IVIVC Assessment of Two Mouse Brain Endothelial Cell Models for Drug Screening

 , , ,
, , ,
Abstract
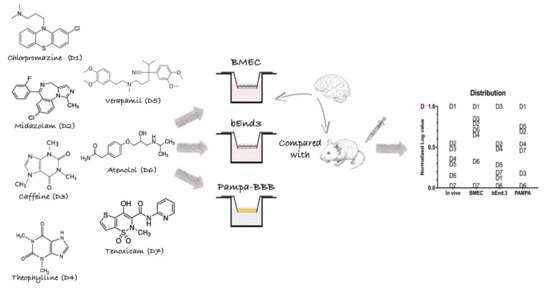
:
1. Introduction
2. Materials and Methods
2.1. Chemical and Supplies
2.2. Animals and Cell Culture
2.2.1. Primary Cell Isolation and Culture
2.2.2. Cell Line Culture
2.2.3. Morphological Cell Characterization
2.2.4. Puromycin Cell Viability Assay
2.3. Model Assembly
2.3.1. Evaluation of the Model Integrity
2.3.2. In vitro Drug Permeability Assay
2.3.3. Permeability Coefficient (Pe) Calculation
2.3.4. Model Characterization
2.4. PAMPA Model
2.5. Animal Studies
In Vitro/In Vivo Correlation (IVIVC)
2.6. LC-MS/MS Drug Analysis
2.7. Statistical Analysis
3. Results
3.1. Endothelial Cells—Basal Characterization
3.1.1. Morphology of Mouse Brain Endothelial Cells
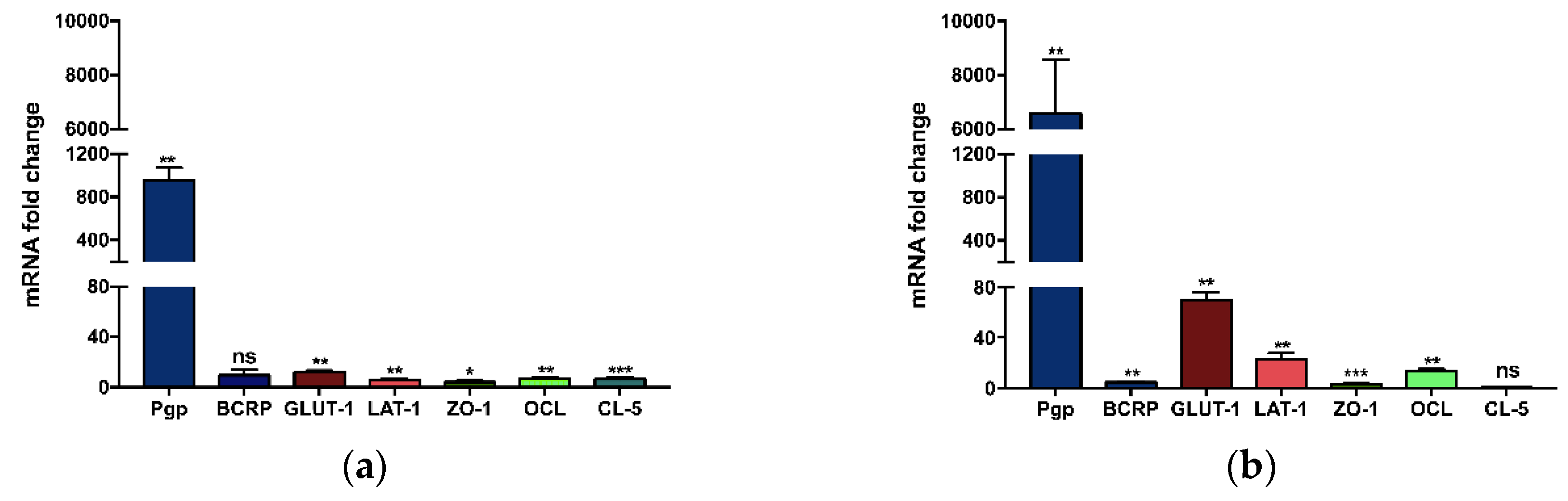
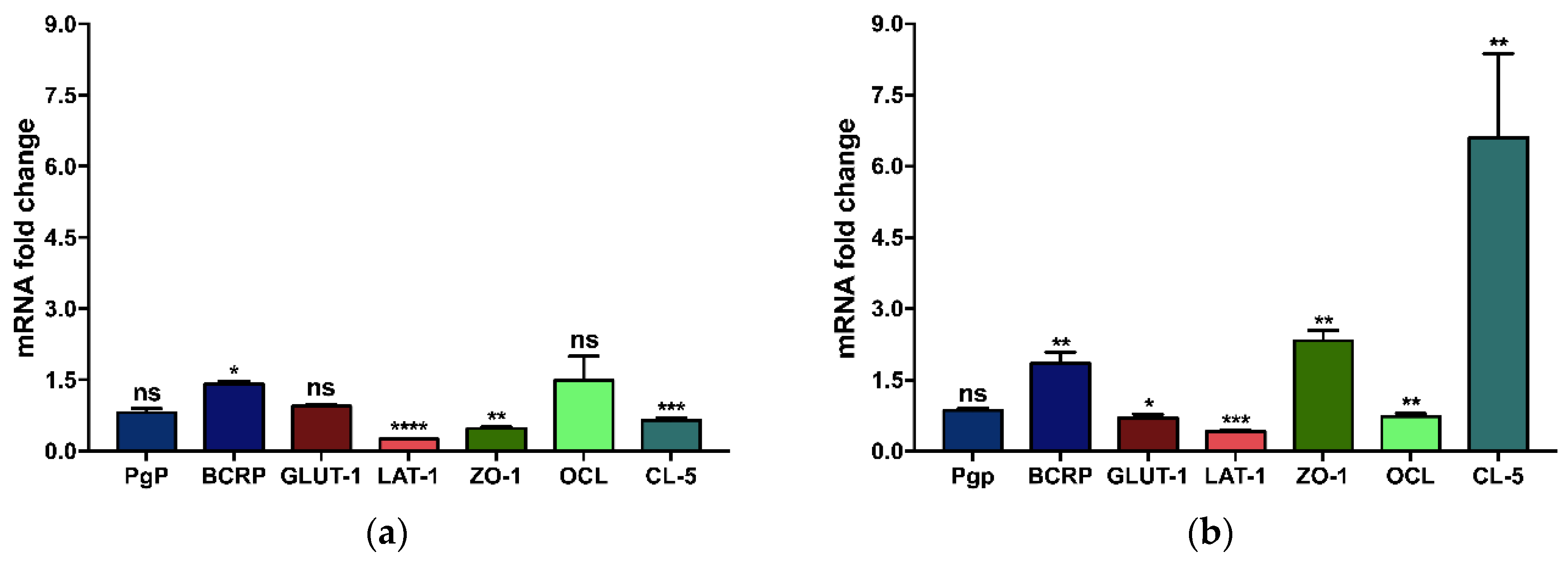
3.1.2. Specific mRNA Gene Expression in Brain Mouse Endothelial Cells
3.1.3. Puromycin Viability Assay
3.2. Monolayer Models and Subsequent Characterization
3.2.1. TEER and Pe of Fluorescent Markers
3.2.2. Influence of the Support Material on the Gene Expression of Brain Endothelial Monolayers
3.2.3. Immunostaining of TJ Proteins
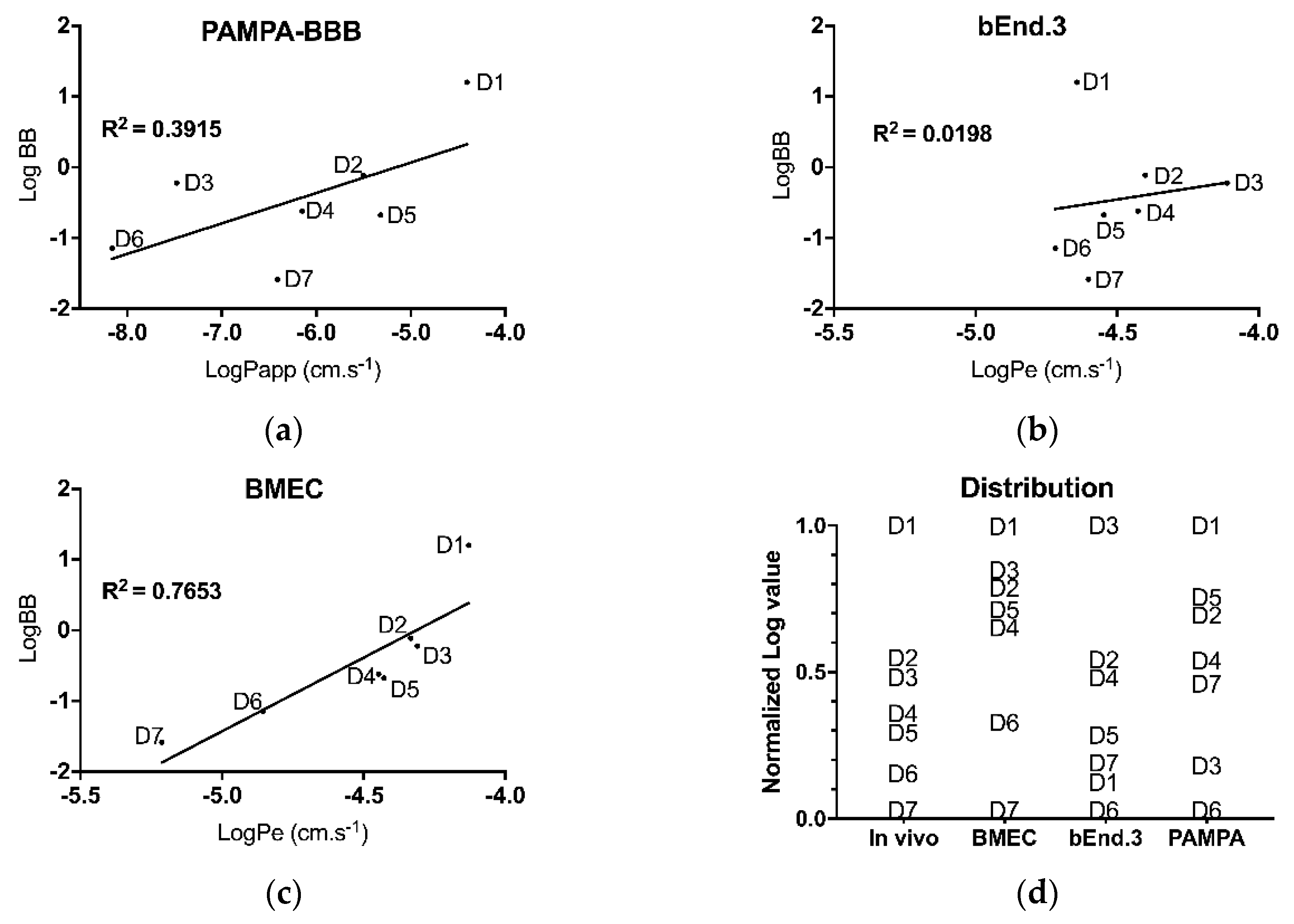
3.3. In Vivo/In Vitro Correlation and Validation
4. Discussion
4.1. Although Morphologically Identical, Primary and Immortalized Brain ECs Display Different Genomic Patterns
4.2. Primary Cells Form a Tighter, More Impermeable Monolayer Model Than Immortalized Cells
4.3. Primary Cells Allow a Better Predictability of Drug Permeation Across the BBB
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alavijeh, M.S.; Chishty, M.; Qaiser, M.Z.; Palmer, A.M. Drug metabolism and pharmacokinetics, the blood-brain barrier, and central nervous system drug discovery. NeuroRx 2005, 2, 554–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, N.J. Blood-brain barrier structure and function and the challenges for CNS drug delivery. J. Inherit. Metab. Dis. 2013, 36, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. Blood-brain barrier delivery. Drug Discov. Today 2007, 12, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Scherrmann, J.M. Drug delivery to brain via the blood-brain barrier. Vasc. Pharmacol. 2002, 38, 349–354. [Google Scholar] [CrossRef]
- Wong, K.H.; Riaz, M.K.; Xie, Y.; Zhang, X.; Liu, Q.; Chen, H.; Bian, Z.; Chen, X.; Lu, A.; Yang, Z. Review of Current Strategies for Delivering Alzheimer’s Disease Drugs across the Blood-Brain Barrier. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Stanimirovic, D.B.; Bani-Yaghoub, M.; Perkins, M.; Haqqani, A.S. Blood-brain barrier models: In vitro to in vivo translation in preclinical development of CNS-targeting biotherapeutics. Expert Opin. Drug Discov. 2015, 10, 141–155. [Google Scholar] [CrossRef]
- Rankovic, Z. CNS drug design: Balancing physicochemical properties for optimal brain exposure. J. Med. Chem. 2015, 58, 2584–2608. [Google Scholar] [CrossRef]
- Wolff, A.; Antfolk, M.; Brodin, B.; Tenje, M. In Vitro Blood-Brain Barrier Models-An Overview of Established Models and New Microfluidic Approaches. J. Pharm. Pharm. Sci. 2015, 104, 2727–2746. [Google Scholar] [CrossRef]
- Palmiotti, C.A.; Prasad, S.; Naik, P.; Abul, K.M.; Sajja, R.K.; Achyuta, A.H.; Cucullo, L. In vitro cerebrovascular modeling in the 21st century: Current and prospective technologies. Pharm Res-Dordr 2014, 31, 3229–3250. [Google Scholar] [CrossRef]
- Cho, C.F.; Wolfe, J.M.; Fadzen, C.M.; Calligaris, D.; Hornburg, K.; Chiocca, E.A.; Agar, N.Y.R.; Pentelute, B.L.; Lawler, S.E. Blood-brain-barrier spheroids as an in vitro screening platform for brain-penetrating agents. Nat. Commun. 2017, 8, 15623. [Google Scholar] [CrossRef]
- Goh, J.-Y.; Weaver, R.J.; Dixon, L.; Platt, N.J.; Roberts, R.A. Development and use of in vitro alternatives to animal testing by the pharmaceutical industry 1980–2013. Toxicol. Res. 2015, 4, 1297–1307. [Google Scholar] [CrossRef]
- Bicker, J.; Alves, G.; Fortuna, A.; Soares-da-Silva, P.; Falcao, A. A new PAMPA model using an in-house brain lipid extract for screening the blood-brain barrier permeability of drug candidates. Int. J. Pharm. 2016, 501, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Helms, H.C. In vitro models of the blood–brain barrier: An overview of commonly used brain endothelial cell culture models and guidelines for their use. J. Cereb. Blood Flow Metab. 2016, 36. [Google Scholar] [CrossRef] [PubMed]
- Mahringer, A.; Fricker, G. ABC transporters at the blood–brain barrier. Expert Opin. Drug Metab. Toxicol. 2016, 12, 499–508. [Google Scholar] [CrossRef]
- Volpe, D.A. Drug-permeability and transporter assays in Caco-2 and MDCK cell lines. Future Med. Chem. 2011, 3, 2063–2077. [Google Scholar] [CrossRef]
- De Bock, M.; Van Haver, V.; Vandenbroucke, R.E.; Decrock, E.; Wang, N.; Leybaert, L. Into rather unexplored terrain-transcellular transport across the blood-brain barrier. Glia 2016, 64, 1097–1123. [Google Scholar] [CrossRef]
- Rahman, N.A.; Rasil, A.; Meyding-Lamade, U.; Craemer, E.M.; Diah, S.; Tuah, A.A.; Muharram, S.H. Immortalized endothelial cell lines for in vitro blood-brain barrier models: A systematic review. Brain Res. 2016, 1642, 532–545. [Google Scholar] [CrossRef]
- Hollmann, E.K.; Bailey, A.K.; Potharazu, A.V.; Neely, M.D.; Bowman, A.B.; Lippmann, E.S. Accelerated differentiation of human induced pluripotent stem cells to blood-brain barrier endothelial cells. Fluids Barriers CNS 2017, 14, 9. [Google Scholar] [CrossRef]
- Yusof, S.R.; Avdeef, A.; Abbott, N.J. In vitro porcine blood-brain barrier model for permeability studies: pCEL-X software pKa(FLUX) method for aqueous boundary layer correction and detailed data analysis. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2014, 65, 98–111. [Google Scholar] [CrossRef]
- Krajcsi, P.; Jani, M.; Tóth, B.; Erdő, F.; Kis, E.; Beéry, E.; Sziráki, I. Efflux transporters in the blood–brain interfaces – in vitro and in vivo methods and correlations. Expert Opin. Drug Metab. Toxicol. 2012, 8, 419–431. [Google Scholar] [CrossRef]
- Lecuyer, M.A.; Saint-Laurent, O.; Bourbonniere, L.; Larouche, S.; Larochelle, C.; Michel, L.; Charabati, M.; Abadier, M.; Zandee, S.; Haghayegh Jahromi, N.; et al. Dual role of ALCAM in neuroinflammation and blood-brain barrier homeostasis. Proc. Natl. Acad. Sci. USA 2017, 114, E524–E533. [Google Scholar] [CrossRef] [PubMed]
- Wuest, D.M.; Wing, A.M.; Lee, K.H. Membrane configuration optimization for a murine in vitro blood-brain barrier model. J. Neurosci. Methods 2013, 212, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Deli, M.A.; Kawaguchi, H.; Shimizudani, T.; Shimono, T.; Kittel, A.; Tanaka, K.; Niwa, M. A new blood-brain barrier model using primary rat brain endothelial cells, pericytes and astrocytes. Neurochem. Int. 2009, 54, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Deli, M.A.; Ábrahám, C.S.; Kataoka, Y.; Niwa, M. Permeability Studies on In Vitro Blood–Brain Barrier Models: Physiology, Pathology, and Pharmacology. Cell. Mol. Neurobiol. 2005, 25, 59–127. [Google Scholar] [CrossRef] [PubMed]
- Balimane, P.V.; Pace, E.; Chong, S.; Zhu, M.; Jemal, M.; Pelt, C.K. A novel high-throughput automated chip-based nanoelectrospray tandem mass spectrometric method for PAMPA sample analysis. J. Pharm. Biomed. Anal. 2005, 39, 8–16. [Google Scholar] [CrossRef]
- Bickel, U. How to measure drug transport across the blood-brain barrier. NeuroRx 2005, 2, 15–26. [Google Scholar] [CrossRef]
- Perriere, N.; Demeuse, P.; Garcia, E.; Regina, A.; Debray, M.; Andreux, J.P.; Couvreur, P.; Scherrmann, J.M.; Temsamani, J.; Couraud, P.O.; et al. Puromycin-based purification of rat brain capillary endothelial cell cultures. Effect on the expression of blood-brain barrier-specific properties. J. Neurochem. 2005, 93, 279–289. [Google Scholar] [CrossRef]
- Brown, R.C.; Morris, A.P.; O’Neil, R.G. Tight junction protein expression and barrier properties of immortalized mouse brain microvessel endothelial cells. Brain Res. 2007, 1130, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Ruck, T.; Bittner, S.; Epping, L.; Herrmann, A.M.; Meuth, S.G. Isolation of primary murine brain microvascular endothelial cells. JOVE-J Vis Exp. 2014, e52204. [Google Scholar] [CrossRef]
- Lundquist, S.; Renftel, M.; Brillault, J.; Fenart, L.; Cecchelli, R.; Dehouck, M.P. Prediction of drug transport through the blood-brain barrier in vivo: A comparison between two in vitro cell models. Pharm. Res. 2002, 19, 976–981. [Google Scholar] [CrossRef]
- Montesano, R.; Pepper, M.S.; Mohle-Steinlein, U.; Risau, W.; Wagner, E.F.; Orci, L. Increased proteolytic activity is responsible for the aberrant morphogenetic behavior of endothelial cells expressing the middle T oncogene. Cell 1990, 62, 435–445. [Google Scholar] [CrossRef]
- Watanabe, T.; Dohgu, S.; Takata, F.; Nishioku, T.; Nakashima, A.; Futagami, K.; Yamauchi, A.; Kataoka, Y. Paracellular barrier and tight junction protein expression in the immortalized brain endothelial cell lines bEND.3, bEND.5 and mouse brain endothelial cell 4. Biol. Pharm. Bull. 2013, 36, 492–495. [Google Scholar] [CrossRef] [PubMed]
- Tehrani, S.F.; Bernard-Patrzynski, F.; Puscas, I.; Leclair, G.; Hildgen, P.; Roullin, V.G. Length of surface PEG modulates nanocarrier transcytosis across brain vascular endothelial cells. Nanomedicine 2018, 16, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Omidi, Y.; Campbell, L.; Barar, J.; Connell, D.; Akhtar, S.; Gumbleton, M. Evaluation of the immortalised mouse brain capillary endothelial cell line, b.End3, as an in vitro blood-brain barrier model for drug uptake and transport studies. Brain Res. 2003, 990, 95–112. [Google Scholar] [CrossRef]
- Demeuse, P.; Fragner, P.; Leroy-Noury, C.; Mercier, C.; Payen, L.; Fardel, O.; Couraud, P.O.; Roux, F. Puromycin selectively increases mdr1a expression in immortalized rat brain endothelial cell lines. J. Neurochem. 2004, 88, 23–31. [Google Scholar] [CrossRef]
- Hamadneh, L.; Al-Majawleh, M.; Jarrar, Y.; Shraim, S.; Hasan, M.; Abu-Irmaileh, B. Culturing conditions highly affect DNA methylation and gene expression levels in MCF7 breast cancer cell line. Vitr. Cell. Dev. Biol.-Anim. 2018, 54, 331–334. [Google Scholar] [CrossRef]
- Wuest, D.M.; Lee, K.H. Optimization of endothelial cell growth in a murine in vitro blood-brain barrier model. Biotechnol. J. 2012, 7, 409–417. [Google Scholar] [CrossRef]
- Thomsen, M.S.; Birkelund, S.; Burkhart, A.; Stensballe, A.; Moos, T. Synthesis and deposition of basement membrane proteins by primary brain capillary endothelial cells in a murine model of the blood-brain barrier. J. Neurochem. 2017, 140, 741–754. [Google Scholar] [CrossRef]
- Tian, X.; Brookes, O.; Battaglia, G. Pericytes from Mesenchymal Stem Cells as a model for the blood-brain barrier. Sci. Rep. 2017, 7, 39676. [Google Scholar] [CrossRef]
- Weidenfeller, C.; Schrot, S.; Zozulya, A.; Galla, H.J. Murine brain capillary endothelial cells exhibit improved barrier properties under the influence of hydrocortisone. Brain Res. 2005, 1053, 162–174. [Google Scholar] [CrossRef]
- Kutuzov, N.; Flyvbjerg, H.; Lauritzen, M. Contributions of the glycocalyx, endothelium, and extravascular compartment to the blood-brain barrier. Proc. Natl. Acad. Sci. USA 2018, 115, E9429–E9438. [Google Scholar] [CrossRef] [PubMed]
- Sahagun, G.; Moore, S.A.; Hart, M.N. Permeability of neutral vs. anionic dextrans in cultured brain microvascular endothelium. Am. J. Physiol. 1990, 259, H162–H166. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Li, G.; Gil, E.S.; Lowe, T.L.; Fu, B.M. Effect of surface charge of immortalized mouse cerebral endothelial cell monolayer on transport of charged solutes. Ann. Biomed. Eng. 2010, 38, 1463–1472. [Google Scholar] [CrossRef] [PubMed]
- Aman, J.; Weijers, E.M.; van Nieuw Amerongen, G.P.; Malik, A.B.; van Hinsbergh, V.W. Using cultured endothelial cells to study endothelial barrier dysfunction: Challenges and opportunities. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2016, 311, L453–L466. [Google Scholar] [CrossRef]
- Hellinger, E.; Veszelka, S.; Toth, A.E.; Walter, F.; Kittel, A.; Bakk, M.L.; Tihanyi, K.; Hada, V.; Nakagawa, S.; Duy, T.D.; et al. Comparison of brain capillary endothelial cell-based and epithelial (MDCK-MDR1, Caco-2, and VB-Caco-2) cell-based surrogate blood-brain barrier penetration models. Eur. J. Pharm. Biopharm. 2012, 82, 340–351. [Google Scholar] [CrossRef]
- Vilar, S.; Chakrabarti, M.; Costanzi, S. Prediction of passive blood-brain partitioning: Straightforward and effective classification models based on in silico derived physicochemical descriptors. J. Mol. Graph. Model. 2010, 28, 899–903. [Google Scholar] [CrossRef]
- Hanak, A.S.; Chevillard, L.; El Balkhi, S.; Risede, P.; Peoc’h, K.; Megarbane, B. Study of blood and brain lithium pharmacokinetics in the rat according to three different modalities of poisoning. Toxicol. Sci. 2015, 143, 185–195. [Google Scholar] [CrossRef]
- Kulkarni, A.D.; Patel, H.M.; Surana, S.J.; Belgamwar, V.S.; Pardeshi, C.V. Brain-blood ratio: implications in brain drug delivery. Expert Opin. Drug Deliv. 2016, 13, 85–92. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviation | Name | MW (g/mol) | log P 1 |
|---|---|---|---|
| D1 | Chlorpromazine hydrochloride (Enzo life Sciences, Lausanne, Switzerland USA) | 355.3 | 5.41 |
| D2 | Midazolam (TRC, Lowell, MA, USA) | 325.8 | 4.33 |
| D3 | Caffeine, anhydrous (Medisca, Montréal, QC, Canada) | 194.2 | −0.07 |
| D4 | Theophylline, anhydrous (Medisca, QC, Canada) | 180.2 | −0.02 |
| D5 | Verapamil hydrochloride (Sigma-Aldrich, Oakville, ON, Canada) | 491.1 | 3.79 |
| D6 | Atenolol (Acros Organics, Springfield, New Jersey, USA) | 266.3 | 0.16 |
| D7 | Tenoxicam (Alfa Aesar, Haverhill, MA, USA) | 337.4 | 1.90 |
| HPLC | Agilent 1100 Series | |||
| MS/MS | MDS Sciex 4000 Qtrap | |||
| Software | Analyst® (v1.6.2) | |||
| Ionisation source, mode | Turbo electrospray, positive ionisation | |||
| Scan mode | Multiple reaction monitoring (MRM) | |||
| Analyte parameters | Compounds | DP (V) | MRM | CE (eV) |
| Verapamil | 110 | 455.3 > 165.0 | 60 | |
| Midazolam | 90 | 326.2 > 291.1 | 42 | |
| Chlorpromazine | 65 | 319.2 > 86.0 | 28 | |
| Caffeine | 90 | 181.1 > 124.2 | 28 | |
| Atenolol | 41 | 267.1 > 145.0 | 45 | |
| Theophilline | 70 | 194.1 > 138.2 | 27 | |
| Tenoxicam | 71 | 337.3 > 121.0 | 33 | |
| Metochlopramide (ISTD) | 70 | 300.1 > 184.3 | 44 | |
| Source parameters | Gas temp (°C) | 550 | ||
| Gas flow (L/min) | 50 | |||
| Curtain gaz (psi) | 25 | |||
| Capillary (V) | 5500 | |||
| Mobile phase | Composition | A: 0.1% FA+ H2O | ||
| B: 0.1% FA + ACN | ||||
| Gradient | 2 to 98% B in 3.5 min | |||
| Flow rate | 0.75 mL·min−1 | |||
| Column temperature | 45 °C | |||
| Injection volume | 4 µL | |||
| Injection temperature | 5 °C | |||
| Column | YMC-Pack ODS-AQ, (50 × 3.0 mm, 5 µm) | |||
| Target Protein/Abbreviation/Gene Symbol | Function | Reference Sequence | Forward/Reverse Primers |
|---|---|---|---|
| P-glycoprotein/Pgp/Abcb1a | Efflux transporter | NM_011076.2 | gggcatttacttcaaacttgtca/tttacaagcttcatttcctaattcaa |
| Breast cancer resistance protein/BCRP/Abcg2 | Efflux transporter | NM_011920.3 | aggtctggaaaaagtagcagattc/ctccatccctatgcttgtcc |
| Glucose transporter-1/GLUT-1/Slc2a1 | Uptake transporter | NM_011400.3 | gtatcctgttgcccttctgc/tcgaagcttcttcagcacac |
| L-type amino acid transporter/LAT-1/Slc7a5 | Uptake transporter | NM_011404.3 | tcagcttcttcaactggctgt/ggagggccagattcacct |
| Zonula occludens-1/ZO-1/Tjp1 | Tight junction | NM_009386.2, NM_001163574 | cgcggagagagacaagatgt/gaagcgtcactgtgtgctgt |
| Occludin/OCL/Ocln | Tight junction | NM_008756.2 | gtccgtgaggccttttga/ggtgcataatgattgggtttg |
| Claudin-5/CL-5/Cldn5 | Tight junction | NM_013805.4 | ttaaggcacgggtagcactc/atgttggcgaaccagcag |
| Hypoxanthine-guanine Phosphoribosyltransferase/HPRT/Hprt | Endogenous protein | NM_013556.2 | tcctcctcagaccgctttt/cctggttcatcatcgctaatc |
| Molecule Name | Molecule Formulation (%) | Administered Dose (mg/kg) 1 |
|---|---|---|
| Chlorpromazine HCl | PEG 400/Water (30:70) | 2.0 |
| Midazolam | PEG 400/Water (30:70) | 3.0 |
| Caffeine | NaCl 0.9% (100) | 12.4 |
| Theophylline | DMSO/PEG 400/Water (10:30:60) | 27.2 |
| Verapamil HCl | PEG 400/Water (30:70) | 1.52 |
| Atenolol | DMSO/PEG 400/NaCl 0.9% (10:30:60) | 10.0 |
| Tenoxicam | DMSO/Solutol HS15 10% in PBS (10:90) | 5.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puscas, I.; Bernard-Patrzynski, F.; Jutras, M.; Lécuyer, M.-A.; Bourbonnière, L.; Prat, A.; Leclair, G.; Roullin, V.G. IVIVC Assessment of Two Mouse Brain Endothelial Cell Models for Drug Screening. Pharmaceutics 2019, 11, 587. https://doi.org/10.3390/pharmaceutics11110587
Puscas I, Bernard-Patrzynski F, Jutras M, Lécuyer M-A, Bourbonnière L, Prat A, Leclair G, Roullin VG. IVIVC Assessment of Two Mouse Brain Endothelial Cell Models for Drug Screening. Pharmaceutics. 2019; 11(11):587. https://doi.org/10.3390/pharmaceutics11110587
Chicago/Turabian StylePuscas, Ina, Florian Bernard-Patrzynski, Martin Jutras, Marc-André Lécuyer, Lyne Bourbonnière, Alexandre Prat, Grégoire Leclair, and V. Gaëlle Roullin. 2019. "IVIVC Assessment of Two Mouse Brain Endothelial Cell Models for Drug Screening" Pharmaceutics 11, no. 11: 587. https://doi.org/10.3390/pharmaceutics11110587