Safety Monitoring in Clinical Trials

Abstract
:1. Introduction
2. Common Practice in Safety Monitoring
2.1. Stakeholders in Safety Monitoring
2.1.1. Sponsor
2.1.2. Subjects
2.1.3. Investigators
2.1.4. Institutional Review Board/Ethics Committee
2.1.5. Data and Safety Monitoring Board
2.1.6. Regulatory Authorities
2.1.7. Medical Community and Patients
2.2. Communicating Safety Information among Stakeholders
3. Statistical Methods in Safety Monitoring
3.1. Methods for Single Arm Trials
- if Λn ≥ Un, conclude π1;
- if Λn ≤ Ln, conclude π0;
- if Ln < Λn < Un and n < nmax, then continue the trial.
- if xi ≥ Ui, conclude π greater than πS;
- if xi ≤ Li, conclude π less than πS + δ;
- if Li < xi < Ui and i < nmax, then continue the trial.
3.2. Methods for Randomized, Controlled Trials
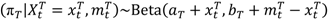
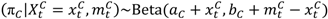
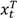 and
and 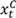 are the numbers of events from the treatment and the control arm and
are the numbers of events from the treatment and the control arm and 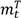 and
and 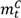 are the numbers of subjects on the treatment arm and the control arm up to time t.
are the numbers of subjects on the treatment arm and the control arm up to time t. 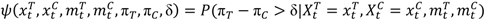
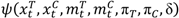 > p, where p is the predetermined upper threshold probability, for example, p = 0.9, the risk is considered alarming to warrant considerations of stopping the trial due to imbalance of toxicities.
> p, where p is the predetermined upper threshold probability, for example, p = 0.9, the risk is considered alarming to warrant considerations of stopping the trial due to imbalance of toxicities. 3.3. A Hypothetical Clinical Trial Example

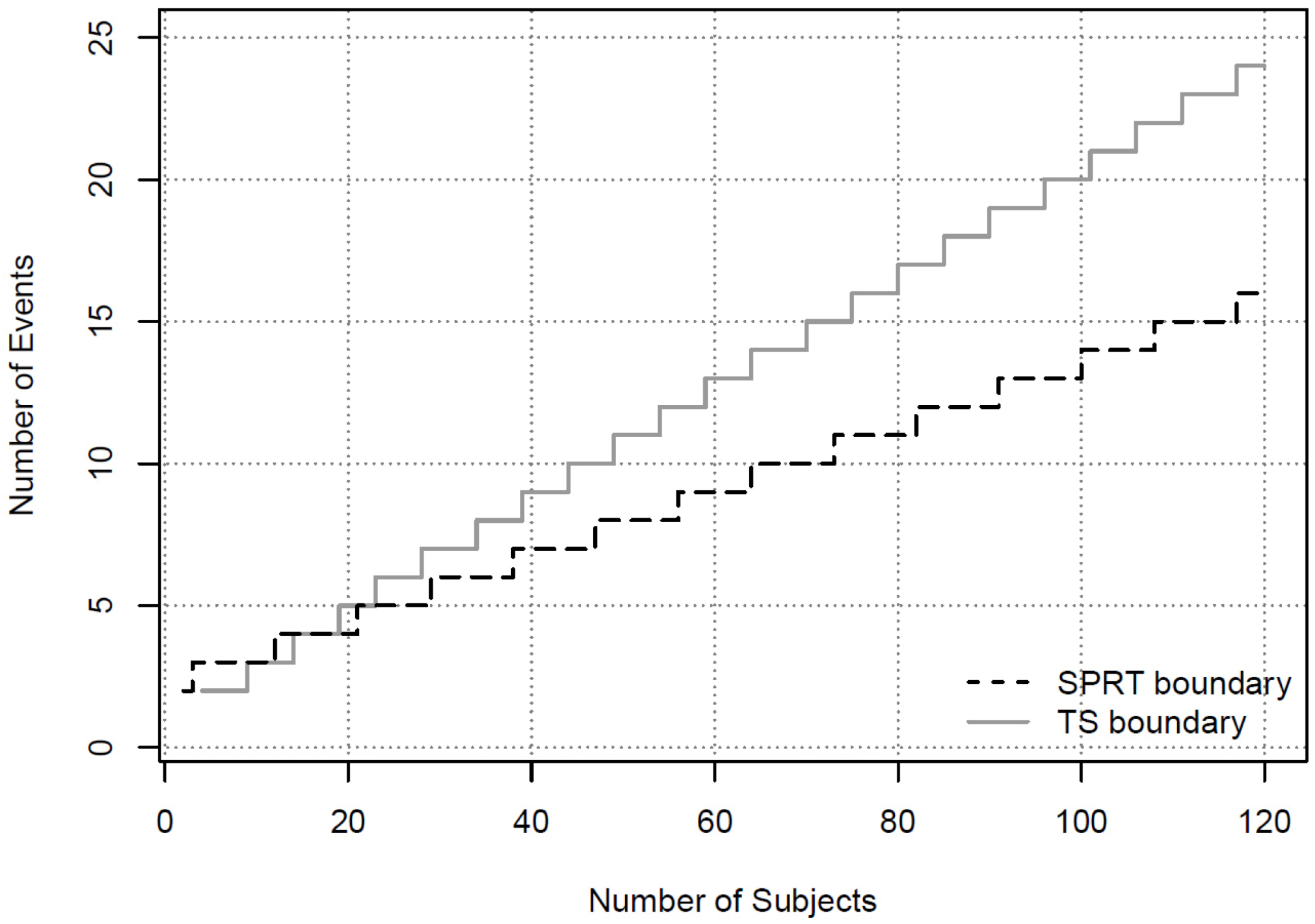{kind=link}
| Number of Subjects | ||
|---|---|---|
| Number of Events | SPRT | TS |
| 1 | n.a. | n.a. |
| 2 | 2 | ≤3 |
| 3 | ≤11 | ≤8 |
| 4 | ≤20 | ≤13 |
| 5 | ≤28 | ≤18 |
| 6 | ≤37 | ≤22 |
| 7 | ≤46 | ≤27 |
| 8 | ≤55 | ≤33 |
| 9 | ≤63 | ≤38 |
| 10 | ≤72 | ≤43 |

 > 0.9, the operating characteristics for such a monitoring guideline can be computed through simulations. under all possible scenarios. In this table, all the bolded numbers highlight the scenarios in which the stopping boundary is crossed. For instance, if at this analysis time there are 3 subjects on the treatment arm who experience the AESI, while none of the subjects on the control arm has the AESI, then = 0.92 and the boundary is crossed.
> 0.9, the operating characteristics for such a monitoring guideline can be computed through simulations. under all possible scenarios. In this table, all the bolded numbers highlight the scenarios in which the stopping boundary is crossed. For instance, if at this analysis time there are 3 subjects on the treatment arm who experience the AESI, while none of the subjects on the control arm has the AESI, then = 0.92 and the boundary is crossed.| Treatment Arm: Events/Subjects | |||||||||
| 1/8 | 2/8 | 3/8 | 4/8 | 5/8 | 6/8 | 7/8 | 8/8 | ||
| Control Arm: Events/Subjects | 0/11 | 0.65 | 0.82 | 0.92 | 0.97 | 0.99 | 1 | 1 | 1 |
| 1/11 | 0.58 | 0.77 | 0.89 | 0.96 | 0.98 | 1 | 1 | 1 | |
| 2/11 | 0.52 | 0.71 | 0.85 | 0.94 | 0.97 | 0.99 | 1 | 1 | |
| 3/11 | 0.45 | 0.65 | 0.81 | 0.91 | 0.96 | 0.99 | 1 | 1 | |
| 4/11 | 0.39 | 0.59 | 0.76 | 0.88 | 0.94 | 0.98 | 0.99 | 1 | |
| 5/11 | 0.34 | 0.53 | 0.71 | 0.84 | 0.93 | 0.97 | 0.99 | 1 | |
| 6/11 | 0.29 | 0.47 | 0.66 | 0.81 | 0.90 | 0.96 | 0.98 | 0.99 | |
| 7/11 | 0.24 | 0.42 | 0.60 | 0.76 | 0.87 | 0.94 | 0.97 | 0.99 | |
| 8/11 | 0.21 | 0.37 | 0.55 | 0.71 | 0.84 | 0.92 | 0.96 | 0.99 | |
| 9/11 | 0.17 | 0.32 | 0.50 | 0.67 | 0.80 | 0.90 | 0.95 | 0.98 | |
| 10/11 | 0.14 | 0.27 | 0.44 | 0.61 | 0.76 | 0.87 | 0.93 | 0.97 | |
| 11/11 | 0.11 | 0.23 | 0.39 | 0.56 | 0.72 | 0.84 | 0.92 | 0.96 | |
4. Conclusions
Conflict of Interest
References
- International Conference on Harmonization (ICH). Guideline for Good Clinical Practice E6(R1), 1996. Available online: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6_R1/Step4/E6_R1__Guideline.pdf (accessed on 8 October 2010).
- Council for International Organizations of Medical Sciences (CIOMS) in collaboration with the World Health Organization (WHO). International Ethical Guidelines for Biomedical Research Involving Human Subjects. CIOMS & WHO: Geneva, Switzerland, 2002.
- The Declaration of Helsinki; World Medical Association: Somerset West, South Africa, 1996.
- European Clinical Trials Directive 2001/20/EC. Available online: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2001:121:0034:0044:en:PDF (accessed on 09 October 2012).
- United States Food and Drug Administration. Guidance for Industry, Premarketing Risk Assessment, 2005. Available online: http://www.fda.gov/downloads/RegulatoryInformation/Guidances/ucm126958.pdf (accessed on 09 October 2012).
- Food and Drug Administration (FDA). Guidance for Industry, Development and Use of Risk Minimization Action Plans, 2005. Available online: http://www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM126830.pdf (accessed on 09 October 2012).
- Food and Drug Administration (FDA). Guidance for Industry, Good Pharmacovigilance Practices and Pharmacoepidemiologic Assessment, 2005. Available online: http://www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM126834.pdf (accessed on 09 October 2012).
- Grady, C. Payment of Clinical Research Subjects. J. Clin. Invest. 2005, 115, 1681–1687. [Google Scholar] [CrossRef]
- Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research. Guidance for industry and investigators: Safety reporting requirements for INDs and BA/BE studies, 2010. Available online: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM227351.pdf (accessed on 09 October 2012).
- Council for International Organizations of Medical Sciences (CIOMS) Working Group VI, Management of safety information from clinical trials. CIOMS: Geneva, Switzerland, 2005.
- Crowe, B.J.; Xia, H.A.; Berlin, J.A.; Watson, D.J.; Shi, H.; Lin, S.L.; Kuebler, J.; Schriver, R.C.; Santanello, N.C.; Rochester, G.; et al. Recommendations for safety planning, data collection, evaluation and reporting during drug, biologic and vaccine development: A report of the safety planning, evaluation andreporting team. Clin. Trials 2009, 6, 430–440. [Google Scholar]
- Xia, H.A.; Crowe, B.J.; Schriver, R.C.; Oster, M.; Hall, D.B. Planning and core analyses for periodic aggregate safety data reviews. Clin. Trials 2011, 8, 175–182. [Google Scholar] [CrossRef]
- Wald, A. Sequential tests of statistical hypotheses. Ann. Math. Stat. 1945, 16, 117–186. [Google Scholar] [CrossRef]
- Wald, A. Sequential Analysis; Wiley: New York, NY, USA, 1947. [Google Scholar]
- Goldman, A.; Hannan, P. Optimal continuous sequential boundaries for monitoring toxicity in clinical trials: A restricted search algorithm. Stat. Med. 2001, 20, 1575–1589. [Google Scholar] [CrossRef]
- Thall, C.P.; Simon, R. Practical Bayesian guidelines for phase IIB clinical trials. Biometrics 1994, 50, 337–349. [Google Scholar] [CrossRef]
- Yin, G. Clinical Trial Design: Bayesian and Frequentist Adaptive Methods. Wiley: Hoboken, NJ, USA, 2012. [Google Scholar]
- Ball, G. Continuous safety monitoring for randomized controlled clinical trials with blinded treatment information. Contemp. Clin. Trials 2011, 32, S11–S17. [Google Scholar] [CrossRef]
- SISA. Wald’s Sequential Probability. Available online: http://www.quantitativeskills.com/sisa/statistics/sprt.htm (accessed on 09 October 2012).
- MD Anderson Cancer Center, Software Download Site, Multc Lean. Available online: https://biostatistics.mdanderson.org/SoftwareDownload (accessed on 09 October 2012).
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yao, B.; Zhu, L.; Jiang, Q.; Xia, H.A. Safety Monitoring in Clinical Trials. Pharmaceutics 2013, 5, 94-106. https://doi.org/10.3390/pharmaceutics5010094
Yao B, Zhu L, Jiang Q, Xia HA. Safety Monitoring in Clinical Trials. Pharmaceutics. 2013; 5(1):94-106. https://doi.org/10.3390/pharmaceutics5010094
Chicago/Turabian StyleYao, Bin, Li Zhu, Qi Jiang, and H. Amy Xia. 2013. "Safety Monitoring in Clinical Trials" Pharmaceutics 5, no. 1: 94-106. https://doi.org/10.3390/pharmaceutics5010094