
Increased/Targeted Brain (Pro)Drug Delivery via Utilization of Solute Carriers (SLCs)

 ,
,  and
and 
Abstract
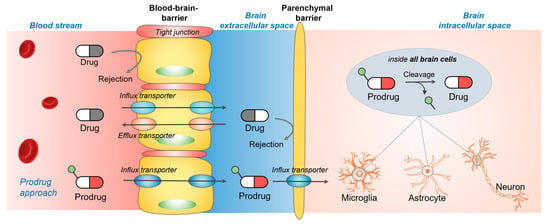
1. Introduction
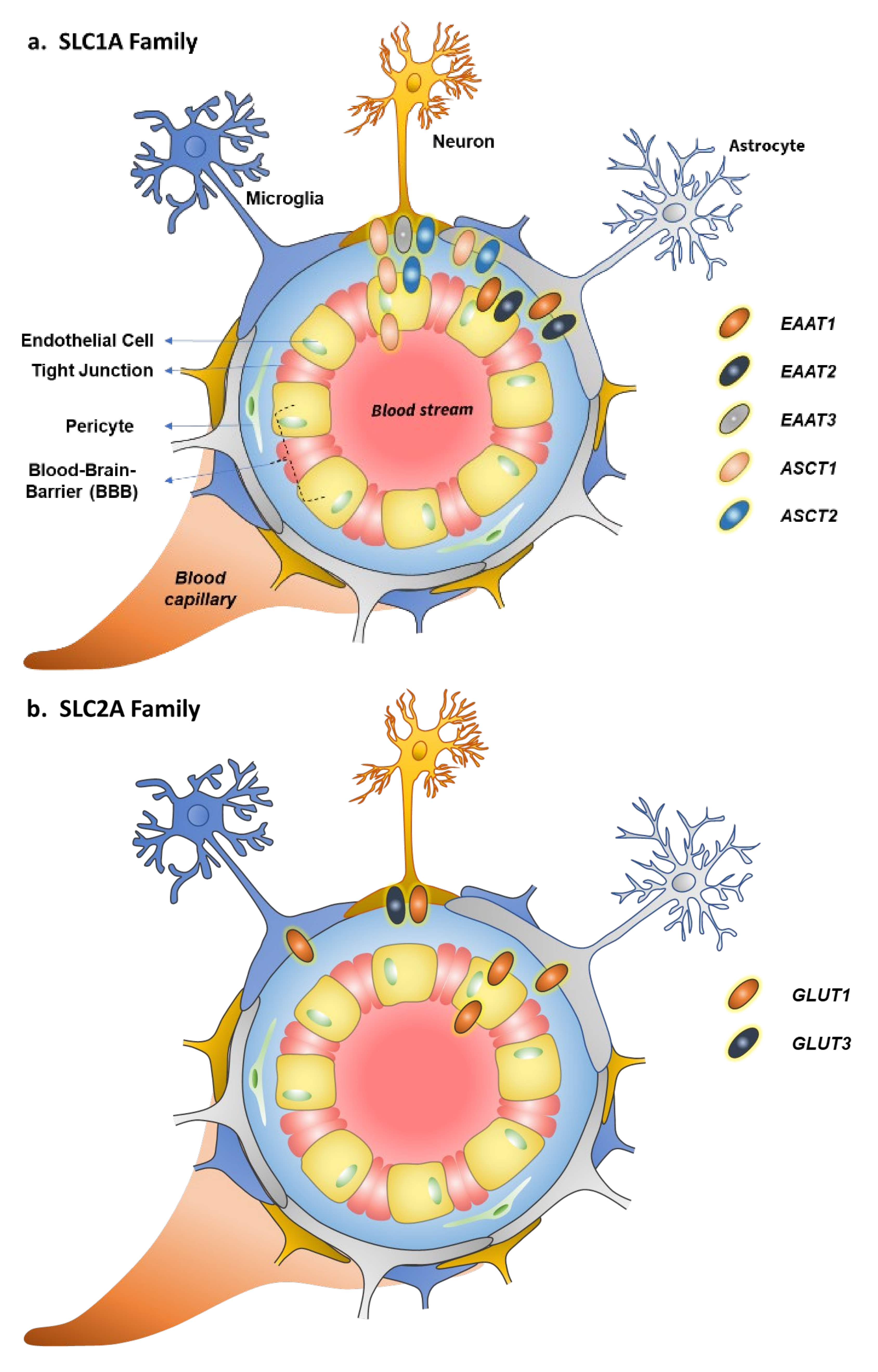
2. Glutamate and Neutral Amino Acid Transporter Family (SLC1A)
3. Glucose Transporter Family (SLC2A)
4. Cationic and Neutral Amino Acid Transporter Family (SLC7A)
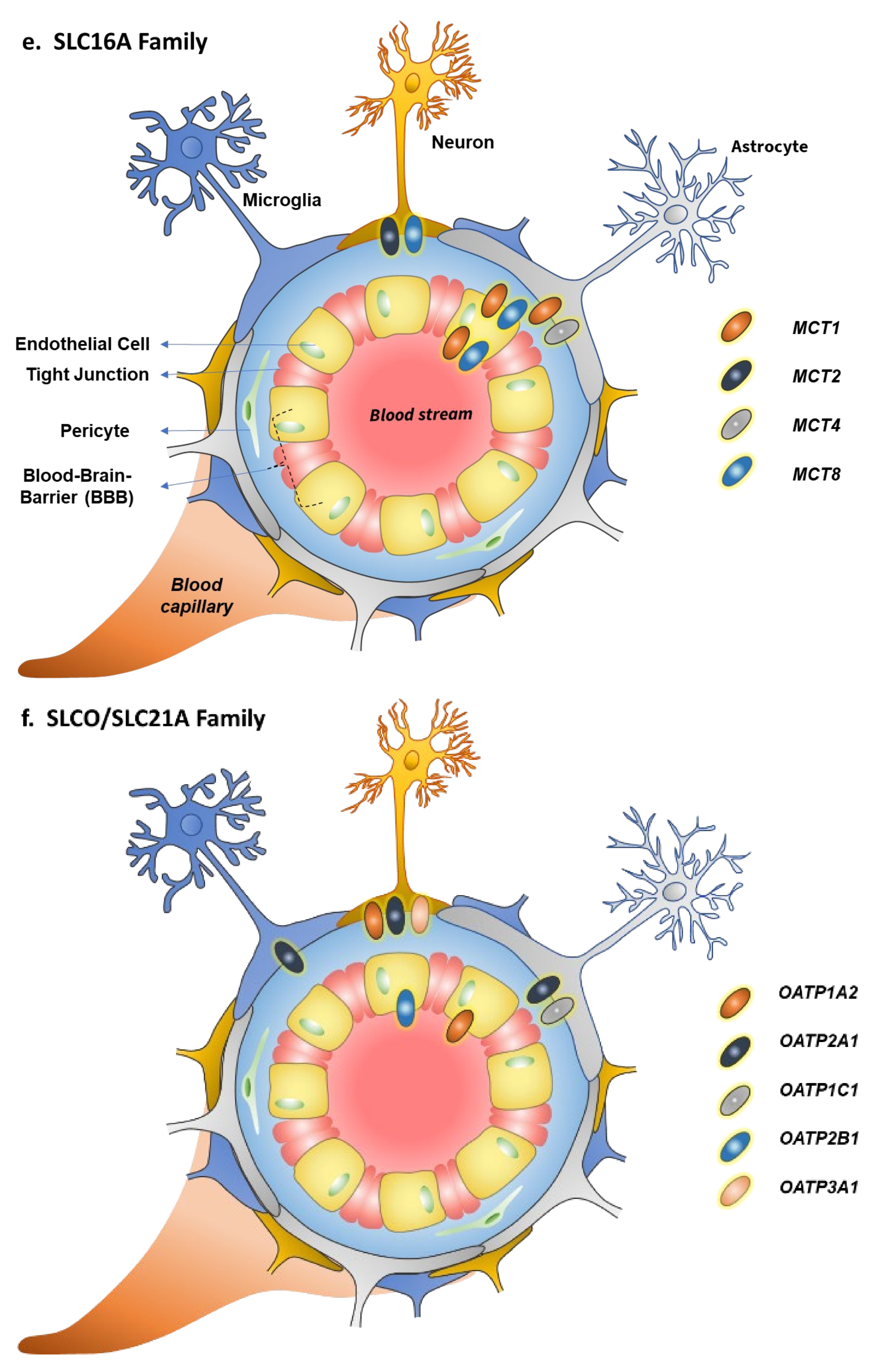
5. Monocarboxylate Transporter Family (SLC16A)
6. Organic Anion Transporting Family (SLCO/SLC21A)
7. Organic Cation/Anion/Zwitterion Transporter Family (SLC22A)
8. Sodium-Coupled Neutral Amino Acid (System N/A) Transporter Family (SLC38A)
9. Other Transporters in Discrete Families
10. Conclusions and Future Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Deuschl, G.; Beghi, E.; Fazekas, F.; Varga, T.; Christoforidi, A.K.; Sipido, E.; Bassetti, C.L.; Vos, T.; Feigin, V.L. The burden of neurological diseases in Europe: An analysis for the Global Burden of Disease Study 2017. Lancet Public Health 2020, 5, e551–e567. [Google Scholar] [CrossRef]
- Olesen, J.; Gustavsson, A.; Svensson, M.; Wittchen, H.-U.; Jönsson, B. The economic cost of brain disorders in Europe. Eur. J. Neurol. 2012, 19, 155–162. [Google Scholar] [CrossRef]
- Pankevich, D.E.; Altevogt, B.M.; Dunlop, J.; Gage, F.H.; Hyman, S.E. Improving and Accelerating Drug Development for Nervous System Disorders. Neuron 2014, 84, 546–553. [Google Scholar] [CrossRef]
- Gribkoff, V.K.; Kaczmarek, L.K. The need for new approaches in CNS drug discovery: Why drugs have failed, and what can be done to improve outcomes. Neuropharmacology 2016, 120, 11–19. [Google Scholar] [CrossRef]
- Uchida, Y.; Yagi, Y.; Takao, M.; Tano, M.; Umetsu, M.; Hirano, S.; Usui, T.; Tachikawa, M.; Terasaki, T. Comparison of Absolute Protein Abundances of Transporters and Receptors among Blood–Brain Barriers at Different Cerebral Regions and the Blood–Spinal Cord Barrier in Humans and Rats. Mol. Pharm. 2020, 17, 2006–2020. [Google Scholar] [CrossRef]
- Pardridge, W.M. Drug Transport across the Blood–Brain Barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1959–1972. [Google Scholar] [CrossRef]
- Dragunow, M. The adult human brain in preclinical drug development. Nat. Rev. Drug Discov. 2008, 7, 659–666. [Google Scholar] [CrossRef]
- Lee, G.; Dallas, S.; Hong, M.; Bendayan, R. Drug transporters in the central nervous system: Brain barriers and brain parenchyma considerations. Pharmacol. Rev. 2001, 53, 569–596. [Google Scholar] [CrossRef]
- Banks, W.A. From blood–brain barrier to blood–brain interface: New opportunities for CNS drug delivery. Nat. Rev. Drug Discov. 2016, 15, 275–292. [Google Scholar] [CrossRef]
- Lin, L.; Yee, S.W.; Kim, R.B.; Giacomini, K.M. SLC transporters as therapeutic targets: Emerging opportunities. Nat. Rev. Drug Discov. 2015, 14, 543–560. [Google Scholar] [CrossRef]
- Huttunen, K.M.; Raunio, H.; Rautio, J. Prodrugs—From Serendipity to Rational Design. Pharmacol. Rev. 2011, 63, 750–771. [Google Scholar] [CrossRef]
- Rautio, J.; Meanwell, N.; Di, L.; Hageman, M.J. The expanding role of prodrugs in contemporary drug design and development. Nat. Rev. Drug Discov. 2018, 17, 559–587. [Google Scholar] [CrossRef]
- Rautio, J.; Kärkkäinen, J.; Sloan, K.B. Prodrugs—Recent approvals and a glimpse of the pipeline. Eur. J. Pharm. Sci. 2017, 109, 146–161. [Google Scholar] [CrossRef]
- Colas, C.; Ung, P.M.-U.; Schlessinger, A. SLC transporters: Structure, function, and drug discovery. MedChemComm 2016, 7, 1069–1081. [Google Scholar] [CrossRef]
- Majumder, P.; Mallela, A.K.; Penmatsa, A. Transporters Through the Looking Glass: An Insight into the Mechanisms of Ion-Coupled Transport and Methods That Help Reveal Them. J. Indian Inst. Sci. 2018, 98, 283–300. [Google Scholar] [CrossRef]
- Januliene, D.; Moeller, A. Cryo-EM of ABC transporters: An ice-cold solution to everything? FEBS Lett. 2020, 594, 3776–3789. [Google Scholar] [CrossRef]
- Schlessinger, A.; Welch, M.A.; Van Vlijmen, H.; Korzekwa, K.; Swaan, P.W.; Matsson, P. Molecular Modeling of Drug-Transporter Interactions-An International Transporter Consortium Perspective. Clin. Pharmacol. Ther. 2018, 104, 818–835. [Google Scholar] [CrossRef]
- Magi, S.; Piccirillo, S.; Amoroso, S.; Lariccia, V. Excitatory Amino Acid Transporters (EAATs): Glutamate Transport and Beyond. Int. J. Mol. Sci. 2019, 20, 5674. [Google Scholar] [CrossRef]
- O’Kane, R.L.; Martínez-López, I.; DeJoseph, M.R.; Viña, J.R.; Hawkins, R.A. Na(+)-dependent glutamate transporters (EAAT1, EAAT2, and EAAT3) of the blood-brain barrier. A mechanism for glutamate removal. J. Biol. Chem. 1999, 274, 31891–31895. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Martin, L.; Levey, A.I.; Dykes-Hoberg, M.; Jin, L.; Wu, D.; Nash, N.; Kuncl, R.W. Localization of neuronal and glial glutamate transporters. Neuron 1994, 13, 713–725. [Google Scholar] [CrossRef]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Malik, A.R.; Willnow, T.E. Excitatory Amino Acid Transporters in Physiology and Disorders of the Central Nervous System. Int. J. Mol. Sci. 2019, 20, 5671. [Google Scholar] [CrossRef]
- Lee, S.-G.; Su, Z.-Z.; Emdad, L.; Gupta, P.; Sarkar, D.; Borjabad, A.; Volsky, D.J.; Fisher, P.B. Mechanism of Ceftriaxone Induction of Excitatory Amino Acid Transporter-2 Expression and Glutamate Uptake in Primary Human Astrocytes. J. Biol. Chem. 2008, 283, 13116–13123. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Patel, S.; Regan, M.R.; Haenggeli, C.; Huang, Y.H.; Bergles, D.E.; Jin, L.; Hoberg, M.D.; Vidensky, S.; Chung, D.S.; et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 2005, 433, 73–77. [Google Scholar] [CrossRef]
- Underhill, S.M.; Wheeler, D.S.; Li, M.; Watts, S.D.; Ingram, S.L.; Amara, S. Amphetamine Modulates Excitatory Neurotransmission through Endocytosis of the Glutamate Transporter EAAT3 in Dopamine Neurons. Neuron 2014, 83, 404–416. [Google Scholar] [CrossRef]
- Jensen, A.A.; Erichsen, M.N.; Nielsen, C.W.; Stensbøl, T.B.; Kehler, J.; Bunch, L. Discovery of the First Selective Inhibitor of Excitatory Amino Acid Transporter Subtype 1. J. Med. Chem. 2009, 52, 912–915. [Google Scholar] [CrossRef]
- Dunlop, J.; McIlvain, H.B.; Carrick, T.A.; Jow, B.; Lu, Q.; Kowal, D.; Lin, S.; Greenfield, A.; Grosanu, C.; Fan, K.; et al. Characterization of Novel Aryl-Ether, Biaryl, and Fluorene Aspartic Acid and Diaminopropionic Acid Analogs as Potent Inhibitors of the High-Affinity Glutamate Transporter EAAT2. Mol. Pharmacol. 2005, 68, 974–982. [Google Scholar] [CrossRef]
- Wu, P.; Bjørn-Yoshimoto, W.E.; Staudt, M.; Jensen, A.A.; Bunch, L. Identification and Structure-Activity Relationship Study of Imidazo[1,2-a]pyridine-3-amines as First Selective Inhibitors of Excitatory Amino Acid Transporter Subtype 3 (EAAT3). ACS Chem. Neurosci. 2019, 10, 4414–4429. [Google Scholar] [CrossRef]
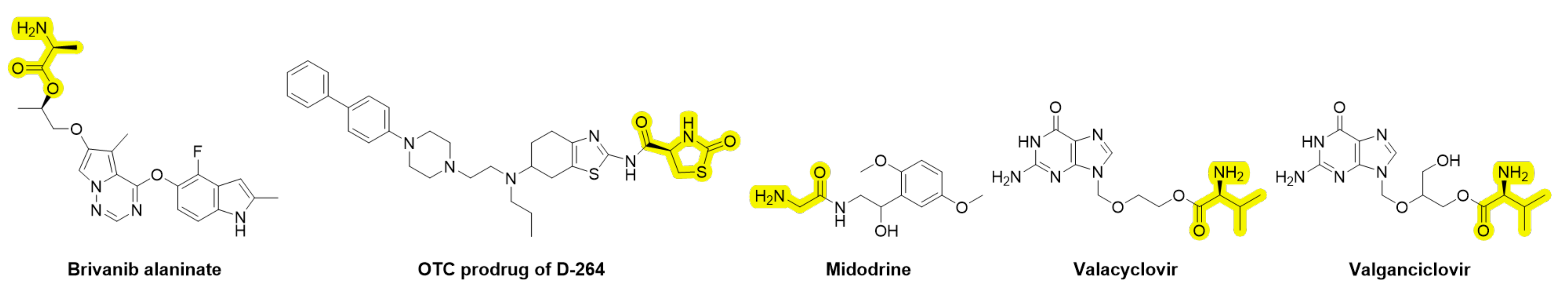
- Dholkawala, F.; Voshavar, C.; Dutta, A.K. Synthesis and characterization of brain penetrant prodrug of neuroprotective D-264: Potential therapeutic application in the treatment of Parkinson’s disease. Eur. J. Pharm. Biopharm. 2016, 103, 62–70. [Google Scholar] [CrossRef]
- Weiss, M.D.; Rossignol, C.; Sumners, C.; Anderson, K.J. A pH-dependent increase in neuronal glutamate efflux in vitro: Possible involvement of ASCT1. Brain Res. 2005, 1056, 105–112. [Google Scholar] [CrossRef]
- Bröer, A.; Brookes, N.; Ganapathy, V.; Dimmer, K.S.; Wagner, C.A.; Lang, F.; Bröer, S. The astroglial ASCT2 amino acid transporter as a mediator of glutamine efflux. J. Neurochem. 1999, 73, 2184–2194. [Google Scholar] [CrossRef]
- Gliddon, C.M.; Shao, Z.; LeMaistre, J.L.; Anderson, C.M. Cellular distribution of the neutral amino acid transporter subtype ASCT2 in mouse brain. J. Neurochem. 2009, 108, 372–383. [Google Scholar] [CrossRef]
- Weiss, M.D.; Derazi, S.; Kilberg, M.S.; Anderson, K.J. Ontogeny and localization of the neutral amino acid transporter ASCT1 in rat brain. Dev. Brain Res. 2001, 130, 183–190. [Google Scholar] [CrossRef]
- Sakai, K.; Shimizu, H.; Koike, T.; Furuya, S.; Watanabe, M. Neutral Amino Acid Transporter ASCT1 Is Preferentially Expressed in l-Ser-Synthetic/Storing Glial Cells in the Mouse Brain with Transient Expression in Developing Capillaries. J. Neurosci. 2003, 23, 550–560. [Google Scholar] [CrossRef]
- Kaplan, E.; Zubedat, S.; Radzishevsky, I.; Valenta, A.C.; Rechnitz, O.; Sason, H.; Sajrawi, C.; Bodner, O.; Konno, K.; Esaki, K.; et al. ASCT1 (Slc1a4) transporter is a physiologic regulator of brain d -serine and neurodevelopment. Proc. Natl. Acad. Sci. USA 2018, 115, 9628–9633. [Google Scholar] [CrossRef]
- Fuchs, B.C.; Bode, B.P. Amino acid transporters ASCT2 and LAT1 in cancer: Partners in crime? Semin. Cancer Biol. 2005, 15, 254–266. [Google Scholar] [CrossRef]
- Scalise, M.; Pochini, L.; Console, L.; Losso, M.A.; Indiveri, C. The Human SLC1A5 (ASCT2) Amino Acid Transporter: From Function to Structure and Role in Cell Biology. Front. Cell Dev. Biol. 2018, 6, 96. [Google Scholar] [CrossRef]
- Vig, B.S.; Huttunen, K.M.; Laine, K.; Rautio, J. Amino acids as promoieties in prodrug design and development. Adv. Drug Deliv. Rev. 2013, 65, 1370–1385. [Google Scholar] [CrossRef]
- Scalise, M.; Console, L.; Cosco, J.; Pochini, L.; Galluccio, M.; Indiveri, C. ASCT1 and ASCT2: Brother and Sister? SLAS Discov. 2021, 26, 1148–1163. [Google Scholar] [CrossRef]
- Scopelliti, A.J.; Font, J.; Vandenberg, R.J.; Boudker, O.; Ryan, R.M. Structural characterisation reveals insights into substrate recognition by the glutamine transporter ASCT2/SLC1A5. Nat. Commun. 2018, 9, 38. [Google Scholar] [CrossRef]
- Scopelliti, A.J.; Ryan, R.; Vandenberg, R.J. Molecular Determinants for Functional Differences between Alanine-Serine-Cysteine Transporter 1 and Other Glutamate Transporter Family Members. J. Biol. Chem. 2013, 288, 8250–8257. [Google Scholar] [CrossRef]
- Li, Y.-X.; Yang, J.-Y.; Alcantara, M.; Abelian, G.; Kulkarni, A.; Staubli, U.; Foster, A.C. Inhibitors of the Neutral Amino Acid Transporters ASCT1 and ASCT2 Are Effective in In Vivo Models of Schizophrenia and Visual Dysfunction. J. Pharmacol. Exp. Ther. 2018, 367, 292–301. [Google Scholar] [CrossRef]
- Grewer, C.; Grabsch, E. New inhibitors for the neutral amino acid transporter ASCT2 reveal its Na+-dependent anion leak. J. Physiol. 2004, 557, 747–759. [Google Scholar] [CrossRef]
- Albers, T.; Marsiglia, W.; Thomas, T.; Gameiro, A.; Grewer, C. Defining Substrate and Blocker Activity of Alanine-Serine-Cysteine Transporter 2 (ASCT2) Ligands with Novel Serine Analogs. Mol. Pharmacol. 2011, 81, 356–365. [Google Scholar] [CrossRef]
- Esslinger, C.S.; Cybulski, K.A.; Rhoderick, J.F. Ngamma-aryl glutamine analogues as probes of the ASCT2 neutral amino acid transporter binding site. Bioorg. Med. Chem. 2005, 13, 1111–1118. [Google Scholar] [CrossRef]
- Singh, K.; Tanui, R.; Gameiro, A.; Eisenberg, G.; Colas, C.; Schlessinger, A.; Grewer, C. Structure activity relationships of benzylproline-derived inhibitors of the glutamine transporter ASCT2. Bioorganic Med. Chem. Lett. 2016, 27, 398–402. [Google Scholar] [CrossRef]
- Patching, S.G. Glucose Transporters at the Blood-Brain Barrier: Function, Regulation and Gateways for Drug Delivery. Mol. Neurobiol. 2016, 54, 1046–1077. [Google Scholar] [CrossRef]
- Buck, A.; Schirrmeister, H.; Mattfeldt, T.; Reske, S.N. Biological characterisation of breast cancer by means of PET. Eur. J. Pediatr. 2004, 31, S80–S87. [Google Scholar] [CrossRef]
- Pliszka, M.; Szablewski, L. Glucose Transporters as a Target for Anticancer Therapy. Cancers 2021, 13, 4184. [Google Scholar] [CrossRef]
- Younes, M.; Lechago, L.V.; Somoano, J.R.; Mosharaf, M.; Lechago, J. Wide expression of the human erythrocyte glucose transporter Glut1 in human cancers. Cancer Res. 1996, 56, 1164–1167. [Google Scholar]
- Macheda, M.L.; Rogers, S.; Best, J. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J. Cell. Physiol. 2004, 202, 654–662. [Google Scholar] [CrossRef]
- Barron, C.C.; Bilan, P.J.; Tsakiridis, T.; Tsiani, E. Facilitative glucose transporters: Implications for cancer detection, prognosis and treatment. Metabolism 2015, 65, 124–139. [Google Scholar] [CrossRef]
- Bonina, F.P.; Arenare, L.; Ippolito, R.; Boatto, G.; Battaglia, G.; Bruno, V.; de Caprariis, P. Synthesis, pharmacokinetics and anticonvulsant activity of 7-chlorokynurenic acid prodrugs. Int. J. Pharm. 2000, 202, 79–88. [Google Scholar] [CrossRef]
- Bonina, F.; Puglia, C.; Rimoli, M.G.; Melisi, D.; Boatto, G.; Nieddu, M.; Calignano, A.; Rana, G.L.; Caprariis, P. Glycosyl derivatives of dopamine and L-dopa as anti-Parkinson prodrugs: Synthesis, pharmacological activity and in vitro stability studies. J. Drug Target. 2003, 11, 25–36. [Google Scholar] [CrossRef]
- Fernández, C.; Nieto, O.; Fontenla, J.A.; Rivas, E.; de Ceballos, M.L.; Fernández-Mayoralas, A. Synthesis of glycosyl derivatives as dopamine prodrugs: Interaction with glucose carrier GLUT-1. Org. Biomol. Chem. 2003, 1, 767–771. [Google Scholar] [CrossRef]
- Halmos, T.; Santarromana, M.; Antonakis, K.; Scherman, D. Synthesis of glucose-chlorambucil derivatives and their recognition by the human GLUT1 glucose transporter. Eur. J. Pharmacol. 1996, 318, 477–484. [Google Scholar] [CrossRef]
- Bilsky, E.J.; Egleton, R.D.; Mitchell, S.A.; Palian, M.M.; Davis, P.; Huber, J.D.; Jones, H.; Yamamura, H.I.; Janders, J.; Davis, T.P.; et al. Enkephalin Glycopeptide Analogues Produce Analgesia with Reduced Dependence Liability. J. Med. Chem. 2000, 43, 2586–2590. [Google Scholar] [CrossRef]
- Gynther, M.; Ropponen, J.; Laine, K.; Leppänen, J.; Haapakoski, P.; Peura, L.; Järvinen, T.; Rautio, J. Glucose Promoiety Enables Glucose Transporter Mediated Brain Uptake of Ketoprofen and Indomethacin Prodrugs in Rats. J. Med. Chem. 2009, 52, 3348–3353. [Google Scholar] [CrossRef]
- Leenders, R.G.G.; Damen, E.W.P.; Bijsterveld, E.J.A.; Scheeren, H.W.; Houba, P.H.J.; Meulen-Muileman, I.H.; Boven, E.; Haisma, H.J. Novel anthracycline-spacer-beta-glucuronide,-beta-glucoside, and -beta-galactoside prodrugs for application in selective chemotherapy. Bioorg. Med. Chem. 1999, 7, 1597–1610. [Google Scholar] [CrossRef]
- Tranoy-Opalinski, I.; Legigan, T.; Barat, R.; Clarhaut, J.; Thomas, M.; Renoux, B.; Papot, S. β-Glucuronidase-responsive prodrugs for selective cancer chemotherapy: An update. Eur. J. Med. Chem. 2014, 74, 302–313. [Google Scholar] [CrossRef]
- Matović, J.; Järvinen, J.; Sokka, I.K.; Imlimthan, S.; Raitanen, J.-E.; Montaser, A.; Maaheimo, H.; Huttunen, K.M.; Peräniemi, S.; Airaksinen, A.J.; et al. Exploring the Biochemical Foundations of a Successful GLUT1-Targeting Strategy to BNCT: Chemical Synthesis and In Vitro Evaluation of the Entire Positional Isomer Library of ortho-Carboranylmethyl-Bearing Glucoconjugates. Mol. Pharm. 2020, 18, 285–304. [Google Scholar] [CrossRef]
- Zhang, K.; Xu, P.; Sowers, J.L.; Machuca, D.F.; Mirfattah, B.; Herring, J.; Tang, H.; Chen, Y.; Tian, B.; Brasier, A.R.; et al. Proteome Analysis of Hypoxic Glioblastoma Cells Reveals Sequential Metabolic Adaptation of One-Carbon Metabolic Pathways. Mol. Cell. Proteom. 2017, 16, 1906–1921. [Google Scholar] [CrossRef]
- Ohnishi, K.; Misawa, M.; Sikano, N.; Nakai, K.; Suzuki, M. Enhancement of Cancer Cell-Killing Effects of Boron Neutron Capture Therapy by Manipulating the Expression of L-Type Amino Acid Transporter 1. Radiat. Res. 2021, 196, 17–22. [Google Scholar] [CrossRef]
- Drew, D.; North, R.A.; Nagarathinam, K.; Tanabe, M. Structures and General Transport Mechanisms by the Major Facilitator Superfamily (MFS). Chem. Rev. 2021, 121, 5289–5335. [Google Scholar] [CrossRef]
- Galochkina, T.; Chong, M.N.F.; Challali, L.; Abbar, S.; Etchebest, C. New insights into GluT1 mechanics during glucose transfer. Sci. Rep. 2019, 9, 998. [Google Scholar] [CrossRef]
- Gonzalez-Resines, S.; Quinn, P.J.; Naftalin, R.J.; Domene, C. Multiple Interactions of Glucose with the Extra-Membranous Loops of GLUT1 Aid Transport. J. Chem. Inf. Model. 2021, 61, 3559–3570. [Google Scholar] [CrossRef]
- Park, M.-S. Molecular Dynamics Simulations of the Human Glucose Transporter GLUT1. PLoS ONE 2015, 10, e0125361. [Google Scholar] [CrossRef]
- Tachikawa, M.; Hirose, S.; Akanuma, S.-I.; Matsuyama, R.; Hosoya, K.-I. Developmental changes of l -arginine transport at the blood-brain barrier in rats. Microvasc. Res. 2018, 117, 16–21. [Google Scholar] [CrossRef]
- Hosokawa, H.; Ninomiya, H.; Sawamura, T.; Sugimoto, Y.; Ichikawa, A.; Fujiwara, K.; Masaki, T. Neuron-specific expression of cationic amino acid transporter 3 in the adult rat brain. Brain Res. 1999, 838, 158–165. [Google Scholar] [CrossRef]
- Braissant, O.; Gotoh, T.; Loup, M.; Mori, M.; Bachmann, C. Differential expression of the cationic amino acid transporter 2(B) in the adult rat brain. Mol. Brain Res. 2001, 91, 189–195. [Google Scholar] [CrossRef]
- Stevens, B.R.; Kakuda, D.K.; Yu, K.; Waters, M.; Vo, C.B.; Raizada, M.K. Induced Nitric Oxide Synthesis Is Dependent on Induced Alternatively Spliced CAT-2 Encoding L-Arginine Transport in Brain Astrocytes. J. Biol. Chem. 1996, 271, 24017–24022. [Google Scholar] [CrossRef]
- Zaragozá, R. Transport of Amino Acids Across the Blood-Brain Barrier. Front. Physiol. 2020, 11, 973. [Google Scholar] [CrossRef]
- Huang, Y.; Kang, B.N.; Tian, J.; Liu, Y.; Luo, H.R.; Hester, L.; Snyder, S.H. The Cationic Amino Acid Transporters CAT1 and CAT3 Mediate NMDA Receptor Activation-Dependent Changes in Elaboration of Neuronal Processes via the Mammalian Target of Rapamycin mTOR Pathway. J. Neurosci. 2007, 27, 449–458. [Google Scholar] [CrossRef]
- Dai, R.; Peng, F.; Xiao, X.; Gong, X.; Jiang, Y.; Zhang, M.; Tian, Y.; Xu, Y.; Ma, J.; Li, M.; et al. Hepatitis B virus X protein-induced upregulation of CAT-1 stimulates proliferation and inhibits apoptosis in hepatocellular carcinoma cells. Oncotarget 2017, 8, 60962–60974. [Google Scholar] [CrossRef]
- Abdelmagid, S.A.; Rickard, J.A.; McDonald, W.J.; Thomas, L.N.; Too, C.K. CAT-1-mediated arginine uptake and regulation of nitric oxide synthases for the survival of human breast cancer cell lines. J. Cell. Biochem. 2011, 112, 1084–1092. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, W.; Wang, J.; Yang, C.; Mao, H.; Fu, X.; Wu, Y.; Cai, J.; Han, J.; Xu, Z.; et al. Overexpression of Arginine Transporter CAT-1 Is Associated with Accumulation of L-Arginine and Cell Growth in Human Colorectal Cancer Tissue. PLoS ONE 2013, 8, e73866. [Google Scholar] [CrossRef]
- Werner, A.; Pieh, D.; Echchannaoui, H.; Rupp, J.; Rajalingam, K.; Theobald, M.; Closs, E.I.; Munder, M. Cationic Amino Acid Transporter-1-Mediated Arginine Uptake Is Essential for Chronic Lymphocytic Leukemia Cell Proliferation and Viability. Front. Oncol. 2019, 9, 1268. [Google Scholar] [CrossRef]
- Jungnickel, K.E.J.; Parker, J.; Newstead, S. Structural basis for amino acid transport by the CAT family of SLC7 transporters. Nat. Commun. 2018, 9, 550. [Google Scholar] [CrossRef]
- Fort, J.; Nicolàs-Aragó, A.; Palacín, M. The Ectodomains of rBAT and 4F2hc Are Fake or Orphan α-Glucosidases. Molecules 2021, 26, 6231. [Google Scholar] [CrossRef]
- Estrach, S.; Lee, S.-A.; Boulter, E.; Pisano, S.; Errante, A.; Tissot, F.S.; Cailleteau, L.; Pons, C.; Ginsberg, M.H.; Féral, C.C. CD98hc (SLC3A2) Loss Protects Against Ras-Driven Tumorigenesis by Modulating Integrin-Mediated Mechanotransduction. Cancer Res. 2014, 74, 6878–6889. [Google Scholar] [CrossRef]
- Feral, C.C.; Nishiya, N.; Fenczik, C.A.; Stuhlmann, H.; Slepak, M.; Ginsberg, M.H. CD98hc (SLC3A2) mediates integrin signaling. Proc. Natl. Acad. Sci. USA 2004, 102, 355–360. [Google Scholar] [CrossRef]
- Kanai, Y.; Segawa, H.; Miyamoto, K.-I.; Uchino, H.; Takeda, E.; Endou, H. Expression Cloning and Characterization of a Transporter for Large Neutral Amino Acids Activated by the Heavy Chain of 4F2 Antigen (CD98). J. Biol. Chem. 1998, 273, 23629–23632. [Google Scholar] [CrossRef]
- Prasad, P.D.; Wang, H.; Huang, W.; Kekuda, R.; Rajan, D.P.; Leibach, F.H.; Ganapathy, V. Human LAT1, a Subunit of System L Amino Acid Transporter: Molecular Cloning and Transport Function. Biochem. Biophys. Res. Commun. 1999, 255, 283–288. [Google Scholar] [CrossRef]
- Boado, R.J.; Li, J.Y.; Nagaya, M.; Zhang, C.; Pardridge, W.M. Selective expression of the large neutral amino acid transporter at the blood–brain barrier. Proc. Natl. Acad. Sci. USA 1999, 96, 12079–12084. [Google Scholar] [CrossRef]
- Scalise, M.; Galluccio, M.; Console, L.; Pochini, L.; Indiveri, C. The Human SLC7A5 (LAT1): The Intriguing Histidine/Large Neutral Amino Acid Transporter and Its Relevance to Human Health. Front. Chem. 2018, 6, 243. [Google Scholar] [CrossRef]
- Huttunen, J.; Peltokangas, S.; Gynther, M.; Natunen, T.; Hiltunen, M.; Auriola, S.; Ruponen, M.; Vellonen, K.-S.; Huttunen, K.M. L-Type Amino Acid Transporter 1 (LAT1/Lat1)-Utilizing Prodrugs Can Improve the Delivery of Drugs into Neurons, Astrocytes and Microglia. Sci. Rep. 2019, 9, 12860. [Google Scholar] [CrossRef]
- Duelli, R.; Enerson, B.E.; Gerhart, D.Z.; Drewes, L.R. Expression of Large Amino Acid Transporter LAT1 in Rat Brain Endothelium. J. Cereb. Blood Flow Metab. 2000, 20, 1557–1562. [Google Scholar] [CrossRef]
- Yanagida, O.; Kanai, Y.; Chairoungdua, A.; Kim, D.K.; Segawa, H.; Nii, T.; Cha, S.H.; Matsuo, H.; Fukushima, J.-I.; Fukasawa, Y.; et al. Human L-type amino acid transporter 1 (LAT1): Characterization of function and expression in tumor cell lines. Biochim. Biophys. Acta Biomembr. 2001, 1514, 291–302. [Google Scholar] [CrossRef]
- Wang, Q.; Holst, J. L-type amino acid transport and cancer: Targeting the mTORC1 pathway to inhibit neoplasia. Am. J. Cancer Res. 2015, 5, 1281–1294. [Google Scholar]
- Häfliger, P.; Charles, R.-P. The L-Type Amino Acid Transporter LAT1—An Emerging Target in Cancer. Int. J. Mol. Sci. 2019, 20, 2428. [Google Scholar] [CrossRef]
- Furuya, M.; Horiguchi, J.; Nakajima, H.; Kanai, Y.; Oyama, T. Correlation of L-type amino acid transporter 1 and CD98 expression with triple negative breast cancer prognosis. Cancer Sci. 2011, 103, 382–389. [Google Scholar] [CrossRef]
- Sakata, T.; Ferdous, G.; Tsuruta, T.; Satoh, T.; Baba, S.; Muto, T.; Ueno, A.; Kanai, Y.; Endou, H.; Okayasu, I. L-type amino-acid transporter 1 as a novel biomarker for high-grade malignancy in prostate cancer. Pathol. Int. 2009, 59, 7–18. [Google Scholar] [CrossRef]
- Hayashi, K.; Anzai, N. Novel therapeutic approaches targeting L-type amino acid transporters for cancer treatment. World J. Gastrointest. Oncol. 2017, 9, 21–29. [Google Scholar] [CrossRef]
- Tărlungeanu, D.C.; Deliu, E.; Dotter, C.P.; Kara, M.; Janiesch, P.C.; Scalise, M.; Galluccio, M.; Tesulov, M.; Morelli, E.; Sonmez, F.M.; et al. Impaired Amino Acid Transport at the Blood Brain Barrier Is a Cause of Autism Spectrum Disorder. Cell 2016, 167, 1481–1494. [Google Scholar] [CrossRef]
- Kärkkäinen, J.; Gynther, M.; Kokkola, T.; Petsalo, A.; Auriola, S.; Lahtela-Kakkonen, M.; Laine, K.; Rautio, J.; Huttunen, K.M. Structural properties for selective and efficient l-type amino acid transporter 1 (LAT1) mediated cellular uptake. Int. J. Pharm. 2018, 544, 91–99. [Google Scholar] [CrossRef]
- Kärkkäinen, J.; Laitinen, T.; Markowicz-Piasecka, M.; Montaser, A.; Lehtonen, M.; Rautio, J.; Gynther, M.; Poso, A.; Huttunen, K.M. Molecular characteristics supporting l-Type amino acid transporter 1 (LAT1)-mediated translocation. Bioorganic Chem. 2021, 112, 104921. [Google Scholar] [CrossRef]
- Lee, Y.; Wiriyasermkul, P.; Jin, C.; Quan, L.; Ohgaki, R.; Okuda, S.; Kusakizako, T.; Nishizawa, T.; Oda, K.; Ishitani, R.; et al. Cryo-EM structure of the human L-type amino acid transporter 1 in complex with glycoprotein CD98hc. Nat. Struct. Mol. Biol. 2019, 26, 510–517. [Google Scholar] [CrossRef]
- Yan, R.; Zhao, X.; Lei, J.; Zhou, Q. Structure of the human LAT1–4F2hc heteromeric amino acid transporter complex. Nature 2019, 568, 127–130. [Google Scholar] [CrossRef]
- Chien, H.-C.; Colas, C.; Finke, K.; Springer, S.; Stoner, L.; Zur, A.A.; Venteicher, B.; Campbell, J.; Hall, C.; Flint, A.; et al. Reevaluating the Substrate Specificity of the L-Type Amino Acid Transporter (LAT1). J. Med. Chem. 2018, 61, 7358–7373. [Google Scholar] [CrossRef]
- Tampio, J.; Löffler, S.; Guillon, M.; Hugele, A.; Huttunen, J.; Huttunen, K.M. Improved l-Type amino acid transporter 1 (LAT1)-mediated delivery of anti-inflammatory drugs into astrocytes and microglia with reduced prostaglandin production. Int. J. Pharm. 2021, 601, 120565. [Google Scholar] [CrossRef]
- Forrest, L.; Rudnick, G. The Rocking Bundle: A Mechanism for Ion-Coupled Solute Flux by Symmetrical Transporters. Physiology 2009, 24, 377–386. [Google Scholar] [CrossRef]
- Gynther, M.; Peura, L.; Vernerová, M.; Leppänen, J.; Kärkkäinen, J.; Lehtonen, M.; Rautio, J.; Huttunen, K.M. Amino Acid Promoieties Alter Valproic Acid Pharmacokinetics and Enable Extended Brain Exposure. Neurochem. Res. 2016, 41, 2797–2809. [Google Scholar] [CrossRef]
- Gynther, M.; Pickering, D.S.; Spicer, J.A.; Denny, W.A.; Huttunen, K.M. Systemic and Brain Pharmacokinetics of Perforin Inhibitor Prodrugs. Mol. Pharm. 2016, 13, 2484–2491. [Google Scholar] [CrossRef]
- Montaser, A.B.; Järvinen, J.; Löffler, S.; Huttunen, J.; Auriola, S.; Lehtonen, M.; Jalkanen, A.; Huttunen, K.M. L-Type Amino Acid Transporter 1 Enables the Efficient Brain Delivery of Small-Sized Prodrug across the Blood–Brain Barrier and into Human and Mouse Brain Parenchymal Cells. ACS Chem. Neurosci. 2020, 11, 4301–4315. [Google Scholar] [CrossRef]
- Peura, L.; Malmioja, K.; Huttunen, K.; Leppänen, J.; Hämäläinen, M.; Forsberg, M.M.; Rautio, J.; Laine, K. Design, Synthesis and Brain Uptake of LAT1-Targeted Amino Acid Prodrugs of Dopamine. Pharm. Res. 2013, 30, 2523–2537. [Google Scholar] [CrossRef]
- Puris, E.; Gynther, M.; Huttunen, J.; Petsalo, A.; Huttunen, K.M. L-type amino acid transporter 1 utilizing prodrugs: How to achieve effective brain delivery and low systemic exposure of drugs. J. Control. Release 2017, 261, 93–104. [Google Scholar] [CrossRef]
- Puris, E.; Gynther, M.; Huttunen, J.; Auriola, S.; Huttunen, K.M. L-type amino acid transporter 1 utilizing prodrugs of ferulic acid revealed structural features supporting the design of prodrugs for brain delivery. Eur. J. Pharm. Sci. 2019, 129, 99–109. [Google Scholar] [CrossRef]
- Hokari, M.; Wu, H.-Q.; Schwarcz, R.; Smith, Q.R. Facilitated brain uptake of 4-chlorokynurenine and conversion to 7-chlorokynurenic acid. NeuroReport 1996, 8, 15–18. [Google Scholar] [CrossRef]
- Okano, N.; Naruge, D.; Kawai, K.; Kobayashi, T.; Nagashima, F.; Endou, H.; Furuse, J. First-in-human phase I study of JPH203, an L-type amino acid transporter 1 inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2020, 38, 1495–1506. [Google Scholar] [CrossRef]
- Segawa, H.; Fukasawa, Y.; Miyamoto, K.-I.; Takeda, E.; Endou, H.; Kanai, Y. Identification and Functional Characterization of a Na+-independent Neutral Amino Acid Transporter with Broad Substrate Selectivity. J. Biol. Chem. 1999, 274, 19745–19751. [Google Scholar] [CrossRef]
- Pineda, M.; Fernández, E.; Torrents, D.; Estevez, R.; López, C.; Camps, M.; Lloberas, J.; Zorzano, A.; Palacín, M. Identification of a Membrane Protein, LAT-2, That Co-expresses with 4F2 Heavy Chain, an L-type Amino Acid Transport Activity with Broad Specificity for Small and Large Zwitterionic Amino Acids. J. Biol. Chem. 1999, 274, 19738–19744. [Google Scholar] [CrossRef]
- Bröer, A.; Wagner, C.A.; Lang, F.; Bröer, S. The heterodimeric amino acid transporter 4F2hc/y+LAT2 mediates arginine efflux in exchange with glutamine. Biochem. J. 2000, 349, 787–795. [Google Scholar] [CrossRef]
- Meier, C.; Ristic, Z.; Klauser, S.; Verrey, F. Activation of system L heterodimeric amino acid exchangers by intracellular substrates. EMBO J. 2002, 21, 580–589. [Google Scholar] [CrossRef]
- Braun, D.; Kinne, A.; Bräuer, A.U.; Sapin, R.; Klein, M.O.; Köhrle, J.; Wirth, E.K.; Schweizer, U. Developmental and cell type-specific expression of thyroid hormone transporters in the mouse brain and in primary brain cells. Glia 2010, 59, 463–471. [Google Scholar] [CrossRef]
- Milewski, K.; Bogacińska-Karaś, M.; Fręśko, I.; Hilgier, W.; Jaźwiec, R.; Albrecht, J.; Zielińska, M. Ammonia Reduces Intracellular Asymmetric Dimethylarginine in Cultured Astrocytes Stimulating Its y+LAT2 Carrier-Mediated Loss. Int. J. Mol. Sci. 2017, 18, 2308. [Google Scholar] [CrossRef]
- Zielińska, M.; Milewski, K.; Skowrońska, M.; Gajos, A.; Zieminska, E.; Beręsewicz, A.; Albrecht, J.K. Induction of inducible nitric oxide synthase expression in ammonia-exposed cultured astrocytes is coupled to increased arginine transport by upregulated y+ LAT2 transporter. J. Neurochem. 2015, 135, 1272–1281. [Google Scholar] [CrossRef]
- Kinne, A.; Wittner, M.; Wirth, E.K.; Hinz, K.M.; Schülein, R.; Köhrle, J.; Krause, G. Involvement of the L-Type Amino Acid Transporter Lat2 in the Transport of 3,3′-Diiodothyronine across the Plasma Membrane. Eur. Thyroid J. 2015, 4, 42–50. [Google Scholar] [CrossRef][Green Version]
- Zevenbergen, C.; Meima, M.E.; Lima de Souza, E.C.; Peeters, R.P.; Kinne, A.; Krause, G.; Visser, W.E.; Visser, T.J. Transport of Iodothyronines by Human L-Type Amino Acid Transporters. Endocrinology 2015, 156, 4345–4355. [Google Scholar] [CrossRef][Green Version]
- Pinto, V.; Pinho, M.J.; Soares-Da-Silva, P. Renal amino acid transport systems and essential hypertension. FASEB J. 2013, 27, 2927–2938. [Google Scholar] [CrossRef]
- Barollo, S.; Bertazza, L.; Fernando, S.W.; Censi, S.; Cavedon, E.; Galuppini, F.; Pennelli, G.; Fassina, A.; Citton, M.; Rubin, B.; et al. Overexpression of L-Type Amino Acid Transporter 1 (LAT1) and 2 (LAT2): Novel Markers of Neuroendocrine Tumors. PLoS ONE 2016, 11, e0156044. [Google Scholar] [CrossRef][Green Version]
- Feng, M.; Xiong, G.; Cao, Z.; Yang, G.; Zheng, S.; Qiu, J.; You, L.; Zheng, L.; Zhang, T.; Zhao, Y. LAT2 regulates glutamine-dependent mTOR activation to promote glycolysis and chemoresistance in pancreatic cancer. J. Exp. Clin. Cancer Res. 2018, 37, 274. [Google Scholar] [CrossRef]
- Yan, R.; Zhou, J.; Li, Y.; Lei, J.; Zhou, Q. Structural insight into the substrate recognition and transport mechanism of the human LAT2–4F2hc complex. Cell Discov. 2020, 6, 82. [Google Scholar] [CrossRef]
- Nakauchi, J.; Matsuo, H.; Kim, D.K.; Goto, A.; Chairoungdua, A.; Cha, S.H.; Inatomi, J.; Shiokawa, Y.; Yamaguchi, K.; Saito, I.; et al. Cloning and characterization of a human brain Na+-independent transporter for small neutral amino acids that transports d-serine with high affinity. Neurosci. Lett. 2000, 287, 231–235. [Google Scholar] [CrossRef]
- Fukasawa, Y.; Segawa, H.; Kim, J.Y.; Chairoungdua, A.; Kim, D.K.; Matsuo, H.; Cha, S.H.; Endou, H.; Kanai, Y. Identification and Characterization of a Na+-independent Neutral Amino Acid Transporter That Associates with the 4F2 Heavy Chain and Exhibits Substrate Selectivity for Small Neutral d- and l-Amino Acids. J. Biol. Chem. 2000, 275, 9690–9698. [Google Scholar] [CrossRef]
- Bassi, M.; Gasol, E.; Manzoni, M.; Pineda, M.; Riboni, M.; Martín, R.; Zorzano, A.; Borsani, G.; Palacín, M. Identification and characterisation of human xCT that co-expresses, with 4F2 heavy chain, the amino acid transport activity system x c–. Pflüg. Arch. 2001, 442, 286–296. [Google Scholar] [CrossRef]
- Sato, H.; Tamba, M.; Ishii, T.; Bannai, S. Cloning and Expression of a Plasma Membrane Cystine/Glutamate Exchange Transporter Composed of Two Distinct Proteins. J. Biol. Chem. 1999, 274, 11455–11458. [Google Scholar] [CrossRef]
- Rutter, A.R.; Fradley, R.L.; Garrett, E.M.; Chapman, K.L.; Lawrence, J.M.; Rosahl, T.W.; Patel, S. Evidence from gene knockout studies implicates Asc-1 as the primary transporter mediating d-serine reuptake in the mouse CNS. Eur. J. Neurosci. 2007, 25, 1757–1766. [Google Scholar] [CrossRef]
- Helboe, L.; Egebjerg, J.; Møller, M.; Thomsen, C. Distribution and pharmacology of alanine-serine-cysteine transporter 1 (asc-1) in rodent brain. Eur. J. Neurosci. 2003, 18, 2227–2238. [Google Scholar] [CrossRef]
- Xie, X.; Dumas, T.; Tang, L.; Brennan, T.; Reeder, T.; Thomas, W.; Klein, R.D.; Flores, J.; O’Hara, B.F.; Heller, H.C.; et al. Lack of the alanine-serine-cysteine transporter 1 causes tremors, seizures, and early postnatal death in mice. Brain Res. 2005, 1052, 212–221. [Google Scholar] [CrossRef]
- Seib, T.M.; Patel, S.A.; Bridges, R.J. Regulation of the System x−C cystine/glutamate exchanger by intracellular glutathione levels in rat astrocyte primary cultures. Glia 2011, 59, 1387–1401. [Google Scholar] [CrossRef]
- Lewerenz, J.; Maher, P.; Methner, A. Regulation of xCT expression and system x (c) (-) function in neuronal cells. Amino Acids 2012, 42, 171–179. [Google Scholar] [CrossRef]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef]
- Sato, M.; Onuma, K.; Domon, M.; Hasegawa, S.; Suzuki, A.; Kusumi, R.; Hino, R.; Kakihara, N.; Kanda, Y.; Osaki, M.; et al. Loss of the cystine/glutamate antiporter in melanoma abrogates tumor metastasis and markedly increases survival rates of mice. Int. J. Cancer 2020, 147, 3224–3235. [Google Scholar] [CrossRef]
- Conrad, M.; Sato, H. The oxidative stress-inducible cystine/glutamate antiporter, system x (c) (-): Cystine supplier and beyond. Amino Acids 2012, 42, 231–246. [Google Scholar] [CrossRef]
- Kutchukian, P.S.; Warren, L.; Magliaro, B.C.; Amoss, A.; Cassaday, J.A.; O’Donnell, G.; Squadroni, B.; Zuck, P.; Pascarella, D.; Culberson, J.C.; et al. Iterative Focused Screening with Biological Fingerprints Identifies Selective Asc-1 Inhibitors Distinct from Traditional High Throughput Screening. ACS Chem. Biol. 2017, 12, 519–527. [Google Scholar] [CrossRef]
- Patel, D.; Kharkar, P.S.; Gandhi, N.S.; Kaur, E.; Dutt, S.; Nandave, M. Novel analogs of sulfasalazine as system x(c) (-) antiporter inhibitors: Insights from the molecular modeling studies. Drug Dev. Res. 2019, 80, 758–777. [Google Scholar] [CrossRef]
- Halestrap, A.P. The monocarboxylate transporter family-Structure and functional characterization. IUBMB Life 2011, 64, 1–9. [Google Scholar] [CrossRef]
- Halestrap, A.P. The SLC16 gene family—Structure, role and regulation in health and disease. Mol. Asp. Med. 2013, 34, 337–349. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Meredith, D. The SLC16 gene family?from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflug. Arch. 2003, 447, 619–628. [Google Scholar] [CrossRef]
- Chiry, O.; Pellerin, L.; Monnet-Tschudi, F.; Fishbein, W.N.; Merezhinskaya, N.; Magistretti, P.J.; Clarke, S. Expression of the monocarboxylate transporter MCT1 in the adult human brain cortex. Brain Res. 2006, 1070, 65–70. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Wilson, M.C. The monocarboxylate transporter family-Role and regulation. IUBMB Life 2011, 64, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, Y.; Nozawa, K.; Yamada, S.; Yokoyama, Y.; Kimura, R. Quantitative evaluation of brain distribution and blood-brain barrier efflux transport of probenecid in rats by microdialysis: Possible involvement of the monocarboxylic acid transport system. J. Pharmacol. Exp. Ther. 1997, 280, 551–560. [Google Scholar] [PubMed]
- Deguchi, Y.; Yokoyama, Y.; Sakamoto, T.; Hayashi, H.; Naito, T.; Yamada, S.; Kimura, R. Brain distribution of 6-mercaptopurine is regulated by the efflux transport system in the blood-brain barrier 1. Life Sci. 2000, 66, 649–662. [Google Scholar] [CrossRef]
- Felmlee, M.A.; Morse, B.L.; Morris, M.E. γ-Hydroxybutyric Acid: Pharmacokinetics, Pharmacodynamics, and Toxicology. AAPS J. 2021, 23, 22. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.-Y.; Kang, Y.-S. In Vivo and In Vitro Evidence for Brain Uptake of 4-Phenylbutyrate by the Monocarboxylate Transporter 1 (MCT1). Pharm. Res. 2016, 33, 1711–1722. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Terasaki, T.; Tsuji, A. Acidic drug transport in vivo through the blood-brain barrier. A role of the transport carrier for monocarboxylic acids. J. Pharm. Dyn. 1990, 13, 158–163. [Google Scholar] [CrossRef]
- Terasaki, T.; Takakuwa, S.; Moritani, S.; Tsuji, A. Transport of monocarboxylic acids at the blood-brain barrier: Studies with monolayers of primary cultured bovine brain capillary endothelial cells. J. Pharmacol. Exp. Ther. 1991, 258, 932–937. [Google Scholar]
- Friesema, E.C.H.; Ganguly, S.; Abdalla, A.; Manning Fox, J.E.; Halestrap, A.P.; Visser, T.J. Identification of Monocarboxylate Transporter 8 as a Specific Thyroid Hormone Transporter. J. Biol. Chem. 2003, 278, 40128–40135. [Google Scholar] [CrossRef]
- Roberts, L.M.; Woodford, K.; Zhou, M.; Black, D.S.; Haggerty, J.E.; Tate, E.H.; Grindstaff, K.K.; Mengesha, W.; Raman, C.; Zerangue, N. Expression of the Thyroid Hormone Transporters Monocarboxylate Transporter-8 (SLC16A2) and Organic Ion Transporter-14 (SLCO1C1) at the Blood-Brain Barrier. Endocrinology 2008, 149, 6251–6261. [Google Scholar] [CrossRef]
- Heuer, H.; Maier, M.K.; Iden, S.; Mittag, J.; Friesema, E.C.H.; Visser, T.J.; Bauer, K. The Monocarboxylate Transporter 8 Linked to Human Psychomotor Retardation Is Highly Expressed in Thyroid Hormone-Sensitive Neuron Populations. Endocrinology 2005, 146, 1701–1706. [Google Scholar] [CrossRef]
- Groeneweg, S.; Geest, F.S.; Peeters, R.P.; Heuer, H.; Visser, W.E. Thyroid Hormone Transporters. Endocr. Rev. 2020, 41, 146–201. [Google Scholar] [CrossRef] [PubMed]
- Di Cosmo, C.; De Marco, G.; Agretti, P.; Ferrarini, E.; Dimida, A.; Falcetta, P.; Benvenga, S.; Vitti, P.; Tonacchera, M. Screening for drugs potentially interfering with MCT8-mediated T3 transport in vitro identifies dexamethasone and some commonly used drugs as inhibitors of MCT8 activity. J. Endocrinol. Investig. 2021, 45, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Bergersen, L.H. Lactate Transport and Signaling in the Brain: Potential Therapeutic Targets and Roles in Body—Brain Interaction. J. Cereb. Blood Flow Metab. 2014, 35, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Bröer, S.; Rahman, B.; Pellegri, G.; Pellerin, L.; Martin, J.-L.; Verleysdonk, S.; Hamprecht, B.; Magistretti, P.J. Comparison of Lactate Transport in Astroglial Cells and Monocarboxylate Transporter 1 (MCT 1) Expressing Xenopus laevis Oocytes. J. Biol. Chem. 1997, 272, 30096–30102. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.-Y.; Vera, J.C.; Chaganti, R.S.K.; Golde, D.W. Human Monocarboxylate Transporter 2 (MCT2) Is a High Affinity Pyruvate Transporter. J. Biol. Chem. 1998, 273, 28959–28965. [Google Scholar] [CrossRef] [PubMed]
- Philp, N.J.; Wang, D.; Yoon, H.; Hjelmeland, L.M. Polarized Expression of Monocarboxylate Transporters in Human Retinal Pigment Epithelium and ARPE-19 Cells. Investig. Ophthalmol. Vis. Sci. 2003, 44, 1716–1721. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Otsuka, Y.; Itagaki, S.; Hirano, T.; Iseki, K. Inhibitory effects of statins on human monocarboxylate transporter 4. Int. J. Pharm. 2006, 317, 19–25. [Google Scholar] [CrossRef]
- Dimmer, K.S.; Friedrich, B.; Lang, F.; Deitmer, J.W.; Bröer, S. The low-affinity monocarboxylate transporter MCT4 is adapted to the export of lactate in highly glycolytic cells. Biochem. J. 2000, 350, 219–227. [Google Scholar] [CrossRef]
- Gandhi, G.K.; Cruz, N.F.; Ball, K.K.; Dienel, G.A. Astrocytes are poised for lactate trafficking and release from activated brain and for supply of glucose to neurons. J. Neurochem. 2009, 111, 522–536. [Google Scholar] [CrossRef]
- Cheng, C.; Edin, N.F.J.; Lauritzen, K.H.; Aspmodal, I.; Christoffersen, S.; Jian, L.; Rasmussen, L.J.; Pettersen, E.O.; Xiaoqun, G.; Bergersen, L.H. Alterations of monocarboxylate transporter densities during hypoxia in brain and breast tumour cells. Cell. Oncol. 2012, 35, 217–227. [Google Scholar] [CrossRef]
- Pinheiro, C.; Longatto-Filho, A.; Azevedo-Silva, J.; Casal, M.; Schmitt, F.C.; Baltazar, F. Role of monocarboxylate transporters in human cancers: State of the art. J. Bioenerg. Biomembr. 2012, 44, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Curtis, N.J.; Mooney, L.; Hopcroft, L.; Michopoulos, F.; Whalley, N.; Zhong, H.; Murray, C.; Logie, A.; Revill, M.; Byth, K.F.; et al. Pre-clinical pharmacology of AZD3965, a selective inhibitor of MCT1: DLBCL, NHL and Burkitt’s lymphoma anti-tumor activity. Oncotarget 2017, 8, 69219–69236. [Google Scholar] [CrossRef] [PubMed]
- Felmlee, M.A.; Jones, R.S.; Rodriguez-Cruz, V.; Follman, K.E.; Morris, M.E. Monocarboxylate Transporters (SLC16): Function, Regulation, and Role in Health and Disease. Pharmacol. Rev. 2020, 72, 466–485. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.; Morris, M.E. In Vitro and In Vivo Efficacy of AZD3965 and Alpha-Cyano-4-Hydroxycinnamic Acid in the Murine 4T1 Breast Tumor Model. AAPS J. 2020, 22, 84. [Google Scholar] [CrossRef]
- Medin, T.; Medin, H.; Hefte, M.B.; Storm-Mathisen, J.; Bergersen, L.H. Upregulation of the lactate transporter monocarboxylate transporter 1 at the blood-brain barrier in a rat model of attention-deficit/hyperactivity disorder suggests hyperactivity could be a form of self-treatment. Behav. Brain Res. 2018, 360, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Fisel, P.; Schaeffeler, E.; Schwab, M. Clinical and Functional Relevance of the Monocarboxylate Transporter Family in Disease Pathophysiology and Drug Therapy. Clin. Transl. Sci. 2018, 11, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Pertega-Gomes, N.; Vizcaíno, J.R.; Felisbino, S.; Warren, A.Y.; Shaw, G.; Kay, J.; Whitaker, H.; Lynch, A.G.; Fryer, L.; Neal, D.E.; et al. Epigenetic and oncogenic regulation of SLC16A7 (MCT2) results in protein over-expression, impacting on signalling and cellular phenotypes in prostate cancer. Oncotarget 2015, 6, 21675–21684. [Google Scholar] [CrossRef]
- Ullah, M.S.; Davies, A.J.; Halestrap, A.P. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J. Biol. Chem. 2006, 281, 9030–9037. [Google Scholar] [CrossRef]
- Lu, W.; Huang, J.; Sun, S.; Huang, S.; Gan, S.; Xu, J.; Yang, M.; Xu, S.; Jiang, X. Changes in lactate content and monocarboxylate transporter 2 expression in Aβ25–35-treated rat model of Alzheimer’s disease. Neurol. Sci. 2015, 36, 871–876. [Google Scholar] [CrossRef]
- Daniele, L.L.; Sauer, B.; Gallagher, S.M.; Pugh, E.N., Jr.; Philp, N.J. Altered visual function in monocarboxylate transporter 3 (Slc16a8) knockout mice. Am. J. Physiol. Cell Physiol. 2008, 295, C451–C457. [Google Scholar] [CrossRef]
- Gallagher-Colombo, S.; Maminishkis, A.; Tate, S.; Grunwald, G.B.; Philp, N.J. Modulation of MCT3 Expression during Wound Healing of the Retinal Pigment Epithelium. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5343–5350. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Goldschmidt-Clermont, P.J.; Dong, C. Inactivation of monocarboxylate transporter MCT3 by DNA methylation in atherosclerosis. Circulation 2005, 112, 1353–1361. [Google Scholar] [CrossRef] [PubMed]
- Friesema, E.C.; Grueters, A.; Biebermann, H.; Krude, H.; Moers, A.; Reeser, M.; Barrett, T.G.; Mancilla, E.E.; Svensson, J.; Kester, M.H.A.; et al. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet 2004, 364, 1435–1437. [Google Scholar] [CrossRef]
- Kersseboom, S.; Kremers, G.-J.; Friesema, E.C.H.; Visser, W.E.; Klootwijk, W.; Peeters, R.P.; Visser, T.J. Mutations in MCT8 in Patients with Allan-Herndon-Dudley-Syndrome Affecting Its Cellular Distribution. Mol. Endocrinol. 2013, 27, 801–813. [Google Scholar] [CrossRef]
- Maranduba, C.M.C.; Friesema, E.C.H.; Kok, F.; Kester, M.H.A.; Jansen, J.; Sertie, A.; Passos-Bueno, M.R.; Visser, T.J. Decreased cellular uptake and metabolism in Allan-Herndon-Dudley syndrome (AHDS) due to a novel mutation in the MCT8 thyroid hormone transporter. J. Med. Genet. 2005, 43, 457–460. [Google Scholar] [CrossRef][Green Version]
- Wittmann, G.; Szabon, J.; Mohácsik, P.; Nouriel, S.S.; Gereben, B.; Fekete, C.; Lechan, R.M. Parallel Regulation of Thyroid Hormone Transporters OATP1c1 and MCT8 During and After Endotoxemia at the Blood-Brain Barrier of Male Rodents. Endocrinology 2015, 156, 1552–1564. [Google Scholar] [CrossRef]
- Sun, Y.; Zhao, D.; Wang, G.; Jiang, Q.; Guo, M.; Kan, Q.; He, Z.; Sun, J. A novel oral prodrug-targeting transporter MCT 1: 5-fluorouracil-dicarboxylate monoester conjugates. Asian J. Pharm. Sci. 2019, 14, 631–639. [Google Scholar] [CrossRef]
- Cundy, K.C.; Branch, R.; Chernov-Rogan, T.; Dias, T.; Estrada, T.; Hold, K.; Koller, K.; Liu, X.; Mann, A.; Panuwat, M.; et al. XP13512 [(+/-)-1-([(alpha-isobutanoyloxyethoxy)carbonyl] aminomethyl)-1-cyclohexane acetic acid], a novel gabapentin prodrug: I. Design, synthesis, enzymatic conversion to gabapentin, and transport by intestinal solute transporters. J. Pharmacol. Exp. Ther. 2004, 311, 315–323. [Google Scholar] [CrossRef]
- Cundy, K.C.; Annamalai, T.; Bu, L.; Vera, J.D.; Estrela, J.; Luo, W.; Shirsat, P.; Torneros, A.; Yao, F.; Zou, J.; et al. XP13512 [(+/-)-1-([(alpha-isobutanoyloxyethoxy)carbonyl] aminomethyl)-1-cyclohexane acetic acid], a novel gabapentin prodrug: II. Improved oral bioavailability, dose proportionality, and colonic absorption compared with gabapentin in rats and monkeys. J. Pharmacol. Exp. Ther. 2004, 311, 324–333. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, G.; Chen, H.; Sun, Y.; Sun, M.; Liu, X.; Jian, W.; He, Z.; Sun, J. A facile di-acid mono-amidation strategy to prepare cyclization-activating mono-carboxylate transporter 1-targeting gemcitabine prodrugs for enhanced oral delivery. Int. J. Pharm. 2019, 573, 118718. [Google Scholar] [CrossRef]
- Kim, E.S.; Deeks, E.D. Gabapentin Enacarbil: A Review in Restless Legs Syndrome. Drugs 2016, 76, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, U.; Johannes, J.; Bayer, D.; Braun, D. Structure and Function of Thyroid Hormone Plasma Membrane Transporters. Eur. Thyroid J. 2014, 3, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Bosshart, P.D.; Kalbermatter, D.; Bonetti, S.; Fotiadis, D. Mechanistic basis of L-lactate transport in the SLC16 solute carrier family. Nat. Commun. 2019, 10, 2649. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.C.; Meredith, D.; Bunnun, C.; Sessions, R.B.; Halestrap, A.P. Studies on the DIDS-binding Site of Monocarboxylate Transporter 1 Suggest a Homology Model of the Open Conformation and a Plausible Translocation Cycle. J. Biol. Chem. 2009, 284, 20011–20021. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, A.; Futagi, Y.; Kobayashi, M.; Narumi, K.; Furugen, A.; Iseki, K. Extracellular lysine 38 plays a crucial role in pH-dependent transport via human monocarboxylate transporter 1. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183068. [Google Scholar] [CrossRef] [PubMed]
- Futagi, Y.; Kobayashi, M.; Narumi, K.; Furugen, A.; Iseki, K. Homology modeling and site-directed mutagenesis identify amino acid residues underlying the substrate selection mechanism of human monocarboxylate transporters 1 (hMCT1) and 4 (hMCT4). Cell. Mol. Life Sci. 2019, 76, 4905–4921. [Google Scholar] [CrossRef] [PubMed]
- Rahman, B.; Schneider, H.-P.; Bröer, A.; Deitmer, J.W.; Bröer, S. Helix 8 and Helix 10 Are Involved in Substrate Recognition in the Rat Monocarboxylate Transporter MCT1. Biochemistry 1999, 38, 11577–11584. [Google Scholar] [CrossRef]
- Galić, S.; Schneider, H.-P.; Bröer, A.; Deitmer, J.W.; Bröer, S. The loop between helix 4 and helix 5 in the monocarboxylate transporter MCT1 is important for substrate selection and protein stability. Biochem. J. 2003, 376, 413–422. [Google Scholar] [CrossRef]
- Wang, N.; Jiang, X.; Zhang, S.; Zhu, A.; Yuan, Y.; Xu, H.; Lei, J.; Yan, C. Structural basis of human monocarboxylate transporter 1 inhibition by anti-cancer drug candidates. Cell 2020, 184, 370–383. [Google Scholar] [CrossRef]
- Groeneweg, S.; De Souza, E.C.L.; Meima, M.E.; Peeters, R.P.; Visser, W.E.; Visser, T.J. Outward-Open Model of Thyroid Hormone Transporter Monocarboxylate Transporter 8 Provides Novel Structural and Functional Insights. Endocrinology 2017, 158, 3292–3306. [Google Scholar] [CrossRef]
- Protze, J.; Braun, D.; Hinz, K.M.; Bayer-Kusch, D.; Schweizer, U.; Krause, G. Membrane-traversing mechanism of thyroid hormone transport by monocarboxylate transporter 8. Cell Mol. Life Sci. 2017, 74, 2299–2318. [Google Scholar] [CrossRef] [PubMed]
- Kinne, A.; Kleinau, G.; Hoefig, C.S.; Grüters, A.; Köhrle, J.; Krause, G.; Schweizer, U. Essential Molecular Determinants for Thyroid Hormone Transport and First Structural Implications for Monocarboxylate Transporter 8. J. Biol. Chem. 2010, 285, 28054–28063. [Google Scholar] [CrossRef] [PubMed]
- Hagenbuch, B.; Meier, P.J. Organic anion transporting polypeptides of the OATP/SLC21 family: Phylogenetic classification as OATP/SLCO superfamily, new nomenclature and molecular/functional properties. Pflüg. Arch. 2004, 447, 653–665. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.; Obaidat, A.; Hagenbuch, B. OATPs, OATs and OCTs: The organic anion and cation transporters of the SLCO and SLC22A gene superfamilies. J. Cereb. Blood Flow Metab. 2012, 165, 1260–1287. [Google Scholar] [CrossRef]
- Ronaldson, P.T.; Davis, T.P. Targeted Drug Delivery to Treat Pain and Cerebral Hypoxia. Pharmacol. Rev. 2013, 65, 291–314. [Google Scholar] [CrossRef]
- Gao, B.; Vavricka, S.R.; Meier, P.J.; Stieger, B. Differential cellular expression of organic anion transporting peptides OATP1A2 and OATP2B1 in the human retina and brain: Implications for carrier-mediated transport of neuropeptides and neurosteriods in the CNS. Eur. J. Physiol. 2014, 467, 1481–1493. [Google Scholar] [CrossRef]
- Schnell, C.; Shahmoradi, A.; Wichert, S.P.; Mayerl, S.; Hagos, Y.; Heuer, H.; Rossner, M.J.; Hülsmann, S. The multispecific thyroid hormone transporter OATP1C1 mediates cell-specific sulforhodamine 101-labeling of hippocampal astrocytes. Anat. Embryol. 2013, 220, 193–203. [Google Scholar] [CrossRef][Green Version]
- Schäfer, A.; zu Schwabedissen, H.M.; Grube, M. Expression and Function of Organic Anion Transporting Polypeptides in the Human Brain: Physiological and Pharmacological Implications. Pharmaceutics 2021, 13, 834. [Google Scholar] [CrossRef]
- Choi, K.; Zhuang, H.; Crain, B.; Doré, S. Expression and localization of prostaglandin transporter in Alzheimer disease brains and age-matched controls. J. Neuroimmunol. 2008, 195, 81–87. [Google Scholar] [CrossRef]
- Huber, R.D.; Gao, B.; Pfändler, M.-A.S.; Zhang-Fu, W.; Leuthold, S.; Hagenbuch, B.; Folkers, G.; Meier, P.J.; Stieger, B. Characterization of two splice variants of human organic anion transporting polypeptide 3A1 isolated from human brain. Am. J. Physiol. Physiol. 2007, 292, C795–C806. [Google Scholar] [CrossRef]
- Leuthold, S.; Hagenbuch, B.; Mohebbi, N.; Wagner, C.A.; Meier, P.J.; Stieger, B. Mechanisms of pH-gradient driven transport mediated by organic anion polypeptide transporters. Am. J. Physiol. Cell Physiol. 2009, 296, C570–C582. [Google Scholar] [CrossRef]
- Kinzi, J.; Grube, M.; Schwabedissen, H.E.M.Z. OATP2B1—The underrated member of the organic anion transporting polypeptide family of drug transporters? Biochem. Pharmacol. 2021, 188, 114534. [Google Scholar] [CrossRef] [PubMed]
- Franke, R.M.; Scherkenbach, L.A.; Sparreboom, A. Pharmacogenetics of the organic anion transporting polypeptide 1A2. Pharmacogenomics 2009, 10, 339–344. [Google Scholar] [CrossRef]
- Westholm, D.E.; Salo, D.R.; Viken, K.; Rumbley, J.N.; Anderson, G.W. The Blood-Brain Barrier Thyroxine Transporter Organic Anion-Transporting Polypeptide 1c1 Displays Atypical Transport Kinetics. Endocrinology 2009, 150, 5153–5162. [Google Scholar] [CrossRef]
- Gose, T.; Nakanishi, T.; Kamo, S.; Shimada, H.; Otake, K.; Tamai, I. Prostaglandin transporter (OATP2A1/SLCO2A1) contributes to local disposition of eicosapentaenoic acid-derived PGE3. Prostaglandins Other Lipid Mediat. 2016, 122, 10–17. [Google Scholar] [CrossRef]
- Bakos, E.; Tusnády, G.E.; Német, O.; Patik, I.; Magyar, C.; Németh, K.; Kele, P.; Özvegy-Laczka, C. Synergistic transport of a fluorescent coumarin probe marks coumarins as pharmacological modulators of Organic anion-transporting polypeptide, OATP3A1. Biochem. Pharmacol. 2020, 182, 114250. [Google Scholar] [CrossRef]
- Bailey, D.G.; Dresser, G.K.; Leake, B.F.; Kim, R.B. Naringin is a Major and Selective Clinical Inhibitor of Organic Anion-Transporting Polypeptide 1A2 (OATP1A2) in Grapefruit Juice. Clin. Pharmacol. Ther. 2007, 81, 495–502. [Google Scholar] [CrossRef]
- Morita, T.; Akiyoshi, T.; Tsuchitani, T.; Kataoka, H.; Araki, N.; Yajima, K.; Katayama, K.; Imaoka, A.; Ohtani, H. Inhibitory Effects of Cranberry Juice and Its Components on Intestinal OATP1A2 and OATP2B1: Identification of Avicularin as a Novel Inhibitor. J. Agric. Food Chem. 2022, 70, 3310–3320. [Google Scholar] [CrossRef]
- Kalliokoski, A.; Niemi, M. Impact of OATP transporters on pharmacokinetics. J. Cereb. Blood Flow Metab. 2009, 158, 693–705. [Google Scholar] [CrossRef]
- Bakos, É.; Német, O.; Patik, I.; Kucsma, N.; Várady, G.; Szakács, G.; Özvegy-Laczka, C. A novel fluorescence-based functional assay for human OATP1A2 and OATP1C1 identifies interaction between third-generation P-gp inhibitors and OATP1A2. FEBS J. 2019, 287, 2468–2485. [Google Scholar] [CrossRef]
- Tikkanen, A.; Pierrot, E.; Deng, F.; Sánchez, V.B.; Hagström, M.; Koenderink, J.B.; Kidron, H. Food Additives as Inhibitors of Intestinal Drug Transporter OATP2B1. Mol. Pharm. 2020, 17, 3748–3758. [Google Scholar] [CrossRef]
- Unger, M.S.; Mudunuru, J.; Schwab, M.; Hopf, C.; Drewes, G.; Nies, A.T.; Zamek-Gliszczynski, M.J.; Reinhard, F. Clinically Relevant OATP2B1 Inhibitors in Marketed Drug Space. Mol. Pharm. 2019, 17, 488–498. [Google Scholar] [CrossRef]
- Chen, M.; Hu, S.; Li, Y.; Gibson, A.A.; Fu, Q.; Baker, S.D.; Sparreboom, A. Role of Oatp2b1 in Drug Absorption and Drug-Drug Interactions. Drug Metab. Dispos. 2020, 48, 420–426. [Google Scholar] [CrossRef]
- Rebello, S.; Zhao, S.; Hariry, S.; Dahlke, M.; Alexander, N.; Vapurcuyan, A.; Hanna, I.; Jarugula, V. Intestinal OATP1A2 inhibition as a potential mechanism for the effect of grapefruit juice on aliskiren pharmacokinetics in healthy subjects. Eur. J. Clin. Pharmacol. 2011, 68, 697–708. [Google Scholar] [CrossRef]
- Kamo, S.; Nakanishi, T.; Aotani, R.; Nakamura, Y.; Gose, T.; Tamai, I. Impact of FDA-Approved Drugs on the Prostaglandin Transporter OATP2A1/SLCO2A1. J. Pharm. Sci. 2017, 106, 2483–2490. [Google Scholar] [CrossRef]
- Lee, W.; Glaeser, H.; Smith, H.; Roberts, R.L.; Moeckel, G.W.; Gervasini, G.; Leake, B.F.; Kim, R.B. Polymorphisms in human organic anion-transporting polypeptide 1A2 (OATP1A2): Implications for altered drug disposition and central nervous system drug entry. J. Biol. Chem. 2005, 280, 9610–9617. [Google Scholar] [CrossRef]
- Zhou, F.; Zheng, J.; Zhu, L.; Jodal, A.; Cui, P.H.; Wong, M.; Gurney, H.; Church, W.; Murray, M. Functional Analysis of Novel Polymorphisms in the Human SLCO1A2 Gene that Encodes the Transporter OATP1A2. AAPS J. 2013, 15, 1099–1108. [Google Scholar] [CrossRef]
- Thompson, B.J.; Sanchez-Covarrubias, L.; Slosky, L.M.; Zhang, Y.; Laracuente, M.-L.; Ronaldson, P.T. Hypoxia/Reoxygenation Stress Signals an Increase in Organic Anion Transporting polypeptide 1a4 (Oatp1a4) at the Blood–Brain Barrier: Relevance to CNS Drug Delivery. J. Cereb. Blood Flow Metab. 2014, 34, 699–707. [Google Scholar] [CrossRef]
- Nies, A.T.; Niemi, M.; Burk, O.; Winter, S.; Zanger, U.M.; Stieger, B.; Schwab, M.; Schaeffeler, E. Genetics is a major determinant of expression of the human hepatic uptake transporter OATP1B1, but not of OATP1B3 and OATP2B1. Genome Med. 2013, 5, 1. [Google Scholar] [CrossRef]
- Tapaninen, T.; Karonen, T.; Backman, J.T.; Neuvonen, P.J.; Niemi, M. SLCO2B1 c.935G>A single nucleotide polymorphism has no effect on the pharmacokinetics of montelukast and aliskiren. Pharm. Genom. 2013, 23, 19–24. [Google Scholar] [CrossRef]
- Schulte, R.R.; Ho, R.H. Organic Anion Transporting Polypeptides: Emerging Roles in Cancer Pharmacology. Mol. Pharmacol. 2019, 95, 490–506. [Google Scholar] [CrossRef]
- Zhu, Q.; Liang, X.; Dai, J.; Guan, X. Prostaglandin transporter, SLCO2A1, mediates the invasion and apoptosis of lung cancer cells via PI3K/AKT/mTOR pathway. Int. J. Clin. Exp. Pathol. 2015, 8, 9175–9181. [Google Scholar]
- Mayerl, S.; Visser, T.J.; Darras, V.M.; Horn, S.; Heuer, H. Impact of Oatp1c1 Deficiency on Thyroid Hormone Metabolism and Action in the Mouse Brain. Endocrinology 2012, 153, 1528–1537. [Google Scholar] [CrossRef]
- Admati, I.; Wasserman-Bartov, T.; Tovin, A.; Rozenblat, R.; Blitz, E.; Zada, D.; Lerer-Goldshtein, T.; Appelbaum, L. Neural Alterations and Hyperactivity of the Hypothalamic–Pituitary–Thyroid Axis in Oatp1c1 Deficiency. Thyroid 2020, 30, 161–174. [Google Scholar] [CrossRef]
- Li, M.; Wang, W.; Cheng, Y.; Zhang, X.; Zhao, N.; Tan, Y.; Xie, Q.; Chai, J.; Pan, Q. Tumor necrosis factor α upregulates the bile acid efflux transporter OATP3A1 via multiple signaling pathways in cholestasis. J. Biol. Chem. 2021, 298. [Google Scholar] [CrossRef]
- Choi, J.H.; Murray, J.W.; Wolkoff, A.W. PDZK1 binding and serine phosphorylation regulate subcellular trafficking of organic anion transport protein 1a1. Am. J. Physiol. Liver Physiol. 2011, 300, G384–G393. [Google Scholar] [CrossRef]
- Hänggi, E.; Grundschober, A.F.; Leuthold, S.; Meier, P.J.; St-Pierre, M.V. Functional Analysis of the Extracellular Cysteine Residues in the Human Organic Anion Transporting Polypeptide, OATP2B1. Mol. Pharmacol. 2006, 70, 806–817. [Google Scholar] [CrossRef]
- Meier-Abt, F.; Mokrab, Y.; Mizuguchi, K. Organic anion transporting polypeptides of the OATP/SLCO superfamily: Identification of new members in nonmammalian species, comparative modeling and a potential transport mode. J. Membr. Biol. 2005, 208, 213–227. [Google Scholar] [CrossRef]
- Betterton, R.D.; Davis, T.P.; Ronaldson, P.T. Organic Cation Transporter (OCT/OCTN) Expression at Brain Barrier Sites: Focus on CNS Drug Delivery. Handb. Exp. Pharmacol. 2021, 266, 301–328. [Google Scholar] [CrossRef]
- Pochini, L.; Galluccio, M.; Scalise, M.; Console, L.; Indiveri, C. OCTN: A Small Transporter Subfamily with Great Relevance to Human Pathophysiology, Drug Discovery, and Diagnostics. SLAS Discov. Adv. Sci. Drug Discov. 2018, 24, 89–110. [Google Scholar] [CrossRef]
- Koepsell, H.; Lips, K.; Volk, C. Polyspecific Organic Cation Transporters: Structure, Function, Physiological Roles, and Biopharmaceutical Implications. Pharm. Res. 2007, 24, 1227–1251. [Google Scholar] [CrossRef] [PubMed]
- Barendt, W.M.; Wright, S.H. The Human Organic Cation Transporter (hOCT2) Recognizes the Degree of Substrate Ionization. J. Biol. Chem. 2002, 277, 22491–22496. [Google Scholar] [CrossRef] [PubMed]
- Sakata, T.; Anzai, N.; Kimura, T.; Miura, D.; Fukutomi, T.; Takeda, M.; Sakurai, H.; Endou, H. Functional Analysis of Human Organic Cation Transporter OCT3 (SLC22A3) Polymorphisms. J. Pharmacol. Sci. 2010, 113, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Kerb, R.; Brinkmann, U.; Chatskaia, N.; Gorbunov, D.; Gorboulev, V.; Mornhinweg, E.; Keil, A.; Eichelbaum, M.; Koepsell, H. Identification of genetic variations of the human organic cation transporter hOCT1 and their functional consequences. Pharmacogenetics 2002, 12, 591–595. [Google Scholar] [CrossRef]
- Fahrmayr, C.; Fromm, M.F.; König, J. Hepatic OATP and OCT uptake transporters: Their role for drug-drug interactions and pharmacogenetic aspects. Drug Metab. Rev. 2010, 42, 380–401. [Google Scholar] [CrossRef]
- Lin, Z.; Nelson, L.; Franke, A.; Poritz, L.; Li, T.-Y.; Wu, R.; Wang, Y.; MacNeill, C.; Thomas, N.J.; Schreiber, S.; et al. OCTN1 variant L503F is associated with familial and sporadic inflammatory bowel disease. J. Crohn’s Colitis 2010, 4, 132–138. [Google Scholar] [CrossRef]
- Lahjouji, K.; Mitchell, G.A.; Qureshi, I.A. Carnitine Transport by Organic Cation Transporters and Systemic Carnitine Deficiency. Mol. Genet. Metab. 2001, 73, 287–297. [Google Scholar] [CrossRef]
- Tsuji, A. Small molecular drug transfer across the blood-brain barrier via carrier-mediated transport systems. NeuroRx 2005, 2, 54–62. [Google Scholar] [CrossRef]
- Nagle, M.A.; Wu, W.; Eraly, S.A.; Nigam, S.K. Organic anion transport pathways in antiviral handling in choroid plexus in Oat1 (Slc22a6) and Oat3 (Slc22a8) deficient tissue. Neurosci. Lett. 2013, 534, 133–138. [Google Scholar] [CrossRef]
- Saidijam, M.; Dermani, F.K.; Sohrabi, S.; Patching, S.G. Efflux proteins at the blood–brain barrier: Review and bioinformatics analysis. Xenobiotica 2017, 48, 506–532. [Google Scholar] [CrossRef]
- Burckhardt, G. Drug transport by Organic Anion Transporters (OATs). Pharmacol. Ther. 2012, 136, 106–130. [Google Scholar] [CrossRef]
- Montaser, A.; Markowicz-Piasecka, M.; Sikora, J.; Jalkanen, A.; Huttunen, K.M. L-type amino acid transporter 1 (LAT1)-utilizing efflux transporter inhibitors can improve the brain uptake and apoptosis-inducing effects of vinblastine in cancer cells. Int. J. Pharm. 2020, 586, 119585. [Google Scholar] [CrossRef] [PubMed]
- U.S. FDA. In Vitro Drug Interaction Studies, Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions, Guidance for Industry. 2020. Available online: https://www.fda.gov/media/134582/download (accessed on 5 April 2022).
- EMA. Guideline on the Investigation of Drug Interactions. 2012. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-drug-interactions-revision-1_en.pdf (accessed on 5 April 2022).
- VanWert, A.L.; Gionfriddo, M.; Sweet, D.H. Organic anion transporters: Discovery, pharmacology, regulation and roles in pathophysiology. Biopharm. Drug Dispos. 2009, 31, 1–71. [Google Scholar] [CrossRef]
- Ciarimboli, G. Regulation Mechanisms of Expression and Function of Organic Cation Transporter 1. Front. Pharmacol. 2021, 11, 2234. [Google Scholar] [CrossRef]
- Dickens, D.; Owen, A.; Alfirevic, A.; Giannoudis, A.; Davies, A.; Weksler, B.; Romero, I.; Couraud, P.-O.; Pirmohamed, M. Lamotrigine is a substrate for OCT1 in brain endothelial cells. Biochem. Pharmacol. 2012, 83, 805–814. [Google Scholar] [CrossRef]
- Sekhar, G.N.; Georgian, A.R.; Sanderson, L.; Vizcay-Barrena, G.; Brown, R.C.; Muresan, P.; Fleck, R.; Thomas, S.A. Organic cation transporter 1 (OCT1) is involved in pentamidine transport at the human and mouse blood-brain barrier (BBB). PLoS ONE 2017, 12, e0173474. [Google Scholar] [CrossRef]
- Dos Santos Pereira, J.N.; Tadjerpisheh, S.; Abu Abed, M.; Saadatmand, A.R.; Weksler, B.; Romero, I.; Couraud, P.-O.; Brockmöller, J.; Tzvetkov, M.V. The Poorly Membrane Permeable Antipsychotic Drugs Amisulpride and Sulpiride Are Substrates of the Organic Cation Transporters from the SLC22 Family. AAPS J. 2014, 16, 1247–1258. [Google Scholar] [CrossRef]
- MacKenzie, B.; Erickson, J.D. Sodium-coupled neutral amino acid (System N/A) transporters of the SLC38 gene family. Pflug. Arch. 2004, 447, 784–795. [Google Scholar] [CrossRef]
- Bröer, S. The SLC38 family of sodium–amino acid co-transporters. Pflug. Arch. 2014, 466, 155–172. [Google Scholar] [CrossRef]
- Schiöth, H.B.; Roshanbin, S.; Hägglund, M.G.; Fredriksson, R. Evolutionary origin of amino acid transporter families SLC32, SLC36 and SLC38 and physiological, pathological and therapeutic aspects. Mol. Asp. Med. 2013, 34, 571–585. [Google Scholar] [CrossRef]
- Solbu, T.T.; Bjørkmo, M.; Berghuis, P.; Harkany, T.; Chaudhry, F.A. SAT1, a glutamine transporter, is preferentially expressed in GABAergic neurons. Front. Neuroanat. 2010, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- González-González, I.; Cubelos, B.; Giménez, C.; Zafra, F. Immunohistochemical localization of the amino acid transporter SNAT2 in the rat brain. Neuroscience 2005, 130, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Melone, M.; Quagliano, F.; Barbaresi, P.; Varoqui, H.; Erickson, J.D.; Conti, F. Localization of the Glutamine Transporter SNAT1 in Rat Cerebral Cortex and Neighboring Structures, With a Note on its Localization in Human Cortex. Cereb. Cortex 2004, 14, 562–574. [Google Scholar] [CrossRef] [PubMed]
- Grewal, S.; Defamie, N.; Zhang, X.; Gois, S.D.; Shawki, A.; Mackenzie, B.; Chen, C.; Varoqui, H.; Erickson, J.D. SNAT2 amino acid transporter is regulated by amino acids of the SLC6 gamma-aminobutyric acid transporter subfamily in neocortical neurons and may play no role in delivering glutamine for glutamatergic transmission. J. Biol. Chem. 2009, 284, 11224–11236. [Google Scholar] [CrossRef]
- Cubelos, B.; González-González, I.M.; Giménez, C.; Zafra, F. Amino acid transporter SNAT5 localizes to glial cells in the rat brain. Glia 2004, 49, 230–244. [Google Scholar] [CrossRef]
- Rubio-Aliaga, I.; Wagner, C.A. Regulation and function of the SLC38A3/SNAT3 glutamine transporter. Channels 2016, 10, 440–452. [Google Scholar] [CrossRef]
- Low, S.Y.; Taylor, P.M.; Ahmed, A.; Pogson, C.I.; Rennie, M.J. Substrate-specificity of glutamine transporters in membrane vesicles from rat liver and skeletal muscle investigated using amino acid analogues. Biochem. J. 1991, 278, 105–111. [Google Scholar] [CrossRef]
- Hägglund, M.G.; Hellsten, S.V.; Bagchi, S.; Philippot, G.; Löfqvist, E.; Nilsson, V.C.; Almkvist, I.; Karlsson, E.; Sreedharan, S.; Tafreshiha, A.; et al. Transport of l-Glutamine, l-Alanine, l-Arginine and l-Histidine by the Neuron-Specific Slc38a8 (SNAT8) in CNS. J. Mol. Biol. 2015, 427, 1495–1512. [Google Scholar] [CrossRef]
- Hägglund, M.G.; Sreedharan, S.; Nilsson, V.C.; Shaik, J.H.; Almkvist, I.M.; Bäcklin, S.; Wrange, Ö.; Fredriksson, R. Identification of SLC38A7 (SNAT7) Protein as a Glutamine Transporter Expressed in Neurons. J. Biol. Chem. 2011, 286, 20500–20511. [Google Scholar] [CrossRef]
- Bagchi, S.; Baomar, H.A.; Al-Walai, S.; Al-Sadi, S.; Fredriksson, R. Histological Analysis of SLC38A6 (SNAT6) Expression in Mouse Brain Shows Selective Expression in Excitatory Neurons with High Expression in the Synapses. PLoS ONE 2014, 9, e95438. [Google Scholar] [CrossRef]
- Gandasi, N.; Arapi, V.; Mickael, M.; Belekar, P.; Granlund, L.; Kothegala, L.; Fredriksson, R.; Bagchi, S. Glutamine Uptake via SNAT6 and Caveolin Regulates Glutamine–Glutamate Cycle. Int. J. Mol. Sci. 2021, 22, 1167. [Google Scholar] [CrossRef] [PubMed]
- Ogura, M.; Taniura, H.; Nakamichi, N.; Yoneda, Y. Upregulation of the glutamine transporter through transactivation mediated by camp/protein kinase a signals toward exacerbation of vulnerability to oxidative stress in rat neocortical astrocytes. J. Cell. Physiol. 2007, 212, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Ogura, M.; Nakamichi, N.; Takano, K.; Oikawa, H.; Kambe, Y.; Ohno, Y.; Taniura, H.; Yoneda, Y. Functional expression of A glutamine transporter responsive to down-regulation by lipopolysaccharide through reduced promoter activity in cultured rat neocortical astrocytes. J. Neurosci. Res. 2006, 83, 1447–1460. [Google Scholar] [CrossRef] [PubMed]
- Rosario, F.J.; Kanai, Y.; Powell, T.L.; Jansson, T. Mammalian target of rapamycin signalling modulates amino acid uptake by regulating transporter cell surface abundance in primary human trophoblast cells. J. Physiol. 2013, 591, 609–625. [Google Scholar] [CrossRef]
- Gu, S.; Villegas, C.J.; Jiang, J.X. Differential Regulation of Amino Acid Transporter SNAT3 by Insulin in Hepatocytes. J. Biol. Chem. 2005, 280, 26055–26062. [Google Scholar] [CrossRef]
- Nissen-Meyer, L.S.H.; Chaudhry, F.A. Protein Kinase C Phosphorylates the System N Glutamine Transporter SN1 (Slc38a3) and Regulates Its Membrane Trafficking and Degradation. Front. Endocrinol. 2013, 4, 138. [Google Scholar] [CrossRef]
- Uchida, Y.; Ito, K.; Ohtsuki, S.; Kubo, Y.; Suzuki, T.; Terasaki, T. Major involvement of Na(+) -dependent multivitamin transporter (SLC5A6/SMVT) in uptake of biotin and pantothenic acid by human brain capillary endothelial cells. J. Neurochem. 2015, 134, 97–112. [Google Scholar] [CrossRef]
- Salazar, K.; Martínez, F.; Pérez-Martín, M.; Cifuentes, M.; Trigueros, L.; Ferrada, L.; Espinoza, F.; Saldivia, N.; Bertinat, R.; Forman, K.; et al. SVCT2 Expression and Function in Reactive Astrocytes Is a Common Event in Different Brain Pathologies. Mol. Neurobiol. 2017, 55, 5439–5452. [Google Scholar] [CrossRef]
- Castro, M.; Caprile, T.; Astuya-Villalón, A.; Millán, C.; Reinicke, K.; Vera, J.C.; Vásquez, O.; Aguayo, L.G.; Nualart, F. High-affinity sodium-vitamin C co-transporters (SVCT) expression in embryonic mouse neurons. J. Neurochem. 2001, 78, 815–823. [Google Scholar] [CrossRef]
- Prasad, P.D.; Wang, H.; Kekuda, R.; Fujita, T.; Fei, Y.-J.; Devoe, L.D.; Leibach, F.H.; Ganapathy, V. Cloning and Functional Expression of a cDNA Encoding a Mammalian Sodium-dependent Vitamin Transporter Mediating the Uptake of Pantothenate, Biotin, and Lipoate. J. Biol. Chem. 1998, 273, 7501–7506. [Google Scholar] [CrossRef]
- Nualart, F.; Mack, L.; Garcia, A.; Cisternas, P.; Bongarzone, E.R.; Heitzer, M.; Jara, N.; Martinez, F.; Ferrada, L.; Espinoza, F.; et al. Vitamin C Transporters, Recycling and the Bystander Effect in the Nervous System: SVCT2 versus Gluts. J. Stem. Cell Res. Ther. 2014, 4, 209. [Google Scholar] [CrossRef]
- Zhao, Y.; Qu, B.; Wu, X.; Li, X.; Liu, Q.; Jin, X.; Guo, L.; Hai, L.; Wu, Y. Design, synthesis and biological evaluation of brain targeting l-ascorbic acid prodrugs of ibuprofen with “lock-in” function. Eur. J. Med. Chem. 2014, 82, 314–323. [Google Scholar] [CrossRef]
- Yue, Q.; Peng, Y.; Zhao, Y.; Lu, R.; Fu, Q.; Chen, Y.; Yang, Y.; Hai, L.; Guo, L.; Wu, Y. Dual-targeting for brain-specific drug delivery: Synthesis and biological evaluation. Drug Deliv. 2018, 25, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, L.; Zhao, Y.; Fu, Q.; Xiao, W.; Lu, R.; Hai, L.; Guo, L.; Wu, Y. Design, synthesis, and neuroprotective effects of dual-brain targeting naproxen prodrug. Arch. Pharm. 2018, 351, e1700382. [Google Scholar] [CrossRef]
- Alam, K.; Crowe, A.; Wang, X.; Zhang, P.; Ding, K.; Li, L.; Yue, W. Regulation of Organic Anion Transporting Polypeptides (OATP) 1B1- and OATP1B3-Mediated Transport: An Updated Review in the Context of OATP-Mediated Drug-Drug Interactions. Int. J. Mol. Sci. 2018, 19, 855. [Google Scholar] [CrossRef]
- Luo, S.; Kansara, V.S.; Zhu, X.; Mandava, N.K.; Pal, D.; Mitra, A.K. Functional Characterization of Sodium-Dependent Multivitamin Transporter in MDCK-MDR1 Cells and Its Utilization as a Target for Drug Delivery. Mol. Pharm. 2006, 3, 329–339. [Google Scholar] [CrossRef][Green Version]
- Inazu, M. Functional Expression of Choline Transporters in the Blood–Brain Barrier. Nutrients 2019, 11, 2265. [Google Scholar] [CrossRef]
- Haga, T. Molecular properties of the high-affinity choline transporter CHT1. J. Biochem. 2014, 156, 181–194. [Google Scholar] [CrossRef]
- Okuda, T.; Haga, T.; Kanai, Y.; Endou, H.; Ishihara, T.; Katsura, I. Identification and characterization of the high-affinity choline transporter. Nat. Neurosci. 2000, 3, 120–125. [Google Scholar] [CrossRef]
- Traiffort, E.; O’Regan, S.; Ruat, M. The choline transporter-like family SLC44: Properties and roles in human diseases. Mol. Asp. Med. 2013, 34, 646–654. [Google Scholar] [CrossRef]
- Iwao, B.; Yara, M.; Hara, N.; Kawai, Y.; Yamanaka, T.; Nishihara, H.; Inoue, T.; Inazu, M. Functional expression of choline transporter like-protein 1 (CTL1) and CTL2 in human brain microvascular endothelial cells. Neurochem. Int. 2015, 93, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, B.A.; Díez-Sampedro, A.; Wright, E.M. Common mechanisms of inhibition for the Na+/glucose (hSGLT1) and Na+/Cl-/GABA (hGAT1) cotransporters. Br. J. Pharmacol. 2001, 134, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Otto, C.; Friedrich, A.; Madunić, I.V.; Baumeier, C.; Schwenk, R.W.; Karaica, D.; Germer, C.-T.; Schürmann, A.; Sabolić, I.; Koepsell, H. Antidiabetic Effects of a Tripeptide That Decreases Abundance of Na+-d-glucose Cotransporter SGLT1 in the Brush-Border Membrane of the Small Intestine. ACS Omega 2020, 5, 29127–29139. [Google Scholar] [CrossRef]
- Tahrani, A.; Barnett, A.H.; Bailey, C.J. SGLT inhibitors in management of diabetes. Lancet Diabetes Endocrinol. 2013, 1, 140–151. [Google Scholar] [CrossRef]
- Boswell-Casteel, R.C.; Hays, F.A. Equilibrative nucleoside transporters—A review. Nucleosides Nucleotides Nucleic Acids 2016, 36, 7–30. [Google Scholar] [CrossRef]
- Arcas, M.M.; Trigueros-Motos, L.; Casado, F.J.; Anglada, M.P. Physiological and Pharmacological Roles of Nucleoside Transporter Proteins. Nucleosides Nucleotides Nucleic Acids 2008, 27, 769–778. [Google Scholar] [CrossRef]
- Chang, C.; Swaan, P.W.; Ngo, L.Y.; Lum, P.Y.; Patil, S.D.; Unadkat, J.D. Molecular Requirements of the Human Nucleoside Transporters hCNT1, hCNT2, and hENT1. Mol. Pharmacol. 2004, 65, 558–570. [Google Scholar] [CrossRef]
- Young, J.D.; Yao, S.Y.; Baldwin, J.M.; Cass, C.E.; Baldwin, S.A. The human concentrative and equilibrative nucleoside transporter families, SLC28 and SLC29. Mol. Asp. Med. 2013, 34, 529–547. [Google Scholar] [CrossRef]
- Kell, D.B.; Dobson, P.D.; Oliver, S.G. Pharmaceutical drug transport: The issues and the implications that it is essentially carrier-mediated only. Drug Discov. Today 2011, 16, 704–714. [Google Scholar] [CrossRef]
- Kell, D.B. Hitchhiking into the cell. Nat. Chem. Biol. 2020, 16, 367–368. [Google Scholar] [CrossRef]
- Superti-Furga, G.; Superti-Furga, G.; Lackner, D.; Wiedmer, T.; Ingles-Prieto, A.; Barbosa, B.; Girardi, E.; Goldmann, U.; Gürtl, B.; Klavins, K.; et al. The RESOLUTE consortium: Unlocking SLC transporters for drug discovery. Nat. Rev. Drug Discov. 2020, 19, 429–430. [Google Scholar] [CrossRef] [PubMed]
- Van de Waterbeemd, H.; Smith, D.A.; Jones, B.C. Lipophilicity in PK design: Methyl, ethyl, futile. J. Comput. Aided Mol. Des. 2001, 15, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Fracassi, A.; Marangoni, M.; Rosso, P.; Pallottini, V.; Fioramonti, M.; Siteni, S.; Segatto, M. Statins and the Brain: More than Lipid Lowering Agents? Curr. Neuropharmacol. 2019, 17, 59–83. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transporter | Gene Name | Tissue Distribution (Expression) | Substrates | Inhibitors | Expression Modulation/Transport Capacity Changes |
|---|---|---|---|---|---|
| EAAT1 | SLC1A3 | Brain: BBB (abluminal), astrocytes | L-Glu, L-Asp | L-Serine-O-sulfate (L-SOS), (R,S)-2-amino- 3-(1-hydroxy-1,2,3-triazol-5-yl)propionate, (4R)-4-methylglutamate (4-Me-Glu), UCPH-101, UCPH-102 | EAAT1 expression ↑ by adenylate cyclase- activating polypeptide (PACAP), transforming growth factor α (TGFα), epidermal growth factor (EGF), estrogen, tamoxifen, raloxifen |
| EAAT2 | SLC1A2 | Brain: BBB (abluminal), astrocytes | L-Glu, L-Asp | Dihydrokainic acid, WAY-213613 | EAAT2 expression ↑ by adenylate cyclase- activating polypeptide (PACAP), transforming growth factor α (TGFα), epidermal growth factor (EGF), estrogen, tamoxifen, raloxifen, glucocorticoids, ceftriaxone |
| EAAT3 | SLC1A1 | Brain: neurons | L-Glu, L-Asp | 2-(Furan-2-yl)- 8-methyl-N-(o-tolyl)imidazo[1,2-a]pyridin-3-amine | Amphetamine induces EAAT3 internalization |
| ASCT1 | SLC1A4 | Ubiquitous, Brain: luminal and abluminal membranes of BBB, neurons, and astrocytes | L-Ala, L-Ser, L-Cys, L-Gly, L-Met, L-Val, L-Leu, L-Ile, L-Thr, D-Ser; L-Glu (efflux) | Phenylglycine analogs | ASCT1 expression ↓ results in neurodevelopmental alterations |
| ASCT2 | SLC1A5 | Ubiquitous, Brain: BBB (abluminal), neurons, and astrocytes | L-Ala, L-Ser, L-Gly, L-Met, L-Val, L-Leu, L-Ile, L-Thr; L-Glu (efflux) | O-Benzyl- L-serine, S-benzyl- cysteine, phenylglycine analogs | ASCT2 expression ↑ in highly proliferative cells, such as cancer cells |
| Transporter | Gene Name | Tissue Distribution (Expression) | Substrates | Inhibitors | Expression Modulation/Transport Capacity Changes |
|---|---|---|---|---|---|
| GLUT1 | SLC2A1 | Ubiquitous, Brain: luminal and abluminal membranes of BBB, astrocytes, (neurons, microglia) | Glucose, galactose, mannose, 2-deoxy-D-glucose, 2-deoxy-2-[18F]-D-glucose, glucosamine and dehydroascorbic acid (vitamin C) | Cytochalasin B, forskolin, phloretin and other flavonoids, WZB117, BAY-876, STF-31, fasentin, apigenin | GLUT1 expression ↑ in numerous cancers and ischemia with poor survival of patients: via hypoxia, p53, PI3K-Akt pathways, Ras or c-Myc oncogenes; GLUT1 expression ↓ in Alzheimer’s disease and GLUT1 deficiency syndrome (G1DS) due to mutations |
| GLUT3 | SLC2A3 | Brain: neurons | D-Glucose, D-galactose, D-mannose, D-xylose, 2-deoxy-D-glucose | Cytochalasin B, forskolin, phloretin, quercetin and other flavonoids, glycogen synthase kinase-3 (GSK-3) inhibitors | GLUT3 expression ↑ in various cancers with poor survival of patients: via hypoxia, p53, PI3K-Akt pathway |
| Transporter | Gene Name | Tissue Distribution (Expression) | Substrates | Inhibitors | Expression Modulation/Transport Capacity Changes |
|---|---|---|---|---|---|
| CAT1 | SLC7A1 | Ubiquitous, Brain: luminal and abluminal membranes of BBB | L-Arg, L-Lys, and L-Orn | Not known | CAT1 expression ↓ via NMDA receptor activation; CAT1 expression ↑ in colorectal and breast cancers, hepatitis B virus-induced hepatocellular carcinoma, and lymphocytic leukemia |
| CAT2B | SLC7A2 | Brain: neurons, oligo-dendrocytes, induced astrocytes | L-Arg, L-Lys, and L-Orn | Not known | CAT2B expression ↑ in different breast cancer cell lines |
| CAT3 | SLC7A3 | Placenta, Brain: neurons | L-Arg, L-Lys, and L-Orn | Not known | CAT3 expression ↓ via NMDA receptor activation: |
| LAT1 | SLC7A5 | Widely distributed, Brain: luminal and abluminal membranes of BBB, neurons, astrocytes, microglia | L-Leu, L-Phe, L-Tyr, L-Trp, L-His, L-Met, L-Ile, L-Val; triiodothyronine (T3) and thyroxine (T4), L-dopa, melphalan, baclofen, gabapentin, pregabalin | JPH203 (unselective) | LAT1 expression ↑ in numerous cancers with poor survival of patients: via hypoxia/HIF-2α, c-Myc or RAS-MEK-ERK pathways |
| y+LAT2 | SLC7A6 | Ubiquitous, Brain: astrocytes | L-Arg, L-Leu, L-glu (efflux) | No specific inhibitor reported | y+LAT2 expression ↑ in the presence of NH4+: via NF-κB pathway |
| LAT2 | SLC7A8 | Ubiquitous, Brain: microglia > neurons > astrocytes | L-Tyr, L-Phe, L-Trp, L-Thr, L-Asn, L-Ile, L-Cys, L-Ser, L-Leu, L-Val, L-Gln, L-His, L-Ala, L-Met; triiodothyronine (T3), 3,3′-diiodothyronine | No specific inhibitor reported | LAT2 expression ↑ in highly proliferative cells, such as cancer cells |
| Asc-1 | SLC7A10 | Adipose tissue, Brain: neurons | L-glycine, L-alanine, D-/L-serine, L-threonine, L-cysteine, α-aminobutyric acid, and β-alanine | Several structures have been proposed, requires more studies | Asc-1 downregulation associated with tremors, ataxia, and seizures |
| xCT | SLC7A11 | Macrophages, Brain: astrocytes, neurons | Cystine (extracellular)/glutamate (intracellular) exchange | S-4-carboxy-3-hydroxy- phenylglycine, erastatin, sorafenib, sulfasalazine | xCT is upregulated in several cancers and its dysfunction is associated with epileptic seizures, neurodegeneration, and brain edema |
| Transporter | Gene Name | Tissue Distribution (Expression) | Substrates | Inhibitors | Expression Modulation/Transport Capacity Changes |
|---|---|---|---|---|---|
| MCT1 | SLC16A1 | Ubiquitous, Brain: luminal and abluminal membranes of BBB, astrocytes | Lactate, pyruvate, ketone bodies; probenecid, 6-mercapto-purine, 4-phenyl-butyrate salicylic acid, nicotinic acid, valproic acid, β-lactams, XP13512, γ-hydroxy butyric acid | 4-Chloro-α-cyanocinnamic acid (non-specific), AZD3965 | MCT1 expression ↑ in numerous cancers, at the BBB of ADHD children, and metabolic active tissues of obese individuals: via MYC, p53 |
| MCT2 | SLC16A7 | Liver, kidneys, Brain: neurons | Lactate, pyruvate, ketone bodies | 4-Chloro-α-cyanocinnamic acid (non-specific), AZD3965 | MCT2 expression ↑ in numerous cancers and metabolic active tissues of obese individuals: via demethylation and hyper-methylation of DNA; MCT2 expression ↓ in hippocampus and cerebral cortex with pathologic progression of Alzheimer’s disease (via reduced energy metabolism?) |
| MCT3 | SLC16A8 | Retinal pigment epithelium, choroid plexus | Lactate | Not reported | MCT3 expression ↓ in retinal pigment epithelium impairs visual functions and wound healing and in smooth muscle cells induces atherosclerosis via DNA methylation |
| MCT4 | SLC16A3 | Skeletal muscles, intestine, kidneys, heart, Brain: astrocytes | Lactate, pyruvate, ketone bodies; fluvastatin, atorvastatin, lovastatin, simvastatin, cerivastatin in their acid form | 4-Chloro-α-cyanocinnamic acid (non-specific) | MCT4 expression ↑ in numerous cancers and in muscles of obese individuals: via hypoxia/HIF-1α |
| MCT8 | SLC16A2 | Liver, endocrine tissues, Brain: luminal and abluminal membranes of BBB, neurons | Thyroid hormones (T3 and T4) | Possibly desipramine, dexa-methasone, buspirone, desethyl- amiodarone, dronedarone, tyrosine kinase inhibitors, and silychristin | MCT8 expression ↓ in Allan–Herndon–Dudley syndrome and during the inflammation |
| Transporter | Gene Name | Tissue Distribution (Expression) | Substrates | Inhibitors | Expression Modulation/Transport Capacity Changes |
|---|---|---|---|---|---|
| OATP1A2 | SLCO1A2 | Ubiquitous, Brain: luminal membranes of BBB, neurons | Anionic, cationic, and neutral amphiphilic compounds of large size with different affinities among the transporters Endogenous substrates, such as bile acids (cholate), steroids (estrone-3-sulfate), thyroid hormones (T3 and T4), prosta-glandins (PGE2); Exogenous substrates, such as statins (fluvastatin), β-blockers (atenolol), and anticancer drugs (methotrexate) | Fruit juices that contain polyphenols and their conjugates, such as hesperidin, naringin, and avicularin; rifampicin, verapamil, elacridar, tariquidar, and zosuquidar (possibly competing substrates) | 11 single-nucleotide polymorphisms (SNPs): transport activity ↓ (substrate-specific); OATP1A2 expression ↑ in several cancers: possible involvement of hypoxia/reoxygenation in the upregulation at the BBB |
| OATP1C1 | SLCO1C1 | Testis Brain: astrocytes, choroid plexus | NSAIDs, (fenamates), phenytoin (competing substrates exhibiting mutual inhibition function) | OATP1C1 expression ↓ during the inflammation | |
| OATP2A1 | SLCO2A1 | Ubiquitous, Brain: neurons, astrocytes, and microglia | Polycyclic aromatic compounds, such as suramin, pranlukast, zafirlukast, olmesartan, losartan, non-steroidal anti-inflammatories | OATP2A1 expression ↓ in AD brain parenchymal cells; OATP2A1 expression ↑ in cancers: via PI3K/AKT/mTOR pathway | |
| OATP2B1 | SLCO2B1 | Ubiquitous, Brain: luminal membranes of BBB | Some of the substrates are also reported as inhibitors due to the drug–drug interactions | 11 single-nucleotide polymorphisms (SNPs): transport activity ↓ with 6 SNPs OATP2B1 expression ↑ in several cancers | |
| OATP3A1 | SLCO3A1 | Ubiquitous, Brain: neurons | OATP3A1 expression ↑ in cholestasis: via TNF-α-activated NF-κB-p65 and ERK-SP1 signaling |
| Transporter | Gene Name | Tissue Distribution (Expression) | Substrates | Inhibitors | Expression Modulation/Transport Capacity Changes |
|---|---|---|---|---|---|
| OCT1 | SLC22A1 | Liver and other peripheral tissues, Brain: BBB | Organic cations with different affinities among the transporters Endogenous substrates: catecholamines, monoamine neurotransmitters Drugs: several cytostatic, antiviral, antibiotic, antioxidant, psycho-stimulant, anti-hypertensive, antiemetic, antidepressan, antidiabetic agents | Transported substrates of the OCTs exhibit mutual inhibition function | 18 single-nucleotide polymorphisms (SNPs): transport activity ↓ with 6 SNPs See more information below (OATs) |
| OCT2 | SLC22A2 | Kidneys and other peripheral tissues, Brain: BBB, neurons, microglia, astrocytes | Cytochalasin B, forskolin, phloretin, quercetin and other flavonoids, glycogensynthase kinase-3 (GSK-3) inhibitors | 10 transporter variants with altered substrate selectivity and transport capacity | |
| OCT3 | SLC22A3 | Abundant, Brain: BBB, neurons | 5 SNPs: transport activity ↓ with 3 SNPs | ||
| OCTN1 | SLC22A4 | Abundant, Brain: BBB, microglia | Acetylcholine, ergothioneine, L-carnitine, TEA, quinidine, pyrilamine, and verapamil | Transported substrates of the OCTNs exhibit mutual inhibition function | OCTN1 variant L503F: familial/ sporadic inflammatory bowel disease |
| OCTN2 | SLC22A5 | Abundant, Brain: BBB, neurons | Acetyl-L- carnitine, D-/L-carnitine, TEA, quinidine, pyrilamine, and verapamil | Transported substrates of the OCTNs exhibit mutual inhibition function | Multiple OCTN2 variants: systemic carnitine deficiency |
| OAT1 | SLC22A6 | Kidneys, Brain: choroid plexus | Overlapping substrate specificities, although not identical, transports several drugs Endogenous substrates: α-ketoglutarate, para-amino-hippuric acid, benzoyl penicillins, indoxyl sulfate, and homovanillic acid, and prostaglandins | Probenecid (inhibitor/ substrate) | Multiple SNPs: decreased function can be compensated with other transporters in the family due to the overlapping substrate specificities; NOTE! OCTs and OATs are all regulated in transcriptional level as well as by post-translational phosphorylation via several protein kinases |
| OAT3 | SLC22A8 | Kidneys, Brain: abluminal side of the BBB, choroid plexus |
| Transporter | Gene Name | Tissue Distribution (Expression) | Substrates | Inhibitors | Expression Modulation/Transport Capacity Changes |
|---|---|---|---|---|---|
| SNAT1 | SLC38A1 | Ubiquitous, Brain: neurons (astrocytes) | L-proline, L-asparagine, L-cysteine, L-glutamine, L-glycine, L-methionine, and L-serine | 2-Methylamino-isobutyric acid (MeAIB, competing substrate); low pH | SNAT1 expression ↑: via protein kinase A (PKA) activation; SNAT1 expression ↓: via inflammation; SNAT1 expression ↑ in many types of cancers |
| SNAT2 | SLC38A2 | Ubiquitous, Brain: abluminal side of the BBB, neurons (astrocytes) | L-proline, L-asparagine, L-cysteine, L-glutamine, L-glycine, L-methionine, and L-serine | 2-Methylamino-isobutyric acid (MeAIB; competing substrate); low pH | Stable SNAT2 expression requires an active mTOR-signaling; SNAT2 expression ↑ in many types of cancers |
| SNAT3 | SLC38A3 | Liver, kidney, muscles, eye, Brain: luminal and abluminal sides of the BBB, astrocytes | L-glutamine, L-histidine, and L-asparagine | Not reported | SNAT3 expression ↓ by insulin: via an mTOR pathway; SNAT3 expression ↑ by calorie restriction: via increased protein kinase C (PKC) activity(?); SNAT3 expression ↑ in many types of cancers |
| SNAT5 | SLC38A5 | Intestinal tract, kidney, retina, lung, Brain: astrocytes | L-glutamine, L-histidine, and L-asparagine | Glutamic acid-γ-hydroxamic acid (GluγHA) | Less studied |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huttunen, J.; Adla, S.K.; Markowicz-Piasecka, M.; Huttunen, K.M. Increased/Targeted Brain (Pro)Drug Delivery via Utilization of Solute Carriers (SLCs). Pharmaceutics 2022, 14, 1234. https://doi.org/10.3390/pharmaceutics14061234
Huttunen J, Adla SK, Markowicz-Piasecka M, Huttunen KM. Increased/Targeted Brain (Pro)Drug Delivery via Utilization of Solute Carriers (SLCs). Pharmaceutics. 2022; 14(6):1234. https://doi.org/10.3390/pharmaceutics14061234
Chicago/Turabian StyleHuttunen, Johanna, Santosh Kumar Adla, Magdalena Markowicz-Piasecka, and Kristiina M. Huttunen. 2022. "Increased/Targeted Brain (Pro)Drug Delivery via Utilization of Solute Carriers (SLCs)" Pharmaceutics 14, no. 6: 1234. https://doi.org/10.3390/pharmaceutics14061234
APA StyleHuttunen, J., Adla, S. K., Markowicz-Piasecka, M., & Huttunen, K. M. (2022). Increased/Targeted Brain (Pro)Drug Delivery via Utilization of Solute Carriers (SLCs). Pharmaceutics, 14(6), 1234. https://doi.org/10.3390/pharmaceutics14061234