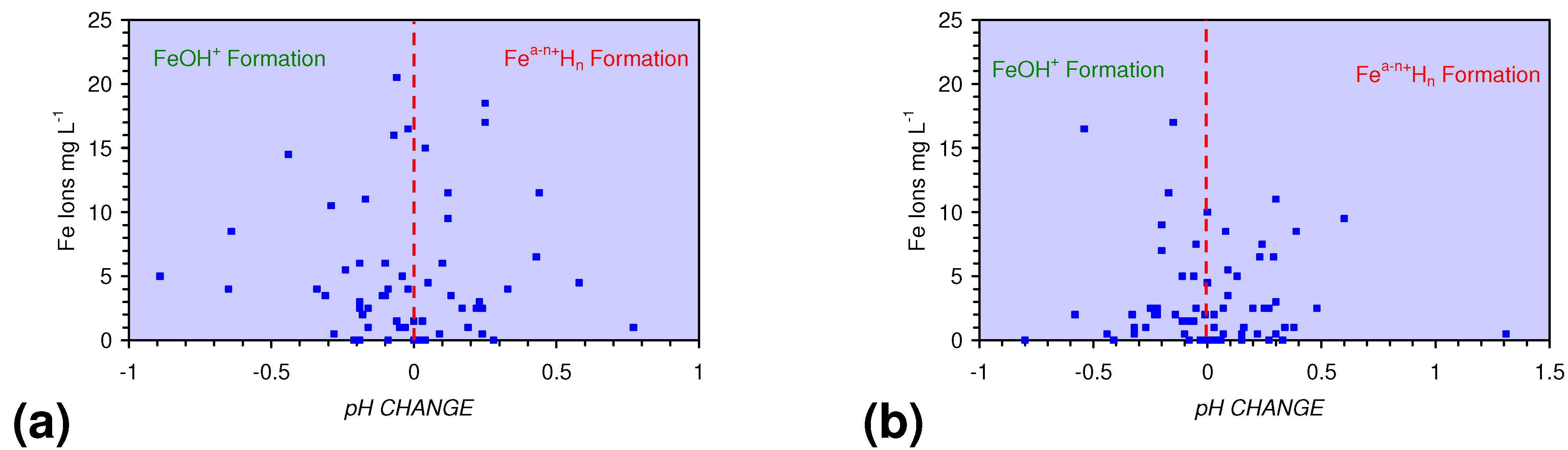
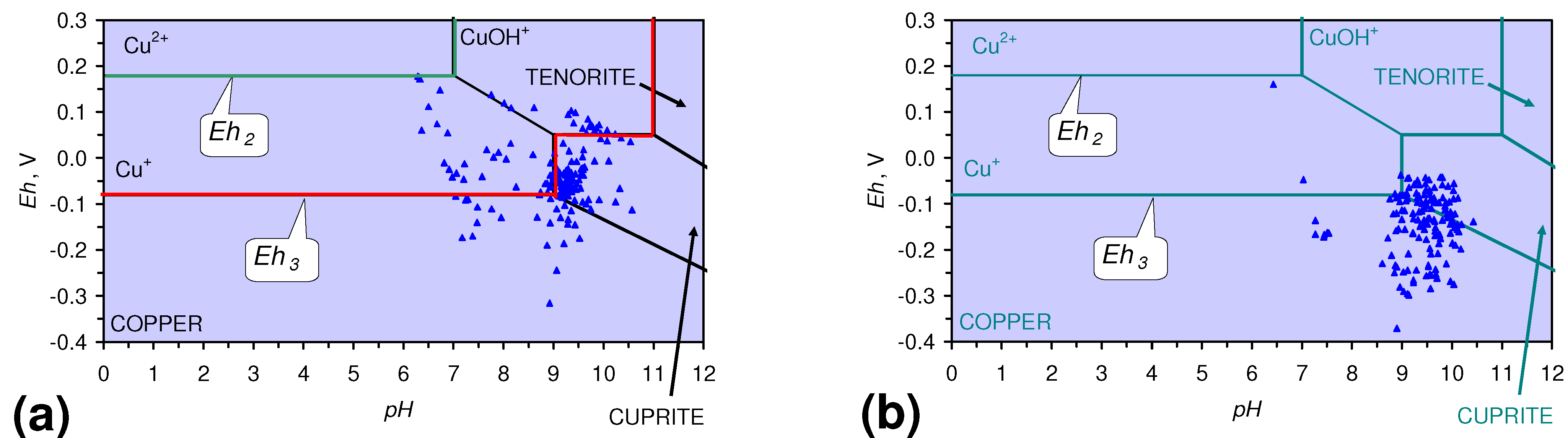
4.1.2. Remediation Implication of Fe Ion Type
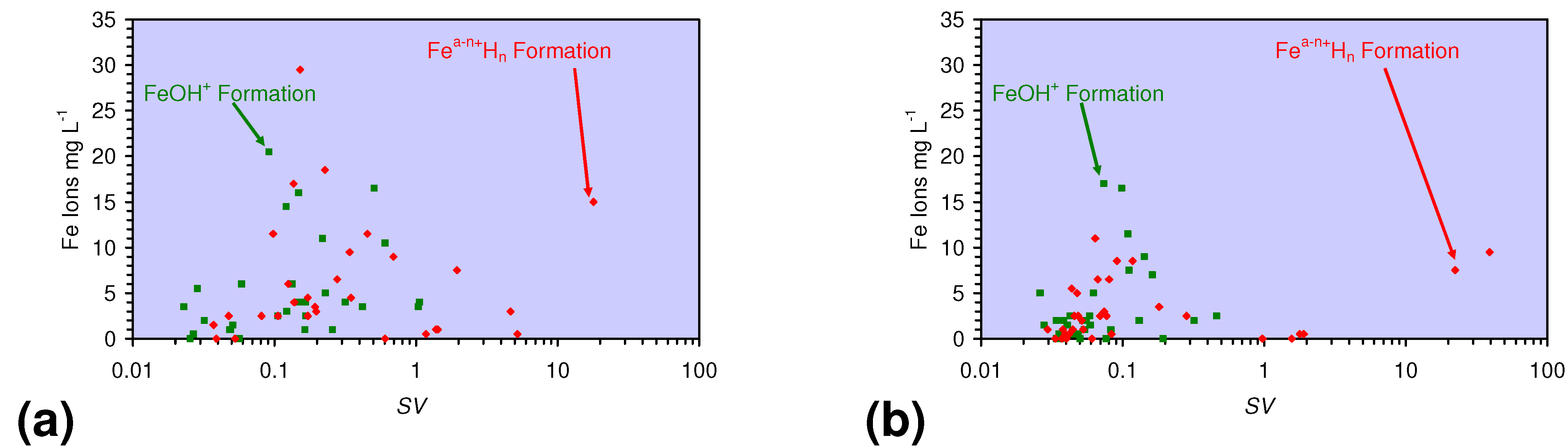
- decreases in pH and increases in Eh (interpreted as indicating FeOH+(aq) formation), or,
- increases in pH associated with increases or decreases in Eh (interpreted as indicating Fe(a−n)+H(aq) formation).
- 3.
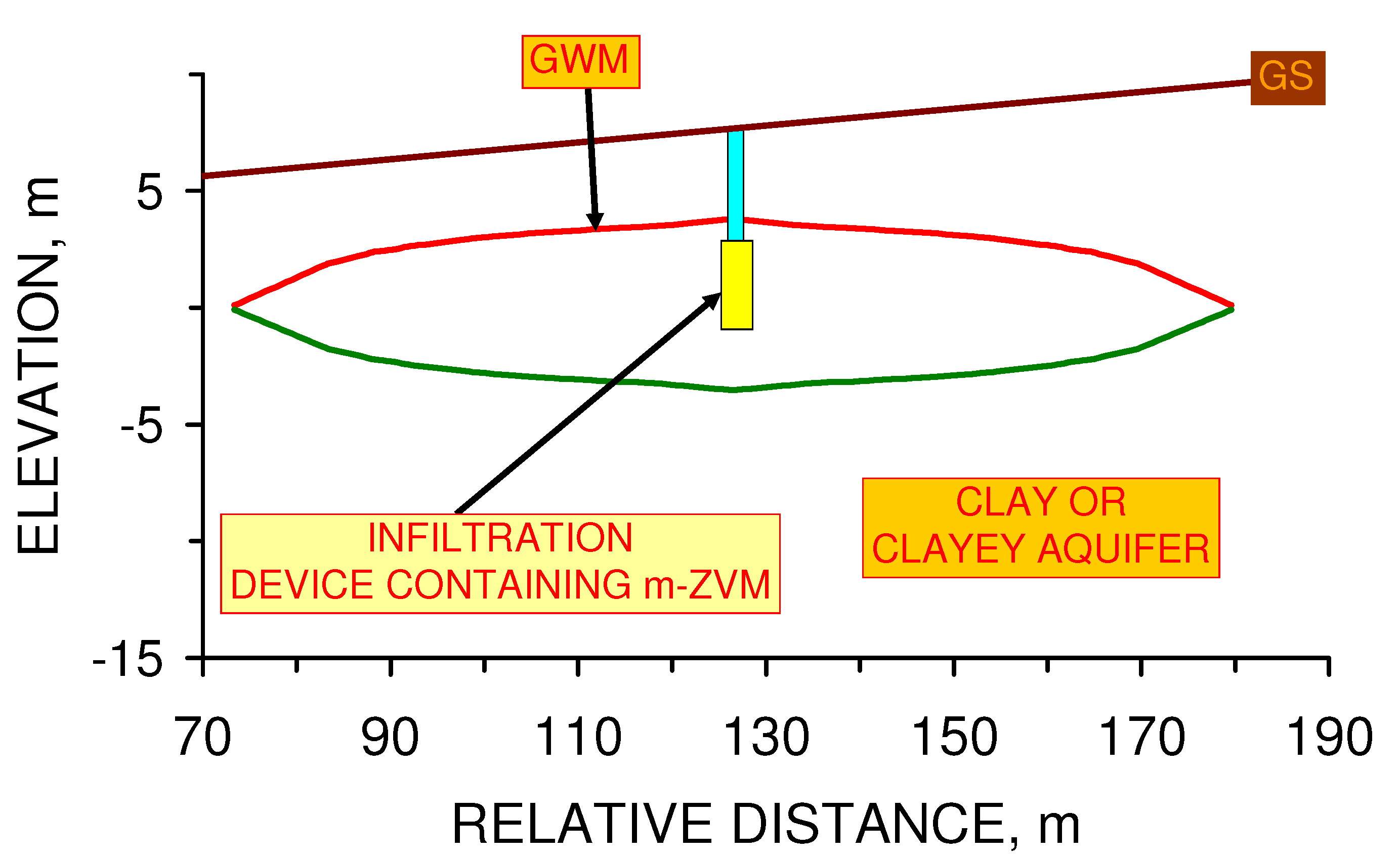
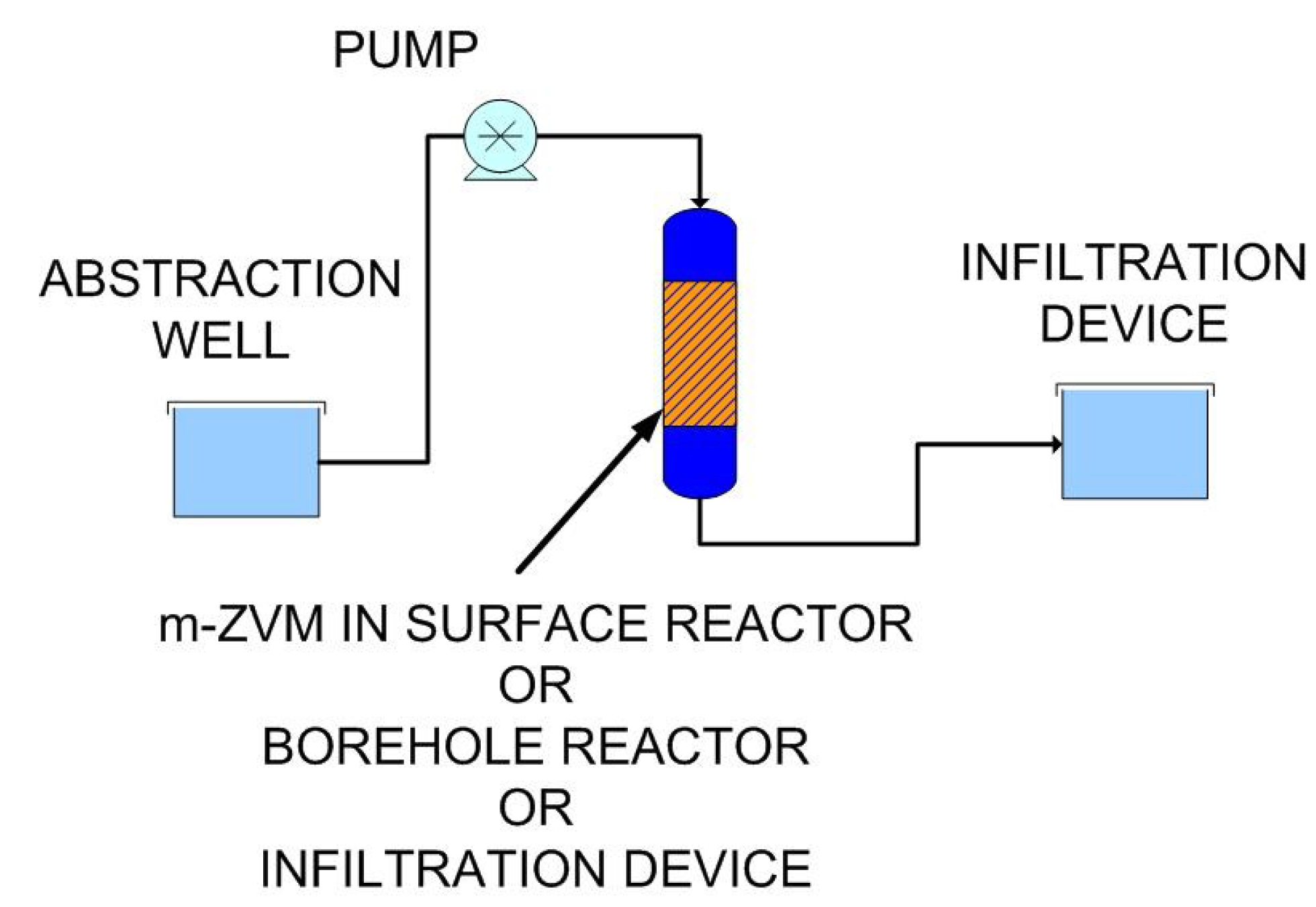
- passing infiltrating water through m-ZVM prior to entering the aquifer,
- 4.
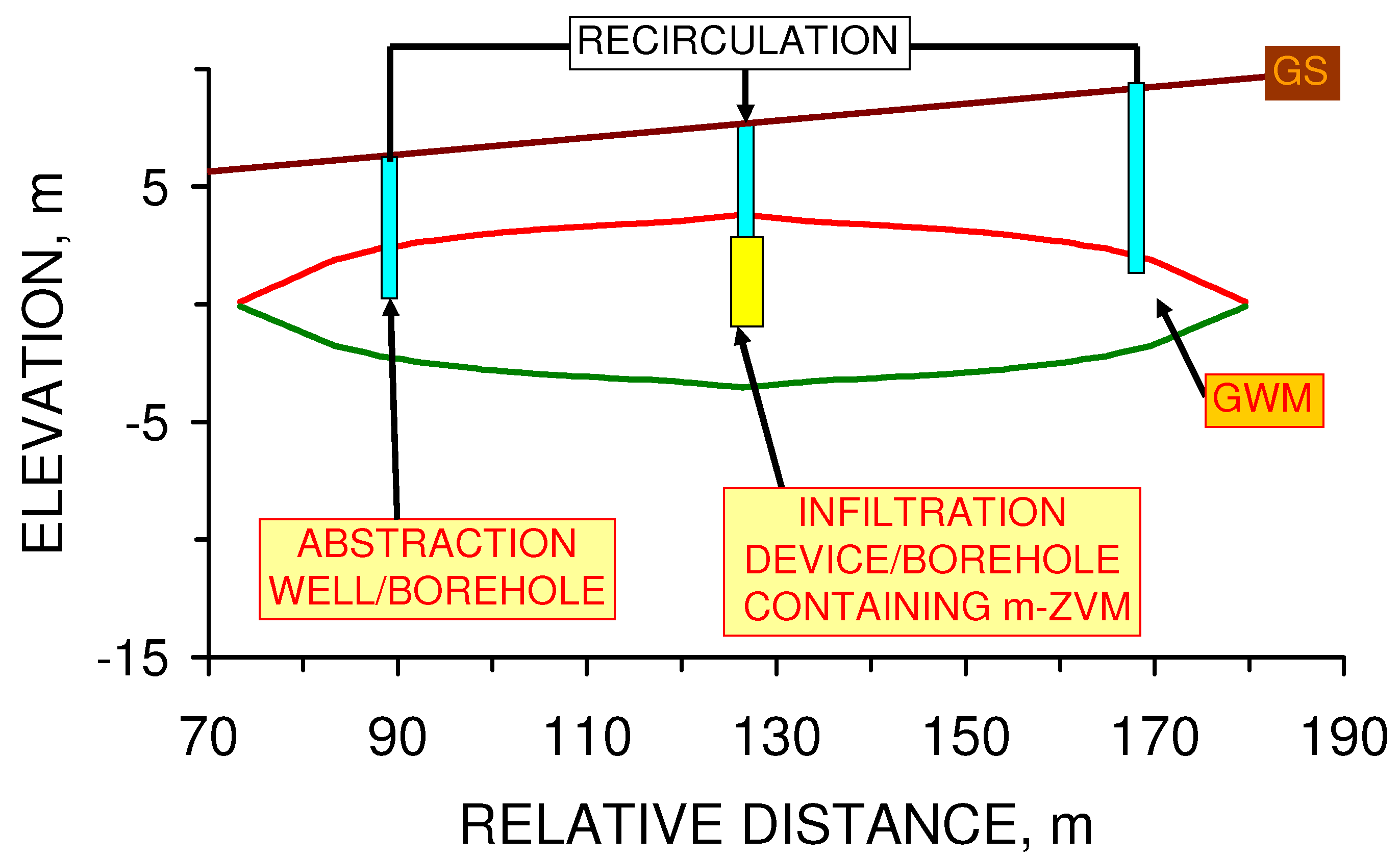
- abstracting water from the aquifer, passing it through m-ZVM, and then infiltrating/injecting the treated water into the aquifer using a process of continuous recirculation.


4.1.3. Implications of Aquifer Equilibrium Redox Oscillations on Fe Ion Type
4.1.4. Implications for Clays within the Aquifer

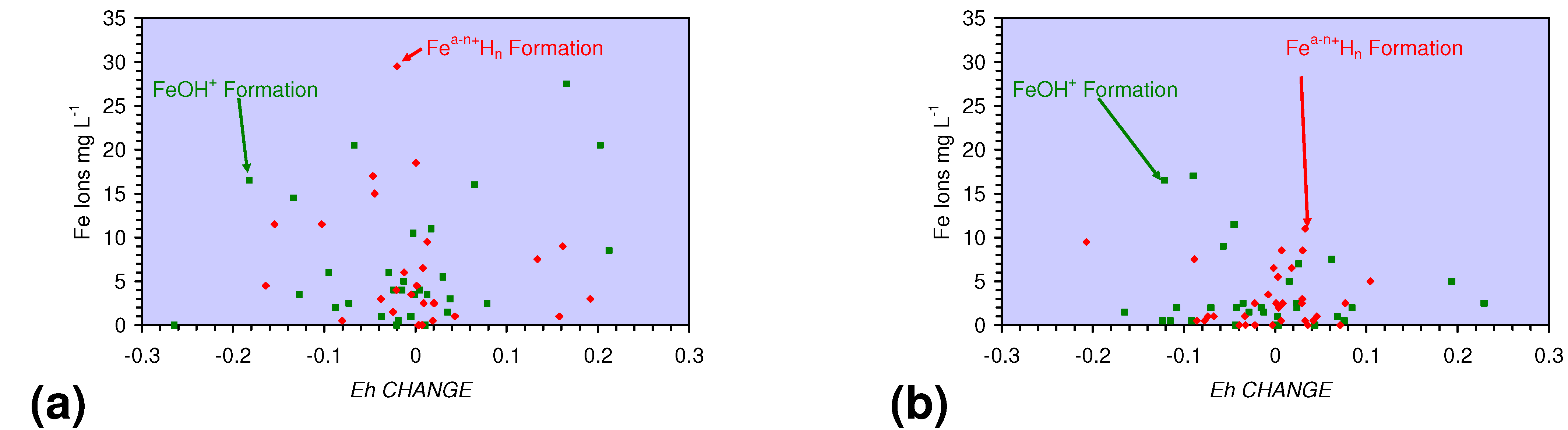
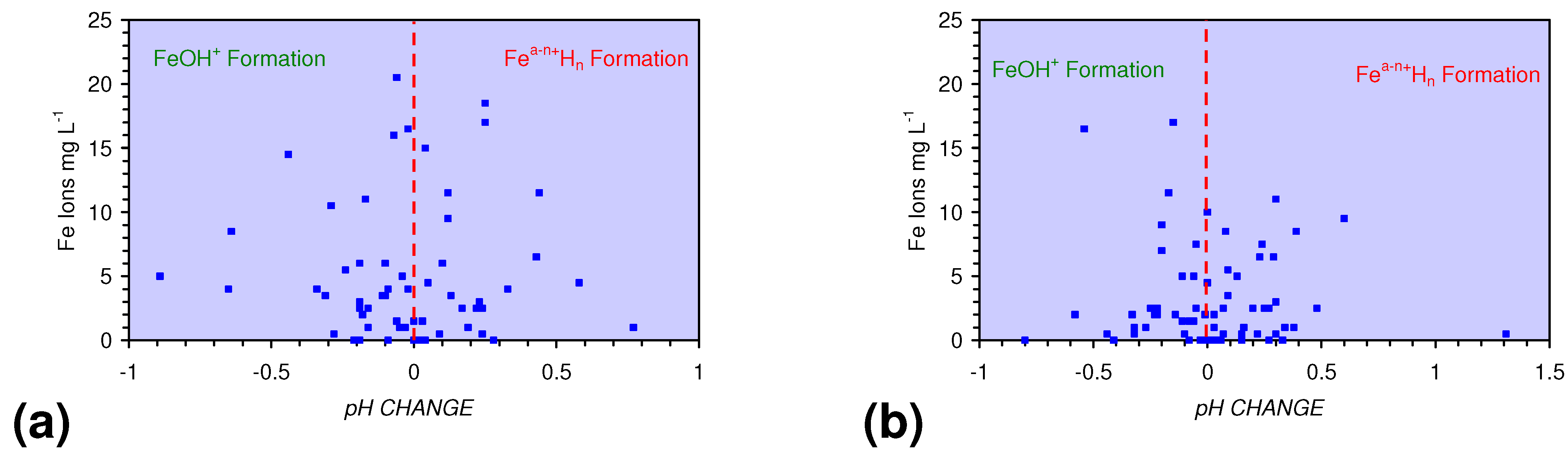

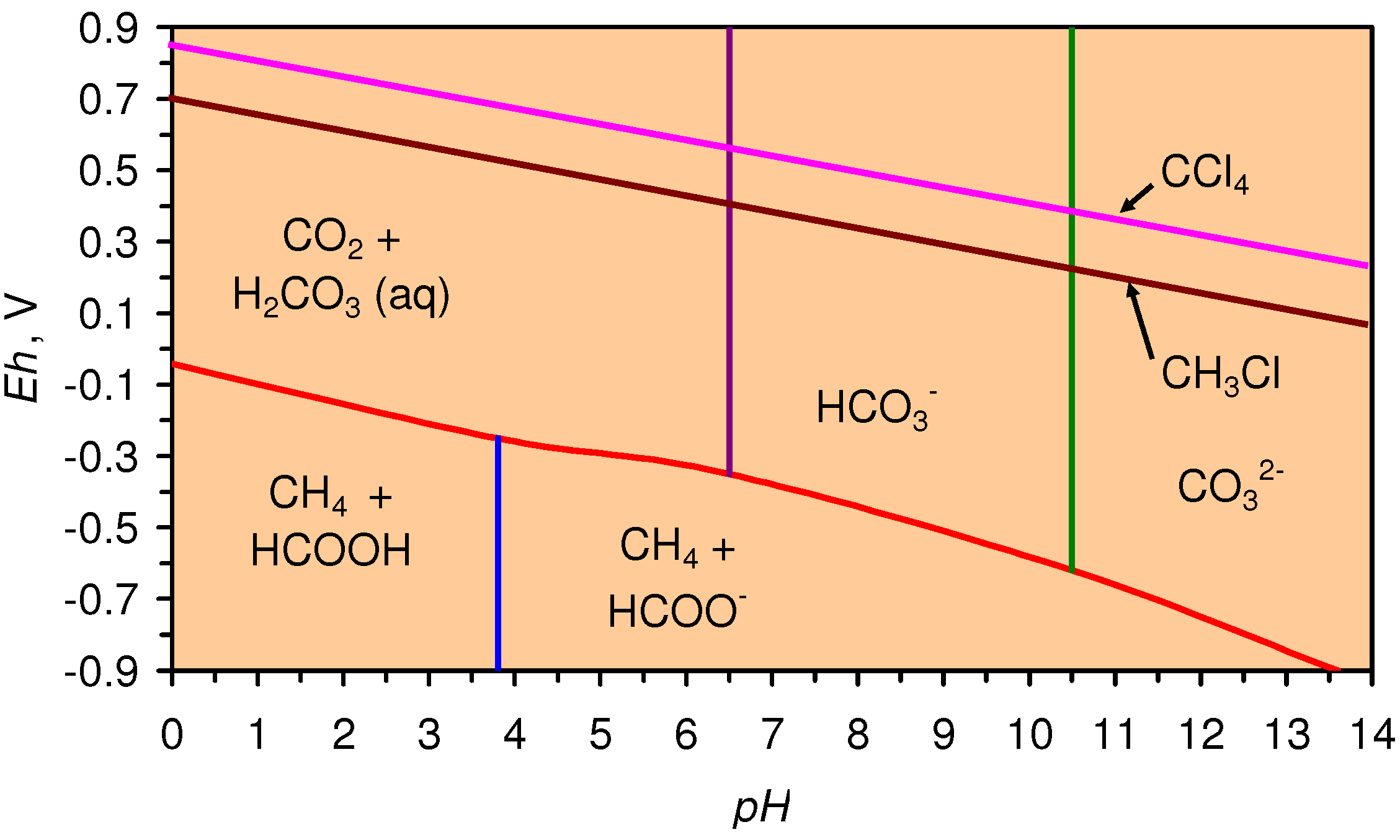
4.2. Applicability of Regression Equations to the Modeling of Pore Water Redox Modification

4.2.1. Reference Pilot n-ZVM Pilot Injection Program

4.2.2. Modeling Static Diffusion: m-ZVM

4.2.3. Modeling Redox Modification by Recirculation: m-ZVM
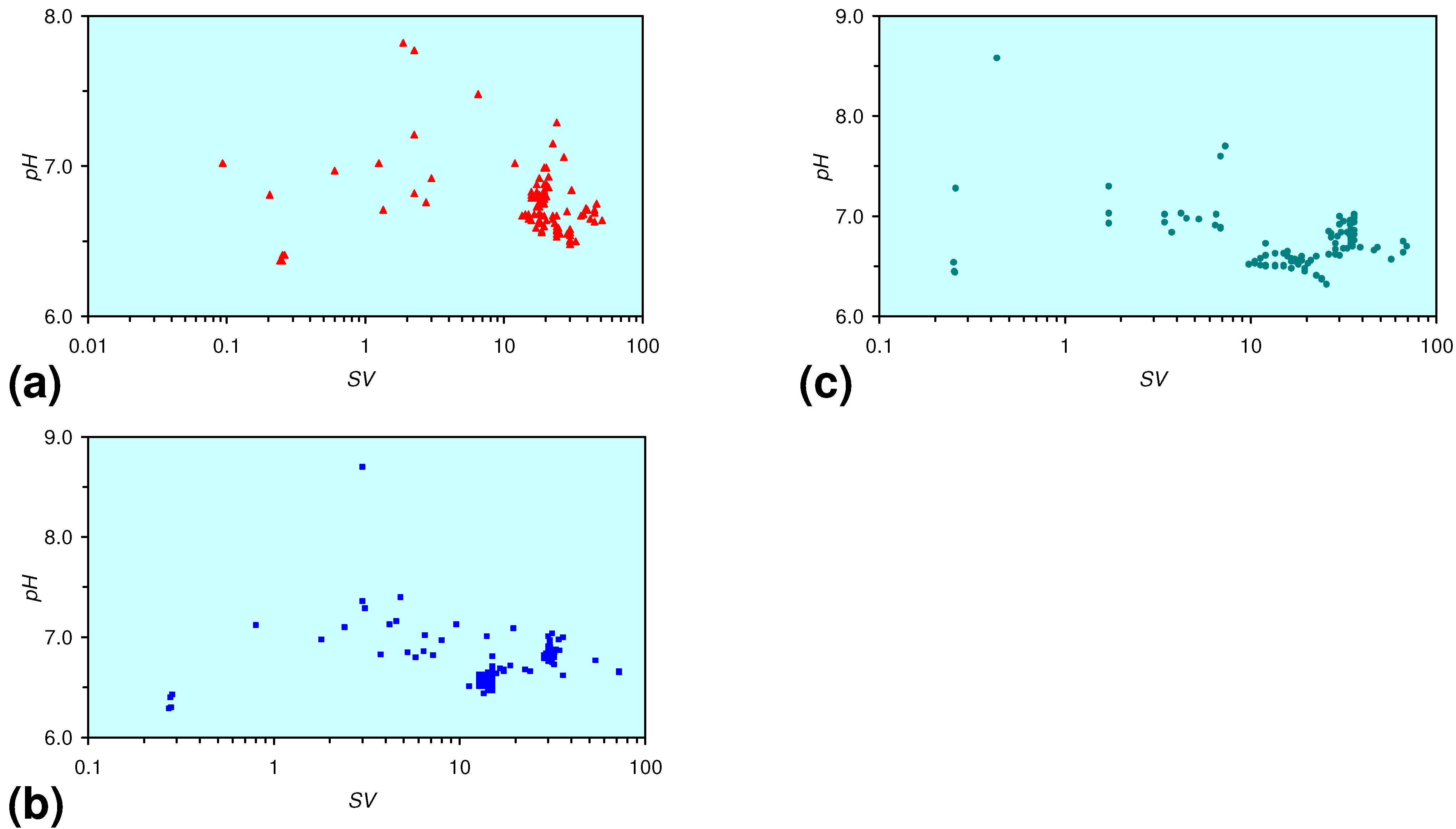
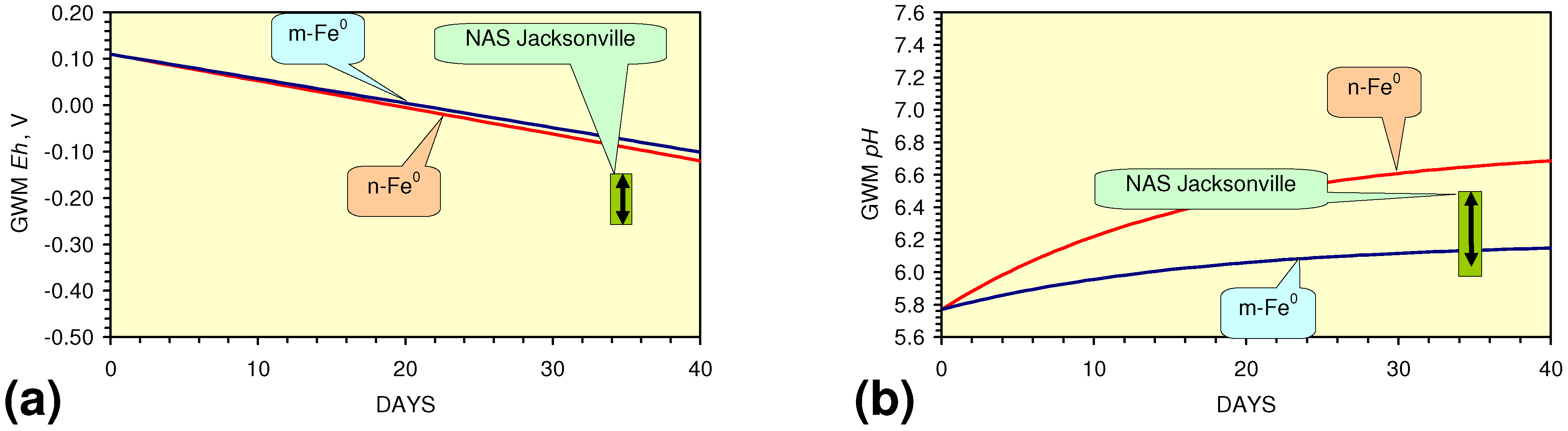
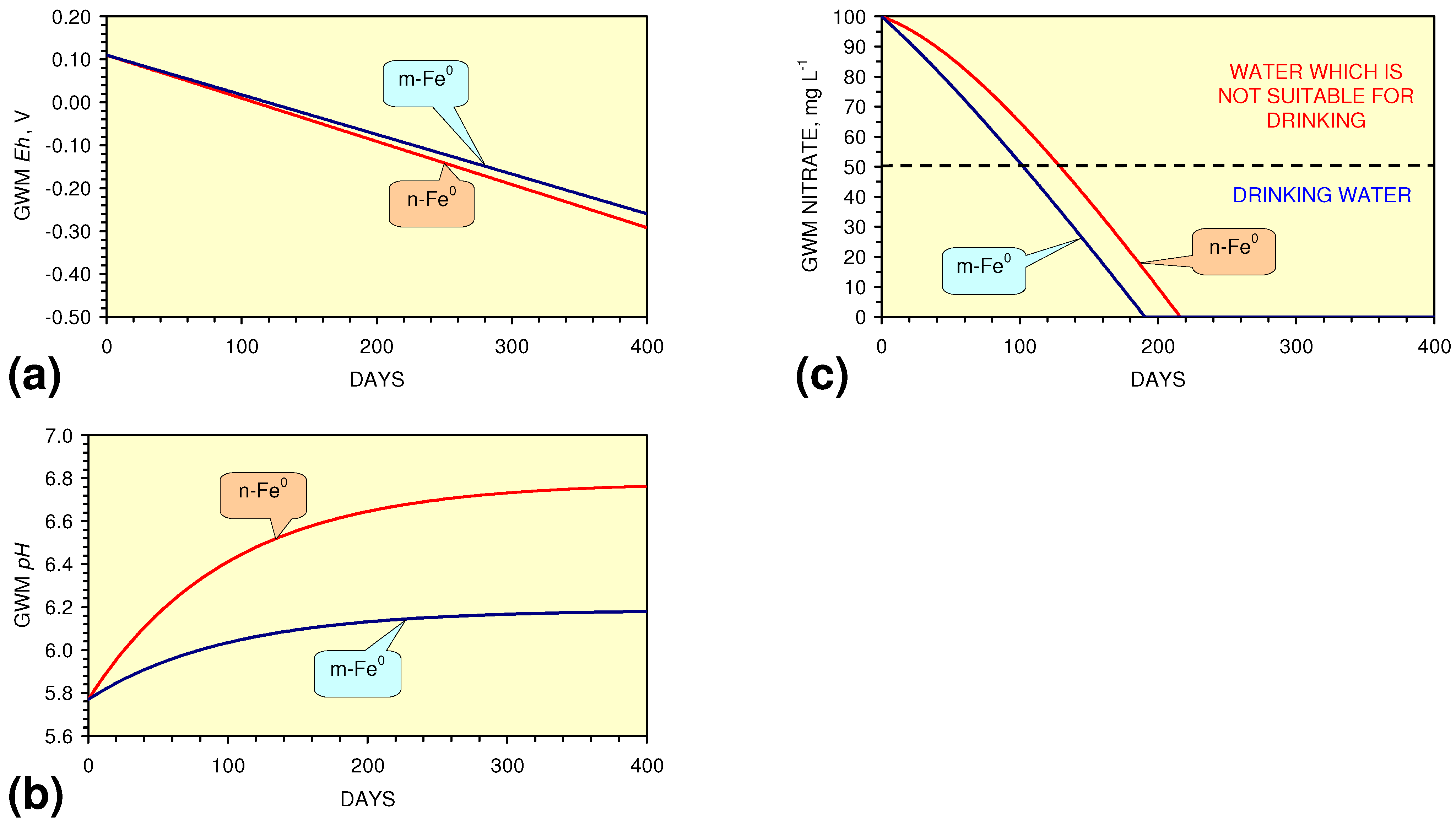
- The continuous flow tests indicate that utilisation of m-Fe0 would require replacement of the m-Fe0 after about 20 m3 of water had been processed.
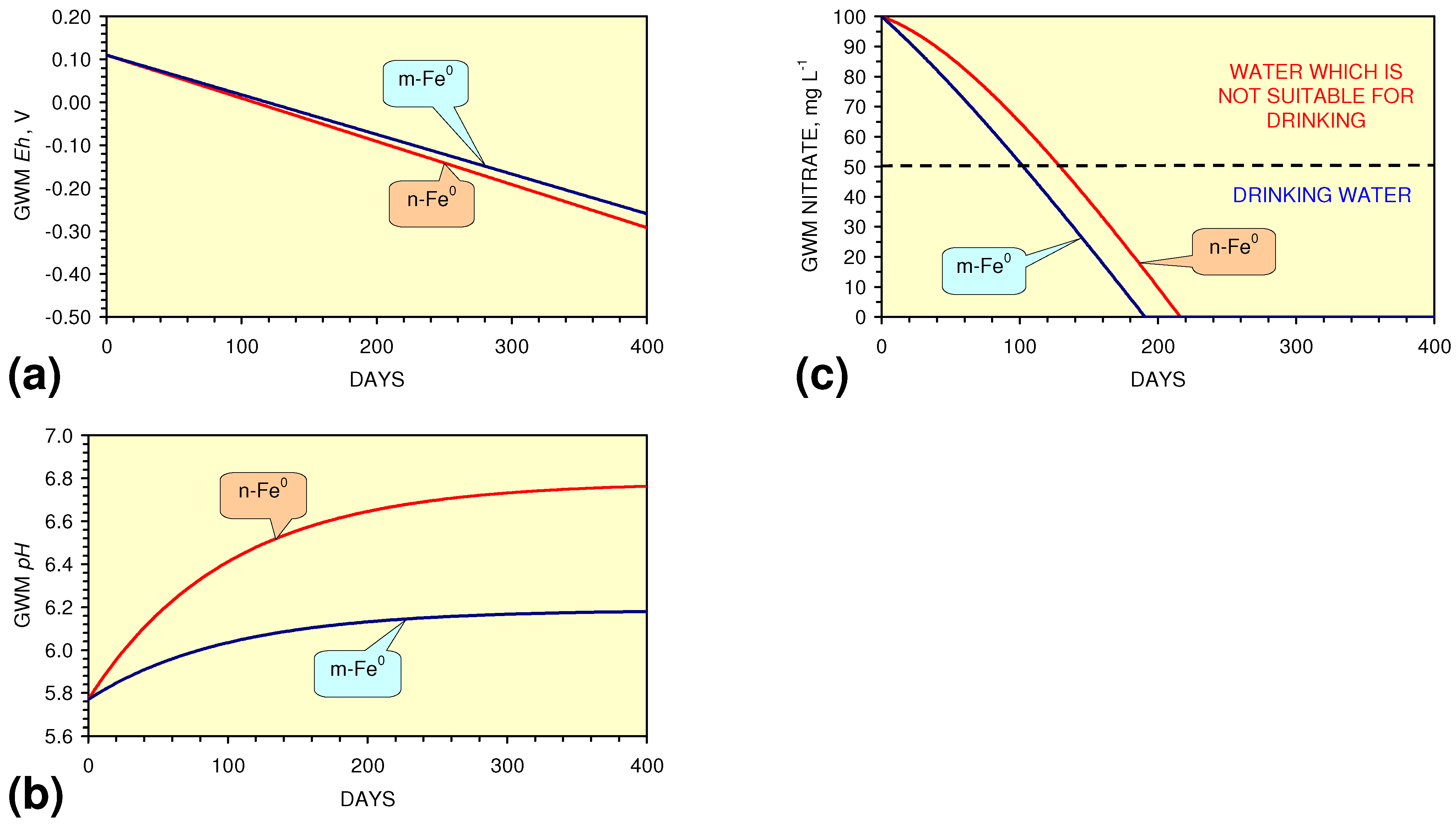
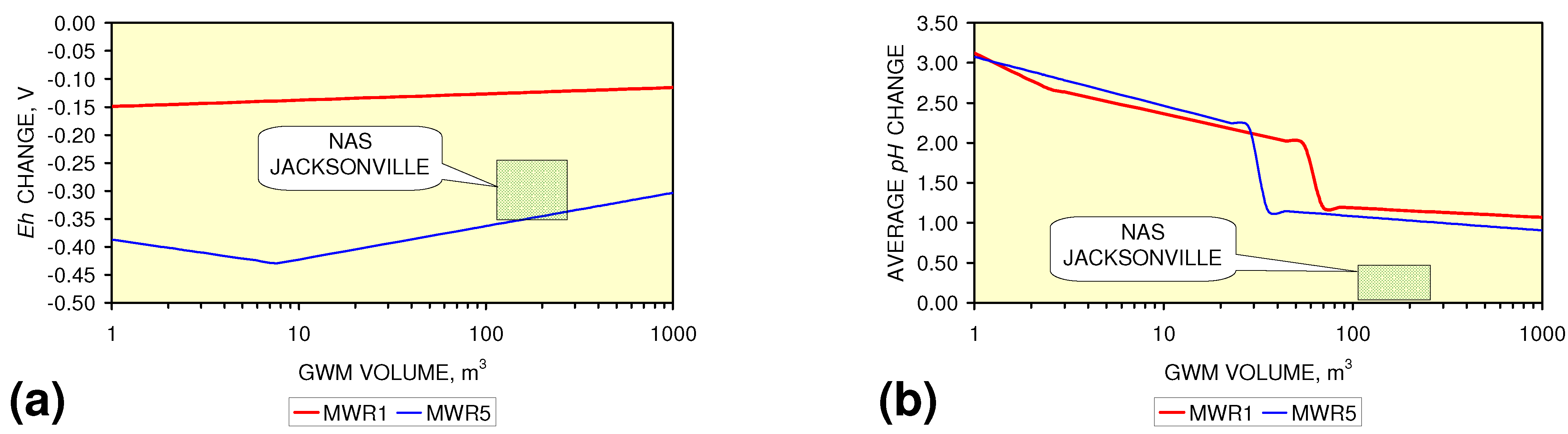
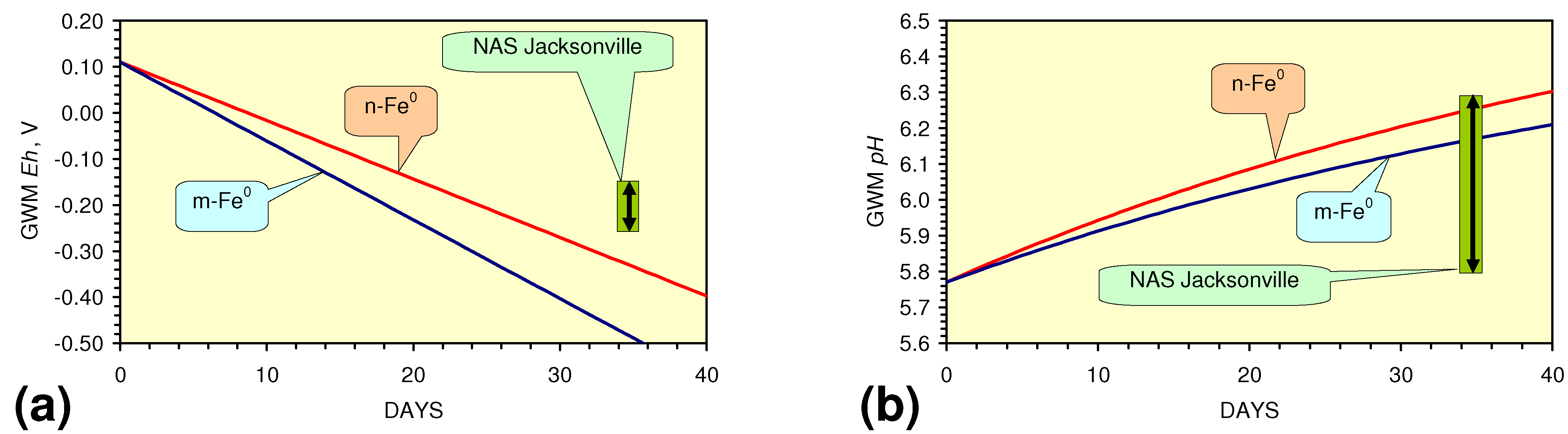
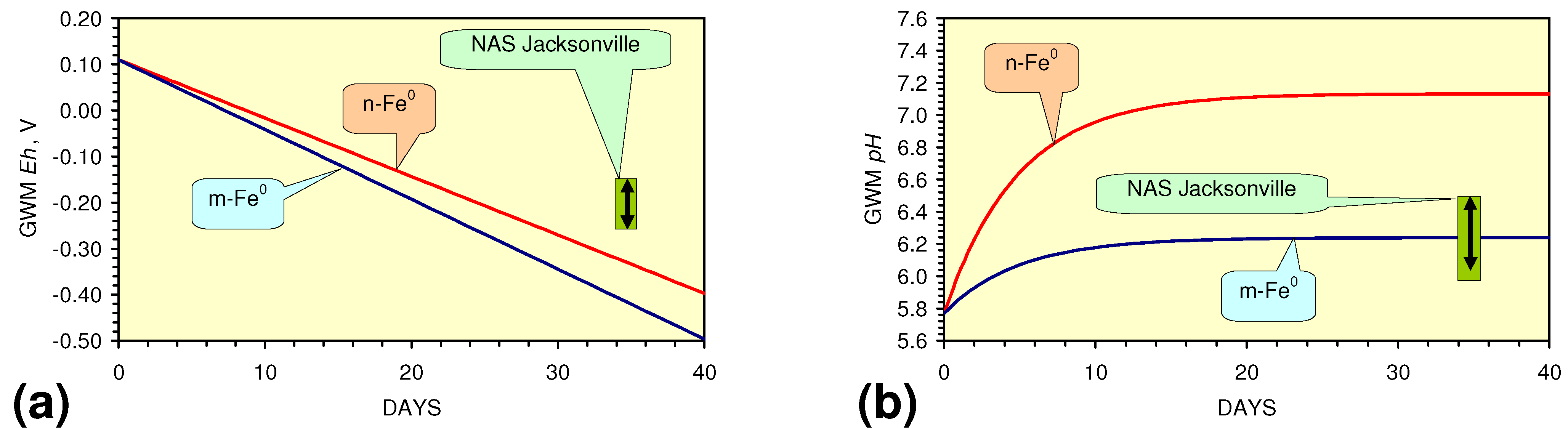
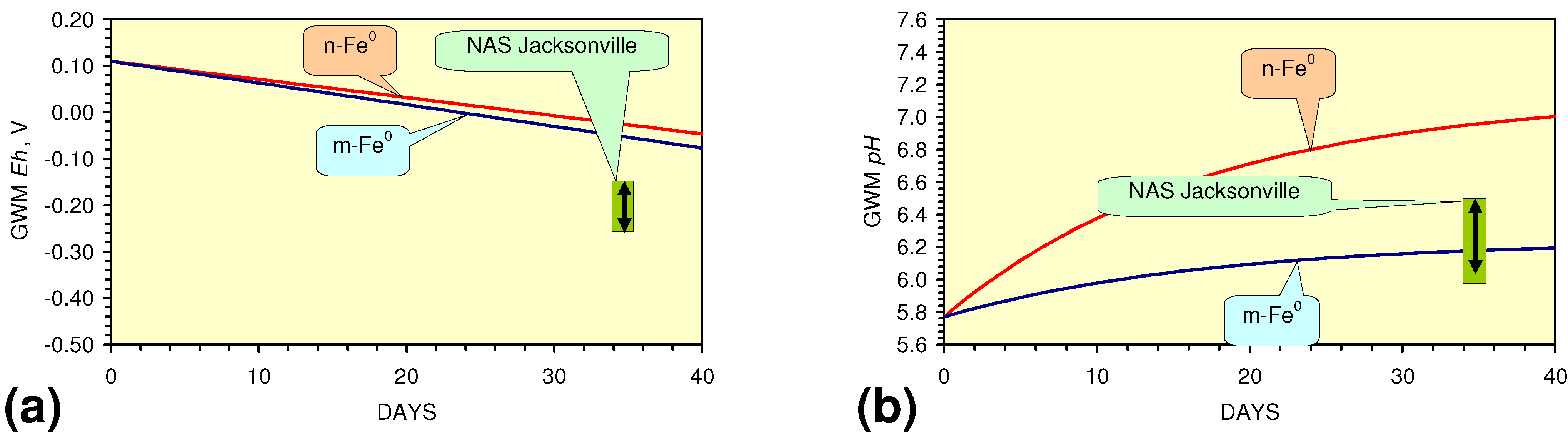
- m-Fe0 will provide a lower Eh in the GWM after 40 days than n-Fe0 (Figure 54)
- m-Fe0 will provide a lower pH in the GWM after 40 days than n-Fe0 (Figure 54)
- Reducing the SV to 10, while maintaining a circulation rate of 0.83 m3 h−1 (i.e., using 83 kg n/m-ZVM), has the effect of establishing an equilibrium pH in the aquifer while reducing the decrease in Eh (Figure 55).
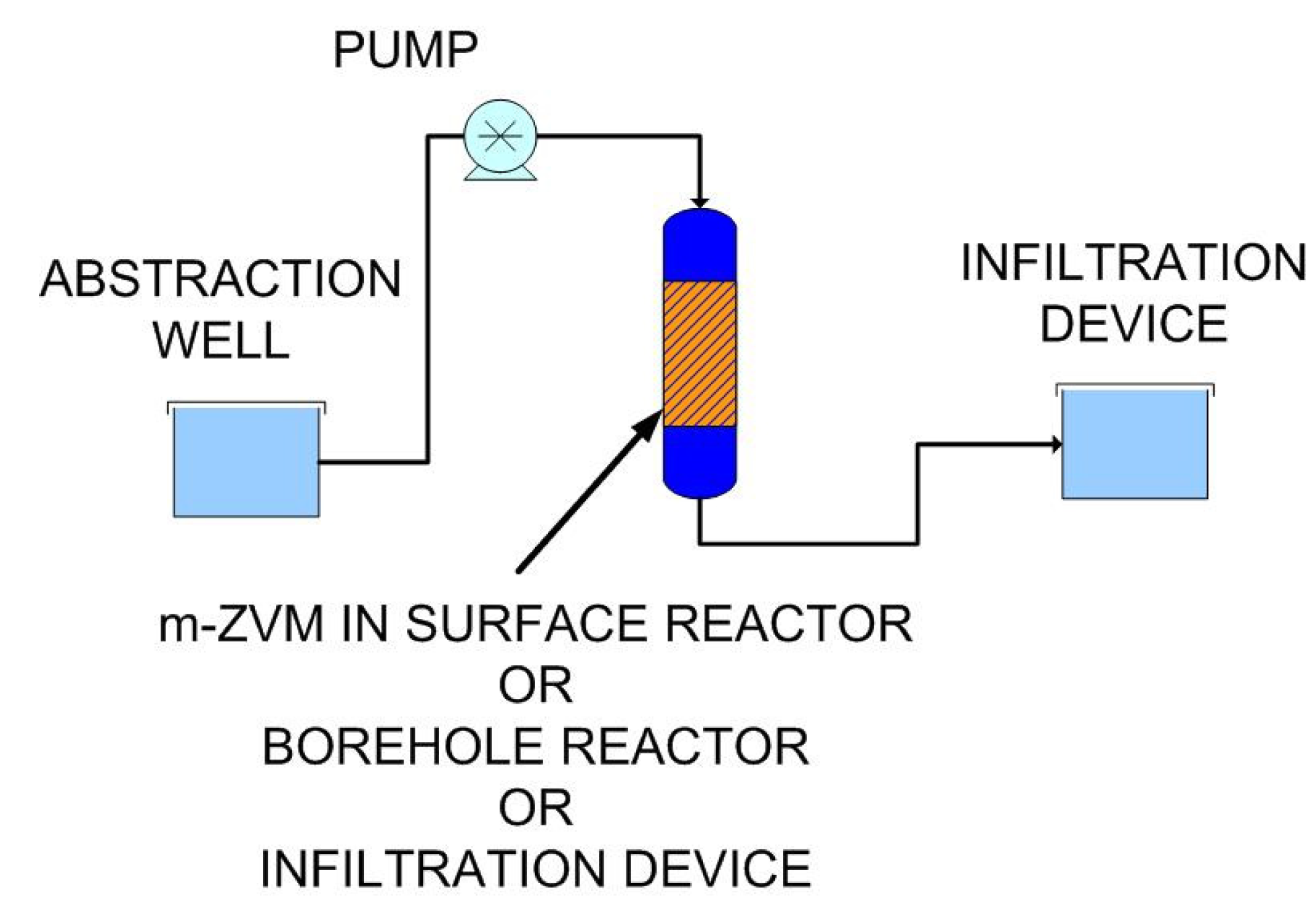




4.3. Potential Applications
- 5.
- 6.
- Remediation of specific anions (e.g., nitrates) to be predicted for static diffusion PRB’s and recirculation remediation programs
- 7.
- Enrichment of specific cations (e.g., ammonia) to be predicted for static diffusion PRB’s and recirculation remediation programs which are designed to convert nitrate enriched groundwater into nitrogen enriched agricultural fertiliser
- 8.
- Remediation of specific cations (e.g., Cu, Pb) to be predicted for static diffusion PRB’s and recirculation remediation programs
- 9.
- Remediation of organic contaminants to be predicted for static diffusion PRB’s and recirculation remediation programs
- 10.
- Aquifer remediation programs to be designed based on recirculation
4.3.1. Remediation of Storm Runoff/Overland Flow
- adjusting the equilibrium ion concentrations, and
- placing Fe(a−n)+H ions (i.e., Fe catalyst) in the infiltrating water.
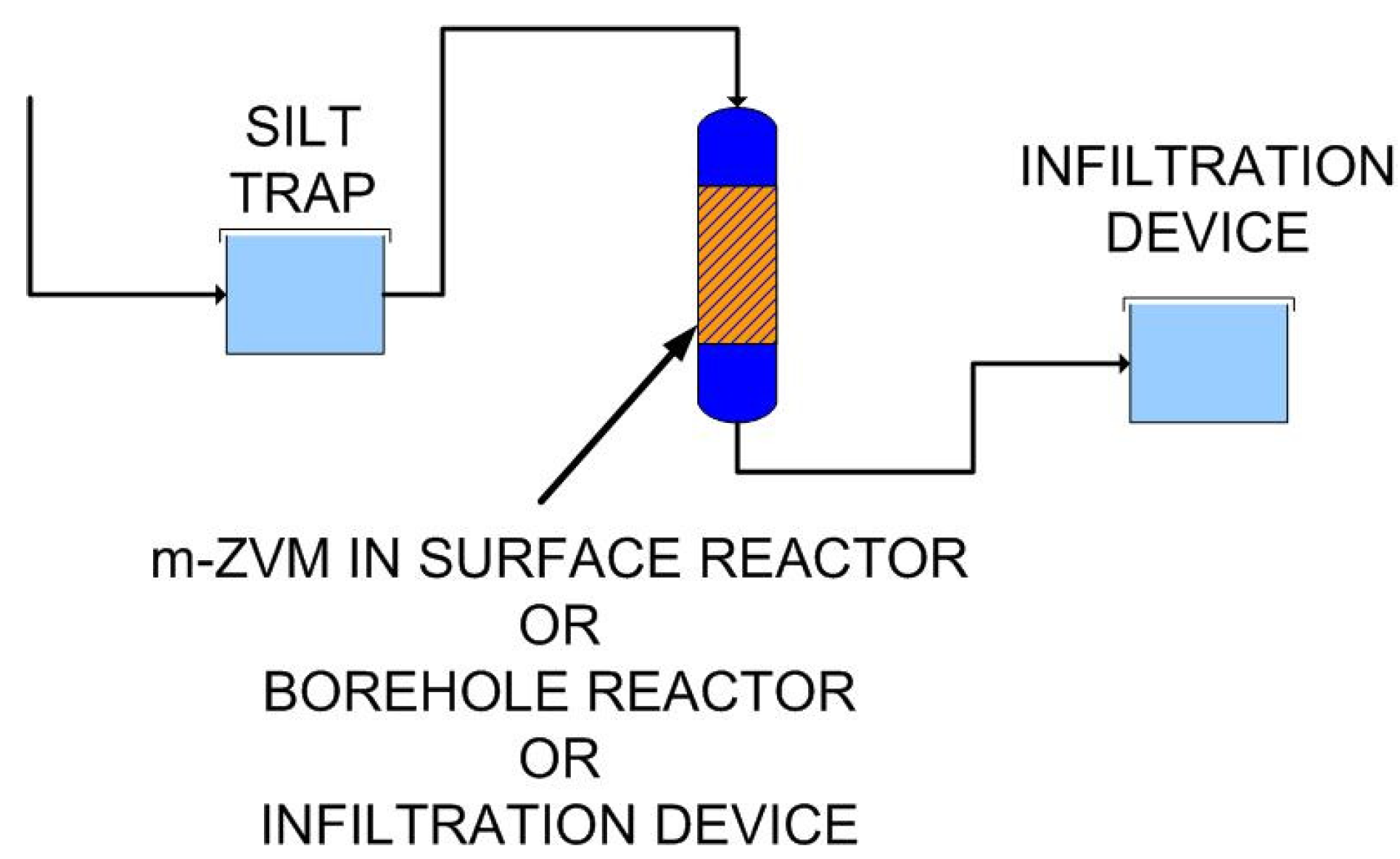
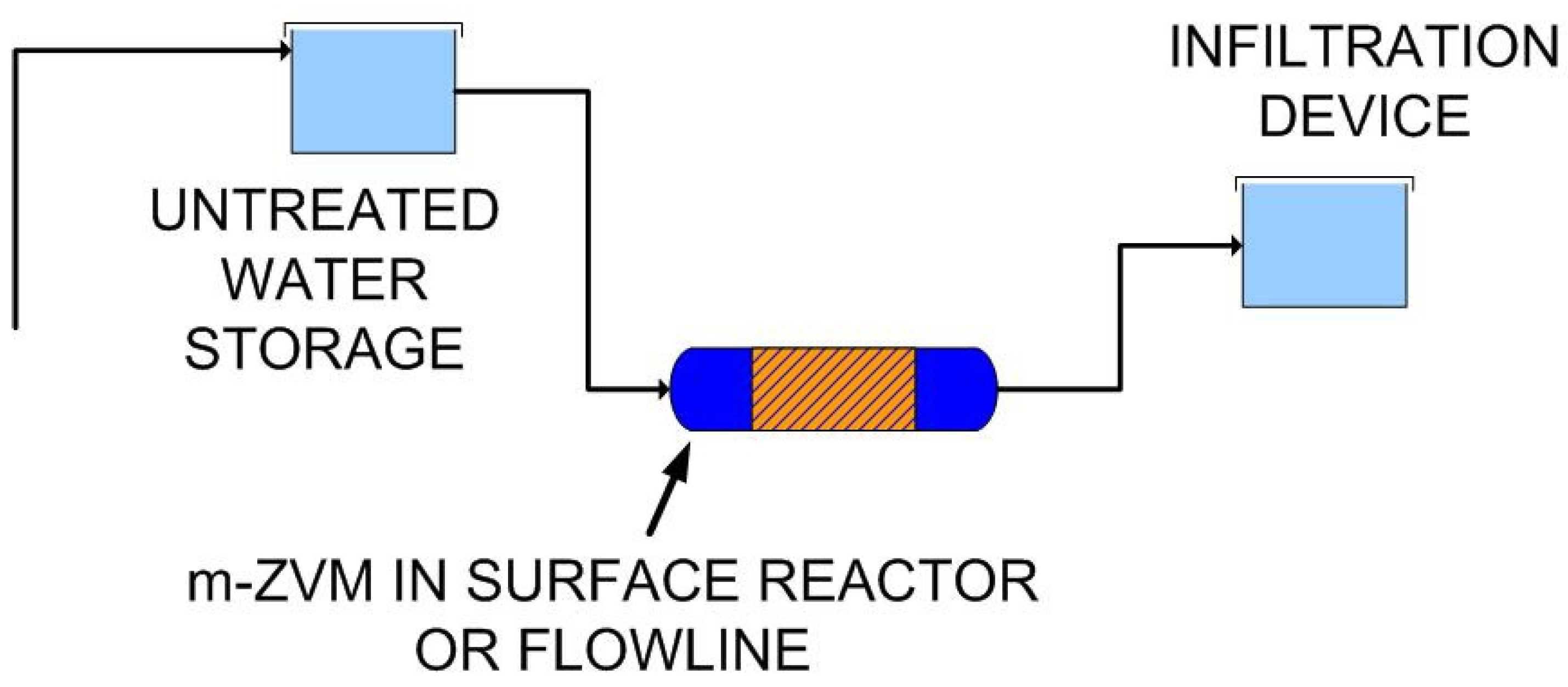

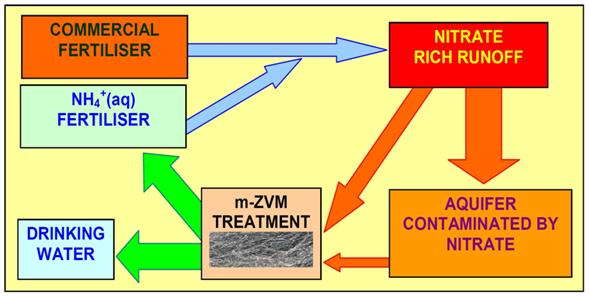
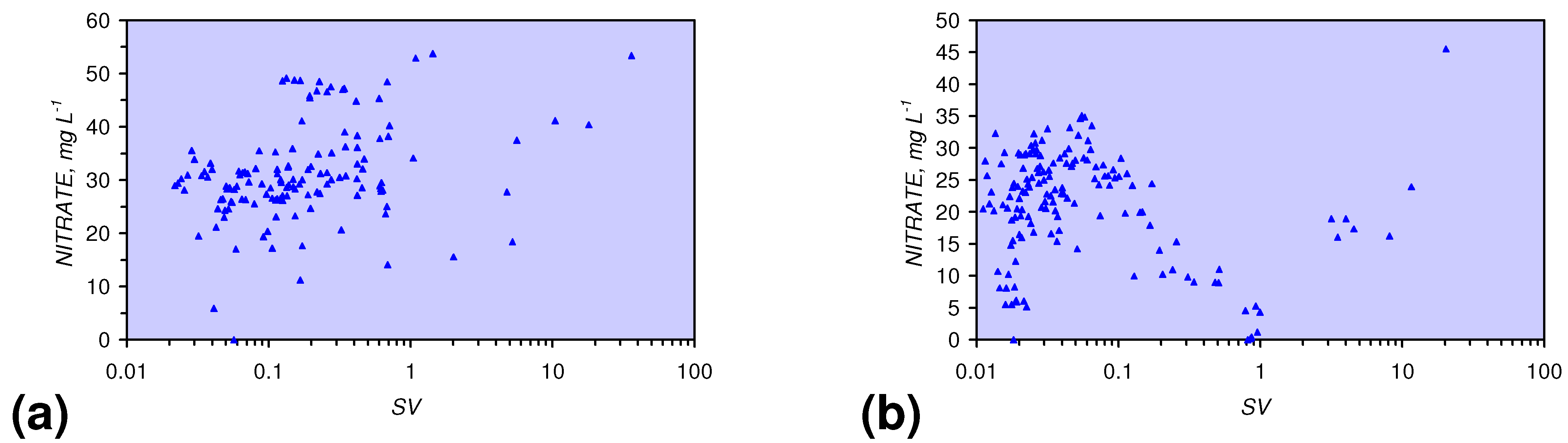
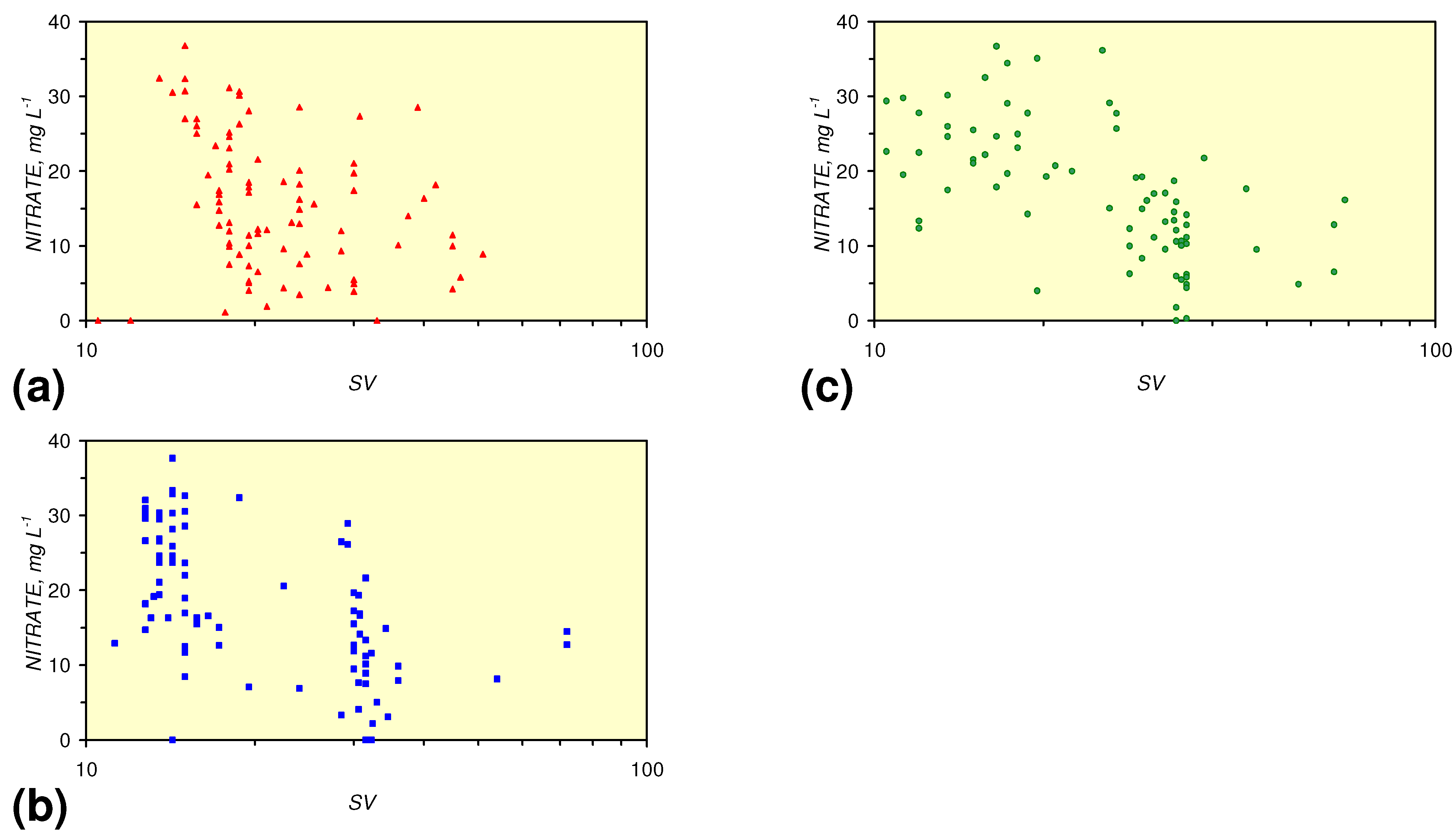
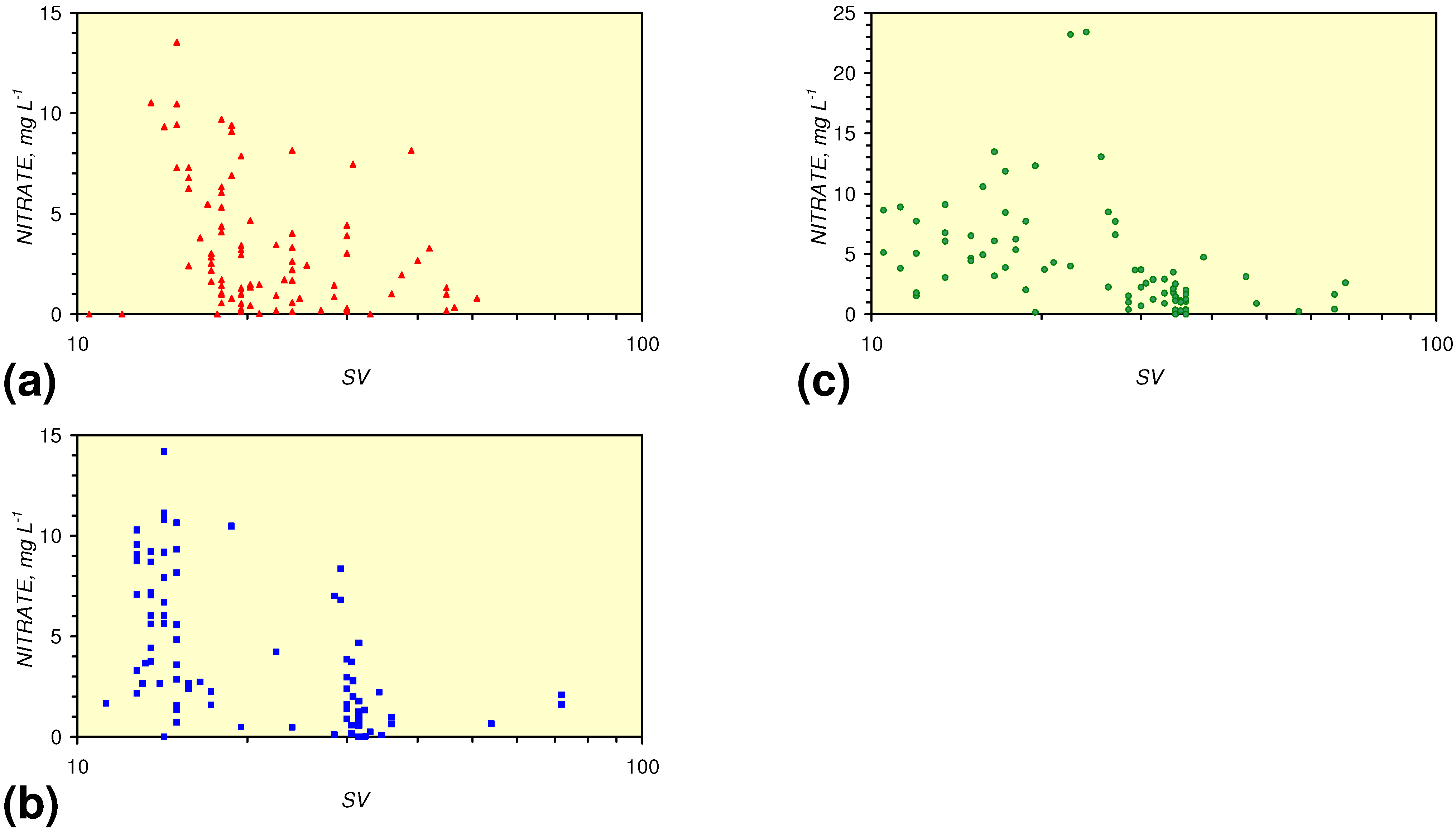
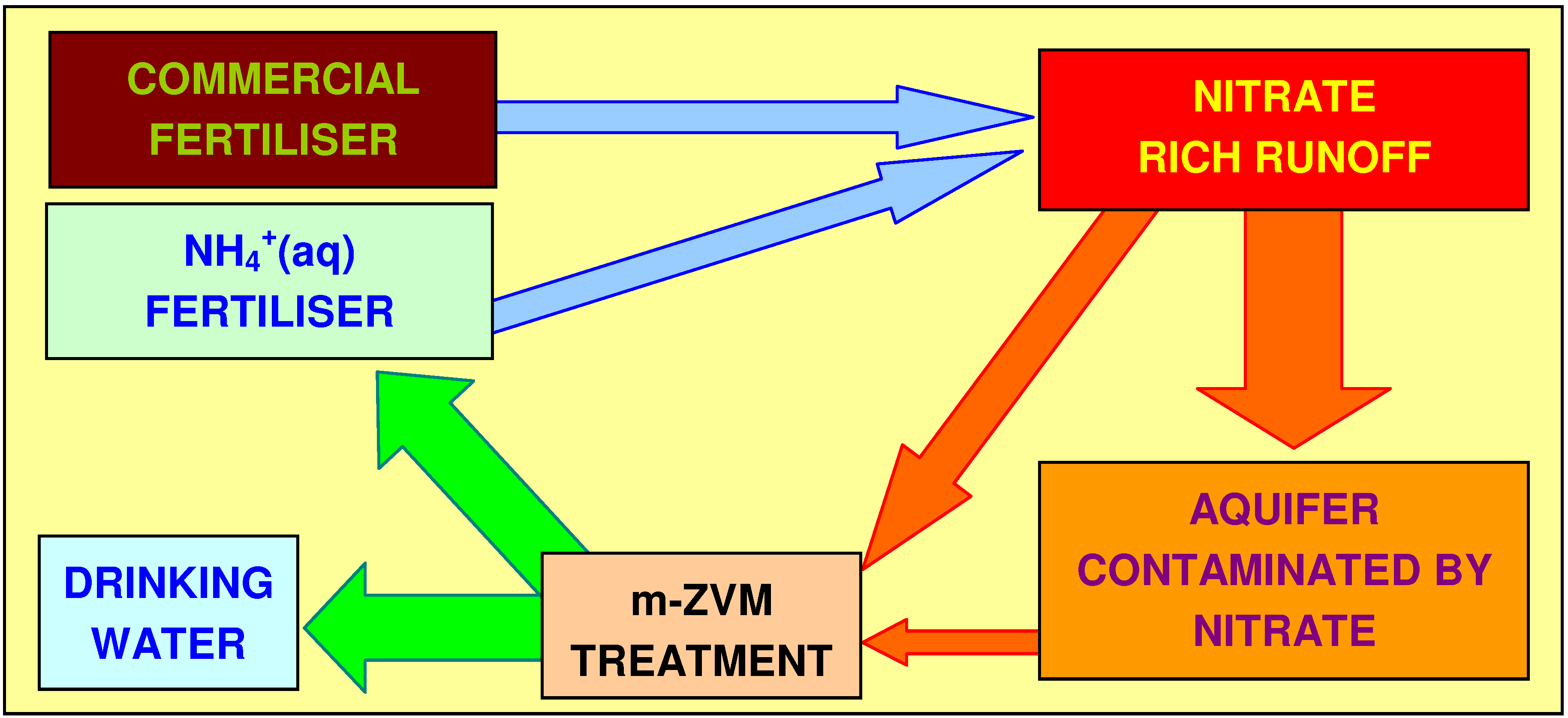
4.3.2. Nitrate Remediation
4.3.2.1. PRB Nitrate Remediation

4.3.2.2. Flowing Water: Nitrate Remediation




4.3.2.3. Regulatory Significance
4.3.2.4. Agricultural Significance

4.3.2.5. Sustainable Nitrate Cycle

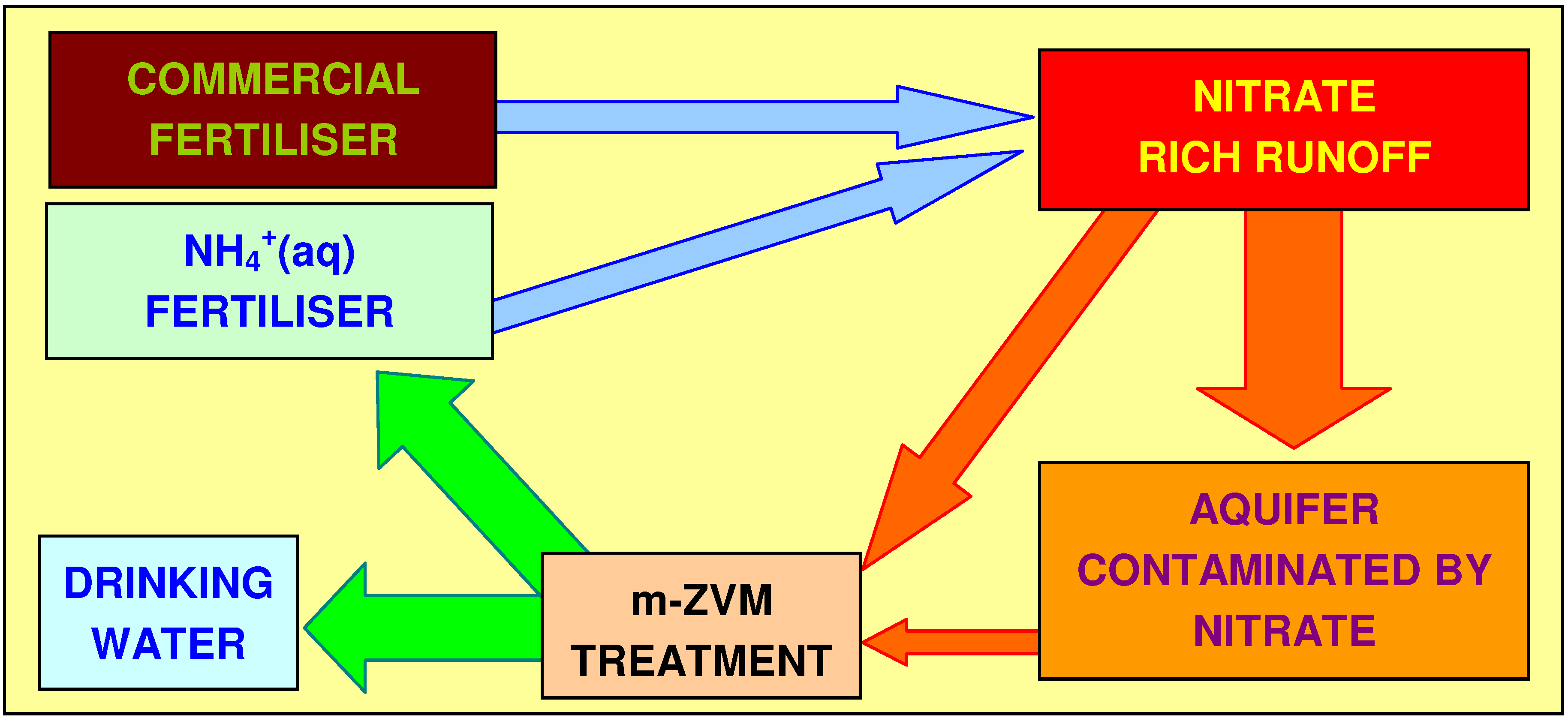
4.3.3. Water Bourne Bacteria and Virus Removal
4.3.4. Metal Remediation
4.3.5. Organic Chemical Remediation
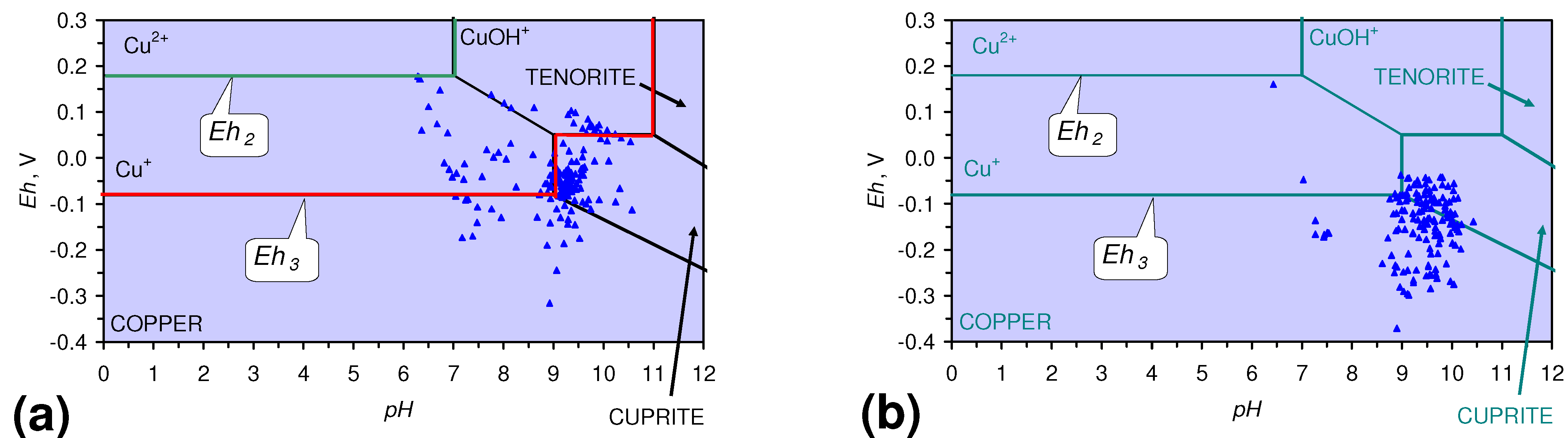

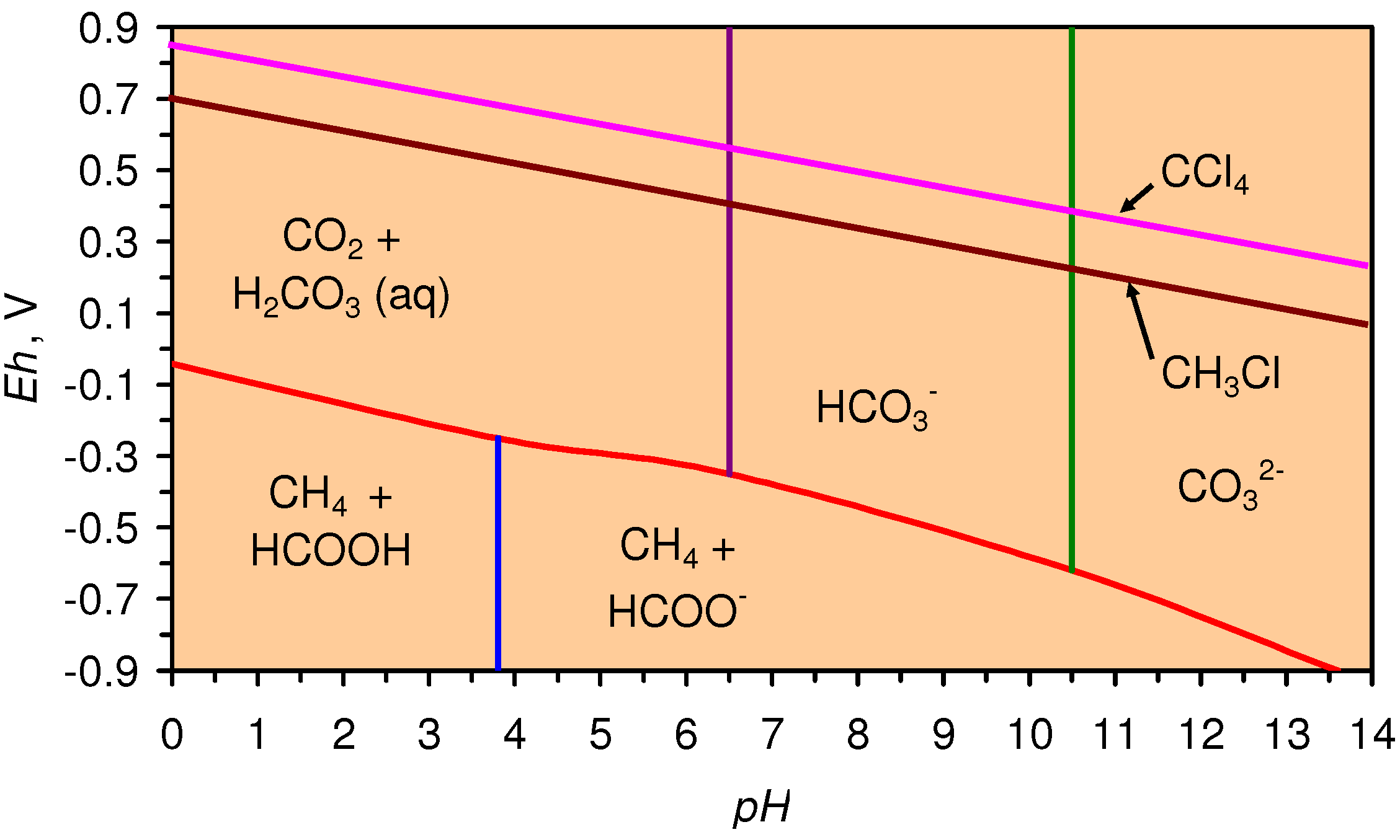
4.3.6. Aquifer, GWM, Plume Nitrate Remediation
4.4. Other Potential Treatment Applications of m-ZVM
- The rapid provision of water treatment in the aftermath of natural/anthropogenic disasters.
- In Situ partial aquifer desalination
- Catalytic use of ZVM to both breakdown organic compounds and form new organic compounds.
4.4.1. The rapid provision of drinking water in the aftermath of natural/anthropogenic disasters
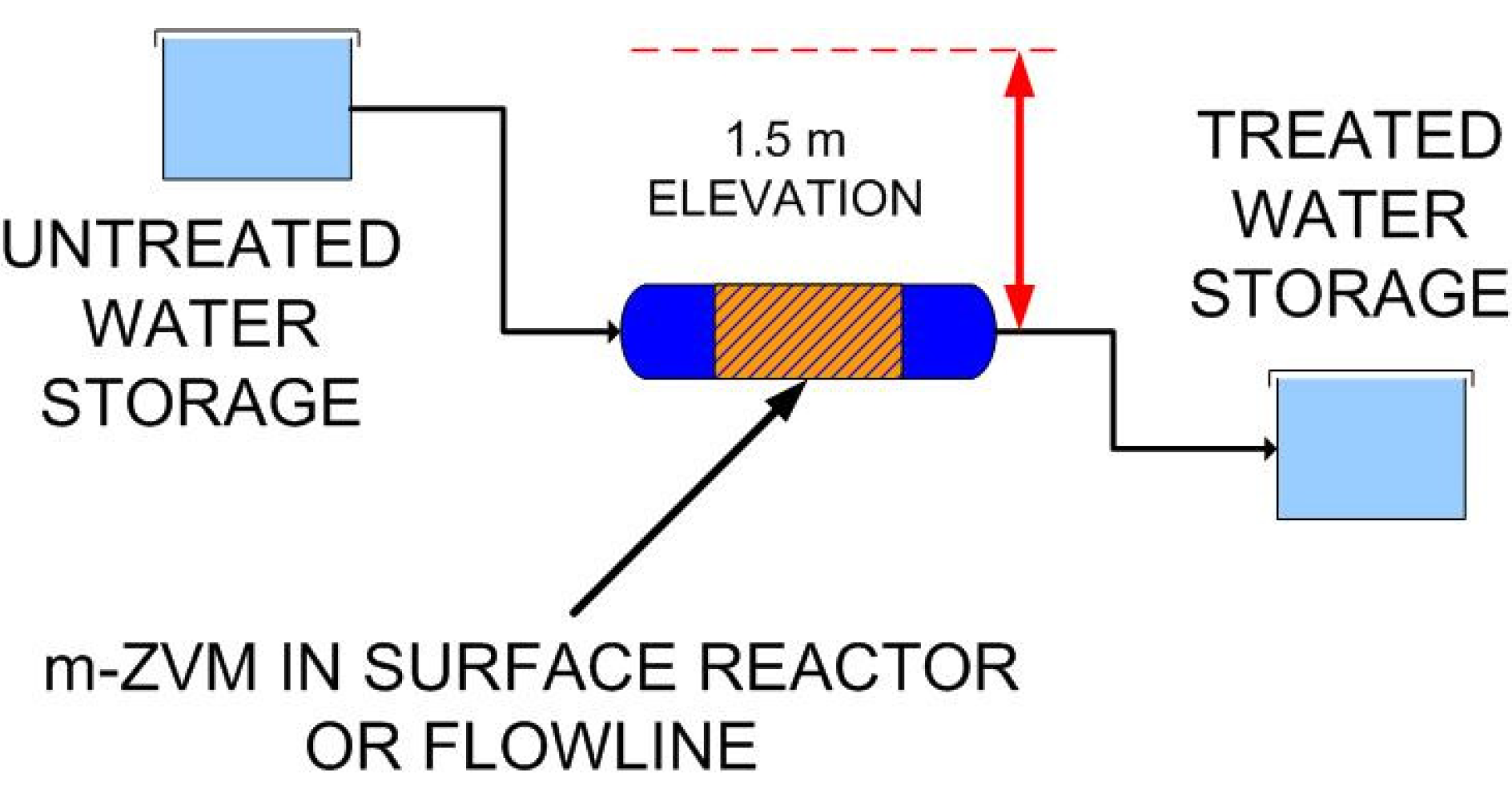
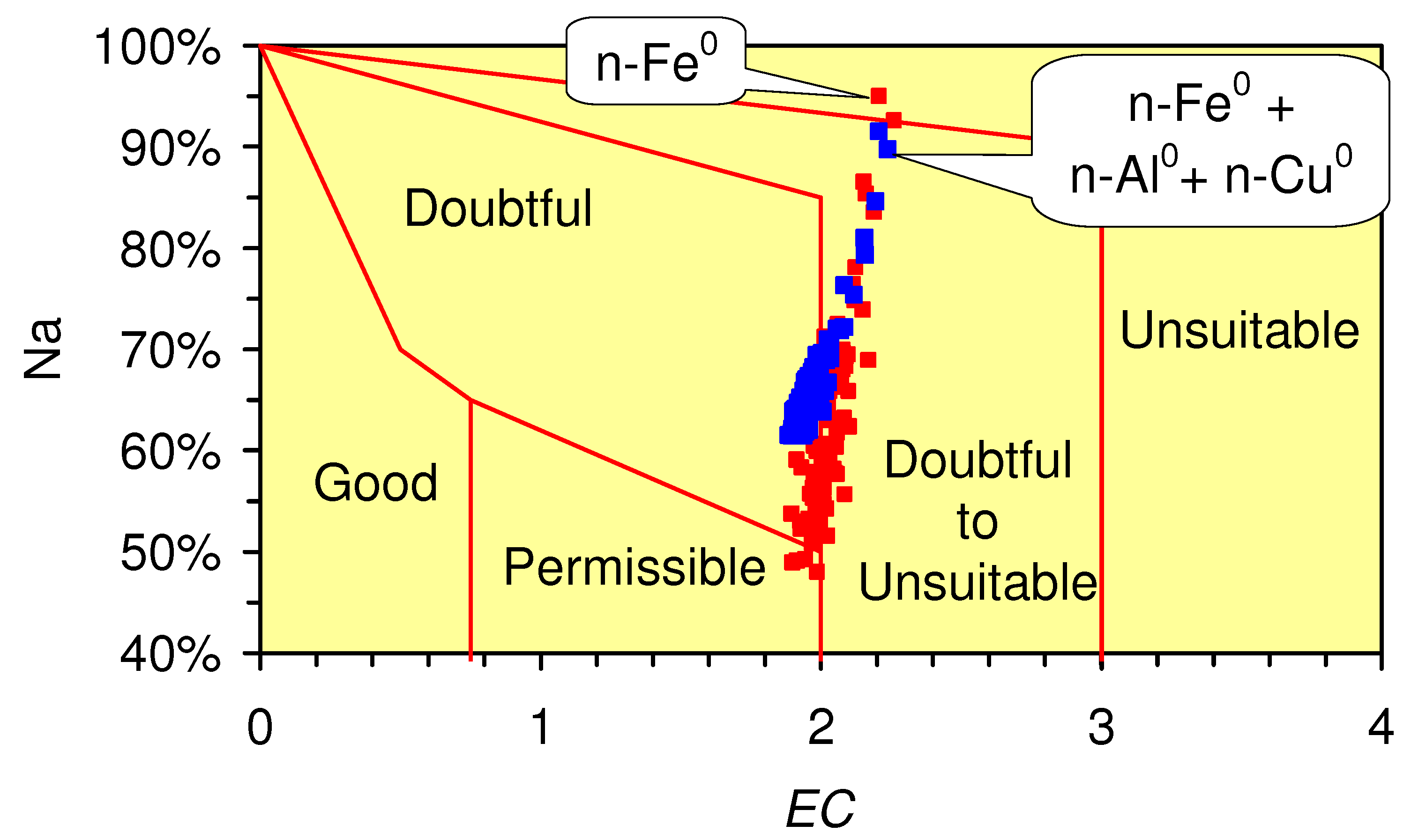
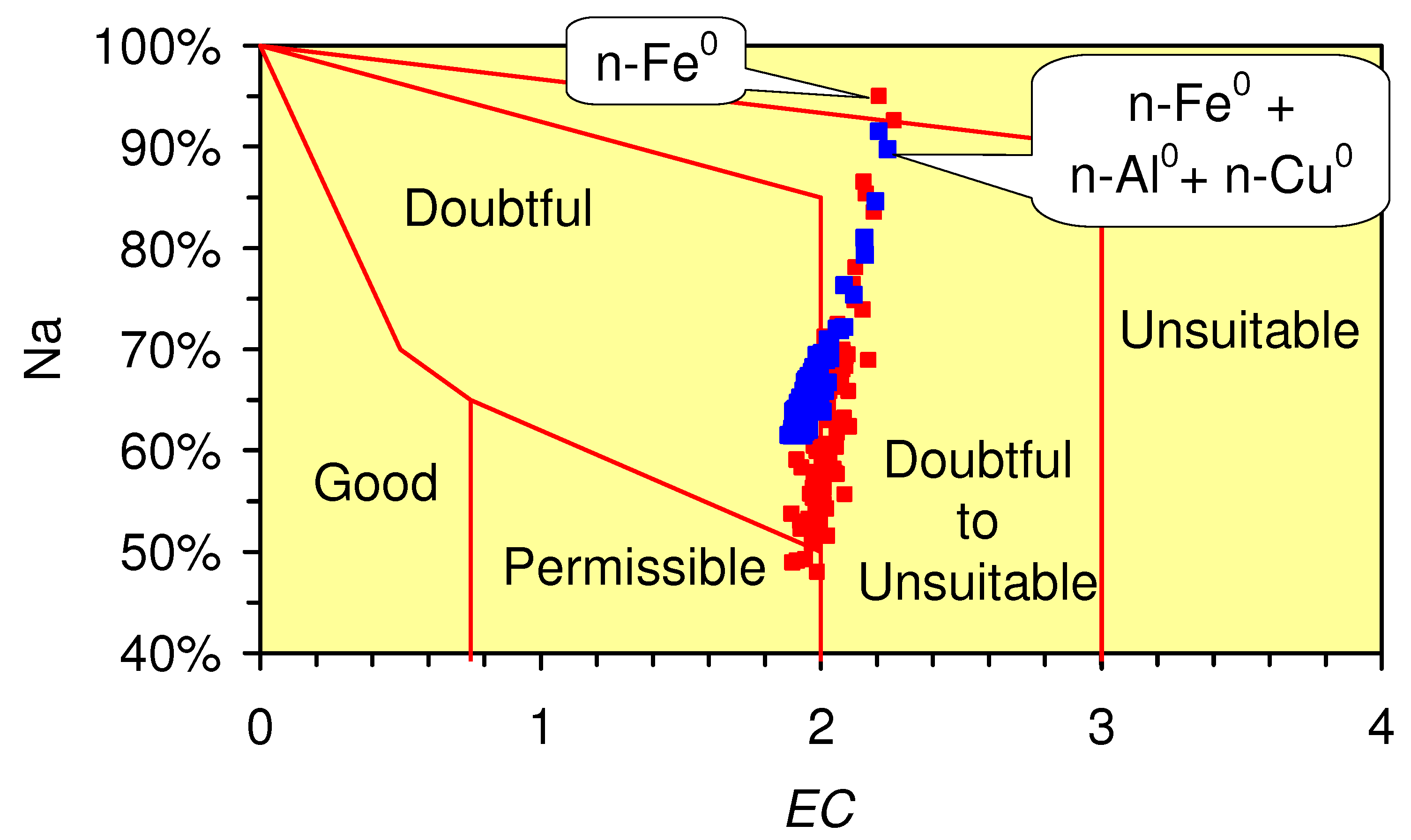
4.4.2. In Situ Aquifer Desalination

4.4.3. Catalytic use of ZVM to both breakdown organic compounds and form new organic compounds
4.4.3.1. General Mechanism
Fe0 + H3O+ = Fe-H+ + H2O – catalytic nuclei formation
Fe-H+ + CO2 + 6H3O+ = Fe-CH3 + 8H2O – chain formation
Fe-CH3+ CO2 + 6H3O+ = Fe-CH2CH3 + 8H2O – chain growth
Fe-CH3+ CO2 + 6H3O+ = Fe-(CH2)2CH3 + 8H2O – chain growth
Fe-(CH2)2CH3 + 2H3O+ = Fe-H+ + 2H2O + CH3CH2CH3– chain termination
ZVM-CCl3 + CH4 = ZVM-CCl2CH3 + Cl− + H+− chain growth
ZVM-CHCl2 + CH4 = ZVM-CH2CH3 + 2Cl−− chain growth
ZVM-CH2CH3 + CH4 = ZVM-CH2CH2CH3 + 2H+− chain growth
ZVM-CHCl2 + 4H+ = ZVM-H+ + CH4 + 2Cl−− chain termination
ZVM-CH2CH2CH3 + 2H+ = ZVM-H+ + CH3CH2CH3− chain termination
ZVM-CH2CH2CH3 + H+ = ZVM+ + CH3CH2CH3− chain termination
ZVM-CH2CH3 + ZVM-CH2CH3 + 2H+ = 2ZVM-H+ + CH3 CH2CH2CH3− chain termination
ZVM-CH2CH3 + ZVM-CH2CH3 = 2ZVM+ + CH3 CH2CH2CH3− chain termination
ZVM-H+ + CO2 = ZVM-COOH+
ZVM-H+ + CO2 + xH+ = ZVM-CHn+ + 2H2O
ZVM-H+ + CH4 = ZVM-CH3 + 2H+
ZVM-H+ + CO2 + 6H+ = ZVM-CH3 + 2H2O
ZVM-CH2CH2CH3 + 2H+ = ZVM-CH2CH3 + CH4 − chain termination
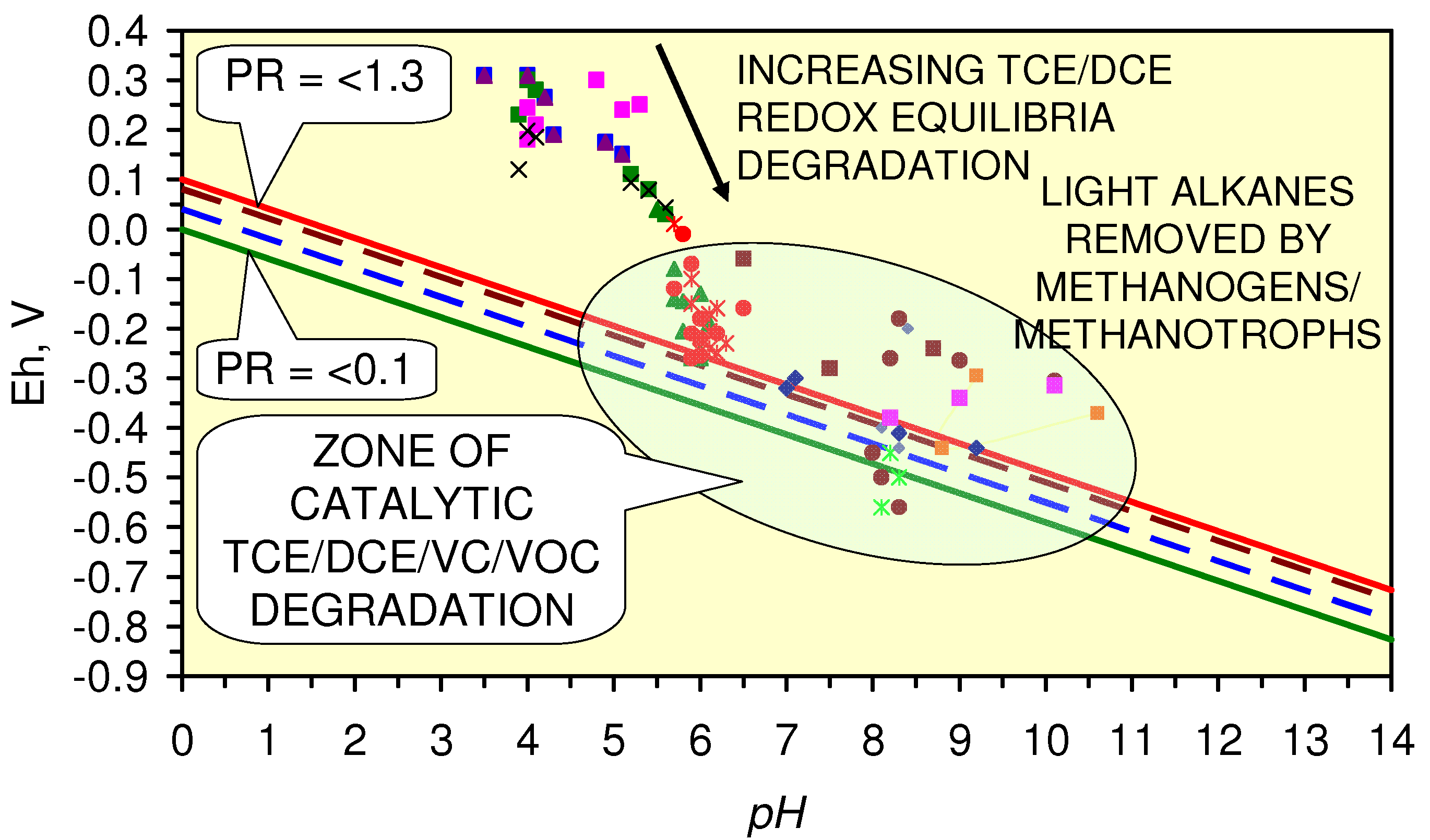
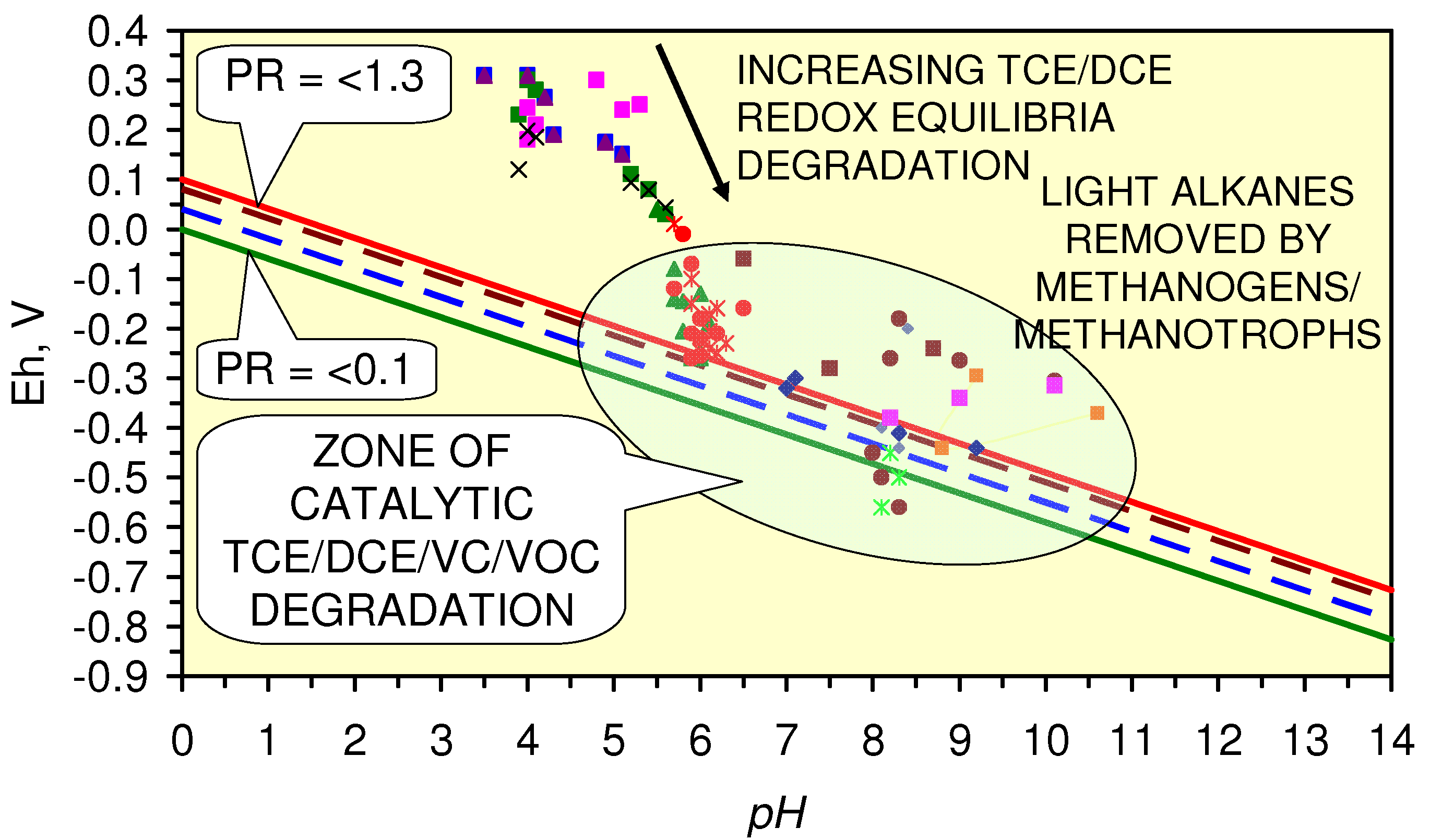
4.4.3.2. TCE, DCE, VC Remediation
4.4.3.3. Optimization of TCE, DCE, VC Remediation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
4.4.3.4. Cathodic Hydrogen Release during TCE, DCE, VC Remediation
H+ ion or H2(g) formation, Eh and pH increase or decrease
4.4.3.5. Relative Reaction Rate for TCE, DCE, VC Remediation
5. Conclusions
Acknowledgements
References
- Antia, D.D.J. Formation and control of self-sealing high permeability groundwater mounds implications for suds and sustainable pressure mound management. Sustainability 2009, 1, 855–923. [Google Scholar] [CrossRef]
- Antia, D.D.J. Interacting Infiltration Devices (Field Analysis, Experimental Observation and Numerical Modeling): Prediction of seepage (overland flow) locations, mechanisms and volumes—Implications for SUDS, groundwater raising projects and carbon sequestration projects. In Hydraulic Engineering: Structural Applications, Numerical Modeling and Environmental Impacts, 1st ed.; Hirsch, G., Kappel, B., Eds.; Nova Science Publishers: Hauppauge, NY, USA, 2010; pp. 85–156. [Google Scholar]
- Merly, C.; Lerner, D.N. Remediation of Chlorinated Organic Contaminants in Fractured Aquifers Using Zero-Valent Metal: Report on Laboratory Trials; R&D Technical Report P2-182/TR; Environment Agency: Bristol, UK, 2002; p. 105. [Google Scholar]
- Dolfing, J.; Van Eekert, M.; Seech, A.; Vogan, J.; Mueller, J. In Situ chemical reduction (ISCR) technologies: Significance of low Eh reactions. Soil Sed. Contam. 2008, 17, 63–73. [Google Scholar] [CrossRef]
- Fiore, S.; Zanetti, M.C. Preliminary tests concerning zero-valent iron efficiency in organic pollutants removal. Am. J. Environ. Sci. 2009, 5, 555–560. [Google Scholar] [CrossRef]
- Gavaskar, A.; Tatar, L.; Condit, W. Cost and Performance Report: Nanoscale Zero-Valent Iron Technologies for Source Remediation; Contract Report CR-05-007-ENV; NAVFAC Naval Facilities Engineering Command, US Navy Engineering Services Center: Port Hueneme, CA, USA, 2005; p. 44. [Google Scholar]
- Gavaskar, A.; Bhargava, M.; Condit, W. Cost and Performance Report for a Zero-valent Iron (ZVI) Treatability Study at Naval Air Station North Island; Technical Report TR-2307-ENV; Naval Facilities Engineering Command (NAVFAC), US Navy Engineering Services Center: Port Hueneme, CA, USA, 2008. [Google Scholar]
- Pula, R.W.; Paul, C.J.; Powell, R.M. The application of In Situ permeable reactive (zero-valent iron) barrier technology for the remediation of chromate-contaminated groundwater: A field test. App. Geochem. 1999, 14, 989–1000. [Google Scholar] [CrossRef]
- Ludwig, R.D.; Smyth, D.J.; Blowes, D.W.; Spink, L.E.; Wilkin, R.T.; Jewett, D.G.; Weisener, C.J. Treatment of arsenic, heavy metals, and acidity using a mixed ZVI-compost PRB. Environ. Sci. Technol. 2009, 43, 1970–1976. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.H.; Van Noolen, T.; Basfianens, L.; Russell, M.I.; Dickson, K.; Plant, S.; Ahad, J.M.; Newton, T.; Elliot, T.; Kalin, R.M. Ten year performance evaluation of a field-scale zero-valent iron permeable reactive barrier installed to remediate trichloroethene contaminated groundwater. Environ. Sci. Technol. 2010, 44, 3061–3069. [Google Scholar]
- Komnitsas, K.; Bartzas, G.; Fytas, K.; Paspaliaris, L. Long-term efficiency and kinetic evaluation of ZVI barriers during clean-up of copper containing solutions. Min. Engrg. 2007, 20, 1200–1209. [Google Scholar]
- Warrender, R. Remediation of Circum-Neutral Metal Mine Drainage Using Laboratory Scale Permeable reactive Barriers; Ph.D. Thesis, University of Aberystwyth, Aberystwyth, UK, 2009.
- Fatemi, F.; Liu, L.; Mahabadi, O.K.; Satish, M. Investigating the effect of implementing heating rods within a ZVI-PRB to enhance performance, improve design and reduce costs. Water Air Soil Poll. 2008, 190, 231–243. [Google Scholar] [CrossRef]
- Higgins, M.R.; Olson, T.M. Life-cycle case study comparison of permeable reactive barrier versus pump-and-treat remediation. Environ. Sci. Technol. 2009, 43, 9432–9438. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.; O’Brien, J.; Nurmi, T. Mineral precipitation up gradient from a zero-valent iron permeable reactive barrier. Ground Water Mon. Remid. 2008, 28, 56–64. [Google Scholar] [CrossRef]
- McElroy, B.; Keith, A.; Glasgow, J.; Dasappa, S. The use of zero-valent iron injection to remediate groundwater: Results of a pilot test at the Marshall Space Flight Center. Remediation J. 2003, 13, 145–153. [Google Scholar] [CrossRef]
- Joo, S.H.; Cheng, F. Nanotechnology for Environmental Remediation; Springer: Hiedelberg, Germany, 2006; p. 166. [Google Scholar]
- Vaughan, P.R. Observations on the behaviour of clay fill containing occluded air bubbles. Geotechnique 2003, 53, 265–272. [Google Scholar] [CrossRef]
- Smoltczyk, V. Geotechnical Engineering Handbook: Fundamentals; Wiley: Chichester, UK, 2002; Volume 1. [Google Scholar]
- Smith, P.G.C. Numerical Analysis of Infiltration into Partially Saturated Soil Slopes ; Ph.D. Thesis, University of London, London, UK, 2003.
- Valdes, J.R.; Santamarina, J.C. Particle clogging in radial flow: Microscale mechanisms. SPE J. 2006, 11, 193–198. [Google Scholar] [CrossRef]
- Wiekowski, A. Interfacial Electrochemistry: Theory, Experiment and Applications; CRC: London, UK, 1999; p. 992. [Google Scholar]
- Greenwood, N.N.; Earnshaw, A. Chemistry of the Elements; Pergamon: Oxford, UK, 1990. [Google Scholar]
- Kim, E.H.; Chung, D.Y.; Park, J.H.; Yoo, J.H. Dissolution of oxylate precipitate and destruction of oxalate ion by hydrogen peroxide in nitric acid solution. J. Nuc. Sci. Technol. 2000, 37, 601–607. [Google Scholar] [CrossRef]
- Astrus, D. Organometallic Chemistry and Catalysis; Springer: London, UK, 2007; p. 608. [Google Scholar]
- Huang, K.H. Catalysis mechanism and kinetic equation of ammonia synthesis on doubly-promoted iron catalysts. Sci. China Ser. A 1981, 24, 800–813. [Google Scholar]
- McKee, M.L. Ab Initio study of the interaction of Fe, Fe+, and HFe with H, CH, CH2, CH3, and C5H5. J. Am. Chem. Soc. 1990, 112, 2601–2607. [Google Scholar] [CrossRef]
- Yamakawa, K.; Nishimuara, R. Hydrogen permeation of carbon steel in weak alkaline solution containing hydrogen sulphide and cyanide ion. Corrosion 1999, 56, 24–30. [Google Scholar] [CrossRef]
- Antia, D.D.J. Oil polymerisation and fluid expulsion from low temperature, low maturity, over pressured sediments. J. Petrol. Geol. 2008, 31, 263–282. [Google Scholar] [CrossRef]
- Antia, D.D.J. Low temperature oil polymerisation and hydrocarbon expulsion from continental shelf and continental slope sediments. Indian J. Petrol. Geol. 2009, 16, 1–30. [Google Scholar]
- Antia, D.D.J. Polymerisation Theory—Formation of hydrocarbons in sedimentary strata (hydrates, clays, sandstones, carbonates, evaporites, volcanoclastics) from CH4 and CO2: Part I: Polymerisation concepts, kinetics, sources of hydrogen, and redox environment. Indian J. Petrol. Geol. 2009, 17, 49–86. [Google Scholar] Part II: Formation and Interpretation of Stage 1 to Stage 5 Oils. Indian J. Petrol. Geol. 2009, 17, 11–70. Part III: Hydrocarbon expulsion from the hydrodynamic flow regimes contained within a generating pressure mound. Indian J. Petrol. Geol. 2010, 18. in press. Part IV: Polymerisation modelling of sequestered carbon dioxide and waste organic liquids to hydrocarbons. Indian J. Petrol. Geol. 2010, 18. in press.
- Antia, D.D.J. Polymerisation Theory (the catalytic formation of oil from CO2 and CH4): Application of an Accelerated Geological Process to Remove Carbon Oxides from Flue Gases and use Carbon Oxide Sequestration to Produce Oil in Sedimentary Sequences. J. Appl. Geochem. 2010, 12. in press. [Google Scholar]
- Lindberg, R.D.; Runnells, D.D.D. Ground Water Redox Reactions: An analysis of the equilibrium state applied to Eh measurements and geochemical modeling. Science 1984, 225, 925–927. [Google Scholar] [CrossRef] [PubMed]
- Stefansson, A.; Arnorsson, S.; Sveinbjornsdorrir, A.E. Redox reactions and potentials in natural waters at disequilibrium. Chem. Geol. 2005, 221, 289–311. [Google Scholar] [CrossRef]
- Grenthe, I.; Stumm, W.; Laaksuharju, M.; Nilsson, A.C.; Wikberg, P. Redox potentials and redox reactions in deep groundwater systems. Chem. Geol. 1992, 98, 131–150. [Google Scholar] [CrossRef]
- Washington, J.W.; Endale, D.M.; Samarkina, L.P.; Chappell, K.E. Kinetic control of oxidation state at thermodynamically buffered potentials in subsurface waters. Geochim. Cosmochim. Acta 2004, 68, 4831–4842. [Google Scholar] [CrossRef]
- Ebbing, D.D.; Gammon, S.D. General Chemistry; Houghton Mifflin: Boston, MA, USA, 1999. [Google Scholar]
- Bokare, A.D.; Choi, W. Zero-valent aluminium for oxidative degradation of aqueous organic pollutants. Environ. Sci. Technol. 2009, 43, 7130–7135. [Google Scholar] [CrossRef] [PubMed]
- Mulder, M. Basic Principles of Membrane Technology; Kluwer Academic Publishers: Dordrecht, Germany, 1996. [Google Scholar]
- Junyapoon, S. Use of zero-valent iron for waste water treatment. KMITL. Sci. Tech. J. 2005, 5, 587–595. [Google Scholar]
- Hao, Z.W.; Xu, X.H.; Wang, D.H. Reductive denitrification of nitrate by scrap iron filings. J. Zhejang Univ. Sci. B 2005, 6, 182–186. [Google Scholar] [CrossRef]
- Hao, Z.W.; Xu, X.H.; Jin, J.; He, P.; Liu, Y.; Wang, D.H. Simultaneous removal of nitrate and heavy metals by iron metal. J. Zhejang Univ. Sci. B 2005, 6, 307–310. [Google Scholar] [CrossRef]
- Ottley, C.J.; Davison, W.; Edmunds, W.M. Chemical catalysis of nitrate reduction by iron (II). Geochim. Cosmochim. Acta 1997, 61, 1819–1828. [Google Scholar] [CrossRef]
- Young, G.K.; Bungay, H.R.; Brown, L.M.; Parsons, W.A. Chemical reduction of nitrate in water. J. Water Poll. Cont. Fed. 1964, 36, 395–398. [Google Scholar]
- Liou, Y.H.; Lo, S.L.; Lin, C.J.; Kuan, W.H.; Weng, S.C. Chemical reduction of unbuffered nitrate solution using catalysed and uncatalyzed nanoscale iron particles. J. Hazard. Mat. 2005, B127, 102–110. [Google Scholar] [CrossRef]
- Liou, Y.H.; Lo, S.L.; Lin, C.J.; Kuan, W.H.; Weng, S.C. Effects of iron surface pretreatment on kinetics of aqueous nitrate reduction. J. Hazard. Mat. 2005, B126, 189–194. [Google Scholar] [CrossRef]
- Liou, Y.H.; Lo, S.L.; Lin, C.J.; Kuan, W.H.; Weng, S.C. Methods for accelerating nitrate reduction using zero-valent iron at near neutral pH: Effects of H2 reducing pretreatment and copper deposition. Environ. Sci. Technol. 2005, 39, 9643–9648. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Batchelor, B. Nitrate reduction by fluoride green rust modified with copper. Chemosphere 2008, 70, 1106–1116. [Google Scholar] [CrossRef]
- Chen, Y.M.; Li, C.W.; Chen, S.S. Fluidised zero-valent iron bed reactor for nitrate removal. Chemosphere 2005, 59, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.P.; Wang, H.W.; Chiu, P.C. Nitrate reduction by metallic iron. Water Res. 1998, 32, 2257–2264. [Google Scholar] [CrossRef]
- Huang, Y.H.; Zhang, T.C.; Shea, P.J.; Comfort, S.D. Effects of oxide coatings on nitrate reduction by iron metal. J. Environ. Qual. 2003, 32, 1306–1315. [Google Scholar] [CrossRef] [PubMed]
- Buresh, R.J.; Moraghan, J.T. Chemical reduction of nitrate by ferrous iron. J. Environ. Qual. 1976, 5, 320–325. [Google Scholar] [CrossRef]
- Rakshit, S.; Matocha, C.J.; Haszler, G.R. Nitrate reduction in the presence of wustite. J. Environ. Qual. 2005, 34, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Westerhoff, P. Reduction of nitrate, bromate, and chlorate by zero-valent iron (Fe0). J. Envir. Engrg. 2003, 129, 10–16. [Google Scholar] [CrossRef]
- Huang, Y.H.; Zhang, T.C. Nitrite reduction and formation of corrosion coatings in zero-valent iron systems. Chemosphere 2006, 64, 937–943. [Google Scholar] [CrossRef] [PubMed]
- Rakshit, S.; Matocha, C.J.; Coyne, M.S. Nitrite reduction by siderite. Soil. Sci. Soc. Am. J. 2008, 72, 1070–1077. [Google Scholar] [CrossRef]
- Cohen, E.L.; Patterson, B.M.; McKinley, A.J.; Prommer, H. Zero-valent iron remediation of a mixed brominated ethene contaminated groundwater. J. Contam. Hydrol. 2009, 103, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Wer, Y.T.; Wu, S.C.; Chou, C.M.; Che, C.H.; Tsai, S.M.; Lien, H.L. Influence of nanoscale zero-valent iron on geochemical properties of groundwater and vinyl chloride degradation: A field case study. Water Res. 2010, 44, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Nurmi, J.T.; Tratnyek, P.G.; Johnson, R.L.; O’Brien, R. Reduction of TNT and RDX by core material from an iron permeable reactive barrier. In Remediation of Chlorinated and Recalcitrant Compounds; Sass, B.M., Ed.; Battelle: Columbus, OH, USA, 2008. [Google Scholar]
- Lee, J.Y.; Lee, K.L.; Youm, S.Y.; Lee, M.R.; Kamala-Kannan, S.; Oh, B.T. Stability of multi-permeable reactive barriers for long term removal of mixed contaminants. Bull. Environ. Cont. Tox. 2010, 84, 250–254. [Google Scholar] [CrossRef]
- Chang, M.C.; Kang, H.Y. Remediation of pyrene-contaminated soil by synthesized nanoscale zero-valent iron particles. J. Environ. Sci. Health 2009, 44, 576–582. [Google Scholar] [CrossRef]
- Katsenovch, Y.P.; Miralles-Wilhelm, F.R. Evaluation of nanoscale zerovalent iron particles for trichloroethene degradation in clayey soils. Sci. Total Environ. 2009, 407, 4986–4993. [Google Scholar] [CrossRef] [PubMed]
- Lo, I.M.C.; Lai, K.C.K. Zero-Valent Iron Reactive Materials for Hazardous Waste and Inorganics Removal; American Society of Civil Engineers: Reston, VA, USA, 2007; p. 340. [Google Scholar]
- Li, W.G.; Zhang, W.X. Sequestration of metal cations with zerovalent iron nanoparticles—A study with high resolution X-ray photoelectron spectroscopy (HR-XPS). J. Phys. Chem. C 2007, 111, 6939–6946. [Google Scholar] [CrossRef]
- Sun, H.; Wang, L.; Zhang, R.; Sui, J.; Xu, G. Treatment of groundwater polluted by arsenic compounds by zero-valent iron. J. Hazard. Mat. 2006, 129, 297–303. [Google Scholar] [CrossRef]
- Alshaebi, F.Y.; Yaacob, W.Z.W.; Samsuldin, A.R. Sorption on zero-valent iron (ZVI) for arsenic removal. Eur. J. Sci. Res. 2009, 33, 214–219. [Google Scholar]
- Zhang, Y.; Wang, J.; Amrhein, C.; Frankenberger, W.T., Jr. Removal of selanate from water by Zerovalent iron. J. Environ. Qual. 2005, 334, 487–495. [Google Scholar] [CrossRef]
- Fiedor, J.N.; Bostick, W.D.; Jarabek, R.J.; Farrell, J. Understanding the mechanism of uranium removal from groundwater by zero-valent iron using X-ray photoelectron spectroscopy. Environ. Sci. Technol. 1998, 32, 1466–1473. [Google Scholar] [CrossRef]
- Rangsivek, R.; Jekel, M.R. Removal of dissolved metals by zero-valent iron (ZVI): Kinetics, equilibria, processes and implications for storm runoff treatment. Water Res. 2005, 39, 4153–4163. [Google Scholar] [CrossRef] [PubMed]
- Stapanajaru, T.; Comfort, S.D.; Shea, P.J. Enhancing metolachlor destruction rates with aluminum and iron salts during zerovalent iron treatment. J. Environ. Qual. 2003, 32, 1726–1734. [Google Scholar] [CrossRef] [PubMed]
- Satapanajaru, T.; Anurakpongsatorn, P.; Pengthamkeerati, P.; Boparai, H. Remediation of atazine-contaminated soil and water by nano zerovalent iron. Water Air Soil Pol. 2008, 192, 349–359. [Google Scholar] [CrossRef]
- Misstear, B.; Banks, D.; Clark, L. Water Wells and Boreholes; Wiley: Chichester, UK, 2006. [Google Scholar]
- Geological Survey Japan. Atlas of Eh-pH Diagrams; Open File Report No. 419; National Institute of Advanced Industrial Science and Technology: Naoto, Takeno, Japan, 2005. [Google Scholar]
- Stephanov, M.F.; Novakovskaya, Y.V. Rate constants of atomic hydrogen formation in H3O+(H2O)n + e = H + (H2O)n gas-phase. Russian J. Phys. Chem. A. Focus Chem. 2009, 83, 1502–1510. [Google Scholar] [CrossRef]
- Yeh, L.I.; Okumura, M.; Myers, J.D.; Price, J.M.; Lee, Y.T. Vibrational spectroscopy of the hydrated hydronium cluster ions H3O+ · (H2O)n (n = 1,2,3). J. Chem. Phys. 1989, 91, 7319–7330. [Google Scholar] [CrossRef]
- Conway, B.E.; Tessier, D.F. Comparison of the kinetics of cathodic H2 evolution from the unhydrated H3O+ ion and its hydrated form H9O4+: Frequency factor effects and modes of activation. J. Chem. Kin. 2004, 13, 925–944. [Google Scholar] [CrossRef]
- Mabourki, R.; Ibrahim, Y.; Xie, E.; Meof-Ner (Mautner), M.; El-Shali, M.S. Clusters of the hydronium ion (H3O+) with H2, N2 and CO molecules. Chem. Phys. Let. 2006, 424, 257–263. [Google Scholar] [CrossRef]
- Zolla, V.; Sethi, R.; Molfetta, A.D. Performance assessment and monitoring of a permeable reactive barrier for the remediation of a contaminated site. Am. J. Environ. Sci. 2007, 3, 158–167. [Google Scholar] [CrossRef]
- Kamer, G.; Osta, C.; Mitsui, S.; Shibata, M.; Shinozaki, T. Fe(II)-Na ion exchange at interlayers of smectite: Adsorption-desorption experiments and a natural analogue. Engrg. Geol. 1999, 54, 15–20. [Google Scholar] [CrossRef]
- Drame, H. Cation exchange and pillaring of smectites by aqueous Fe nitrate solutions. Clays Clay Min. 2005, 53, 335–347. [Google Scholar] [CrossRef]
- Frini, N.; Crespin, M.; Trabelsi, M.; Messad, D.; Van Damme, H.; Bergaya, F. Preliminary results on the properties of pillared clays by mixed Al-Cu solutions. Appl. Clay Sci. 1997, 12, 281–292. [Google Scholar] [CrossRef]
- Fujita, S.I.; Bhanage, B.M.; Aoki, D.; Ochial, Y.; Iwasa, N.; Arai, M. Mesoporous smectites incorporated with alkali metal cations as solid base catalysts. Appl. Catal. A: Gen. 2006, 313, 151–159. [Google Scholar] [CrossRef]
- Kloprogge, J.T. Synthesis of smectites and porous pillared clay catalysts: A review. J. Por. Mat. 1998, 5, 5–41. [Google Scholar] [CrossRef]
- Martin-Luengo, M.A.; Martins-Carvalho, H.; Ladriere, J.; Grange, P. Fe(III)-pillared montmorillonites: Preparation and characterisation. Clay Min. 1989, 24, 495–504. [Google Scholar] [CrossRef]
- Rightor, E.G.; Tzou, M.S.; Pinnavaia, T.J. Iron oxide pillared clay with large gallery height: Synthesis and properties as a Fischer-Tropsch catalyst. J. Catal. 1991, 130, 29–40. [Google Scholar] [CrossRef]
- Zhu, H.Y.; Yamanaka, S. Molecular recognition of Na-loaded alumina pillared clay. J. Chem. Soc. Faraday Trans. 1997, 93, 477–480. [Google Scholar] [CrossRef]
- Chatterjee, A. Application of localized reactivity index in combination with periodic DFT calculation to rationalise the swelling mechanism of clay type in-organic material. J. Chem. Sci. 2005, 117, 533–539. [Google Scholar] [CrossRef]
- Soakaway Design. Digest 365; Building Research Establishment: Watford, UK, 1991.
- Flood Studies Report; Institute of Hydrology: Wallingford, UK, 1993; Volume 6.
- Flood Estimation Handbook; Institute of Hydrology: Wallingford, UK, 1999; Volume 5.
- Martin, P.; Turner, B.; Waddington, K.; Pratt, C.; Campbell, N.; Payne, J.; Reed, B. Sustainable Urban Drainage Systems: Design Manual for Scotland and Northern Ireland; CIRIA Report No. C521; CIRIA: London, UK, 2000. [Google Scholar]
- Wilson, S.; Bray, R.; Cooper, P. Sustainable Drainage Systems: Hydraulic, Structural and Water Quality Advice; CIRIA Report No. C609; CIRIA: London, UK, 2004. [Google Scholar]
- Woods Ballard, B.; Kellagher, R.; Martin, P.; Jefferies, C.; Bray, R.; Shaffer, P. The SUDS Manual; CIRIA Report No. C697; CIRIA: London, UK, 2007. [Google Scholar]
- Design Manual for Roads and Bridges; Design of Soakaways, Highways Agency, Stationary Office: London, UK, 2006; Volume 4.
- Youngs, E.G. The analysis of groundwater seepage in heterogenous aquifers. Hydrol. Sci. 1980, 25, 155–165. [Google Scholar]
- Antia, D.D.J. Prediction of overland flow and seepage zones associated with the interaction of multiple Infiltration Devices (Cascading Infiltration Devices). Hydrol. Proc. 2008, 22, 2595–2614. [Google Scholar] [CrossRef]
- Sarkar, R.; Dutta, S.; Panigrahy, S. Effect of scale on infiltration in a macropore-dominated hillside. Cur. Sci. 2008, 94, 490–494. [Google Scholar]
- Council Directive of 3 November 1998 on the quality of water intended for human consumption. Offic. J. Euro. Commun 1998, I.330, 32–54.
- Page, E.R. Aqueous ammonia as a nitrogen fertilizer for summer cauliflowers, compared with ammonium nitrate (broadcast) and urea (broadcast and injected). J. Agr. Sci. 1979, 92, 251–254. [Google Scholar] [CrossRef]
- Smith, C.J.; Chalk, P.M. Comparison of the efficiency of urea, aqueous ammonia and ammonium sulphate as nitrogen fertilizers. Plant Soil 2005, 55, 33–337. [Google Scholar]
- Zhang, L. Removal and Inactivation of Waterborne Viruses Using Zerovalent Iron; Ph.D. Thesis, University of Delaware, Newark, DE, USA, 2008.
- Lee, C.; Kim, J.Y.; Lee, W.L.; Nielson, K.L.; Yoon, J.; Sedlak, D.L. Bactericida effect of zerovalent iron nanoparticles on Escherichia coli. Environ. Sci. Technol. 2008, 42, 4927–4933. [Google Scholar] [CrossRef] [PubMed]
- Diao, M.; Tao, M. Use of zero-valent iron nanoparticles in inactivating microbes. Water Res. 2009, 43, 5243–5251. [Google Scholar] [CrossRef] [PubMed]
- Rada, T.; Plugge, C.M.; Abram, N.J.; Hirst, J. Reversible interconversion of carbon dioxide and formate by an electroactive enzyme. PNAS 2008, 105, 10654–10658. [Google Scholar] [CrossRef] [PubMed]
- Rozic, L.; Novakovic, T.; Petrovic, S. Process improvement approach to the acid activation of smectite using factorial and orthogonal central composite design methods. J. Serb. Chem. Soc. 2008, 73, 487–497. [Google Scholar] [CrossRef]
- Wilcox, L.V. Classification and Use of Irrigation Waters; US Department of Agriculture: Washington, DC, USA, 1955. [Google Scholar]
- Yesilnacar, M.I.; Uyanik, S. Worlds largest irrigation tunnel system, the Sanliurfa tunnels in Turkey. Fresenius Environ. Bul. 2005, 14, 300–306. [Google Scholar]
- Suresh, M.; Gurugnanam, B.; Vasudevan, S.; Rao, S.V.L.; Kumaravel, S. Groundwater quality assessment for irrigation uses in Upper Thirumanimuthar sub basin, Cauvery River, Tamil Nadu, India. J. Appl. Geochem. 2010, 12, 95–105. [Google Scholar]
- Hardy, L.I.; Gillham, R.W. Formation of hydrocarbons from the reduction of aqueous CO2 by zero-valent iron. Environ. Sci. Technol. 1995, 30, 57–65. [Google Scholar] [CrossRef]
- Deng, B.; Campbell, T.J.; Burris, D.R. Hydrocarbon formation in metallic iron/water systems. Environ. Sci. Technol. 1997, 31, 1185–1190. [Google Scholar] [CrossRef]
- Orth, W.S.; Gillham, R.W. Dechlorination of trichloroethene in aqueous solution using Fe0. Environ. Sci. Technol. 1995, 30, 66–71. [Google Scholar] [CrossRef]
- Kang, S.H.; Choi, W. Oxidative degradation of organic compounds using zero-valent iron in the presence of natural organic matter serving as an electron shuttle. Environ. Sci. Technol. 2009, 43, 878–883. [Google Scholar] [CrossRef] [PubMed]
- Xiu, Z.M.; Jin, Z.H.; Li, T.L.; Mahendra, S.; Lowry, G.V.; Alvarez, P.J.J. Effects of nano-scale zero-valent iron particles on a mixed culture dechlorinating trichloroethylene. Biores. Technol. 2010, 101, 1141–1146. [Google Scholar] [CrossRef]
- Weathers, L.J.; Parkin, G.F.; Alvarez, P.J. Utilisation of cathodic hydrogen as electron donor for chloroform cometabolism by a mixed, methanogenic culture. Environ. Sci. Technol. 1997, 31, 880–885. [Google Scholar] [CrossRef]
- Lien, H.L.; Zhang, W.X. Nanoscale Pd/Fe bimetallic particles: Catalytic effects of palladium on hydrodechlorination. Appl. Cat. B: Environ. 2007, 77, 110–116. [Google Scholar] [CrossRef]
- Yang, B.; Yu, G.; Huang, J. Electrocatalytic hydrodechlorination of 2,4,5-trichlorobiphenyl on a palladium-modified nickel foam cathode. Environ. Sci. Technol. 2007, 41, 7503–7508. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, M.L.B.; Johnson, R.L.; Alvarez, P.J.J. Microbial characterization of groundwater undergoing treatment with a permeable reactive iron barrier. Envron. Engrg. Sci. 2007, 24, 1121–1126. [Google Scholar]
- Gutsev, G.L.; Mochena, M.D.; Bauschilicher, C.W., Jr. Interaction of water with small Fen clusters. Chem. Phy. 2005, 314, 291–298. [Google Scholar] [CrossRef]
- Pilling, M.J.; Seakins, P.W. Reaction Kinetics; Oxford Press: Oxford, UK, 1995. [Google Scholar]
- Vilcu, R.; Danciu, T.; Bala, D. The study of Bray-Liebhasky reaction over a wide range of temperatures. II. Modelling. Dis. Dyn. Nat. Soc. 2000, 4, 55–62. [Google Scholar]
- Li, X.Q.; Zhang, W.X. Iron nanoparticles: The core-shell structure and unique properties for Ni(ii) sequestration. Langmuir 2006, 22, 4638–4642. [Google Scholar] [CrossRef] [PubMed]
- Polson, E.J.; Buckman, J.O.; Bowen, D.; Todd, A.C.; Gaw, M.M.; Cuthbert, S.J. An environmental-scanning-electron microscope investigation into the effect of biofilm on the wettability of quartz. SPE J. 2010, 15, 223–227. [Google Scholar] [CrossRef]
- Storch, H.H.; Golumbic, N.; Anderson, R.B. The Fischer Tropsch and Related Synthesis; Wiley: New York, NY, USA, 1951. [Google Scholar]
- Addiego, W.P.; Estrada, C.A.; Goodman, D.W.; Rosynek, M.P. An infrared study of the dehydrogenation of ethylbenzene to styrene over iron based catalysts. J. Catal. 1994, 146, 407–414. [Google Scholar] [CrossRef]
- Adler, R.W. Drought, sustainability, and law. Sustainability 2010, 2, 2176–2196. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Antia, D.D.J. Sustainable Zero-Valent Metal (ZVM) Water Treatment Associated with Diffusion, Infiltration, Abstraction, and Recirculation. Sustainability 2010, 2, 2988-3073. https://doi.org/10.3390/su2092988
Antia DDJ. Sustainable Zero-Valent Metal (ZVM) Water Treatment Associated with Diffusion, Infiltration, Abstraction, and Recirculation. Sustainability. 2010; 2(9):2988-3073. https://doi.org/10.3390/su2092988
Chicago/Turabian StyleAntia, David D.J. 2010. "Sustainable Zero-Valent Metal (ZVM) Water Treatment Associated with Diffusion, Infiltration, Abstraction, and Recirculation" Sustainability 2, no. 9: 2988-3073. https://doi.org/10.3390/su2092988