
Investigation of Plasma Treatment on Micro-Injection Moulded Microneedle for Drug Delivery


Abstract
:
1. Introduction
2. Experimental Section
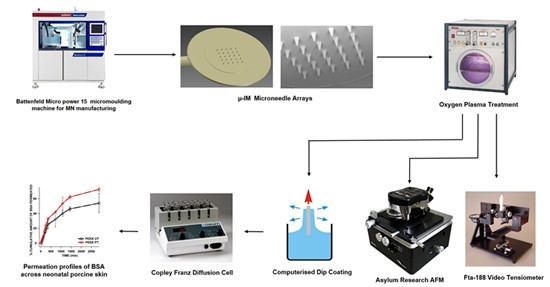
2.1. Manufacture of Microneedles and Flat Polymer Samples for Surface Evaluation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | PEEK | PC |
|---|---|---|
| Melt temperature (°C) | 420 | 320 |
| Injection velocity (mm/s) | 800 | 500 |
| Clamping force (kN) | 150 | 100 |
| Time of holding pressure (s) | 5 | 3 |
| Mould base temperature (°C) | 200 | 120 |
2.2. Oxygen Plasma Treatment
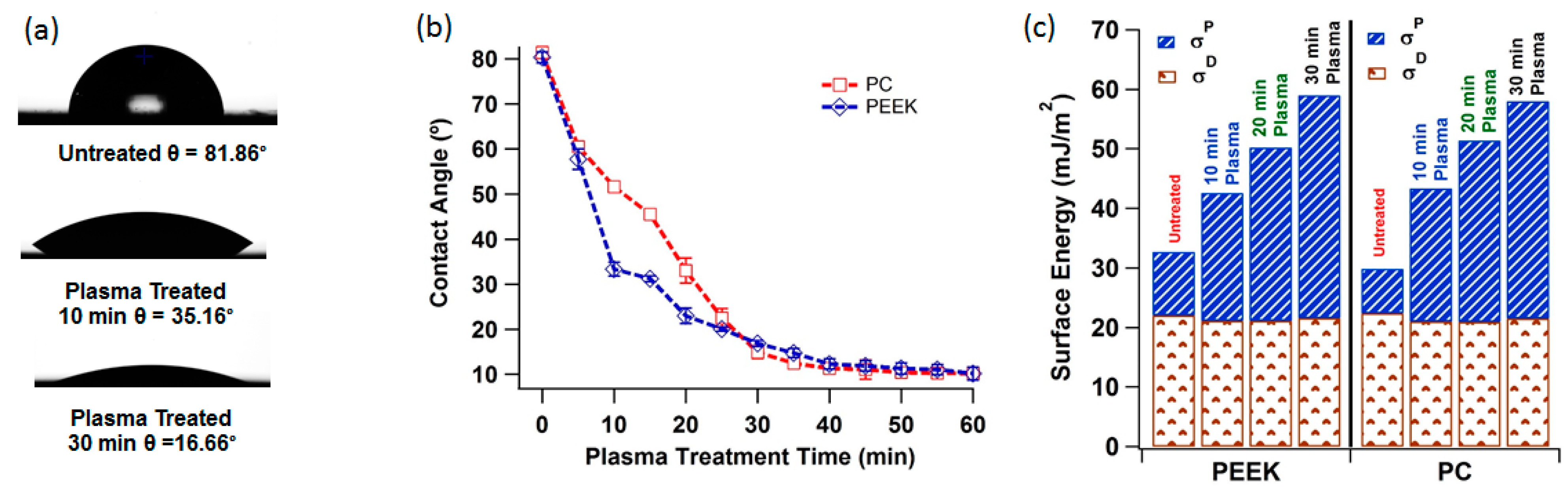
2.3. Contact Angle and Surface Energy Analysis
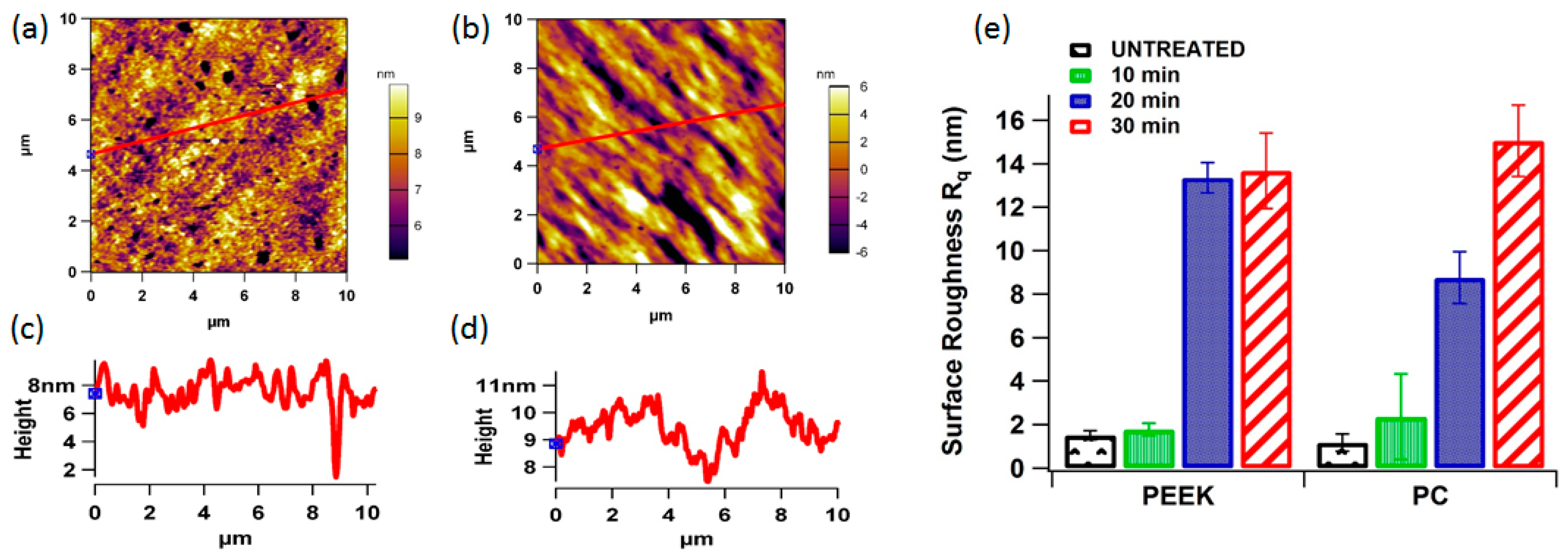
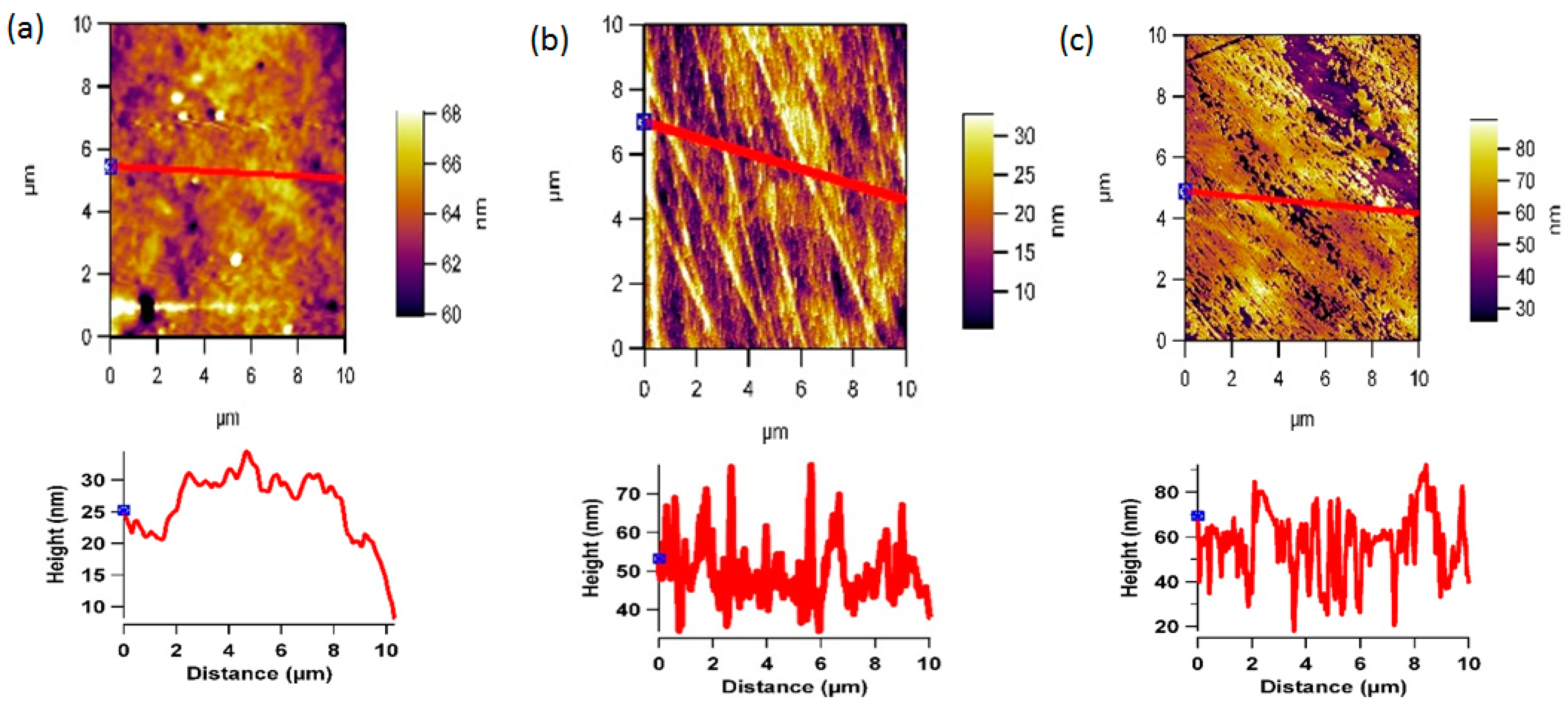
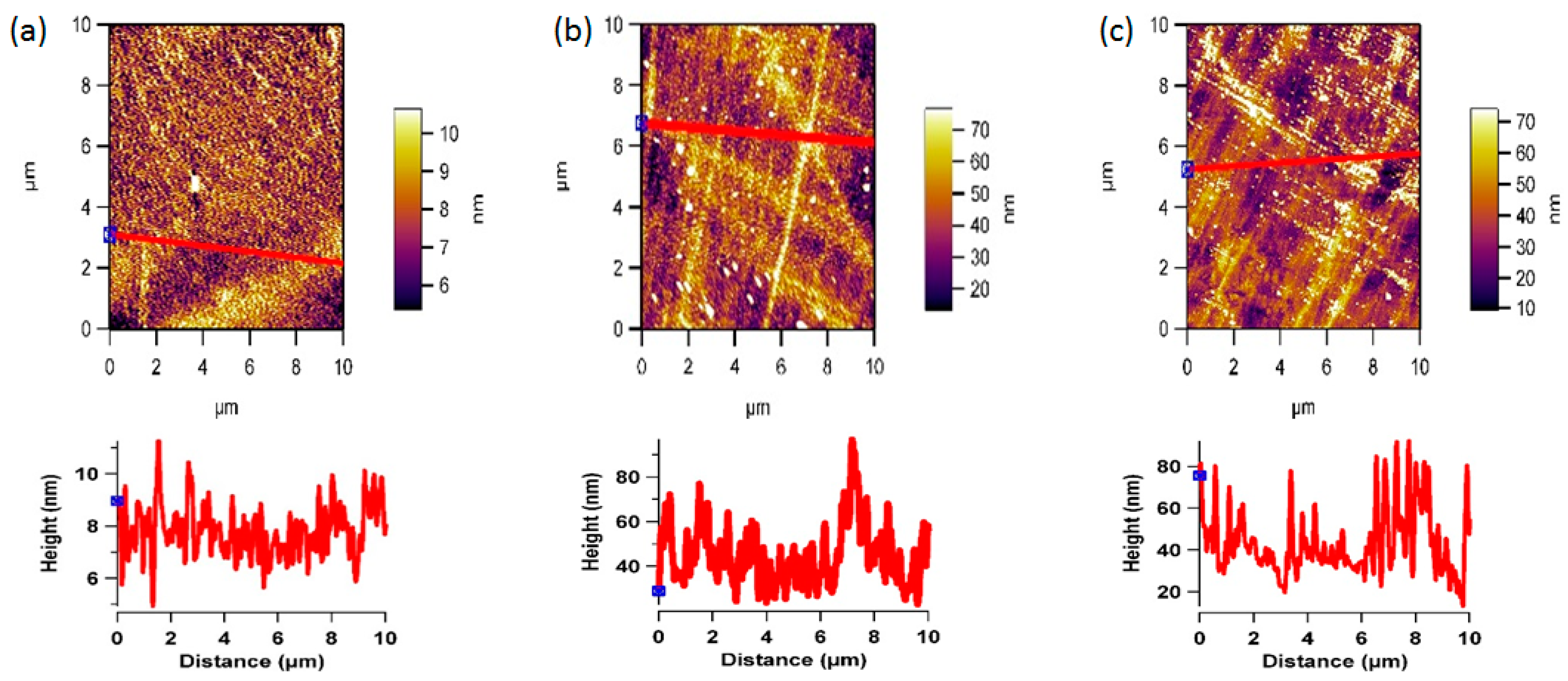
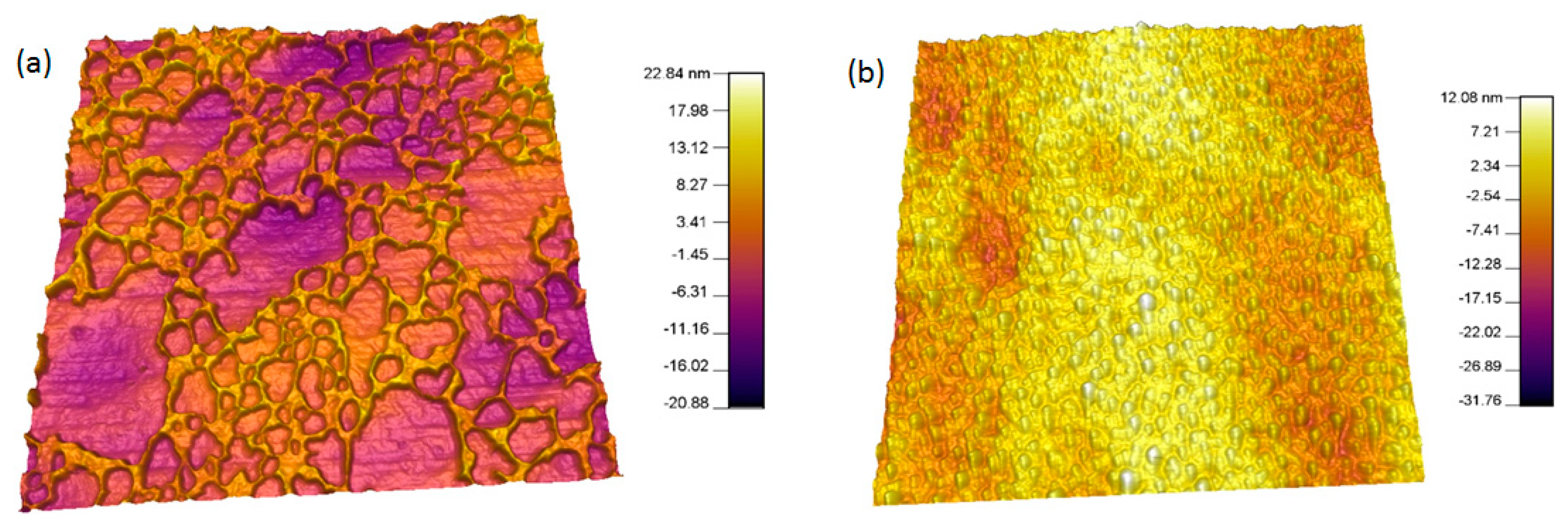
2.4. AFM Analysis
2.5. Surface Morphology after Protein Adsorption
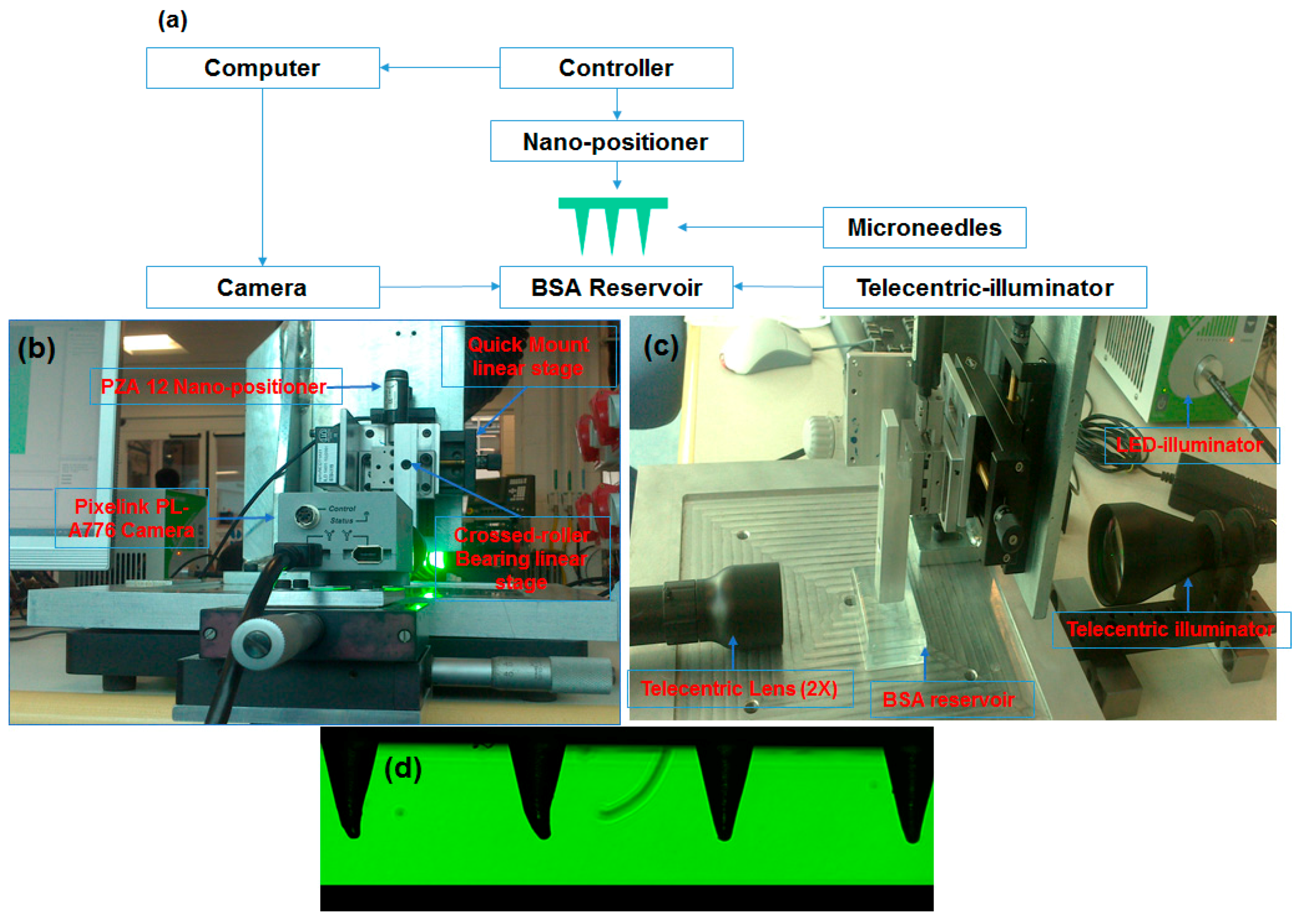
2.6. Coating Arrays of Microneedles
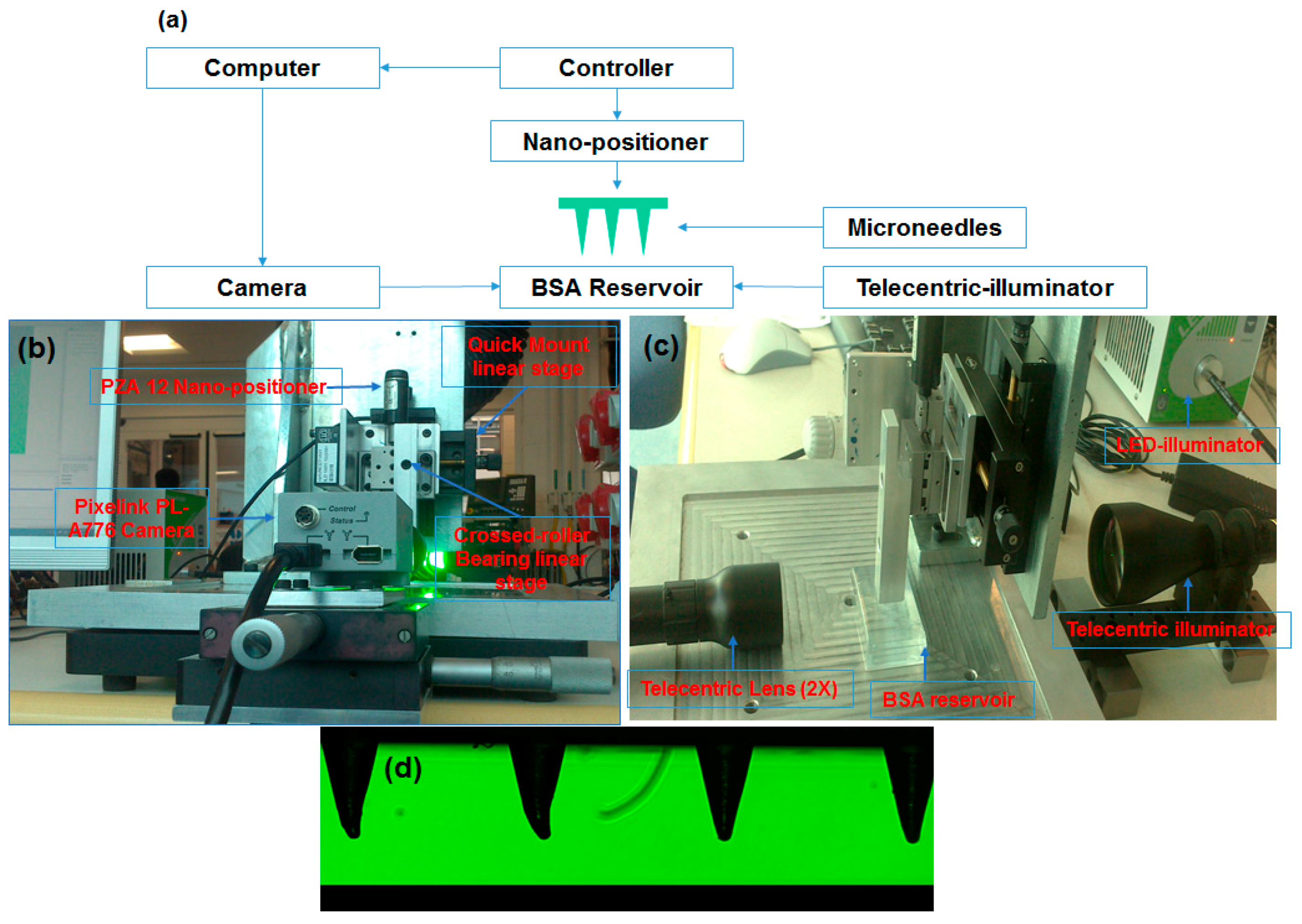
2.7. Analysis of BSA in Solution
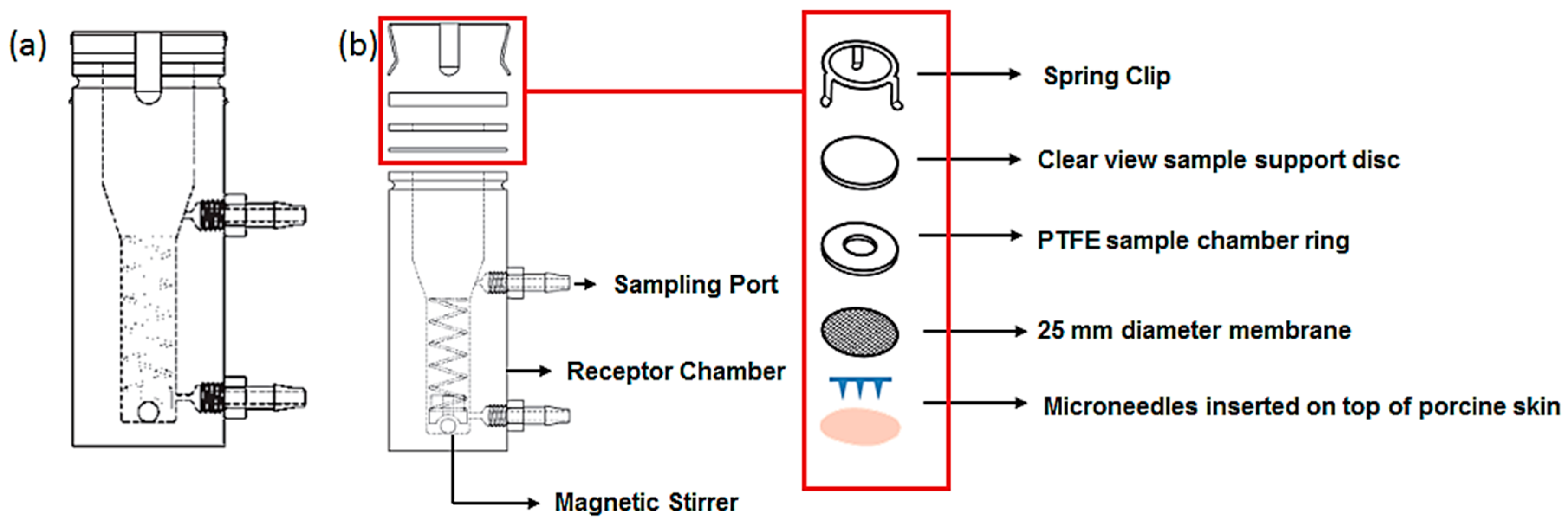
2.8. Drug Release Studies from Microneedles and in Vitro Diffusion Studies
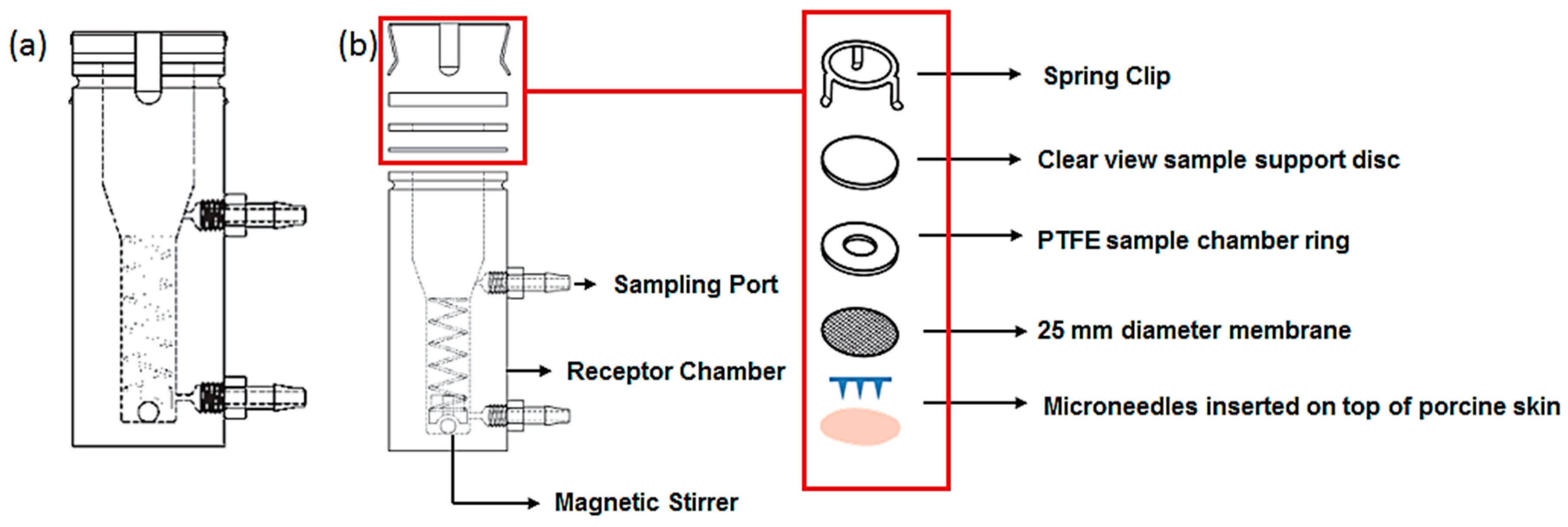
2.9. Quantification of the Residual BSA from the MN Shaft after Insertion
3. Results and Discussion
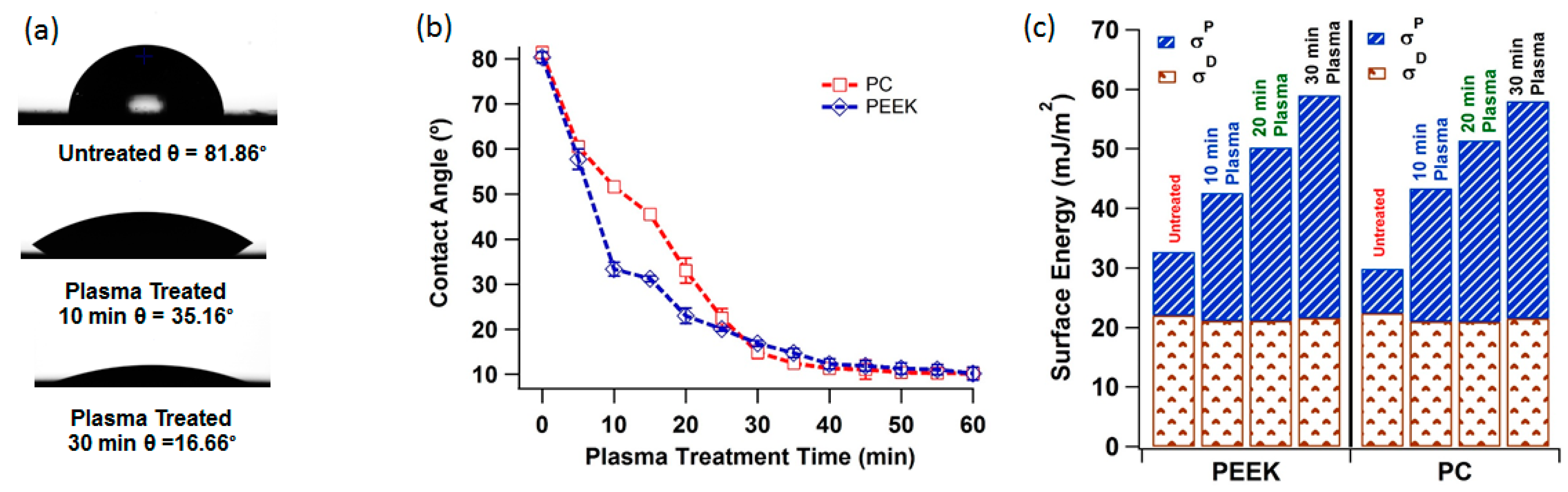
3.1. Plasma Treatment, Contact Angle and Surface Energy
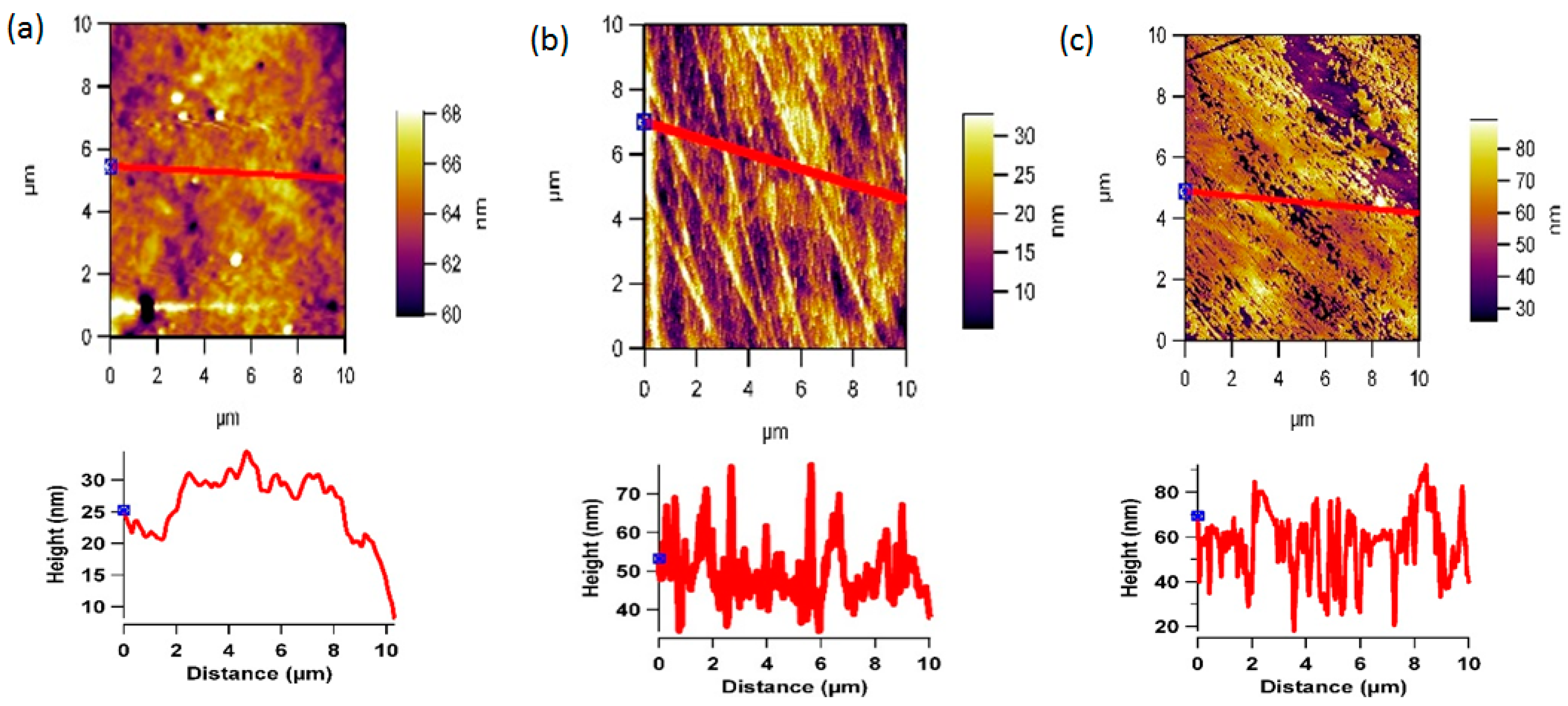
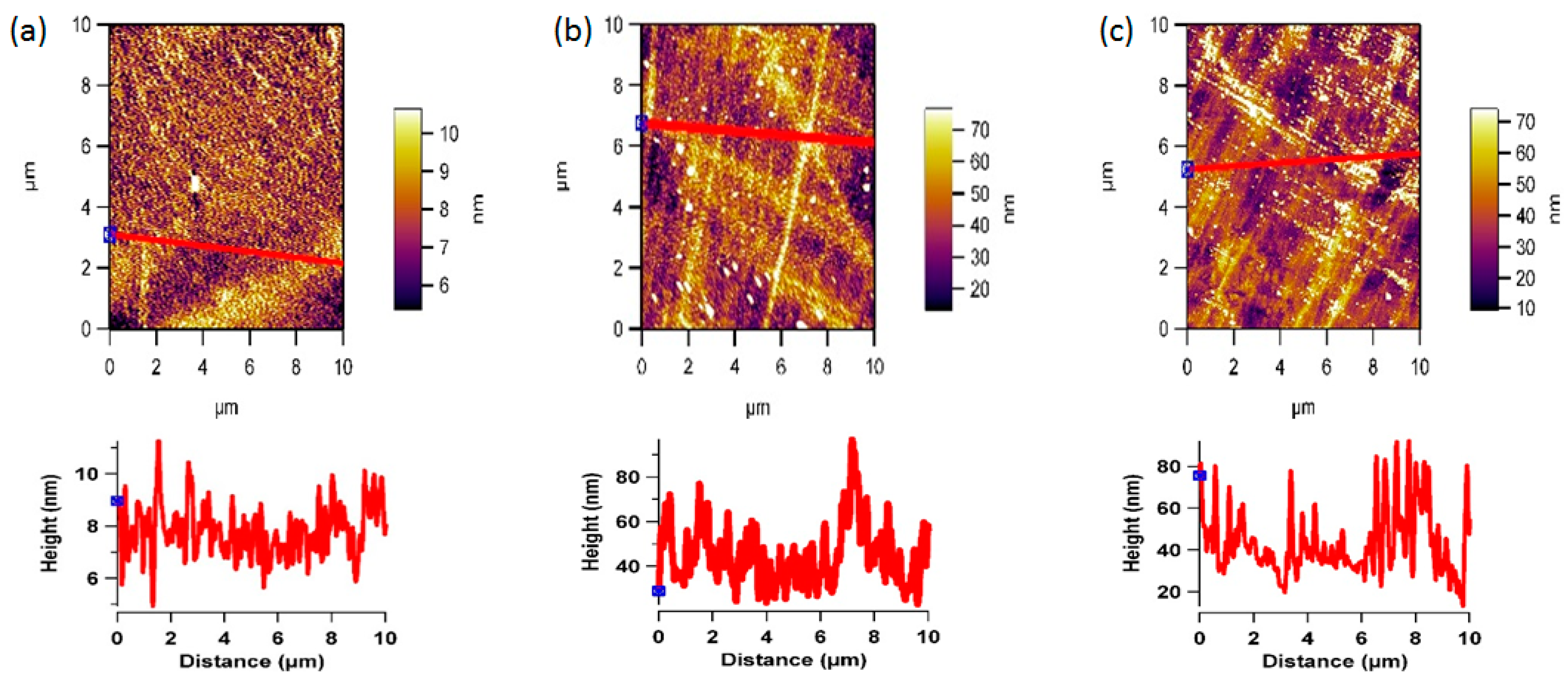
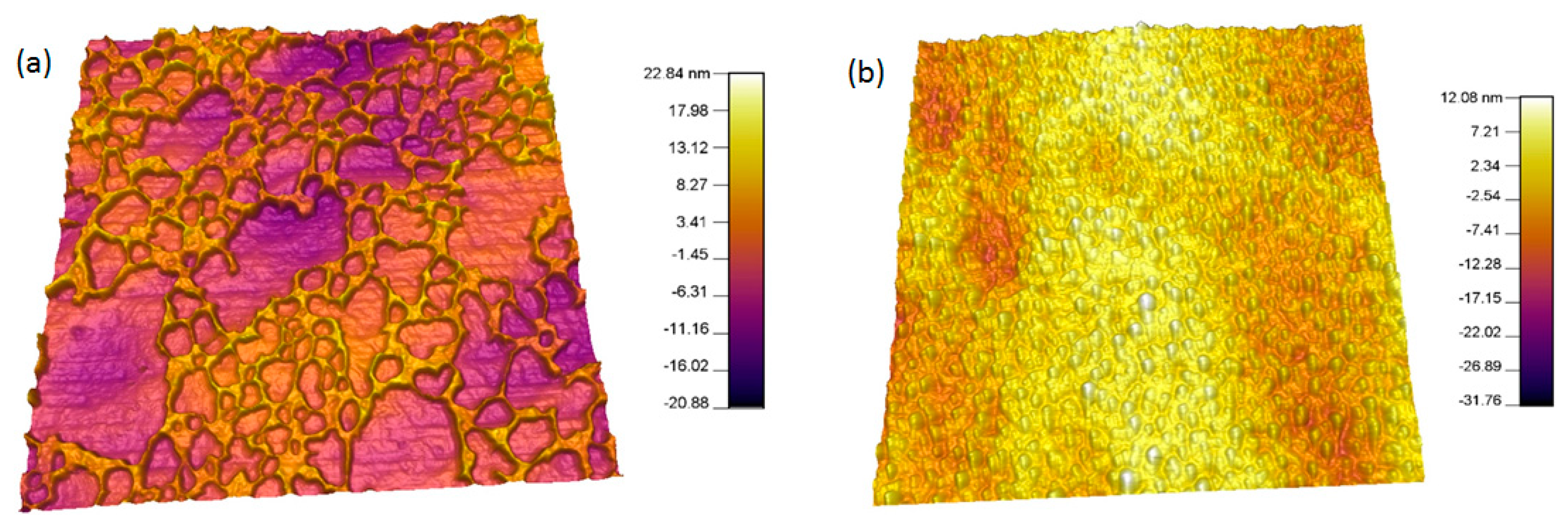
3.2. Plasma Treatment and Surface Topography

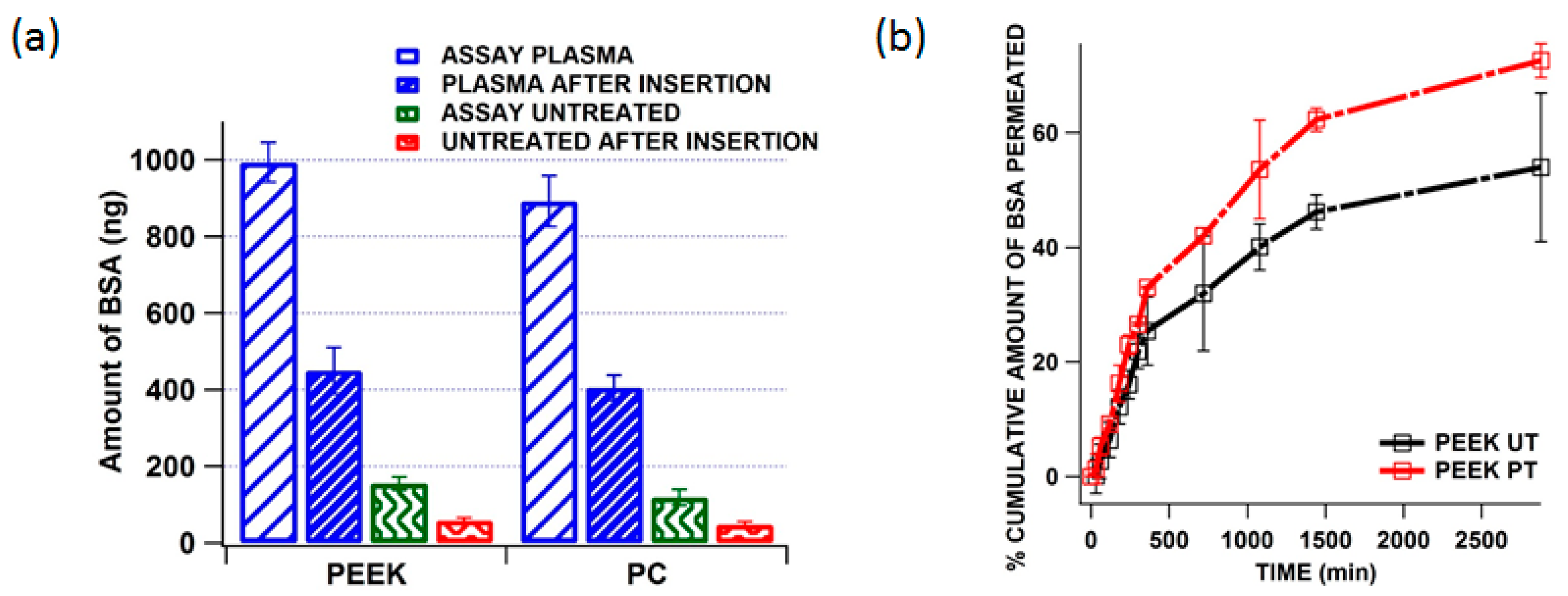
3.3. Plasma Treatment and Protein Adsorption
3.4. Analysis of BSA
| Selected concentration (µg/mL) | Mean concentration found (µg/mL) | SD | Accuracy (%) |
|---|---|---|---|
| Inter-day | |||
| 1 (low) | 0.98 | 0.02 | 98.62 |
| 50 (medium) | 46.45 | 4.67 | 92.91 |
| 300 (high) | 297.57 | 1.92 | 99.19 |
| Intra-day | |||
| 1 (low) | 1.034 | 0.11 | 96.66 |
| 50 (medium) | 48.91 | 6.84 | 97.83 |
| 300 (high) | 295.13 | 7.41 | 98.37 |
3.5. Quantification of Residual BSA from the MN Shaft after Insertion and in Vitro Delivery of BSA across the Neonatal Porcine Skin
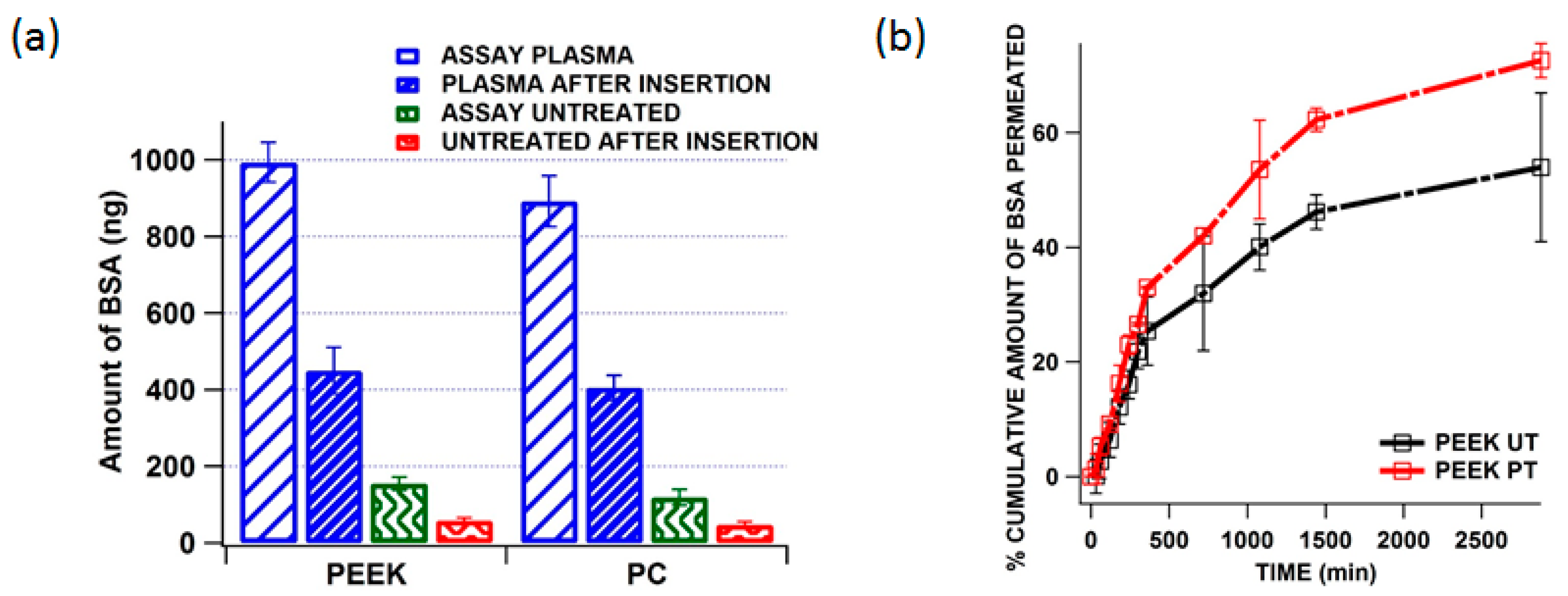
4. Conclusions
Acknowledgment
Author Contributions
Conflicts of Interest
References
- Donnelly, R.F.; Raj Singh, T.R.; Woolfson, D.A. Microneedle-based drug delivery systems: Microfabrication drug delivery and safety. Drug Deliv. 2010, 4, 187–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAllister, D.V.; Allen, M.G.; Prausnitz, M.R. Microfabricated microneedles for gene and drug delivery. Annu. Rev. Biomed. Eng. 2000, 2, 289–313. [Google Scholar] [CrossRef] [PubMed]
- Arduino, M.J.; Bland, L.A.; Danzig, L.E.; McAllister, S.K.; Aguero, S.M. Microbiologic evaluation of needleless and needle-access devices. Am. J. Infect Control 1997, 25, 377–380. [Google Scholar] [CrossRef]
- Ebah, L.M. Extraction and analysis of interstitial fluid and characterisation of the interstitial compartment in kidney disease. Ph.D. Thesis, University of Manchester, Manchester, UK, 2012. [Google Scholar]
- Hamilton, J. Needle Phobia—A Neglected Diagnosis. J. Fam. Pr. 1995, 2, 169–175. [Google Scholar]
- Al-Tabakha, M.M.; Arida, A.L. Recent challenges in insulin delivery systems: A Review. Indian J. Pharm. Sci. 2008, 3, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, G.; Tiwari, R.; Sriwastawa, B.; Bhati, L.; Pnadey, S.; Bannerjee, S.K. Drug delivery systems: An updated review. Inter. J. Pharm. Investig. 2012, 2, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Barry, B.W. Novel mechanism and devices to enable successful transdermal drug delivery. Eur. J. Pharm. Sci. 2001, 14, 101–114. [Google Scholar] [CrossRef]
- Birchall, J.C. Overcoming the stratum corneum barrier: Microneedle delivery of macromolecules and vaccines. Inter. J. Cosmet. Sci. 2012, 4, 361–372. [Google Scholar]
- Cleary, G.W. Microneedles for drug delivery. Pharm. Res. 2011, 28, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Prausnitz, M.R.; Langer, R. Transdermal drug delivery. Nature Biotechnol. 2008, 11, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Prausnitz, M.R. Microneedle for transdermal drug delivery. Adv. Drug Deliv. Rev. 2004, 5, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Attia, U.M.; Marsona, S.; Alcock, J.R. Micro-injection moulding of polymer microfluidic devices. Microfluid. Nano Fluid. 2009, 7, 1–28. [Google Scholar] [CrossRef] [Green Version]
- Birchall, J.C.; Clemo, R.; Anstey, A.; John, D.N. Microneedles in clinical practice—An exploratory study into the opinions of healthcare professionals and the public. Pharm. Res. 2011, 28, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Maaden, K.V.; Jiskoot, W.; Bouwstra, J. Microneedle technologies for transdermal drug and vaccine delivery. J. Control. Release 2012, 161, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Bora, P.; Kumar, L.; Bansal, K.A. Microneedle technology for advanced drug delivery: Evolving vistas. Curr. Res. Inf. Pharm. Sci. 2009, 9, 7–10. [Google Scholar]
- Finn, K.H. The measurement of surface energy of polymer by means of contact angles of liquids on solid surfaces. Master’s Thesis, University of Oslo, Oslo, Norway, 2007. [Google Scholar]
- Hildebrand, H.F.; Blanchemain, N.; Mayer, G.; Chai, F.; Lefebvre, M.; Boschin, F. Surface coatings for biological activation and functionalization of medical devices. Surf. Coat. Technol. 2006, 200, 6318–6324. [Google Scholar] [CrossRef]
- D’Sa, R.A.; Burke, G.A.; Meenan, B.J. Protein adhesion and cell response on atmospheric pressure dielectric barrier discharge-modified polymer surfaces. Acta Biomater. 2010, 6, 2609–2620. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Courty, C.; Xanthopoulos, N.; Mathieu, H.J. Surface treatment for adhesive bonding on carbon fibre-poly(etherether ketone) composites. J. Mater. Sci. Lett. 1991, 10, 335–338. [Google Scholar] [CrossRef]
- Ma, R.; Tang, T. Current Strategies to Improve the Bioactivity of PEEK. Inter. J. Mol. Sci. 2014, 15, 5426–5445. [Google Scholar] [CrossRef] [PubMed]
- Subedi, D.; Zajickova, L.; Bursikova, V.; Janca, J. Surface modification of polycarbonate (bisphenol A) by low pressure of plasma. Himal. J. Sci. 2003, 1, 115–118. [Google Scholar] [CrossRef]
- Vijayalakshmi, K.A.; Mekala, M.; Yoganand, C.P.; Pandiyaraj, K.N. Studies on modification of surface properties in polycarbonate (pc) film induced by DC glow discharge plasma. Inter. J. Polym. Sci. 2011, 2011, 426057. [Google Scholar] [CrossRef]
- Garcia, T. Plasma processes as a cleaner alternative for cleaning, corrosion resistance, and functionalization of metallic Surfaces. In Proceedings of the 2nd International Workshop Advances in Cleaner Production, Sao Paulo, Brazil, 20–22 May, 2009; pp. 21–27.
- Heimenz, P. Principles of Colloid and Surface Chemistry; Marcel Dekker: New York, NY, USA, 1997. [Google Scholar]
- Rulison, C. So you what to measure surface energy? Technical Note; Kruss GmbH: Hamburg, Germany, 2003; pp. 1–16. [Google Scholar]
- Vasilev, K. Nano engineered plasma polymer films for biomaterial applications. Plasma Chem. Plasma Process. 2014, 34, 545–558. [Google Scholar] [CrossRef]
- Yeo, S.H.; Choi, C.R.; Jung, D. Investigation of protein adsorption using plasma treatment for protein chips. J. Korean Phys. Soc. 2006, 48, 1325–1328. [Google Scholar]
- Pelagade, S.; Singh, N.L.; Shahb, S.; Qureshi, A.; Rane, R.S.; Mukherjee, S.; Deshpande, U.P.; Ganesan, V.; Shripathi, T. Surface free energy analysis for bipolar pulsed argon plasma treated polymer films. J. Phys. 2010, 208, 012107. [Google Scholar] [CrossRef]
- Kim, S. Polyetheretherketone Surface Functionalization by Low-Energy Ion-Beam Irradiation under a Reactive O2 Environment and Its Effect on the PEEK/Copper Adhesives. Langmuir. 2004, 20, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Rochford, E.T.J. Effect of Plasma Treatment of PEEK on Bacterial Adhesion. In Proceedings of the 56th Annual Meeting of the Orthopaedic Research Society, New Orleans, LA, USA, 6–9 March 2010.
- Mcintyre, N.S.; Walzak, M.J. New UV/ozone treatment improves adhesiveness of polymer surfaces. Modern Plast. 1995, 72, 79–81. [Google Scholar]
- Finson, E.S.; Kaplan, L.; Wood, L. Plasma treatment of webs and films. In Proceedings of the 38th Annual Technical Conference Proceedings for the Society of Vacuum Coaters, Chicago, IL, USA; 2–7 April 1995. [Google Scholar]
- Powles, R.C. Nanoindentation response of PEEK modified by mesh-assisted plasma immersion ion implantation. Surf. Coat. Technol. 2007, 201, 7961–7969. [Google Scholar] [CrossRef]
- Labay, C.; Canal, C.; Garcia-Celma, M.J. Influence of corona plasma treatment on polypropylene and polyamide 6.6 on the release of a model drug. Plasma Chem. Plasma Process. 2010, 30, 885–896. [Google Scholar] [CrossRef]
- Liu, Y.C.; Lu, D.N. Surface Energy and Wettability of Plasma-treated Polyacrylonitrile Fibers. Plasma Chem. Plasma Process. 2006, 26, 119–126. [Google Scholar] [CrossRef]
- Kasemo, B. Biological surface science. Surf. Sci. 2002, 500, 656–677. [Google Scholar] [CrossRef]
- Kuzuya, M.; Kondo, S.; Sasai, Y. Plasma techniques for preparation of controlled drug release systems. Plasmas Polym. 2001, 6, 145–162. [Google Scholar] [CrossRef]
- Comyn, J.; Mascia, L.; Xiao, G.; Parker, B.M. Plasma-treatment of polyetheretherketone (PEEK) for adhesive bonding. Inter. J. Adhes. Adhes. 1996, 16, 97–104. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nair, K.; Whiteside, B.; Grant, C.; Patel, R.; Tuinea-Bobe, C.; Norris, K.; Paradkar, A. Investigation of Plasma Treatment on Micro-Injection Moulded Microneedle for Drug Delivery. Pharmaceutics 2015, 7, 471-485. https://doi.org/10.3390/pharmaceutics7040471
Nair K, Whiteside B, Grant C, Patel R, Tuinea-Bobe C, Norris K, Paradkar A. Investigation of Plasma Treatment on Micro-Injection Moulded Microneedle for Drug Delivery. Pharmaceutics. 2015; 7(4):471-485. https://doi.org/10.3390/pharmaceutics7040471
Chicago/Turabian StyleNair, Karthik, Benjamin Whiteside, Colin Grant, Rajnikant Patel, Cristina Tuinea-Bobe, Keith Norris, and Anant Paradkar. 2015. "Investigation of Plasma Treatment on Micro-Injection Moulded Microneedle for Drug Delivery" Pharmaceutics 7, no. 4: 471-485. https://doi.org/10.3390/pharmaceutics7040471