Design, Synthesis and in Vitro Degradation of a Novel Co-Drug for the Treatment of Psoriasis


Abstract
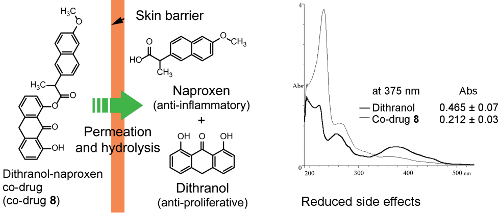
:
1. Introduction
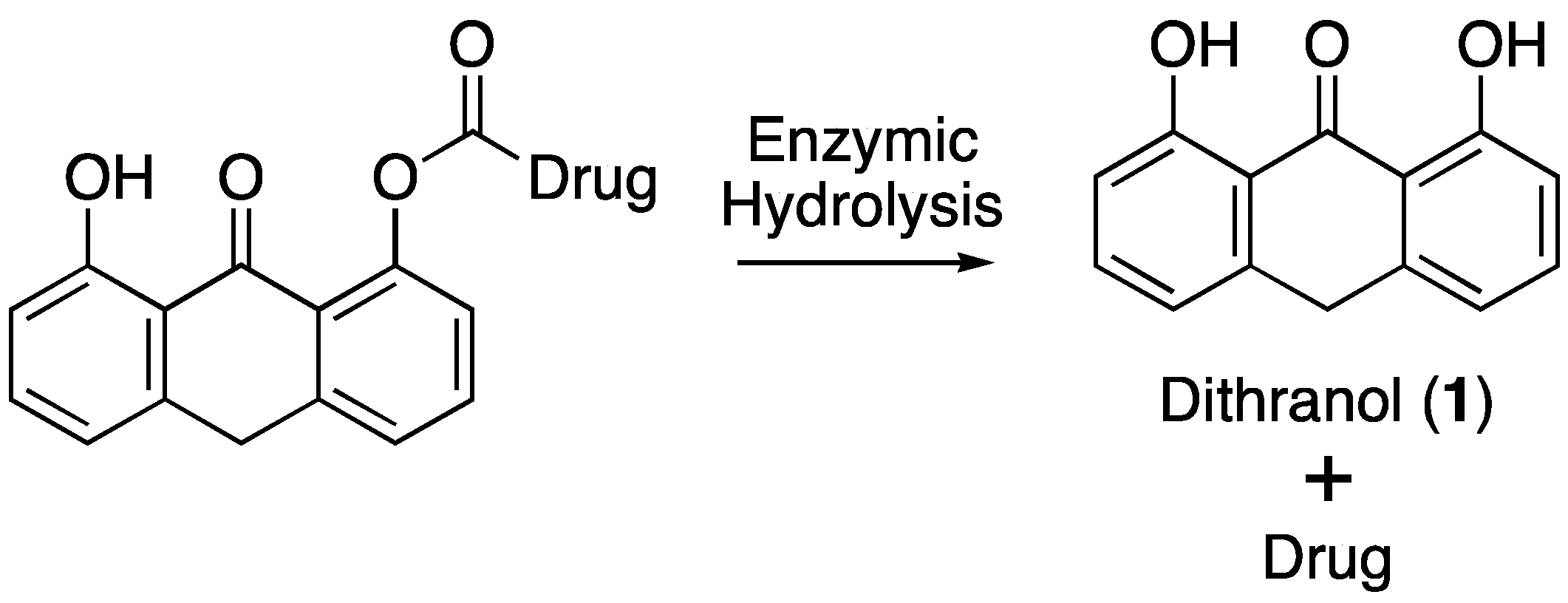
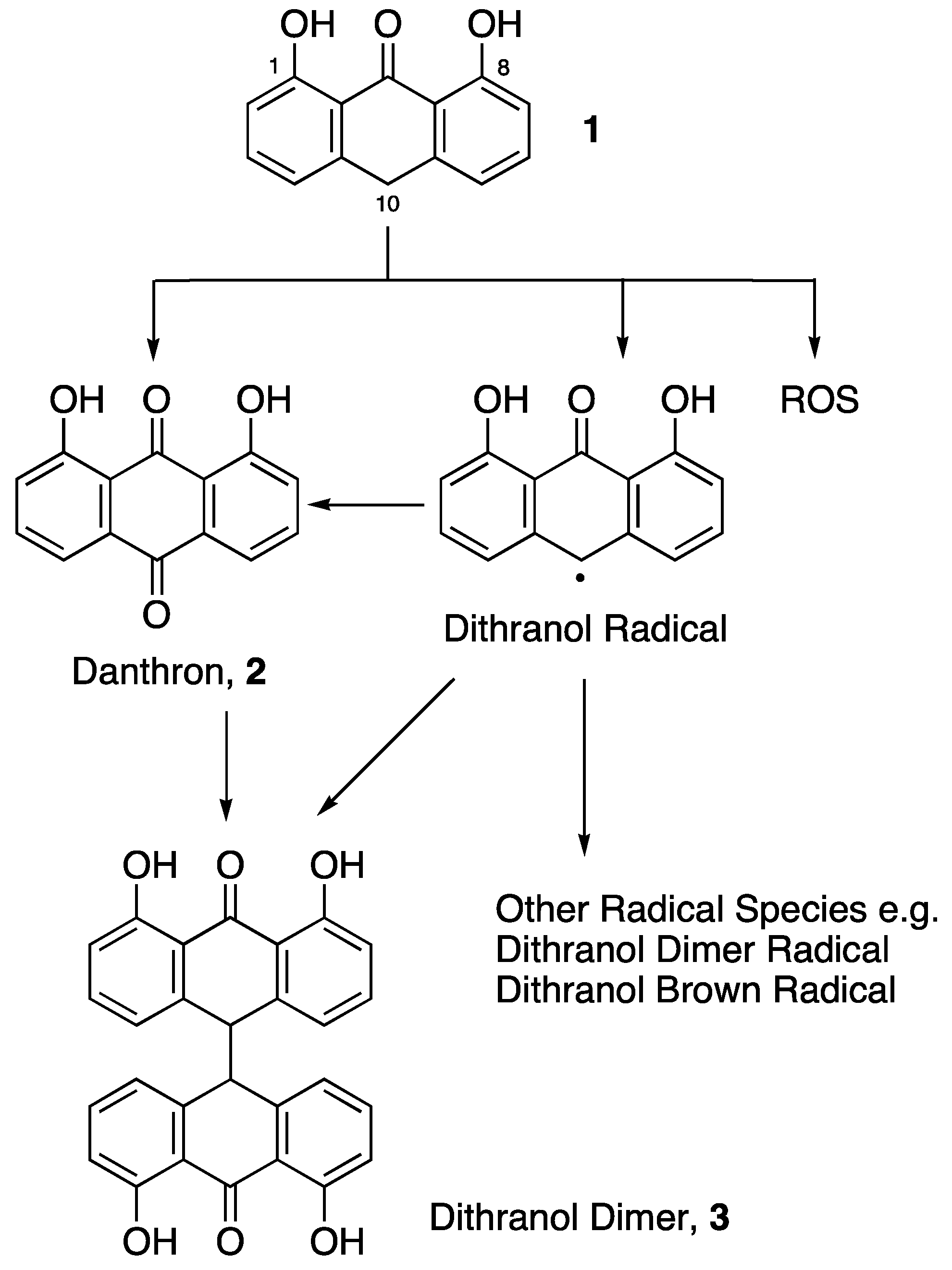
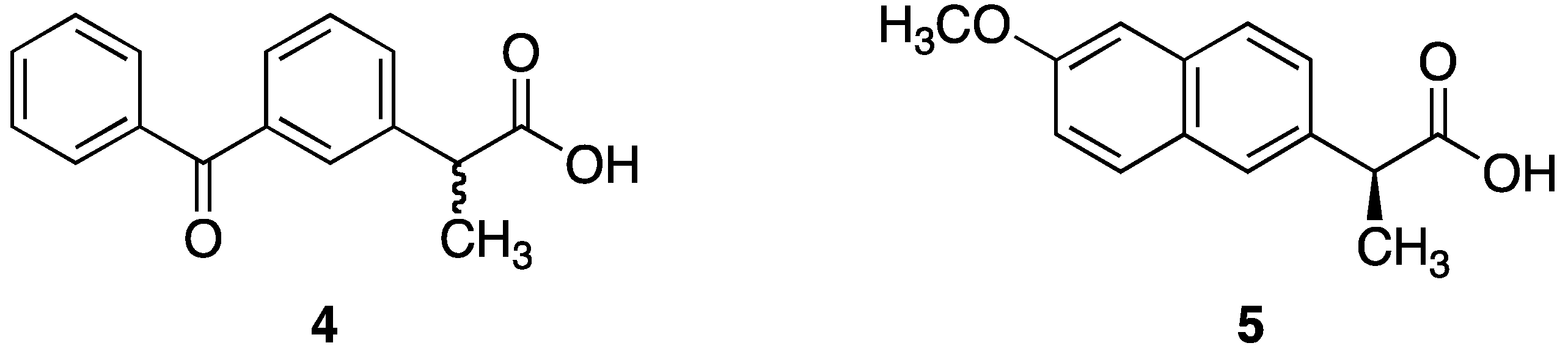
2. Materials and Methods
2.1. General Chemical Procedures
2.1.1. Method 1: Acid Chloride Synthesis
2.1.2. Method 2: Dithranol Di-Ester Co-Drug Synthesis
2.1.3. Method 3: Dithranol Mono-Ester Co-Drug Synthesis
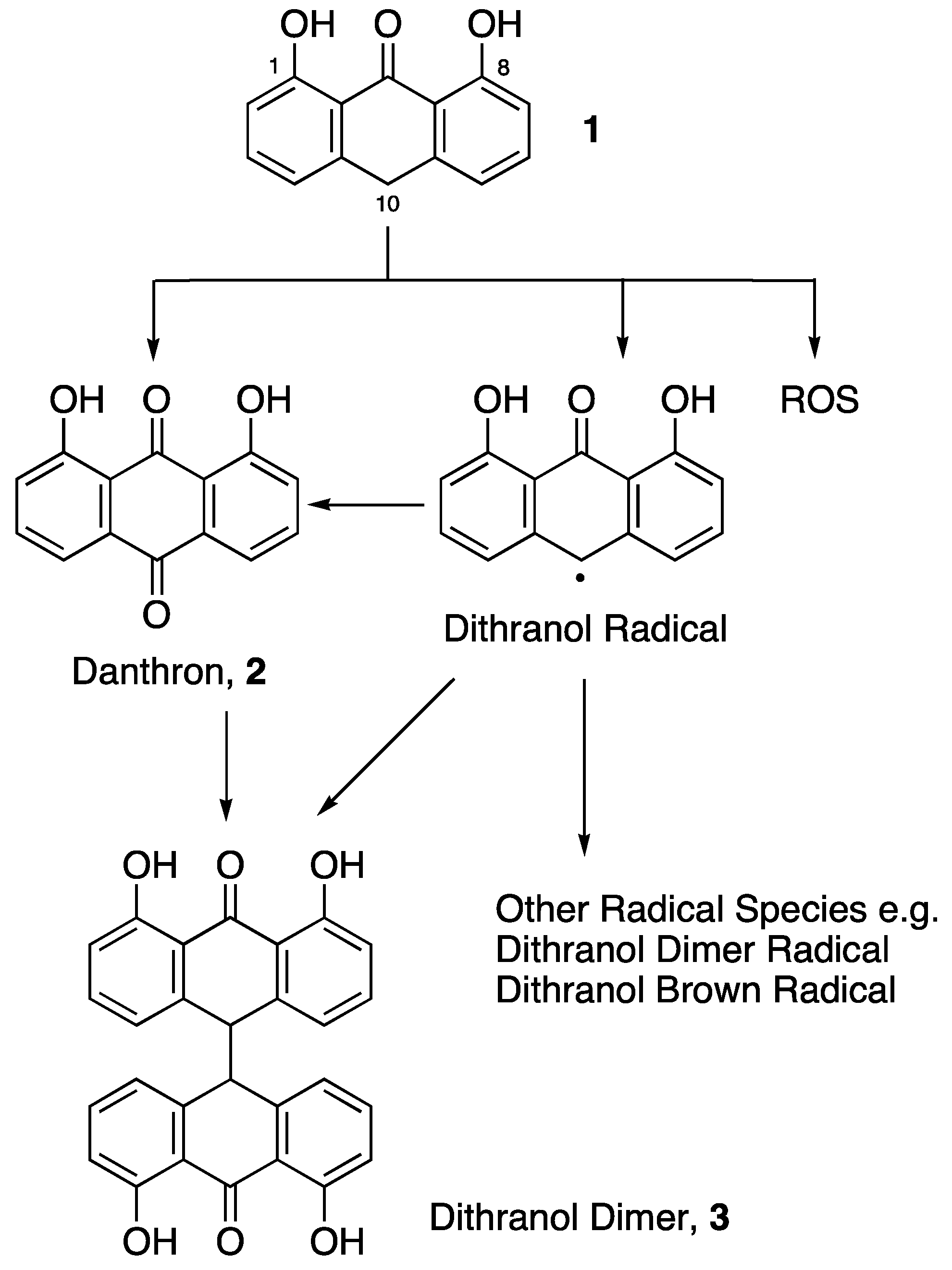
2.1.4. Dithranol Dimer Synthesis (3)
2.2. Co-Drug Synthesis
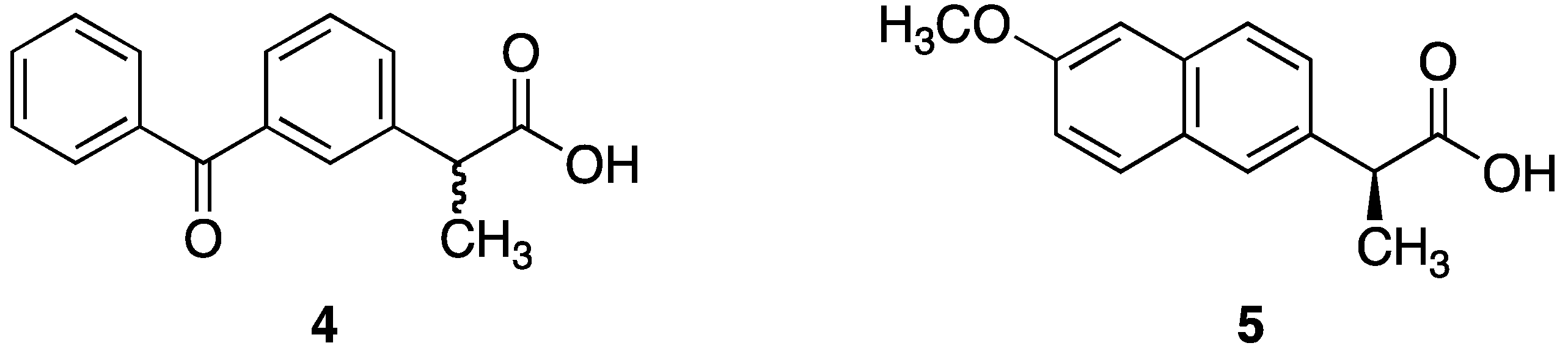
2.2.1. Synthesis of Dithranol Di-Naproxen Co-Drug (6): 9-Oxo-9,10-dihydroanthracene-1,8-diyl bis(2-(6-methoxy-naphthalen-2-yl)propionate)
2.2.2. Synthesis of Dithranol Di-Ketoprofen Co-Drug (7): 9-Oxo-9,10-dihydroanthracene-1,8-diyl bis(2-(3-benzoylphenyl)propionate)
2.2.3. Synthesis of Dithranol Mono-Naproxen Co-Drug (8): 8-Hydroxy-9-oxo-9,10-dihydroanthracen-1-yl 2-(6-methoxynaphthalen-2-yl)propanoate
2.2.4. Synthesis of Dithranol Mono-Ketoprofen Co-Drug (9): 8-Hydroxy-9-oxo-9,10-dihydroanthracen-1-yl 2-(3-benzoylphenyl)propanoate
2.3. HPLC Analysis
2.4. Spectrophotometric Analysis
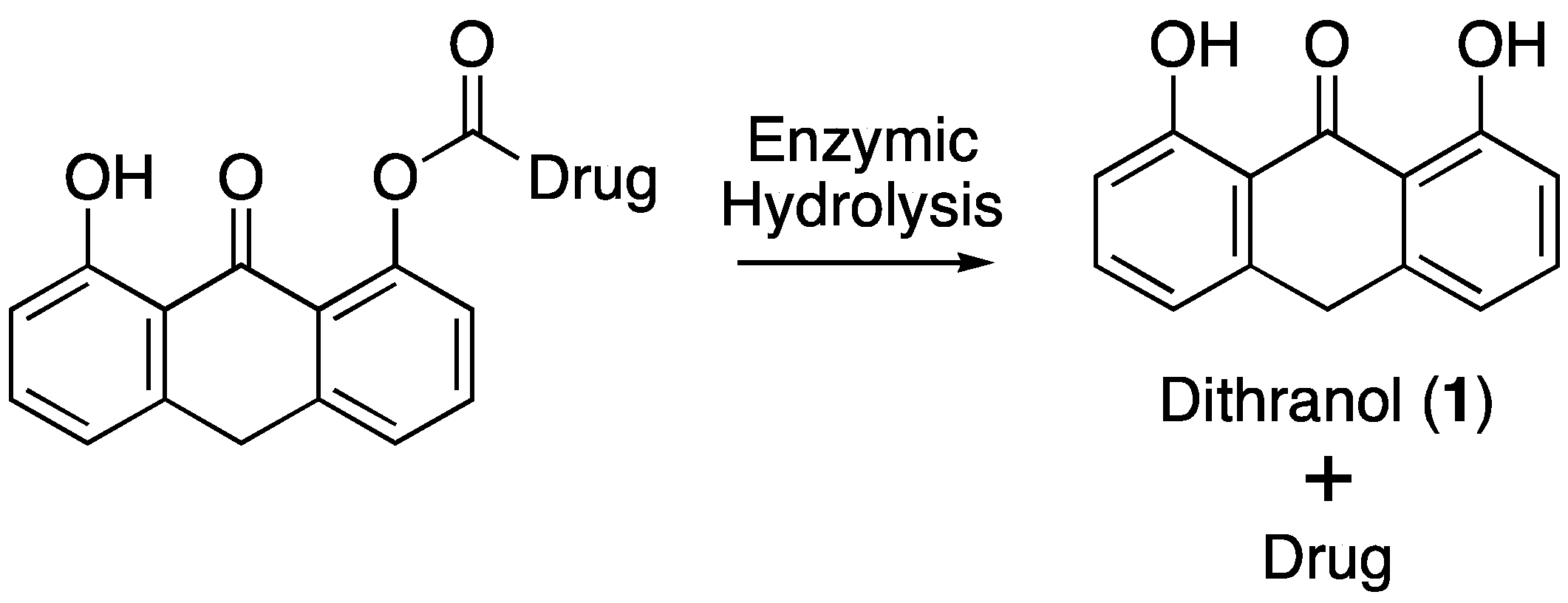
2.5. Enzymatic Co-Drug Hydrolysis
2.5.1. Hydrolysis Using Porcine Liver Esterase
2.5.2. Hydrolysis Using Porcine Skin Homogenate
3. Results and Discussion
3.1. Co-Drug Synthesis
3.2. Co-Drug Selection

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Cpd. | MW a | R1 | R2 | Synthetic Yield (%) | ClogP b |
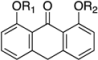
| 6 | 650 | I 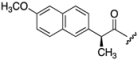 |  | 30 | 8.54 |
| 7 | 698 |  | 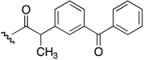 | 39 | 9.74 |
| 8 | 438 | 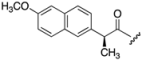 | H | 46 | 5.45 |
| 9 | 462 |  | H | 37 | 5.56 |
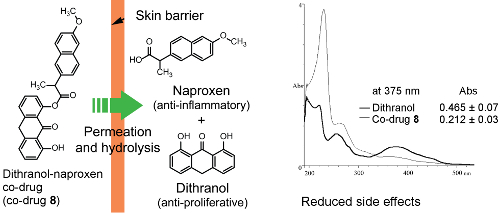
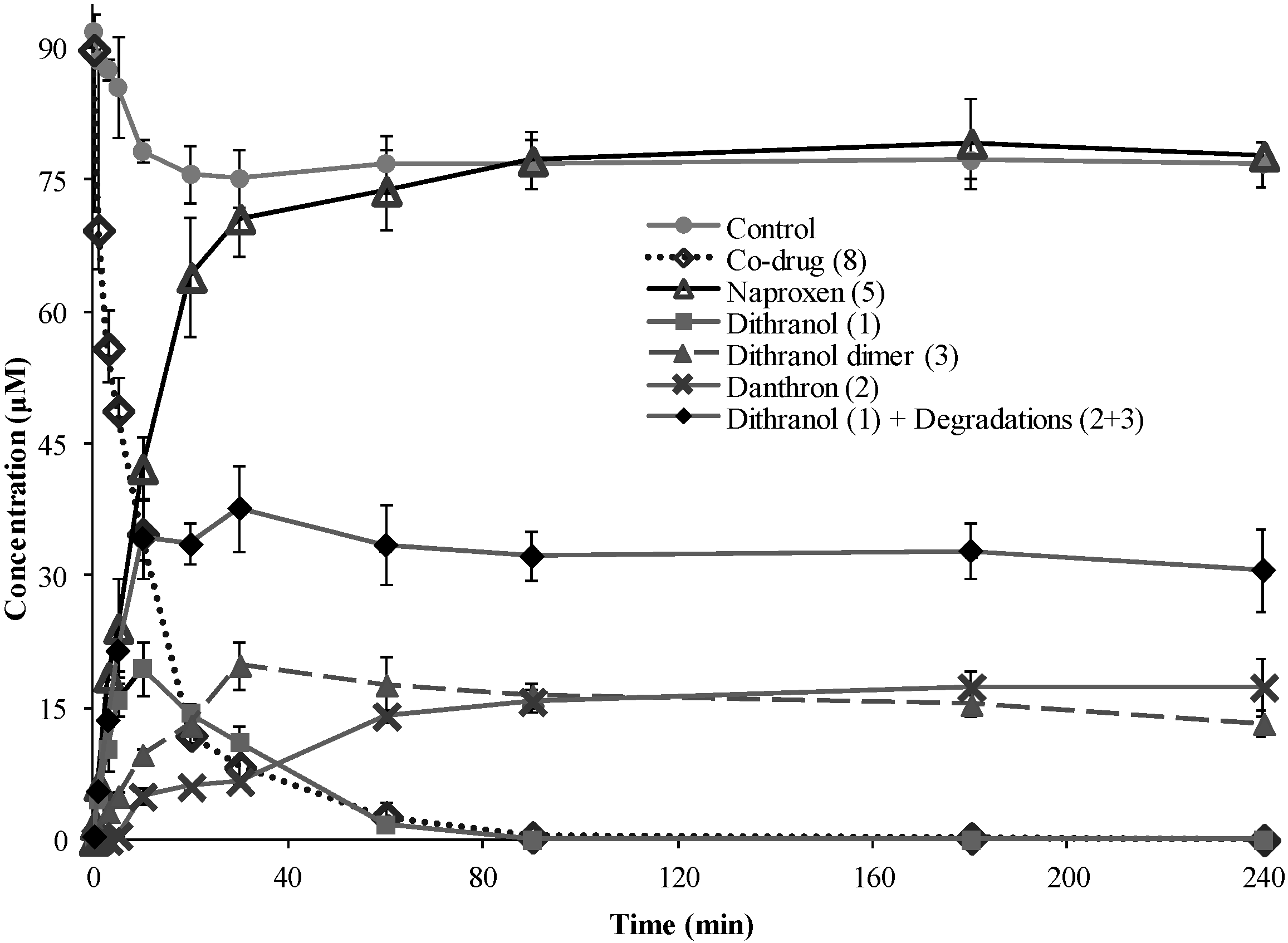
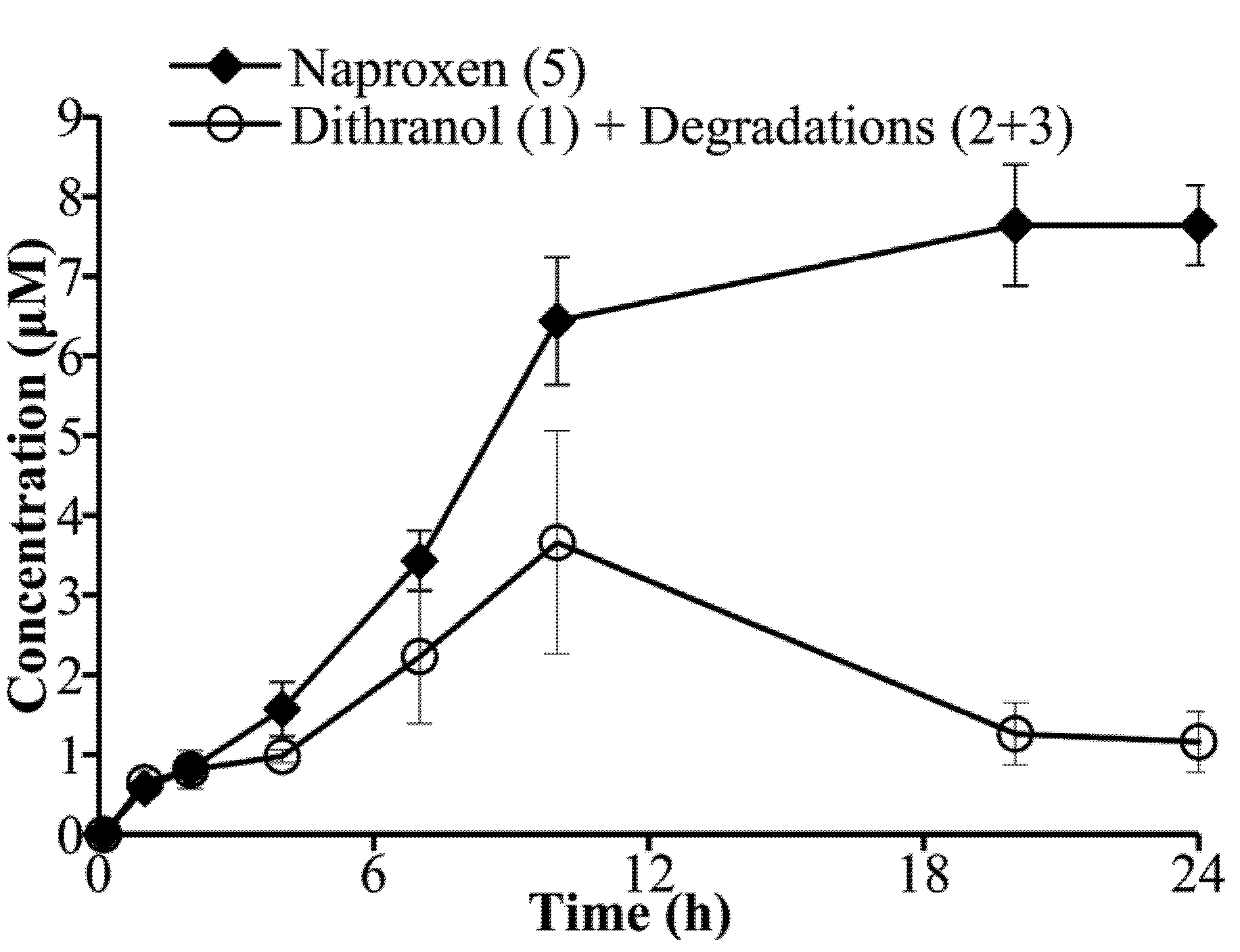
3.3. Hydrolysis of Dithranol-Naproxen Co-Drug (8)
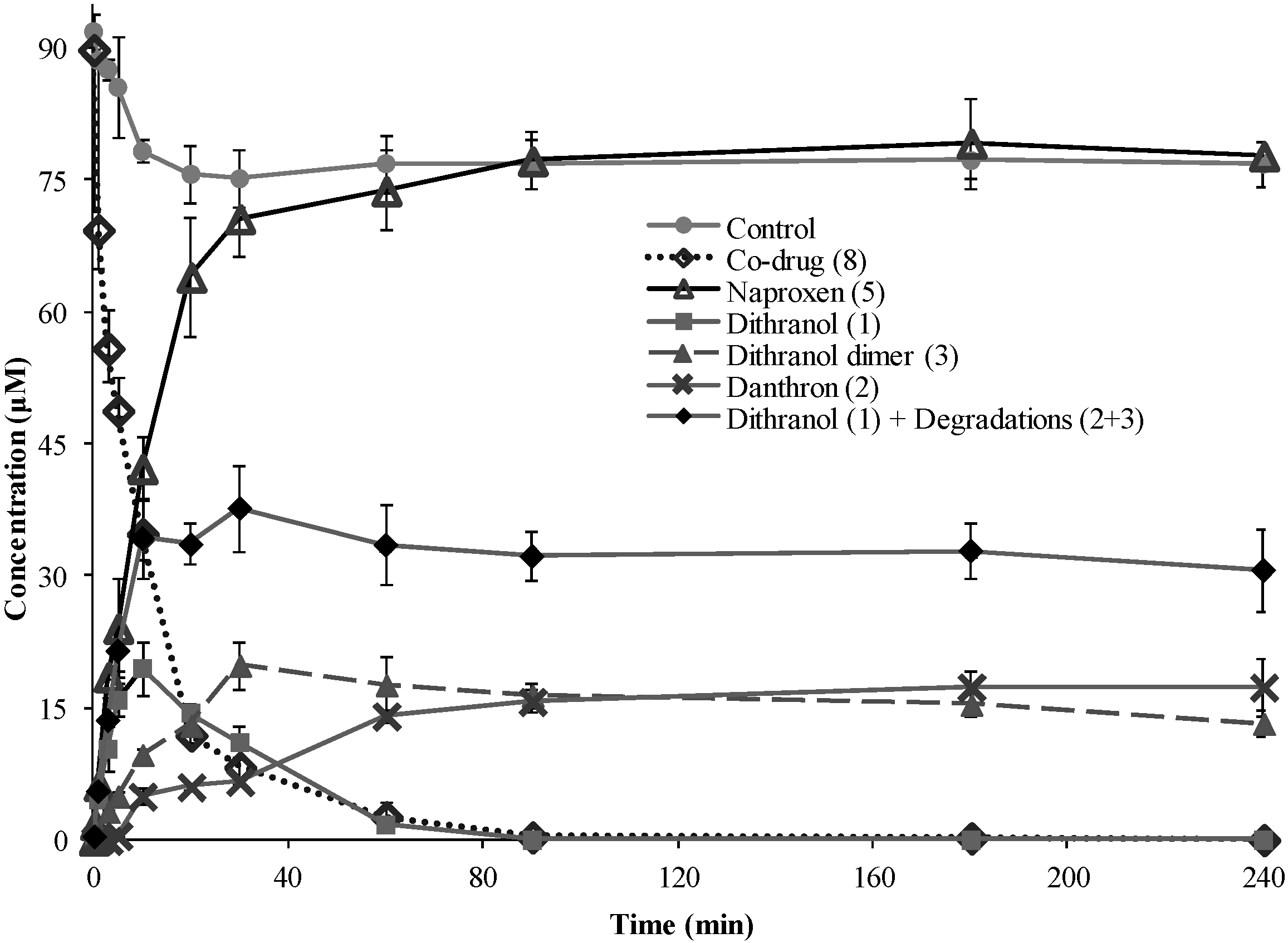
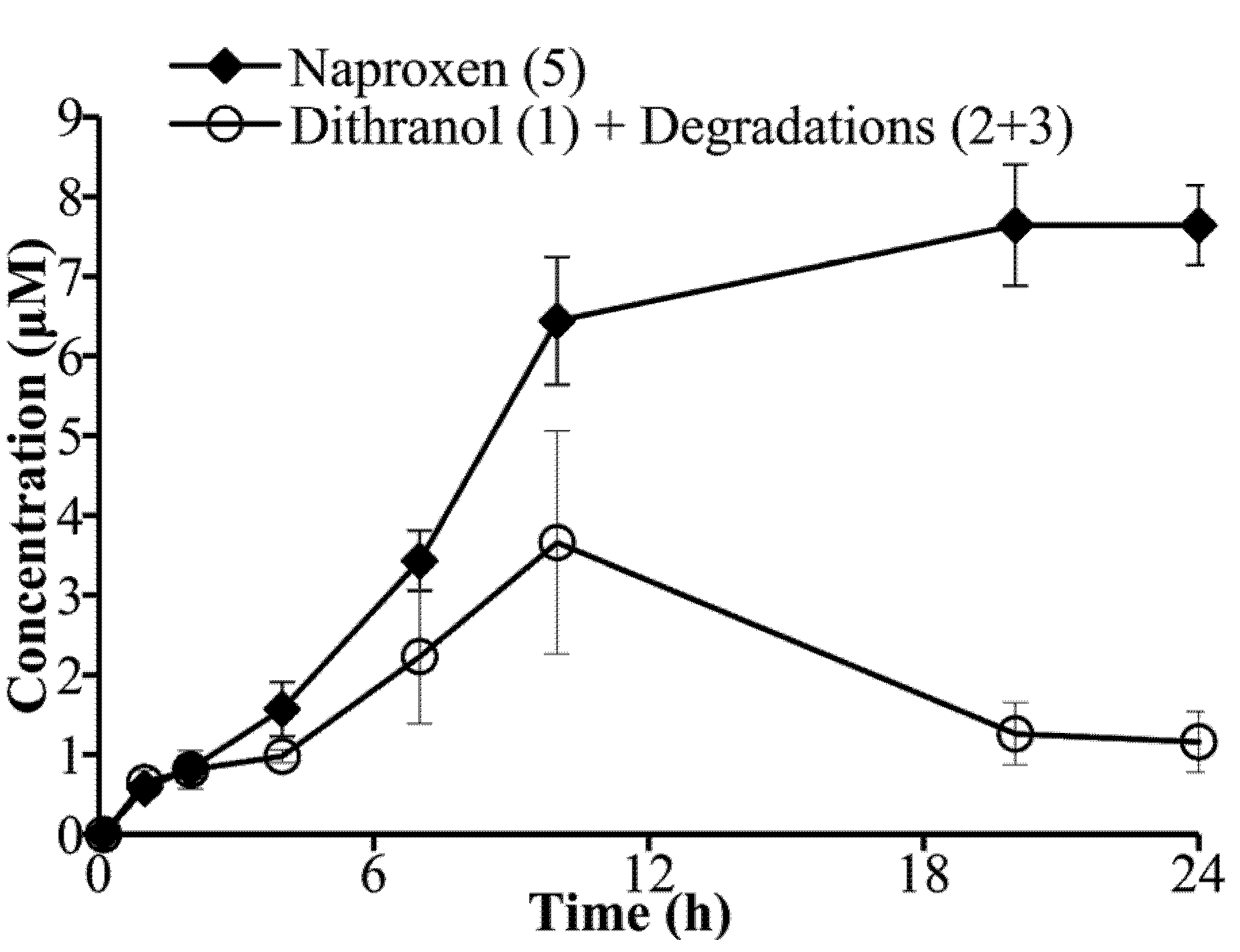
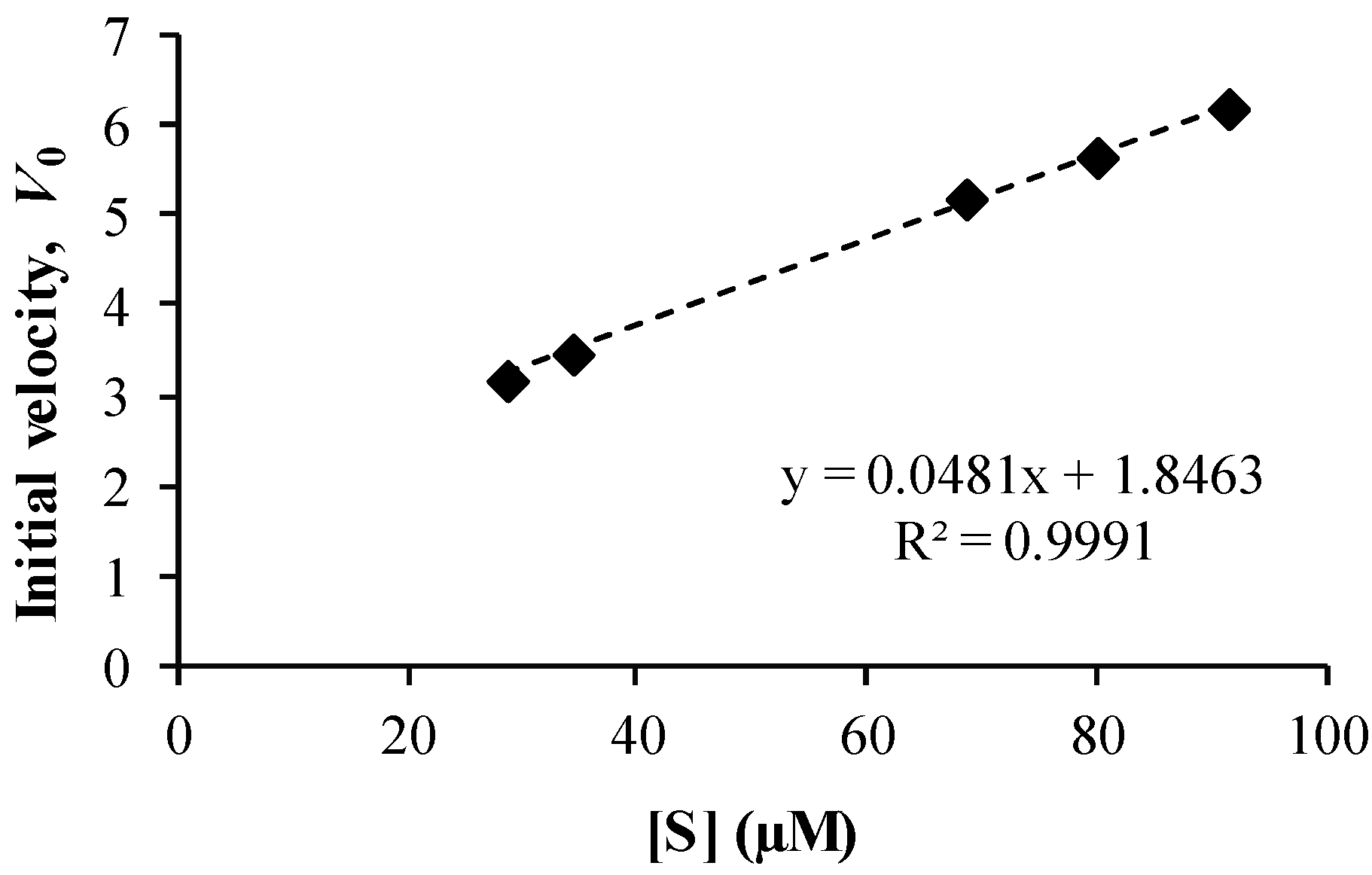
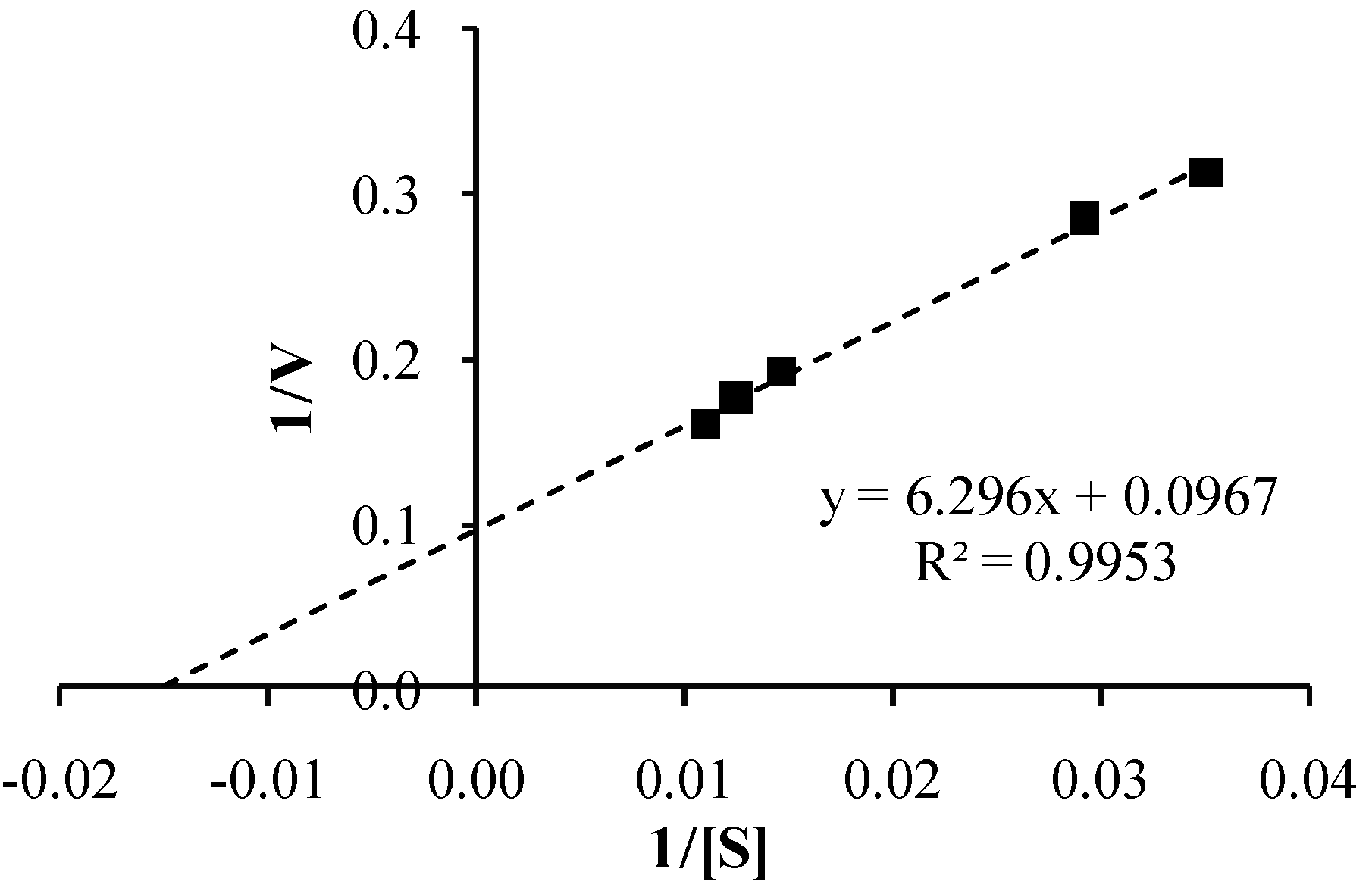
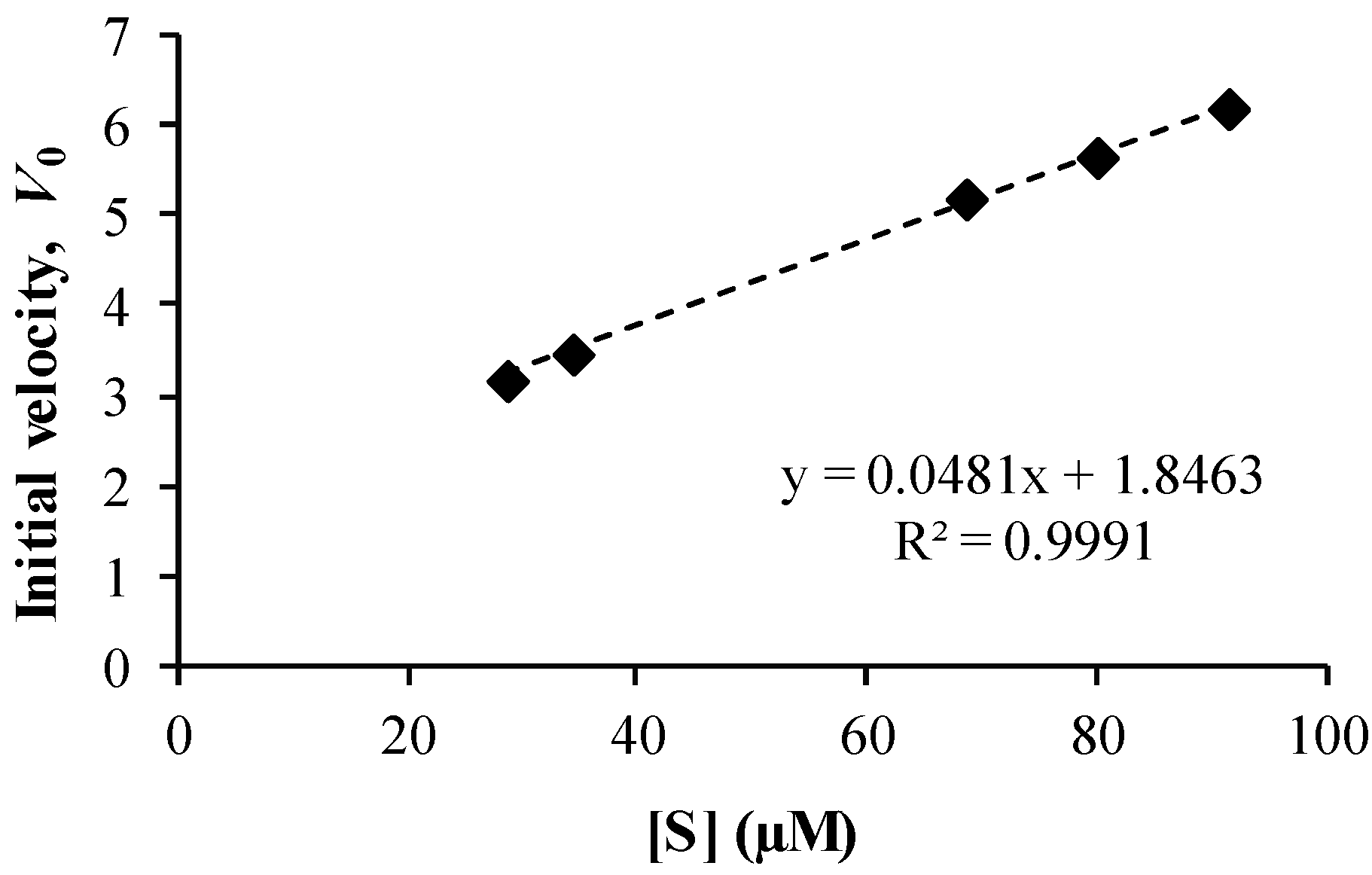
3.4. Co-Drug Hydrolysis Kinetics
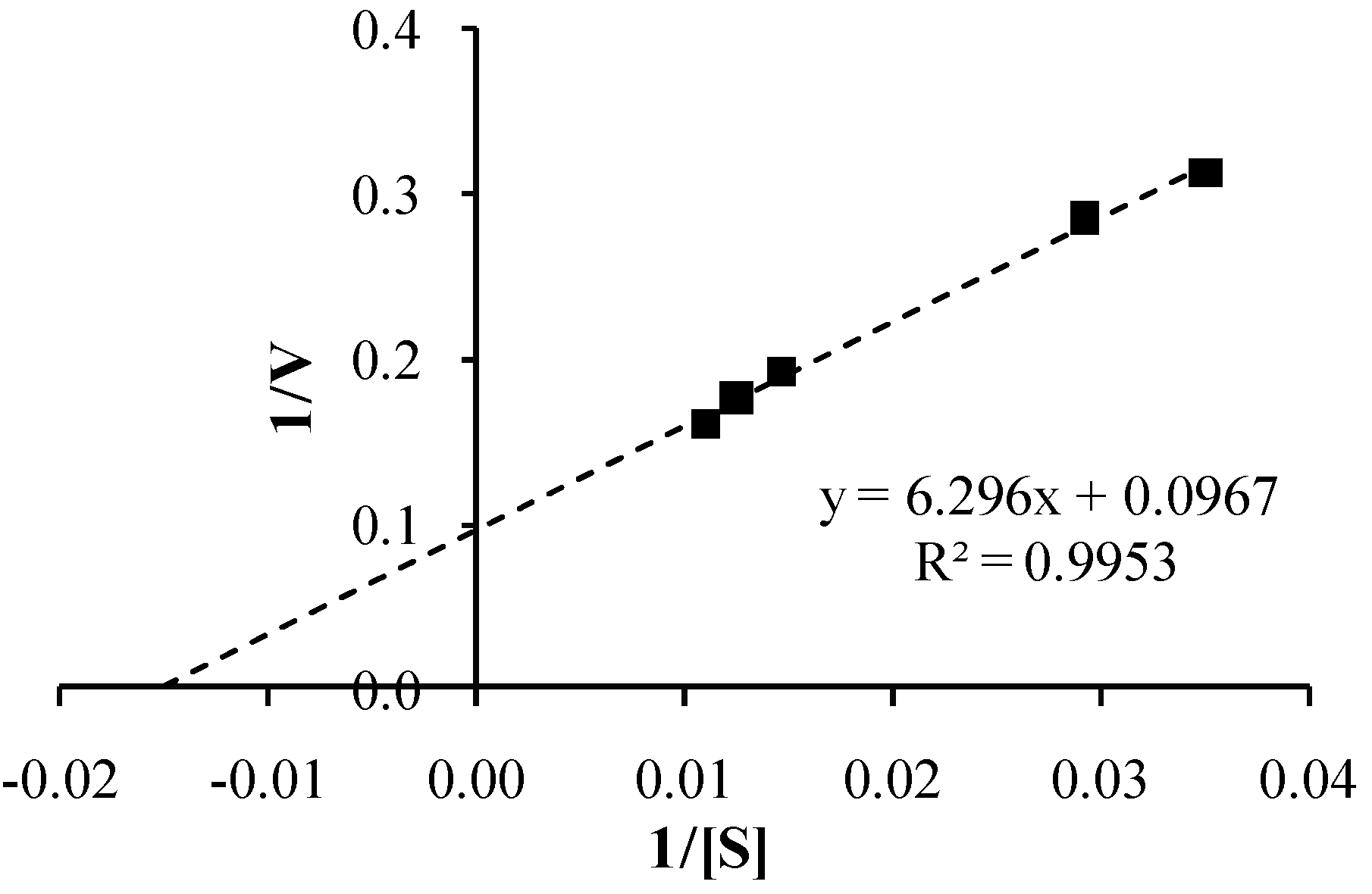
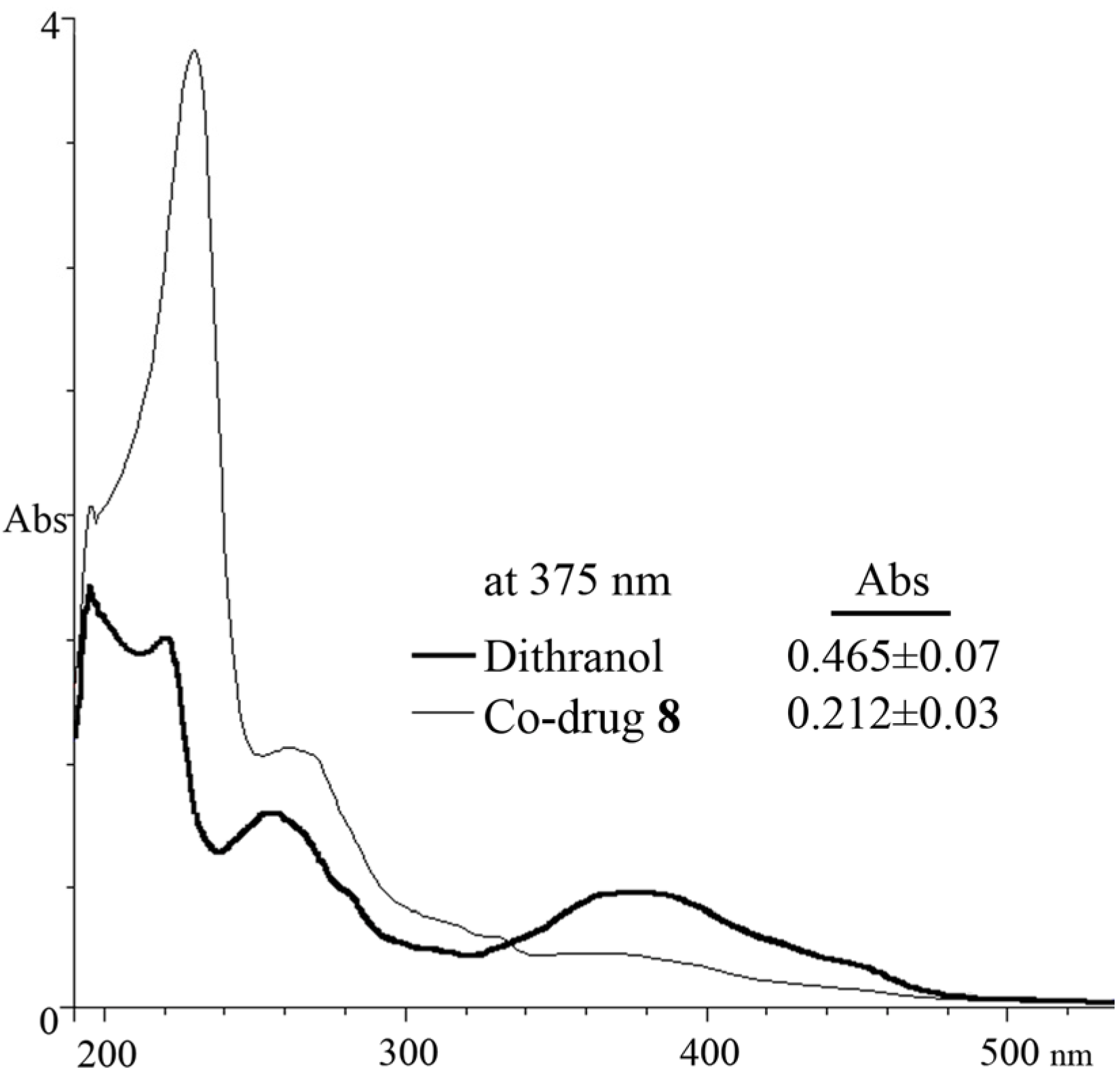
3.5. Spectrophotometric Analysis
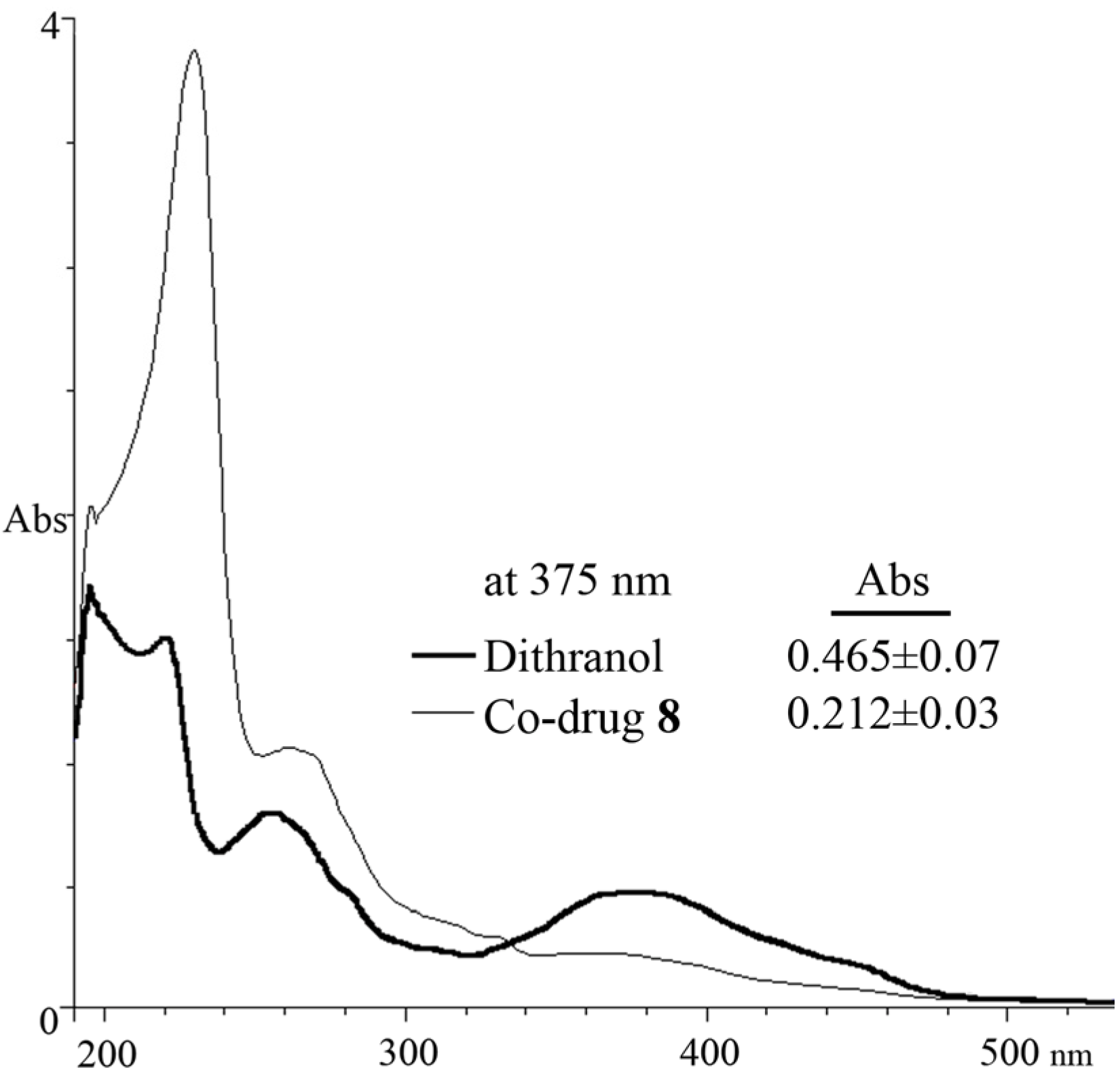
4. Conclusion
Acknowledgments
References
- Christophers, E. Psoriasis—Epidemiology and clinical spectrum. Clin. Exp. Dermatol. 2001, 26, 314–320. [Google Scholar] [CrossRef]
- Gordon, K.B.; Ruderman, E.M. The treatment of psoriasis and psoriatic arthritis: An interdisciplinary approach. J. Am. Acad. Dermatol. 2006, 54, S86–S91. [Google Scholar]
- Rácz, E.; Prens, E.P. Molecular pathophysiology of psoriasis and molecular targets of antipsoriatic therapy. Expert Rev. Mol. Med. 2009, 11, e38. [Google Scholar] [CrossRef]
- Clark, A.R. Topical noncorticosteroid therapies for psoriasis. Curr. Probl. Dermatol. 2000, 12, 230–232. [Google Scholar]
- Ryan, C.; Abramson, A.; Patel, M.; Menter, A. Current investigational drugs in psoriasis. Expert Opin. Investig. Drugs 2012, 21, 473–487. [Google Scholar] [CrossRef]
- Gillard, S.E.; Finlay, A.Y. Current management of psoriasis in the united kingdom: Patterns of prescribing and resource use in primary care. Int. J. Clin. Pract. 2005, 59, 1260–1267. [Google Scholar] [CrossRef]
- Lau, W.M.; White, A.W.; Gallagher, S.J.; Donaldson, M.; McNaughton, G.; Heard, C.M. Scope and limitations of the co-drug approach to topical drug delivery. Curr. Pharm. Des. 2008, 14, 794–802. [Google Scholar]
- Müller, K.; Leukel, P.; Mayer, K.K.; Wiegrebe, W. Modification of DNA bases by anthralin and related compounds. Biochem. Pharmacol. 1995, 49, 1607–1613. [Google Scholar] [CrossRef]
- McGill, A.; Frank, A.; Emmett, N.; Turnbull, D.M.; Birch-Machin, M.A.; Reynolds, N.J. The antipsoriatic drug anthralin accumulates in keratinocyte mitochondria, dissipates mitochondrial membrane potential, and induces apoptosis through a pathway dependent on respiratory competent mitochondria. FASEB J. 2005, 19, 1012–1014. [Google Scholar]
- Peus, D.; Beyerle, A.; Vasa, M.; Pott, M.; Meves, A.; Pittelkow, M.R. Antipsoriatic drug anthralin induces egf receptor phosphorylation in keratinocytes: Requirement for H2O2 generation. Exp. Dermatol. 2004, 13, 78–85. [Google Scholar]
- Reichert, U.; Jacques, Y.; Grangeret, M.; Schmidt, R. Antirespiratory and antiproliferative activity of anthralin in cultured human keratinocytes. J. Invest. Dermatol. 1985, 84, 130–134. [Google Scholar] [CrossRef]
- Van Duuren, B.L.; Segal, A.; Tseng, S.S.; Rusch, G.M.; Loewengart, G.; Mate, U.; Roth, D.; Smith, A.; Melchionne, S.; Seidman, I. Structure and tumor-promoting activity of analogs of anthralin (1,8-dihydroxy-9-anthrone). J. Med. Chem. 1978, 21, 26–31. [Google Scholar] [CrossRef]
- Mahrle, G. Dithranol. Clin. Dermatol. 1997, 15, 723–737. [Google Scholar] [CrossRef]
- Müller, K.; Breu, K.; Reindl, H. 10-Phenylbutyryl-substituted anthracenones as inhibitors of keratinocyte growth and ltb4 biosynthesis. Eur. J. Med. Chem. 2001, 36, 179–184. [Google Scholar] [CrossRef]
- Müller, K.; Altmann, R.; Prinz, H. 10-Benzoyl-1,8-dihydroxy-9(10H)-anthracenones: Synthesis and biological properties. Eur. J. Med. Chem. 1998, 33, 209–214. [Google Scholar] [CrossRef]
- Prinz, H.; Wiegrebe, W.; Müller, K. Syntheses of anthracenones. 3. Revised preparative route to 10-benzoyl-1,8-dihydroxy-9(10H)-anthracenones. J. Org. Chem. 1996, 61, 2861–2864. [Google Scholar] [CrossRef]
- Lau, W.M.; White, A.W.; Heard, C.M. Topical delivery of a naproxen-dithranol co-drug: In vitro skin penetration, permeation, and staining. Pharm. Res. 2010, 27, 2734–2742. [Google Scholar] [CrossRef]
- Bos, J.D.; Meinardi, M. The 500 dalton rule for the skin penetration of chemical compounds and drugs. Exp. Dermatol. 2000, 9, 165–169. [Google Scholar]
- Lau, W.M.; Ng, K.W.; White, A.W.; Heard, C.M. Therapeutic and cytotoxic effects of the novel antipsoriasis codrug, naproxyl-dithranol, on hacat cells. Mol. Pharm. 2011, 8, 2398–2407. [Google Scholar] [CrossRef]
- Lebwohl, M. The role of salicylic acid in the treatment of psoriasis. Int. J. Dermatol. 1999, 38, 16–24. [Google Scholar] [CrossRef]
- Carroll, C.; Clarke, J.; Camacho, F.; Balkrishnan, R.; Feldman, S.R. Topical tacrolimus ointment combined with 6% salicylic acid gel for plaque psoriasis treatment. Arch. Dermatol. 2005, 141, 43–46. [Google Scholar] [CrossRef]
- Rautio, J.; Nevalainen, T.; Taipale, H.; Vepsalainen, J.; Gynther, J.; Laine, K.; Jarvinen, T. Piperazinylalkyl prodrugs of naproxen improve in vitro skin permeation. Eur. J. Pharm. Sci. 2000, 11, 157–163. [Google Scholar] [CrossRef]
- Bonina, F.P.; Puglia, C.; Barbuzzi, T.; de Caprariis, P.; Palagiano, F.; Rimoli, M.G.; Saija, A. In vitro and in vivo evaluation of polyoxyethylene esters as dermal prodrugs of ketoprofen, naproxen and diclofenac. Eur. J. Pharm. Sci. 2001, 14, 123–134. [Google Scholar] [CrossRef]
- Auterhoff, H.; Scherff, F.C. Die dianthrone der pharmazeutisch interessierenden hydroxyanthrachinone. Arch. Pharm. (Weinheim) 1960, 293, 918–925. [Google Scholar] [CrossRef]
- Liang, W.M.; Du, C.J. Potent antipsoriatic agents: A facile preparation of acylated derivatives from dithranol in a mild basic reaction. J. Chin. Chem. Soc. 2004, 51, 115–118. [Google Scholar]
- Abdulmajed, K.; McGuigan, C.; Heard, C.M. Topical delivery of retinyl ascorbate co-drug: 5. In vitro degradation studies. Skin Pharmacol. Physiol. 2006, 19, 248–258. [Google Scholar] [CrossRef]
- Thomas, C.P.; Heard, C.M. Probing the skin permeation of eicosapentaenoic acid and ketoprofen: 2. Comparative depth profiling and metabolism of eicosapentaenoic acid. Eur. J. Pharm. Biopharm. 2007, 67, 156–165. [Google Scholar] [CrossRef]
- Vallet, V.; Cruz, C.; Josse, D.; Bazire, A.; Lallement, G.; Boudry, I. In vitro percutaneous penetration of organophosphorus compounds using full-thickness and split-thickness pig and human skin. Toxicol. Vitro 2007, 21, 1182–1190. [Google Scholar] [CrossRef]
- Simon, G.A.; Maibach, H.I. The pig as an experimental animal model of percutaneous permeation in man: Qualitative and quantitative observations—An overview. Skin Pharmacol. Appl. Skin Physiol. 2000, 13, 229–234. [Google Scholar] [CrossRef]
- Brandt, H.; Mustakallio, K. Irritation and staining by dithranol (anthralin) and related compounds. III. Cumulative irritancy and staining during repeated chamber testing. Acta Derm. Venereol. 1983, 63, 237–240. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lau, W.M.; Heard, C.M.; White, A.W. Design, Synthesis and in Vitro Degradation of a Novel Co-Drug for the Treatment of Psoriasis. Pharmaceutics 2013, 5, 232-245. https://doi.org/10.3390/pharmaceutics5020232
Lau WM, Heard CM, White AW. Design, Synthesis and in Vitro Degradation of a Novel Co-Drug for the Treatment of Psoriasis. Pharmaceutics. 2013; 5(2):232-245. https://doi.org/10.3390/pharmaceutics5020232
Chicago/Turabian StyleLau, Wing Man, Charles M. Heard, and Alex W. White. 2013. "Design, Synthesis and in Vitro Degradation of a Novel Co-Drug for the Treatment of Psoriasis" Pharmaceutics 5, no. 2: 232-245. https://doi.org/10.3390/pharmaceutics5020232
APA StyleLau, W. M., Heard, C. M., & White, A. W. (2013). Design, Synthesis and in Vitro Degradation of a Novel Co-Drug for the Treatment of Psoriasis. Pharmaceutics, 5(2), 232-245. https://doi.org/10.3390/pharmaceutics5020232