Diclofenac Salts, VIII. Effect of the Counterions on the Permeation through Porcine Membrane from Aqueous Saturated Solutions

Abstract
:1. Introduction
2. Results and Discussion
2.1. Nature of the Salts

{kind=link}
{kind=link}
| Diclofenac Salts | J (μg cm−2 h−1) | Q8 (μg cm−2) × 103 | S1 (μg cm−3) × 103 | D (cm h−1) × 103 | logP (salt) | logP (free base) | S2 (mM) | pKa (base) | pH * (sat. sol.) |
|---|---|---|---|---|---|---|---|---|---|
| MEtA | 12 | 9.3 | 6.1 | 2 | 0.01 | −0.13 | 17.9 | 10.84 | 7.62 |
| MEA | 6.9 | 5.6 | 9.9 | 0.7 | 0.08 | −1.31 | 26.4 | 9.50 | 6.95 |
| DEtA | 48 | 1.9 | 13.7 | 3.7 | 0.17 | 0.58 | 35.4 | 10.75 | 7.58 |
| DEA | 51 | 1.8 | 18.0 | 2.8 | 0.08 | −1.43 | 44.9 | 8.97 | 6.68 |
| TEtA | 23 | 0.6 | 6.7 | 3.4 | 0.85 | 1.45 | 16.1 | 10.75 | 7.58 |
| TEA | 20 | 0.6 | 3.4 | 3.0 | 0.64 | −1.59 | 7.6 | 7.76 | 6.08 |
| M | 26 | 1.1 | 6.9 | 3.8 | 0.35 | −0.72 | 18.0 | 8.50 | 6.45 |
| HEM | 21 | 0.3 | 4.4 | 4.8 | 1.04 | −0.489 * | 9.7 | 6.89 | 5.64 |
| Pz | 18 | 0.1 | 0.4 | 45 | 0.67 | −1.17 | 0.59 | 4.19 | 4.29 |
| HEPz | 160 | 0.4 | 12.5 | 13 | 0.24 | −0.957 * | 29.3 | 9.05 | 6.72 |
| PP | 87 | 0.6 | 4.3 | 20 | 0.97 | 0.80 | 11.3 | 11.12 | 7.75 |
| HEPp | 82 | 0.9 | 10.7 | 7.7 | 0.29 | 0.449 * | 25.2 | 9.66 | 7.03 |
| Py | 42 | 0.3 | 2.0 | 21 | 0.21 | 0.46 | 5.1 | 11.32 | 7.85 |
| HEPy | 194 | 1.5 | 20.2 | 9.6 | 0.17 | 0.076 * | 45.2 | 9.72 | 7.06 |
| Na | 0.58 ** | ||||||||
| 1.3 *** | |||||||||
| Acid | 3500 *** |
2.2. Solubility of the Salts
2.3. Partition Coefficient of the Salts
2.4. The Choice of Animal Membrane
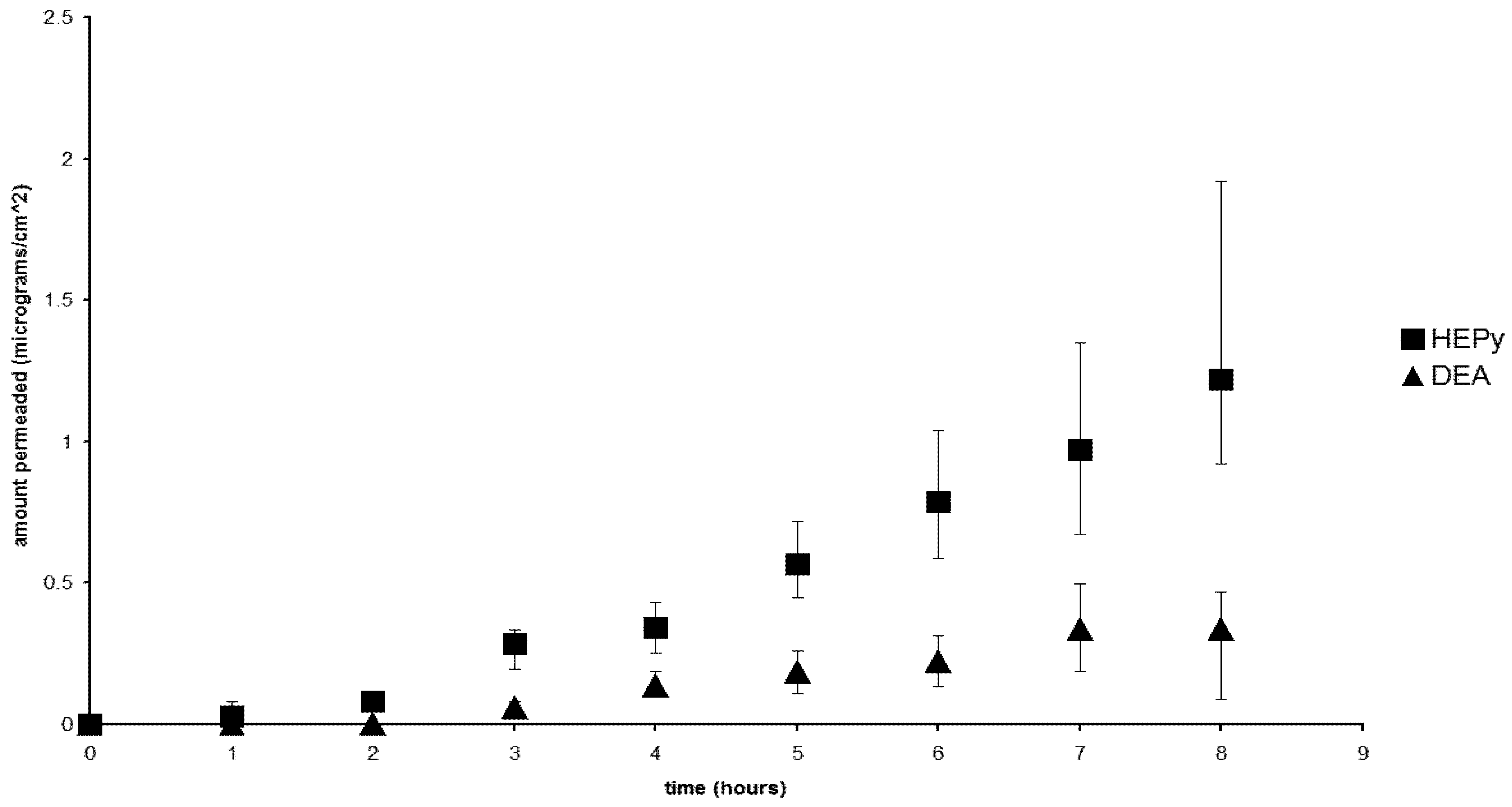
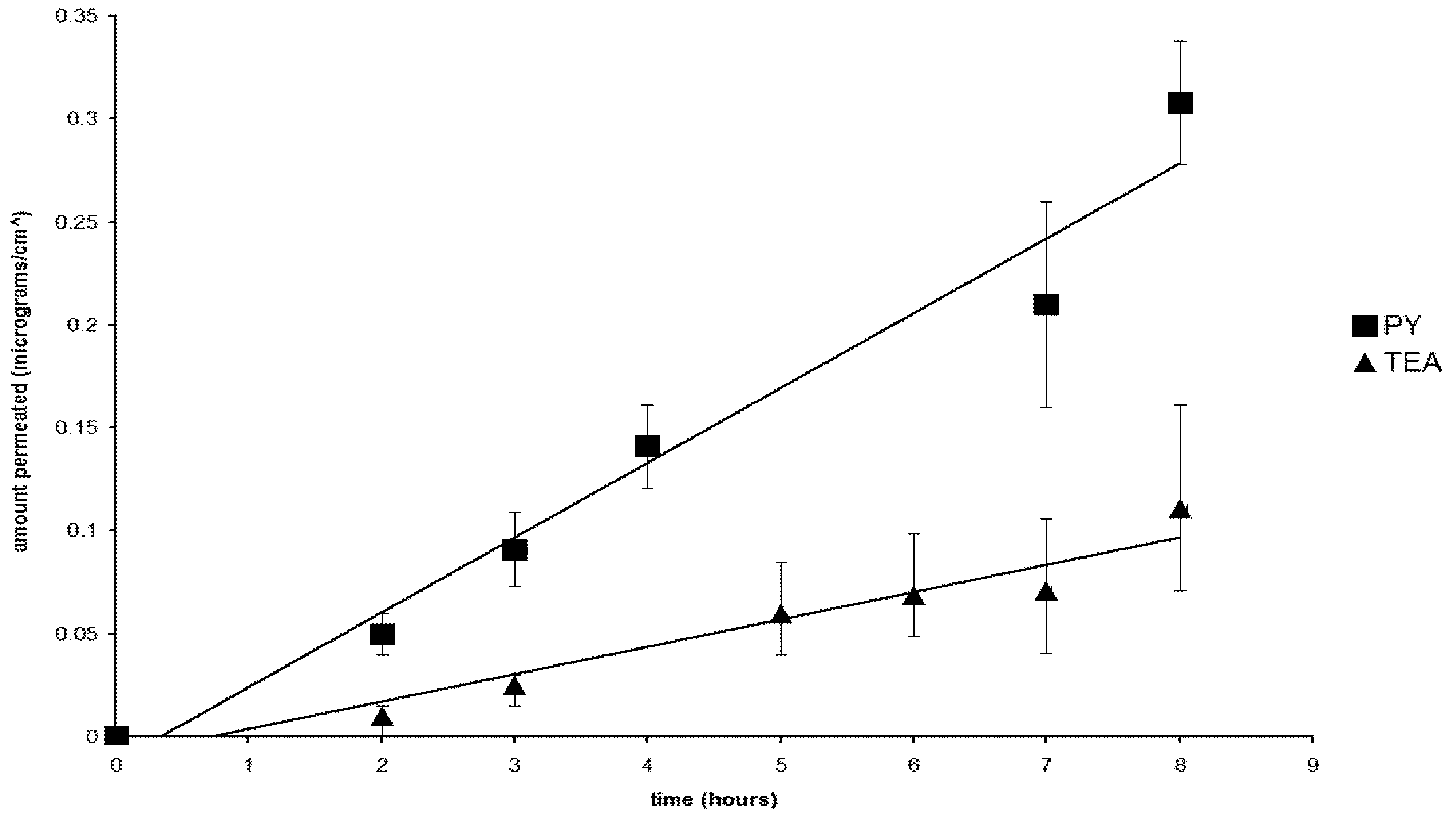
2.5. Permeation of the Salts


2.6. The Nature of the Permeant Species
2.7. The Effect of the Counterions
3. Experimental Section
3.1. Materials
3.2. Preparation of the Diclofenac Salts
3.3. Solubility Studies
3.4. Determination of n-Octanol-Distilled Water Partition Coefficient of the Salts
3.5. Permeation Experiments
3.6. Permeation Parameters
3.7. Analytical Method
3.8. In vitro Data Treatment
4. Conclusions
- • Diclofenac permeates porcine membrane when in the form of salts with simple aliphatic amines and interesting fluxes could be measured when the salts are released from saturated aqueous solution;
- • Permeation coefficient was found higher when the counterion contains a ring; while hydroxy groups alone do not appear to play an important role, the ring could sustain permeation, disrupting the organized domains of the membrane;
- • Different chemical species (acid, anion, ion-pair) contribute to permeation of the anti-inflammatory agent, even though ion-pairs could be hypothesized to operate to a greater extent;
- • Potential toxicity of the aliphatic amines, since their cation accompanies the active moiety during the absorption of the ion pairs, cannot be ignored in evaluating the permeation results.
Acknowledgments
Conflict of interest
References
- Fang, J.Y.; Wang, R.J.; Huang, Y.B.; Wu, P.C.; Tsai, Y.H. Influence of electrical and chemical factors on transdermal iontophoretic delivery of three diclofenac salts. Biol. Pharm. Bull. 2001, 24, 390–394. [Google Scholar] [CrossRef]
- Kamal, M.A.; Nabekura, T.; Kitagawa, S. Permeability of ionized salicylate derivatives through guinea pig dorsal skin. Chem. Pharm. Bull. (Tokyo) 2005, 53, 441–443. [Google Scholar] [CrossRef]
- Van der Bijl, P.; van Eyk, A.D.; Seifart, H.I.; Viljoen, I.; Jooste, M. Transmucosal permeation of topically applied diclofenac and piroxicam. J. App. Res. 2003, 3, 505–511. [Google Scholar]
- Nokhodchi, A.; Sharabiani, K.; Rashidi, M.R.; Ghafourian, T. The effect of terpene concentrations on the skin penetration of diclofenac sodium. Int. J. Pharm. 2007, 335, 97–105. [Google Scholar] [CrossRef]
- Sarıgüllü Özgüney, I.; Yeşim Karasulu, H.; Kantarcı, G.; Sözer, S.; Güneri, T.; Ertan, G. Transdermal delivery of diclofenac sodium through rat skin from various formulations. AAPS PharmSciTech 2006, 7, E39–E45. [Google Scholar] [CrossRef]
- Fini, A.; Fazio, G.; Gonzalez-Rodriguez, M.; Cavallari, C.; Passerini, N.; Rodriguez, L. Formation of ion-pairs in aqueous solutions of diclofenac salts. Int. J. Pharm. 1999, 187, 163–173. [Google Scholar] [CrossRef]
- Minghetti, P.; Cilurzo, F.; Casiraghi, A.; Montanari, L.; Fini, A. Ex vivo study of transdermal permeation of four diclofenac salts from different vehicles. J. Pharm. Sci. 2007, 96, 814–823. [Google Scholar] [CrossRef]
- Parsaee, S.; Sarbolouki, M.N.; Parnianpour, M. In vitro release of diclofenac diethylammonium from lipid-based formulations. Int. J. Pharm. 2002, 241, 185–190. [Google Scholar] [CrossRef]
- Arora, P.; Mukherjee, B. Design, development, physicochemical, and in vitro and in vivo evaluation of transdermal patches containing diclofenac diethylammonium salt. J. Pharm. Sci. 2002, 91, 2076–2089. [Google Scholar] [CrossRef]
- Kweon, J.H.; Chi, S.C.; Park, E.S. Transdermal delivery of diclofenac using microemulsions. Arch. Pharm. Res. 2004, 27, 351–356. [Google Scholar] [CrossRef]
- Manosroi, A.; Jantrawut, P.; Manosroi, J. Anti-inflammatory activity of gel containing novel elastic niosomes entrapped with diclofenac diethylammonium. Int. J. Pharm. 2008, 360, 156–163. [Google Scholar] [CrossRef]
- O’Connor, K.M.; Corrigan, O.I. Preparation and characterisation of a range of diclofenac salts. Int. J. Pharm. 2001, 226, 163–179. [Google Scholar] [CrossRef]
- Fini, A.; Fazio, G.; Benetti, L. Diclofenac salts with alkyl and alkylhydroxy amines. Thermochim. Acta 2007, 464, 65–74. [Google Scholar] [CrossRef]
- Fini, A.; Cavallari, C.; Rabasco Alvarez, A.M.; Rodriguez, M.L. Diclofenac salts, part 6: Release from lipid microspheres. J. Pharm. Sci. 2011, 100, 3482–3494. [Google Scholar] [CrossRef]
- Fini, A.; Cavallari, C.; Bassini, G.; Ospitali, F.; Morigi, R. Diclofenac salts, part 7: Are the pharmaceutical salts with aliphatic amines stable? J. Pharm. Sci. 2012, 101, 3157–3168. [Google Scholar] [CrossRef]
- Simon, G.A.; Maibach, H.I. The pig as an experimental animal model of percutaneous permeation in man: Qualitative and quantitative observations-An overview. Skin Pharmacol. Appl. Skin Physiol. 2000, 13, 229–234. [Google Scholar]
- Jacobi, U.; Taube, H.; Schaefer, U.F.; Sterry, W.; Lademann, J. Comparison of four different in vitro systems to study the reservoir capacity of the stratum corneum. J. Control. Rel. 2005, 103, 61–71. [Google Scholar] [CrossRef]
- Jacobi, U.; Kaiser, M.; Toll, R.; Mangelsdorf, S.; Audring, H.; Otberg, N.; Sterry, W.; Lademann, J. Porcine ear skin: An in vitro model for human skin. Skin Res. Technol. 2007, 13, 19–24. [Google Scholar] [CrossRef]
- Cordero, J.A.; Alarcon, L.; Escribano, E.; Obach, R.; Domènech, J. A comparative study of the transdermal penetration of a series of non steroidal anti-inflammatory drugs. J. Pharm. Sci. 1997, 86, 503–508. [Google Scholar] [CrossRef]
- Vincent, C.-M.; Laugel, C.; Marty, J.-P. In vitro topical delivery of non-steroidal anti-inflammatory drugs through human skin. Arzneimittelforschung Drug Res. 1999, 49, 509–513. [Google Scholar]
- Patel, K.N.; Patel, H.K.; Patel, V.A. Formulation and characterization of drug in adhesive transdermal patches of diclofenac acid. Int. J. Pharm. Pharm. Sci. 2012, 4, 296–299. [Google Scholar]
- Kantarcı, G.; Özgüney, I.; Karasulu, H.Y.; Güneri, T.; Başdemir, G. In vitro permeation of diclofenac sodium from novel microemulsion formulations through rabbit skin. Drug Dev. Res. 2005, 65, 17–25. [Google Scholar] [CrossRef]
- Escribano, E.; Calpena, A.C.; Queralt, J.; Obach, R.; Doménech, J. Assessment of diclofenac permeation with different formulations: Anti-inflammatory study of a selected formula. Eur. J. Pharm. Sci. 2003, 19, 203–210. [Google Scholar] [CrossRef]
- Rana, V.; Rai, P.; Tiwary, A.K.; Gupta, S. Enhanced in vitro percutaneous permeation of diclofenac sodium with primary amine and pyrrolidone ion-pairs. Indian Drugs 1999, 36, 21–28. [Google Scholar]
- Sintov, A.; Botner, S. Transdermal drug delivery using microemulsion and aqueous systems: Influence of skin storage conditions on the in vitro permeability of diclofenac from aqueous vehicle systems. Int. J. Pharm. 2006, 311, 55–62. [Google Scholar] [CrossRef]
- Fini, A.; Fazio, G.; Orienti, I.; Zecchi, V.; Rapaport, I. Chemical properties-dissolution relationship. IV. Behaviour in solution of the diclofenac/N-(2-hydroxyethyl) pyrrolidine salt (DHEP). Pharm. Acta Helv. 1991, 66, 201–203. [Google Scholar]
- Fini, A.; Fazio, G.; Feroci, G. Solubility and solubilization properties of non-steroidal anti-inflammatory drugs. Int. J. Pharm. 1995, 126, 95–102. [Google Scholar] [CrossRef]
- Singh, U.V.; Pandey, S.; Udupa, N. Preparation and evaluation of flurbiprofen and diclofenac sodium transdermal films. Indian J. Pharm. Sci. 1993, 54, 145–147. [Google Scholar]
- Balon, K.; Riebesehl, B.U.; Müller, B.W. Drug liposome partitioning as a tool for the prediction of human passive intestinal absorption. Pharm. Res. 1999, 16, 882–888. [Google Scholar] [CrossRef]
- Sarveiya, V.; Templeton, J.F.; Benson, H.A.E. Ion-pairs of ibuprofen: Increased membrane diffusion. J. Pharm. Pharmacol. 2004, 56, 717–724. [Google Scholar]
- Fritz, P.; Schmook, J.G.; Meingasner, A.B. Comparison of human skin or epidermis models with human and animal skin in in-vitro percutaneous absorption. Int. J. Pharm. 2001, 215, 51–56. [Google Scholar] [CrossRef]
- Sanna, V.; Peana, A.T.; Moretti, M.D. Effect of vehicle on diclofenac sodium permeation from new topical formulations: In vitro and in vivo studies. Curr. Drug Deliv. 2009, 6, 93–100. [Google Scholar] [CrossRef]
- Rainsford, K.D.; Kean, W.F.; Ehrlich, G.E. Review of the pharmaceutical properties and clinical effects of the topical NSAID formulation, diclofenac epolamine. Curr. Med. Res. Opin. 2008, 24, 2967–2992. [Google Scholar] [CrossRef]
- Mohammed, F.A. Topical permeation characteristics of diclofenac sodium from NaCMC gels in comparison with conventional gel formulations. Drug Dev. Ind. Pharm. 2001, 27, 1083–1097. [Google Scholar] [CrossRef]
- Stahl, P.H.; Wermuth, C.G. Handbook of Pharmaceutical Salts: Properties, Selection, and Use; Wiley-VCH: Zürich, Switzerland, 2008. [Google Scholar]
- Wang, M.; Fang, L. Percutaneous absorption of diclofenac acid and its salts from emulgel. Asian J. Pharm. Sci. 2008, 3, 131–141. [Google Scholar]
- Hadgraft, J.; du Plessis, J.; Goosen, C. The choice of non-steroidal anti-inflammatory agents for dermal delivery. Int. J. Pharm. 2000, 207, 31–37. [Google Scholar] [CrossRef]
- Cheong, H.-A.; Choi, H.-K. Enhanced percutaneous absorption of piroxicam via salt formation with ethanolamines. Pharm. Res. 2002, 19, 1372–1377. [Google Scholar]
- Han, H.-K.; Choi, H.-K. Improved absorption of meloxicam via salt formation with ethanolamines. Eur. J. Pharm. Biopharm. 2007, 65, 99–103. [Google Scholar] [CrossRef]
- Chandra, A.; Sharma, P.K.; Irchhiaya, R. Effect of alcohols and enhancers on permeation enhancement of ketorolac. Asian J. Pharm. 2009, 3, 37–42. [Google Scholar] [CrossRef]
- Oliveira, G.; Beezer, A.E.; Hadgraft, J.; Lane, M.E. Alcohol enhanced permeation in model membranes. Part I. Thermodynamic and kinetic analyses of membrane permeation. Int. J. Pharm. 2010, 393, 61–67. [Google Scholar] [CrossRef]
- Castellari, C.; Sabatino, P. Anti-Inflammatory Drugs. III. Salts of Diclofenac with N-(2-Hydroxyethyl)-piperidine, N-(2-Hydroxyethyl)-morpholine and N-(2-Hydroxyethyl)-piperazine. Acta Cryst. 1996, C52, 1708–1712. [Google Scholar]
- Warner, K.S.; Li, S.K.; Higuchi, W.I. Influences of alkyl group chain length and polar head Group on chemical skin permeation enhancement. J.Pharm. Sci. 2001, 90, 1143–1153. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fini, A.; Bassini, G.; Monastero, A.; Cavallari, C. Diclofenac Salts, VIII. Effect of the Counterions on the Permeation through Porcine Membrane from Aqueous Saturated Solutions. Pharmaceutics 2012, 4, 413-429. https://doi.org/10.3390/pharmaceutics4030413
Fini A, Bassini G, Monastero A, Cavallari C. Diclofenac Salts, VIII. Effect of the Counterions on the Permeation through Porcine Membrane from Aqueous Saturated Solutions. Pharmaceutics. 2012; 4(3):413-429. https://doi.org/10.3390/pharmaceutics4030413
Chicago/Turabian StyleFini, Adamo, Glenda Bassini, Annamaria Monastero, and Cristina Cavallari. 2012. "Diclofenac Salts, VIII. Effect of the Counterions on the Permeation through Porcine Membrane from Aqueous Saturated Solutions" Pharmaceutics 4, no. 3: 413-429. https://doi.org/10.3390/pharmaceutics4030413
APA StyleFini, A., Bassini, G., Monastero, A., & Cavallari, C. (2012). Diclofenac Salts, VIII. Effect of the Counterions on the Permeation through Porcine Membrane from Aqueous Saturated Solutions. Pharmaceutics, 4(3), 413-429. https://doi.org/10.3390/pharmaceutics4030413