Co-Amorphous Solid Dispersions for Solubility and Absorption Improvement of Drugs: Composition, Preparation, Characterization and Formulations for Oral Delivery



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
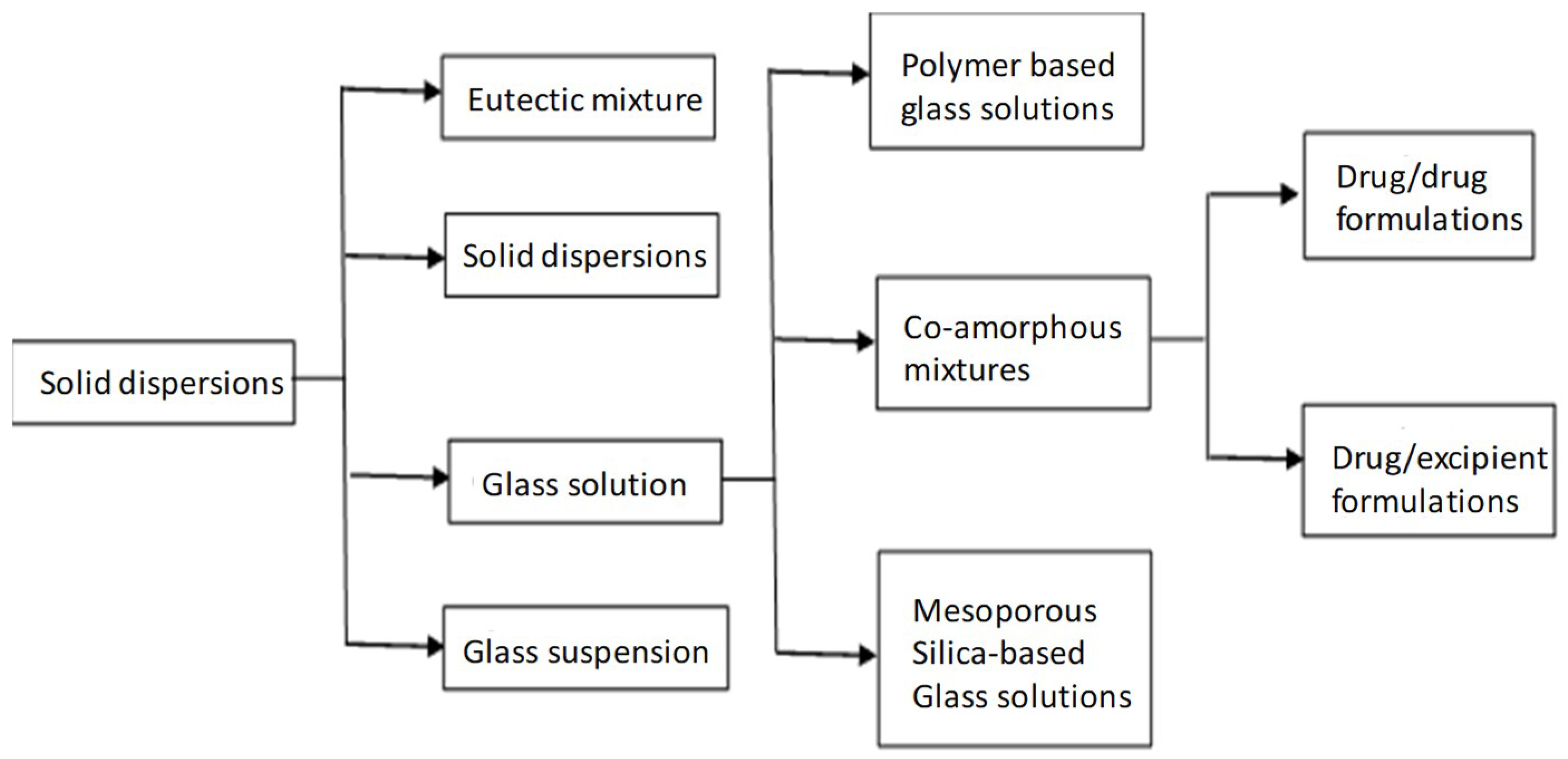
2. Solid Dispersions—Glass Solutions
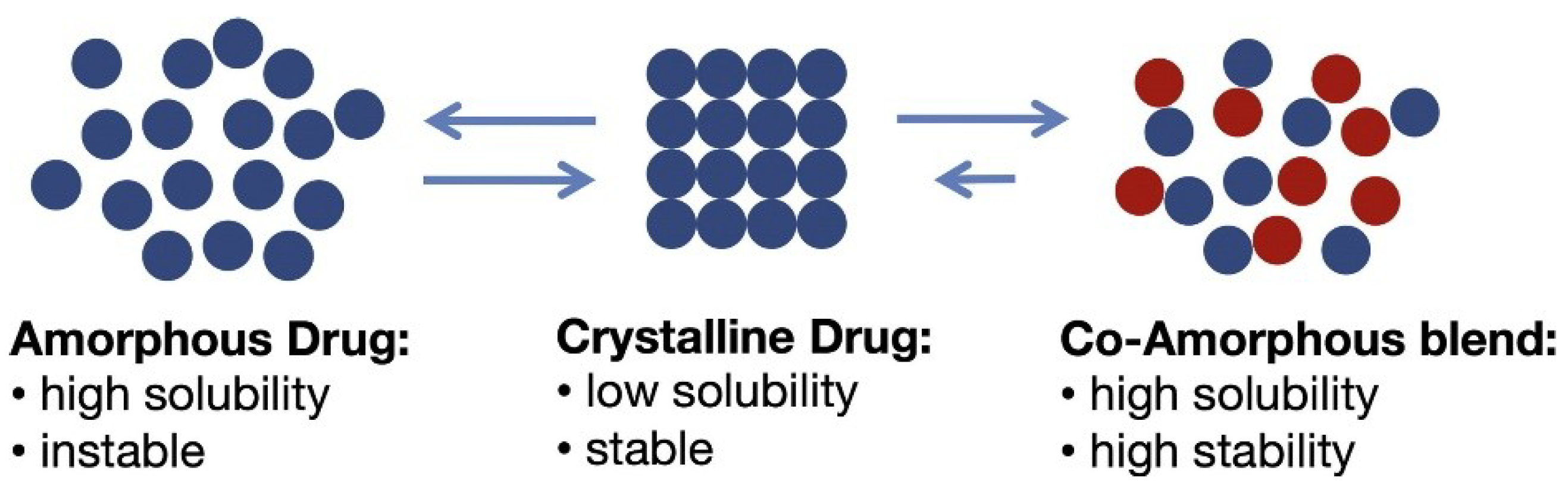
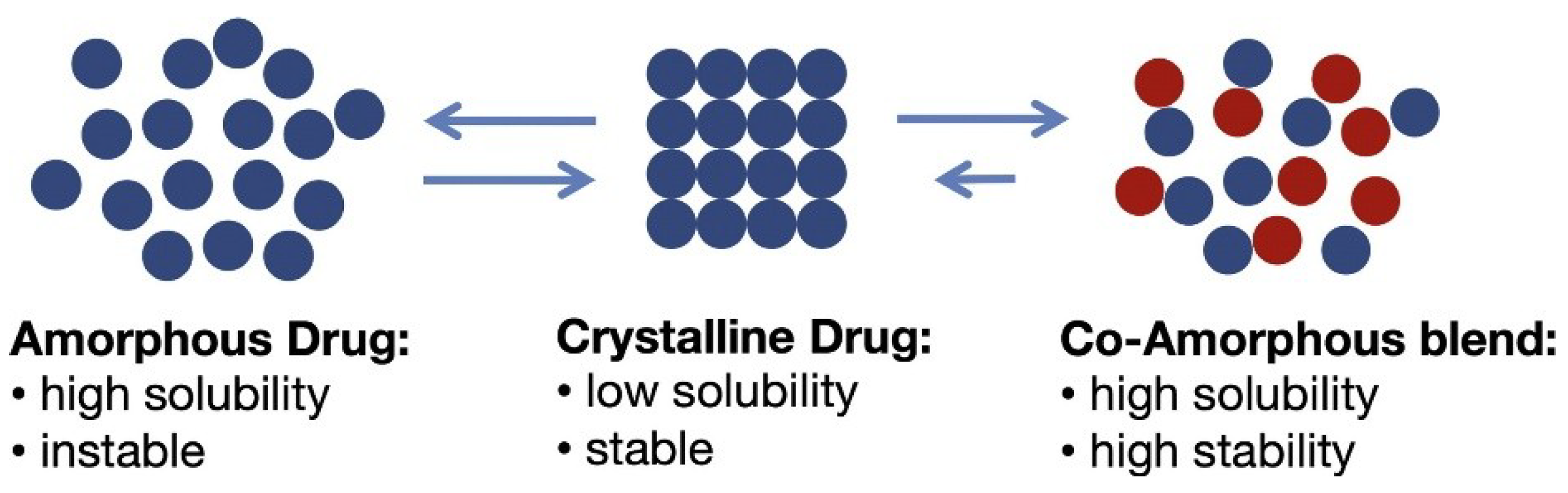
3. Co-Amorphous Dispersions
3.1. Recrystallization in Co-Amorphous Dispersions
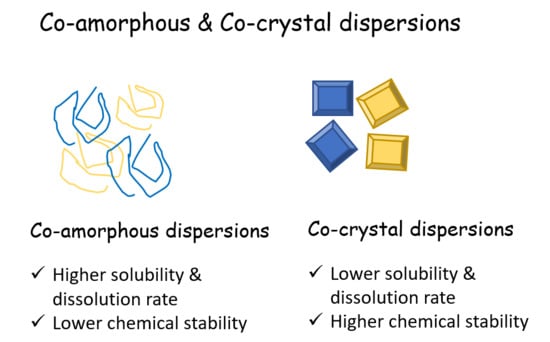
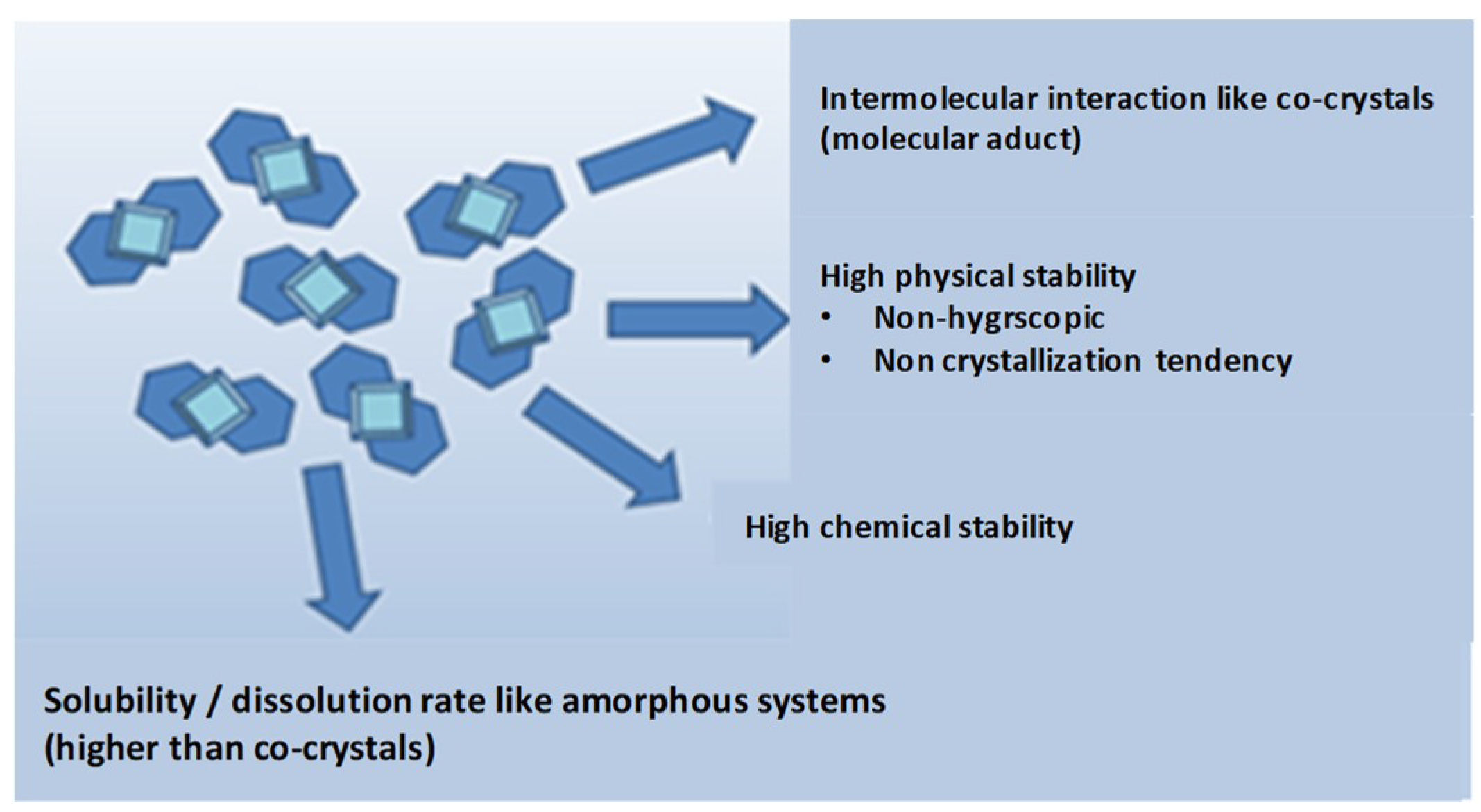
3.2. Co-Amorphous vs. Co-Crystals
3.3. Classification of Co-Amorphous Solid Dispersions
3.3.1. Drug-Drug Compositions
3.3.2. Drug-Amino Acid Co-Amorphous Compositions and Co-Amorphous Salts
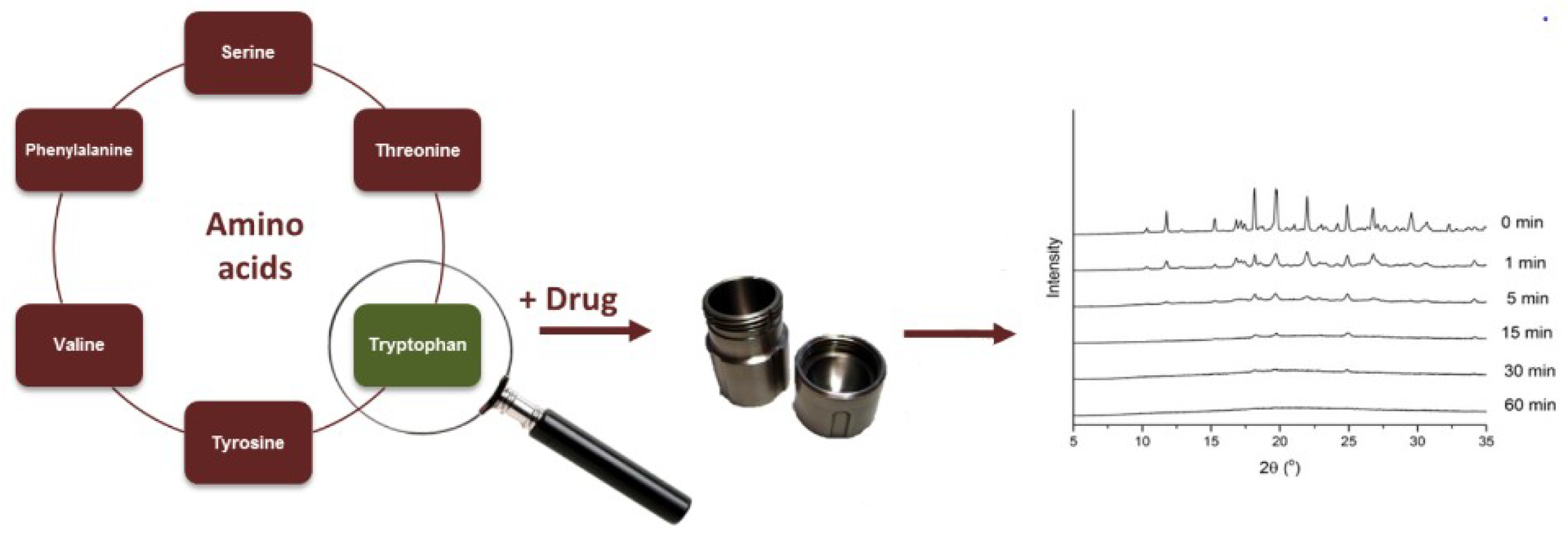
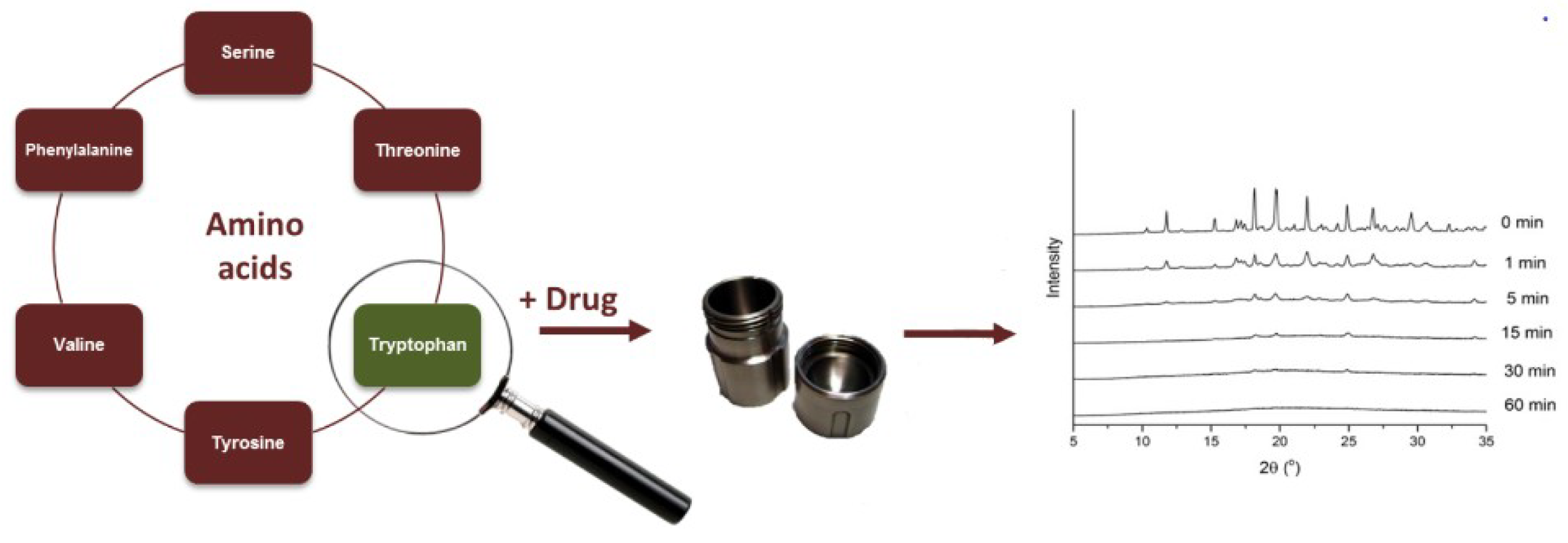
3.4. Selection of Amino Acid Co-Formers
3.5. Dissolution Rate from Co-Amorphous Solid Dispersions
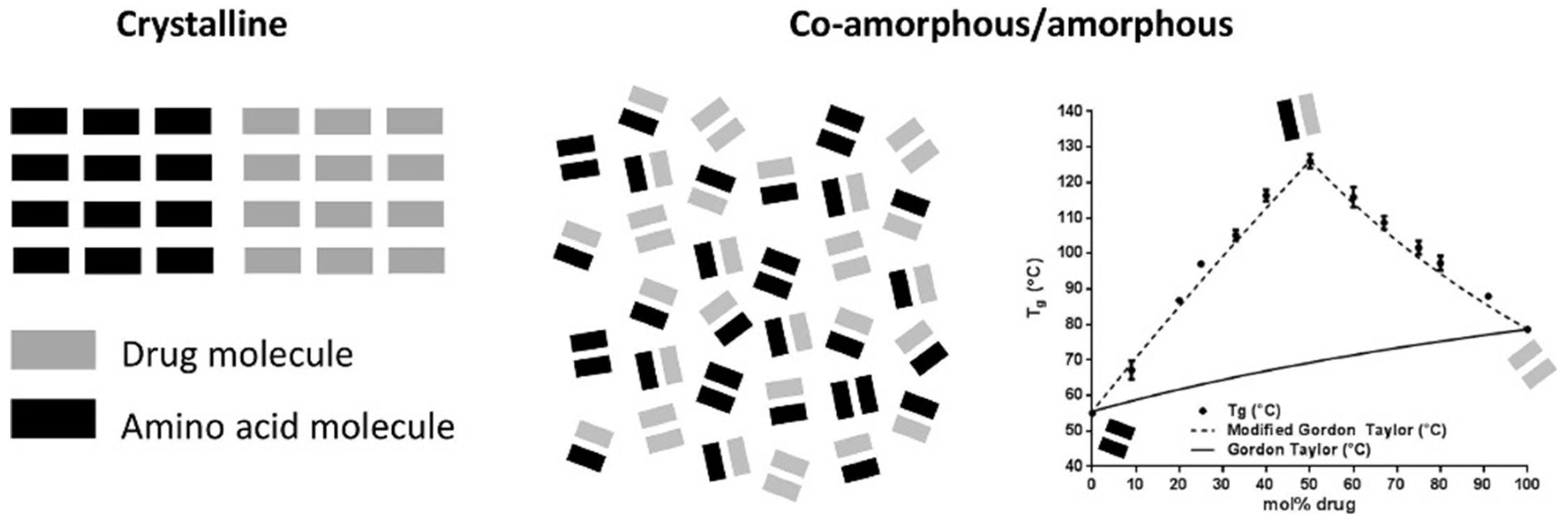
3.6. Molecular Proportion in Co-Amorphous Solid Dispersions
4. Solid State Characterization
4.1. XRPD and DSC
4.2. Fourier-Transform Infrared Spectroscopy—FTIR
4.3. Newer Techniques
4.4. Studies Using Combinations of Analytical Methods
- ➢
- Chieng et al. [15] studied amorphous c-indomethacin and ranitidine hydrochloride form 2 at various weight ratios (1:2, 1:1 and 2:1). During co-milling, the XRPD characteristic peaks of indomethacin were found to decrease faster than those of ranitidine hydrochloride and both drugs were fully converted into the amorphous state after 60 min. DSC results were in agreement with XRPD and Tgs of the amorphous mixtures were in good agreement with the predicted Tgs using the Gordon–Taylor equation. DRIFTS spectra showed interaction at the carboxylic acid carbonyl (HO–C=O) and benzonyl amide (NC=O) of the indomethacin molecule with the aci-nitro (C=N) of ranitidine hydrochloride.
- ➢
- Alleso et al. [17] studied the binary systems of naproxen and cimetidine. DSC showed that the 1:1 sample was the most physically stable even though it did not have the highest Tg when compared to the 1:2 sample. Raman spectroscopy suggested a 1:1 solid-state interaction between the imidazole ring of cimetidine and the carboxylic acid of naproxen in the amorphous sample, indicating that the stabilization of the amorphous binary system was dictated by molecular-level interactions.
- ➢
- Lobman et al. [19] studied naproxen with γ-indomethacin. Tgs were comparable with those from the Gordon Taylor equation. XRPD results showed that naproxen could be made amorphous in combination with indomethacin while this was not possible with naproxen alone. Peak shifts in the FTIR spectra indicated molecular interactions between both drugs, and it is suggested that the two drugs formed a heterodimer.
- ➢
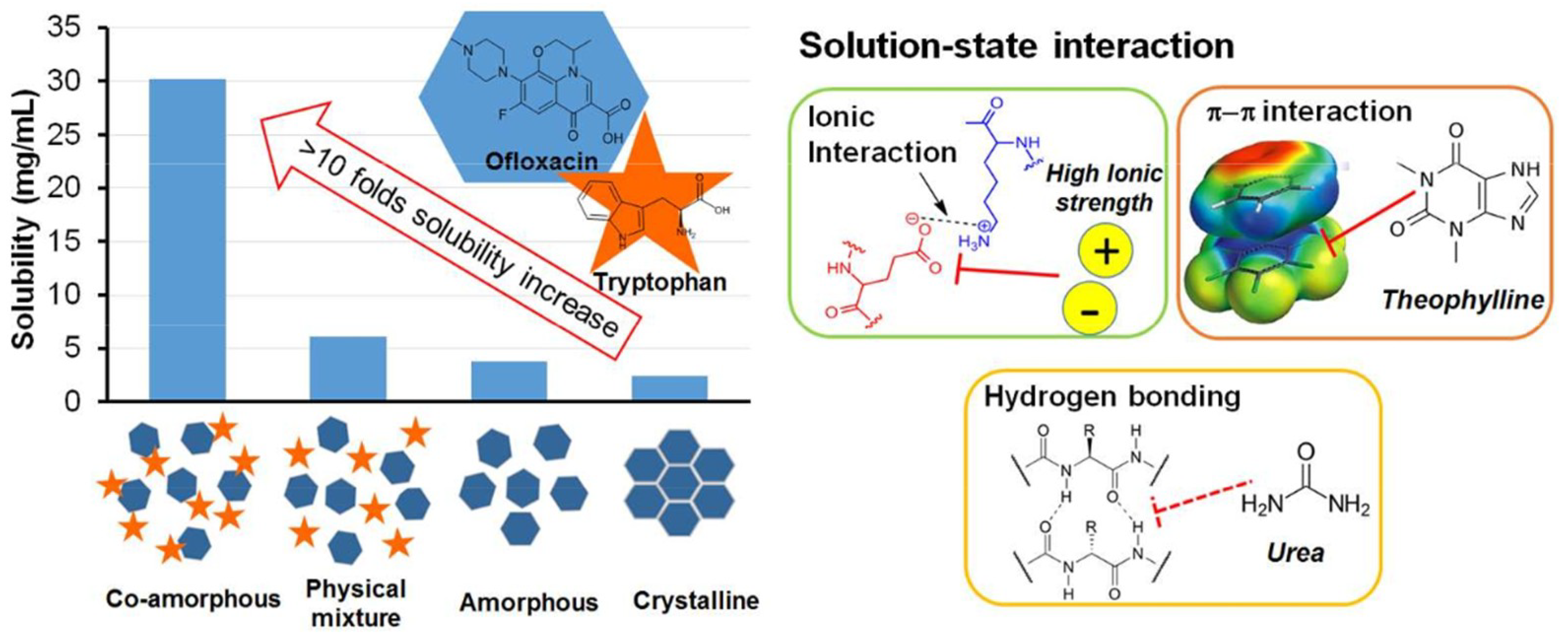
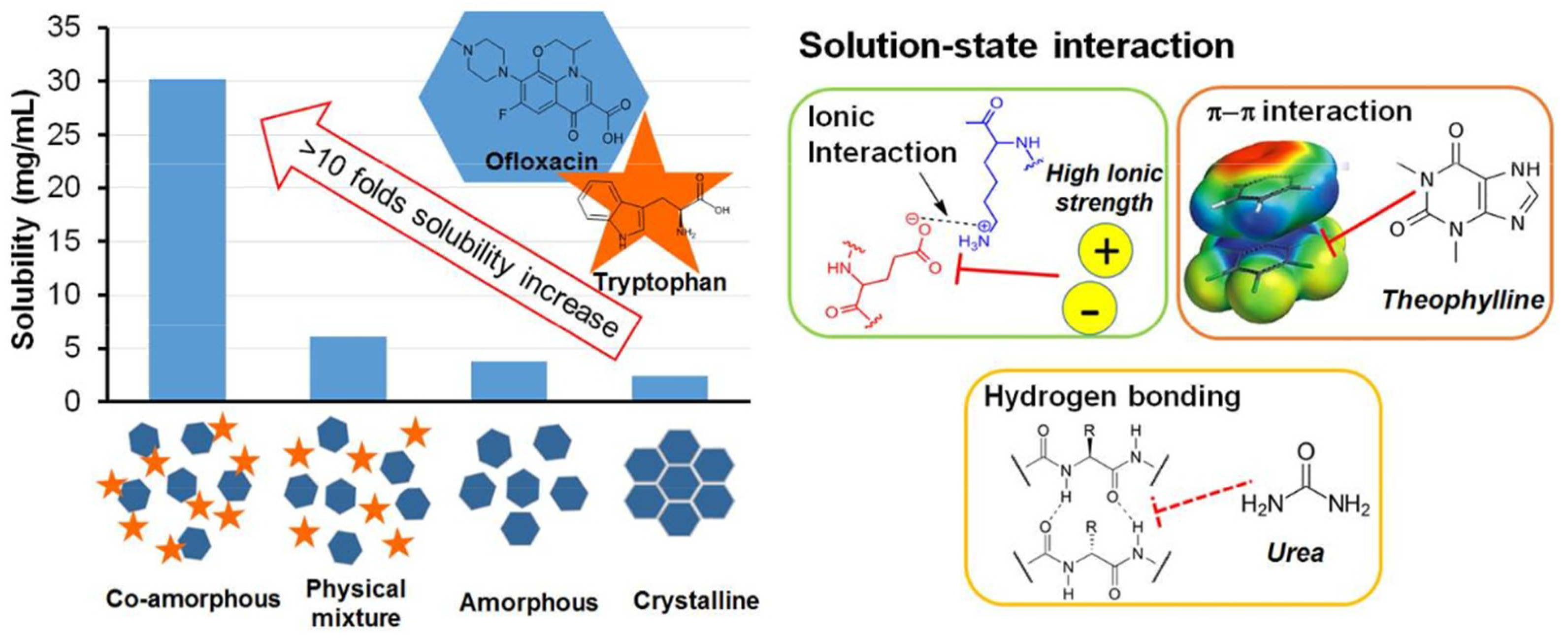
- Zhu et al. [22] studied Tryptophan—ofloxacin at a 1:1 weight ratio. XRPD showed complete co-amorphization and DSC showed an elevated Tg compared with the theoretical value. FTIR revealed that hydrogen bonding and π−π stacking were possibly involved in the formation of co-amorphous system.
- ➢
- Lobman et al. [24] studied mixtures of simvastatin (SVS) and glipizide. Formation of single phase co-amorphous mixtures with mixture ratios of 2:1, 1:1 and 1:2 was detected by DSC. The observed single, concentration-dependent Tgs were found to be lower than those predicted (Gordon–Taylor equation), indicating the absence of intermolecular interactions between the two drugs, which was verified by FTIR spectral data analysis.
- ➢
- Yamamoto et al. [27] studied interactions of itraconazole (ITZ) with fumaric (FA) or l-tartaric acid (TA) by Raman spectroscopy. ITZ-FA co-amorphous was found physically and chemically stable, whereas the ITZ-TA co-amorphous, was physically stable but chemically unstable.
- ➢
- Sovago et al. [28] studied the mixture sodium naproxen-lactose-terahydrate. XRPD revealed the amorphous nature of the mixtures and DSC showed that the blends were single-phase co-amorphous systems as indicated by a single Tg.
- ➢
- Ueda et al. [35] studied tranilast (TRL) and diphenhydramine hydrochloride (DPH) system. The Tg of the co-amorphous TRL-DPH deviated from the theoretical value and the enthalpy relaxation rate of the amorphous drugs (reflecting molecular mobility) was reduced due to the formation of a co-amorphous system. The intermolecular interactions between TRL and DPH in the co-amorphous system were measured from changes in the IR spectra.
- ➢
- Teja et al. [37] studied amorphous forms of Talinolol (TLN) using Naringin (NRG) as a stabilizer. The temperature stability of prepared amorphous forms was attributed to strong intermolecular hydrogen bonding between TLN and NRG, which was confirmed by FTIR.
- ➢
- Moinuddin et al. [38] studied the system of atenolol (ATE) with hydrochlorothiazide (HCT). The co-amorphous ATE-HCT sample with 1:1 molar ratio showed excellent physical stability, which could be attributed to the formation of strong molecular interactions between ATE and HCT as evidenced by FT-IR spectra.
- ➢
- Gao et al. [39] studied repaglinide (REP) with saccharin (SAC). Coamorphous REP–SAC at 1:1 stoichiometry had unique thermal behavior, distinct FTIR shifts and an absence of a sharp diffraction peak, suggesting the formation of a co-amorphous material and the interaction of REP with SAC through hydrogen bonds formed between REP’s secondary amine and SAC’s carbonyl group.
- ➢
- Shayanfar et al. [40] studied atorvastatin (ATC) and nicotinamide (ATC-NIC) characterized using DSC, FT-IR and PXRD. Crystalline ATC was converted to a co-amorphous form due to molecular interactions between ATC and NIC.
- ➢
- Jensen et al. [44] studied indomethacin (IND)–amino acid dispersions. The mixtures were characterized for solid-state properties by DSC, thermogravimetric analysis (TGA) and for molecular interactions by XRPD and FTIR. Elevated TGs were found for all mixtures compared with the pure amorphous drug due to co-amorphization with the amino acids. Furthermore, strong intermolecular interactions in the form of salt/partial salt formation between the drug and amino acids were seen for all blends.
- ➢
- Russo et al. [47] studied mixtures of omeprazole–amoxicillin trihydrate (CGM samples) and omeprazole–anhydrous amoxicillin (CGMa samples) at 3:7, 1:1 and 7:3 molar ratios, respectively. These systems were characterized by DSC, FTIR, PXRD, SEM and solid state uclear Magnetic Resonance (ssNMR). The co-grinding process that was used for the preparation was not useful to get a co-amorphous system but it led to disordered phase at the 1:1 CGMa ratio. Moreover, in this system both FTIR and ssNMR analysis strongly suggested intermolecular interactions between the sulfoxide group of omeprazole and the primary amine of anhydrous amoxicillin.
- ➢
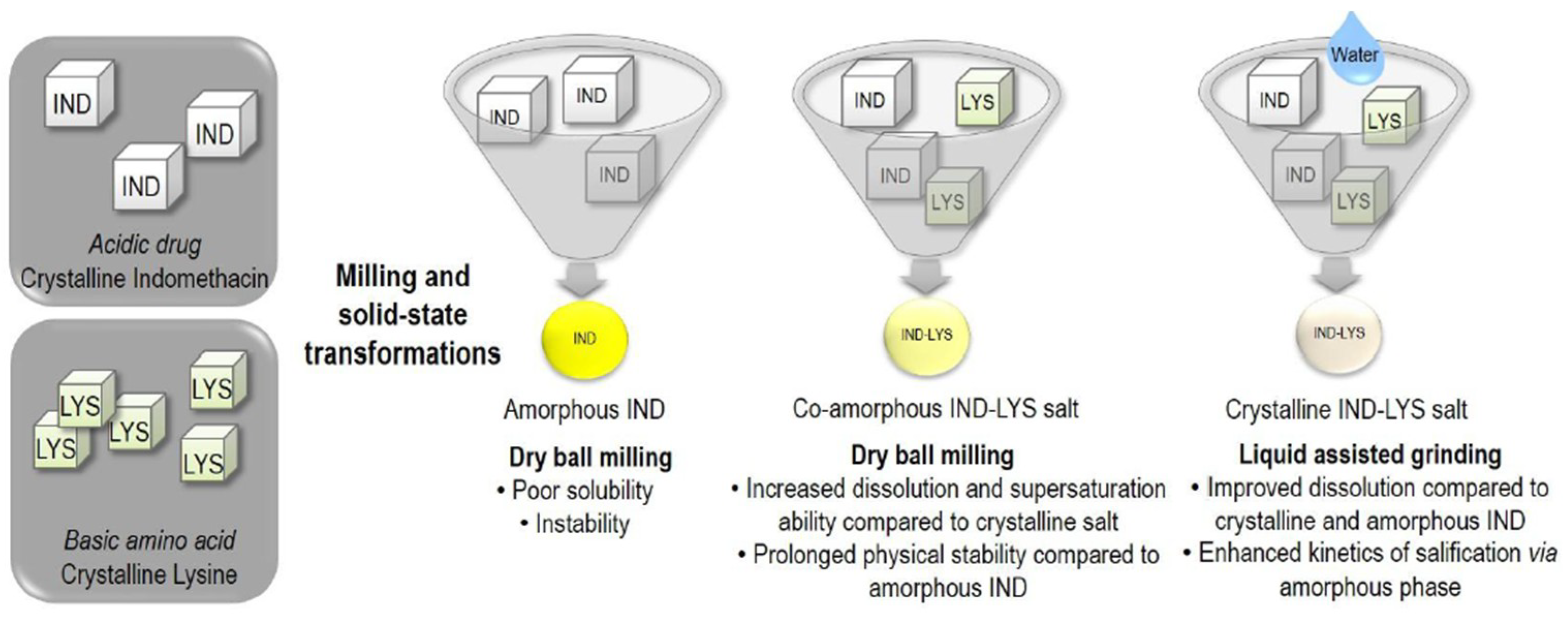
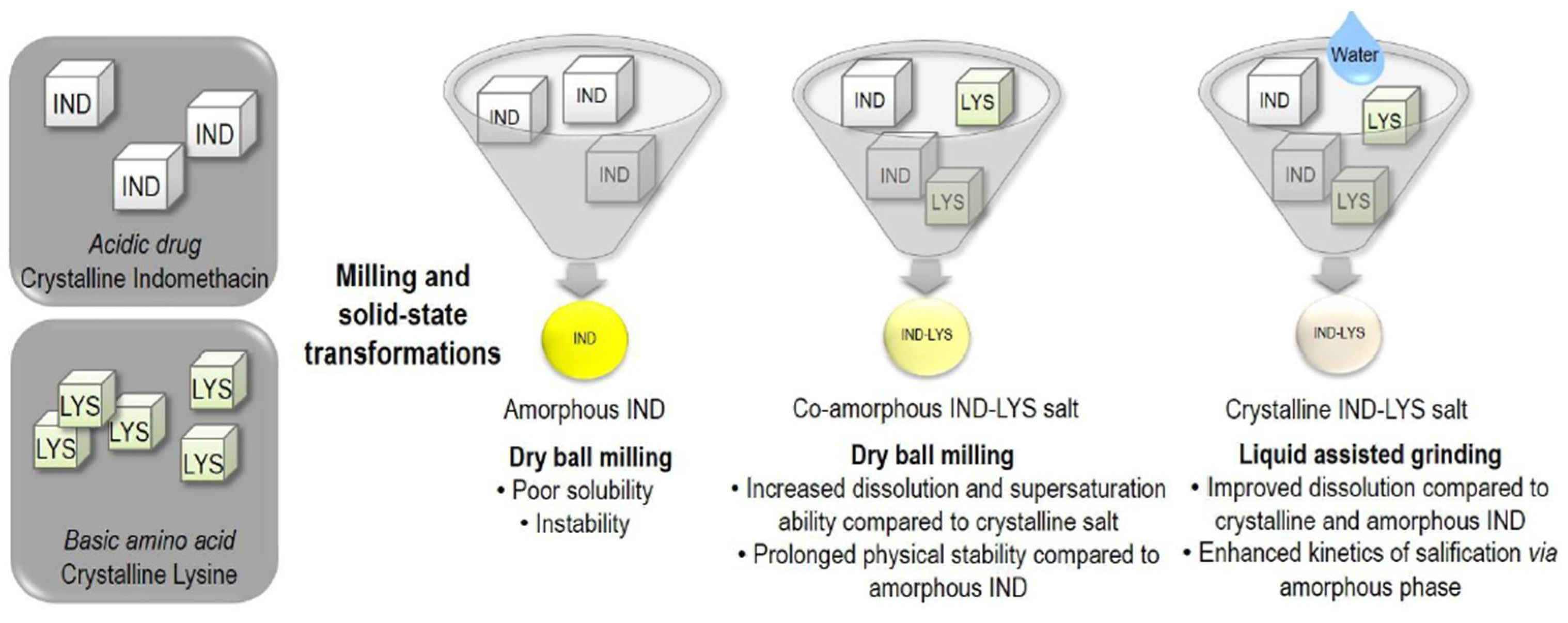
- Kasten et al. [48] prepared different physical forms of salts of indomethacin (IND) with the amino acid lysine (LYS). XRPD, FTIR and DSC showed that dry ball milling led to the formation of a fully co-amorphous salt with a single glass transition temperature (Tg), while liquid-assisted grinding resulted in a crystalline salt while a new melting point (223 °C).
- ➢
- Jensen et al. [54] evaluated four co-amorphous systems (Ind-Trp, Fur-Trp, Ind-Arg, and Fur-Arg) mixed at different molar ratios by determination of the deviation between the experimentally determined and the calculated Tg (Gordon-Taylor). They confirmed that the excess component was amorphous within the co-amorphous mixture. FTIR and ssNMR data did not support the presence of additional intermolecular drug-amino acid interactions.
- ➢
- Orajinta et al. [55] studied mixtures of ibuprofen-arginine and indomethacin-arginine mixtures at two drug-arginine molar ratios (1:1 and 1:2) using DSC, XRPD and FTIR. All mixtures exhibited a single Tg, indicating the formation of homogenous single-phase systems. FTIR revealed strong interactions (mainly salt formation) that accounted for the positive deviation between measured and estimated Tg.
- ➢
- Beyer et al. [56] studied different naproxen-indomethacin (NAP/IND) ratios prepared by melt-quenching at three cooling rates using XRPD and FTIR, both directly after preparation and during storage. All cooling methods led to amorphous samples, but with different physical stabilities.
- ➢
- Beyer et al. [57] developed a quantification model based on XRPD and multivariate partial least squares regression approach that enabled the simultaneous determination of up to four solid state fractions: crystalline naproxen, γ-indomethacin, α-indomethacin as well as co-amorphous naproxen-indomethacin.
- ➢
- Lobman et al. [65] studied naproxen (NAP) and indomethacin (IND) using FTIR and quantum mechanical calculations. The structures of both drugs were optimized as monomer, homodimer and heterodimer using density functional theory. Vibrational modes were computed and compared with experimentally observed spectra. It was shown that both drugs existed as homodimers in their respective individual amorphous form, but as heterodimers in the co-amorphous mixture when quench cooled together from the melt in a 1:1 molar ratio.
- ➢
- Dengale et al. [66] studied mixtures of Ritonavir (RTV) with the known amorphous stabilizing small molecule (IND). Co-amorphous RTV-IND samples were prepared at various molar ratios (2:1, 1:1 and 1:2) and their amorphicity was confirmed by XRPD, DSC and FTIR. The Tg values were in agreement with the theoretical (Gordon–Taylor equation), which also agreed with the FT-IR results that showed no evidence of intermolecular interactions.
- ➢
- Wairkar et al. [70] studied Nateglinide (120 mg) and Metformin hydrochloride (500 mg) mixture as a dose combination. XRPD and DSC confirmed amorphism and FTIR gave evidence of hydrogen bonding interactions (proton exchange between Nateglinide and Metformin hydrochloride).
- ➢
- Laitinen et al. [74] studied mixtures of simvastatin and glibenclamide with the amino acids aspartic acid, lysine, serine and threonine. XRPD, DSC and FTIR revealed that the 1:1 molar combinations of simvastatin-lysine, glibenclamide-serine and glibenclamide-threonine and the 1:1:1 molar combination glibenclamide-serine-threonine formed co-amorphous mixtures.
- ➢
- Qian et al. [75] studied lurasidone hydrochloride (LH) (pH-dependent solubility) with saccharin (SAC) at a 1:1 molar ratio. DSC and XPRD showed formation of the co-amorphous system and peak shifts in the FTIR spectra indicated the formation of a charge-assisted hydrogen bond between the NH group of LH and the C=O group of SAC.
- ➢
- Lim et al. [76] studied indomethacin–cimetidine and naproxen–cimetidine co-amorphous systems (1:1 molar ratio) by PXRD and DSC. Structural relaxation was estimated from the Kohlrausch–Williams–Watts (KWW) relationship. The Tg was about 20 °C higher than that predicted from the Fox equation.
- ➢
- Cray et al. [77] used sodium lauryl sulfate (SLS) as a solubilizer, adding in the co-amorphous simvastatin–lysine (SVS-LYS) 1:1 molar mixtures. Energy-dispersive X-ray spectroscopy (EDS) revealed that SLS coated the SVS and SVS-LYS particles upon spray drying. XRPD and DSC showed that in the spray-dried formulations the remaining crystallinity was due to SLS.
- ➢
- Beyer et al. [78] optimised the physicochemical properties of co-amorphous naproxen-indomethacin dispersion by the incorporation of naproxen sodium. The influence of naproxen-sodium on the resulting physicochemical properties was analyzed by XRPD, FTIR, DSC. Fully amorphous samples with single glass transition temperatures could be prepared with naproxen molar fractions up to 0.7 which lasted for 270 days of storage.
- ➢
- Huang et al. [79] studied combinations of valsartan (VAL) with l-histidine, l-arginine, and l-lysine with XRPD and DSC. The Tg values of the co-amorphous mixtures were higher than those of the single components and the predicted Tg values of the combinations (Gordon Taylor equation) suggesting a strong interaction between VAL and the amino acids.
- ➢
- Pan et al. [80] studied the effect of different polymorphs of azelnidipine (AZE) on the micro-structure of the complexes with oxalic acid (OXA) using XRPD, modulated DSC and TGA, cryo-field emission, SEM, FTIR and solid-state nuclear magnetic resonance spectroscopy. AZE-OXA co-crystal was produced at a molar ratio of 2:1 when grinding with a solvent, while co-amorphous forms were obtained under dry grinding.
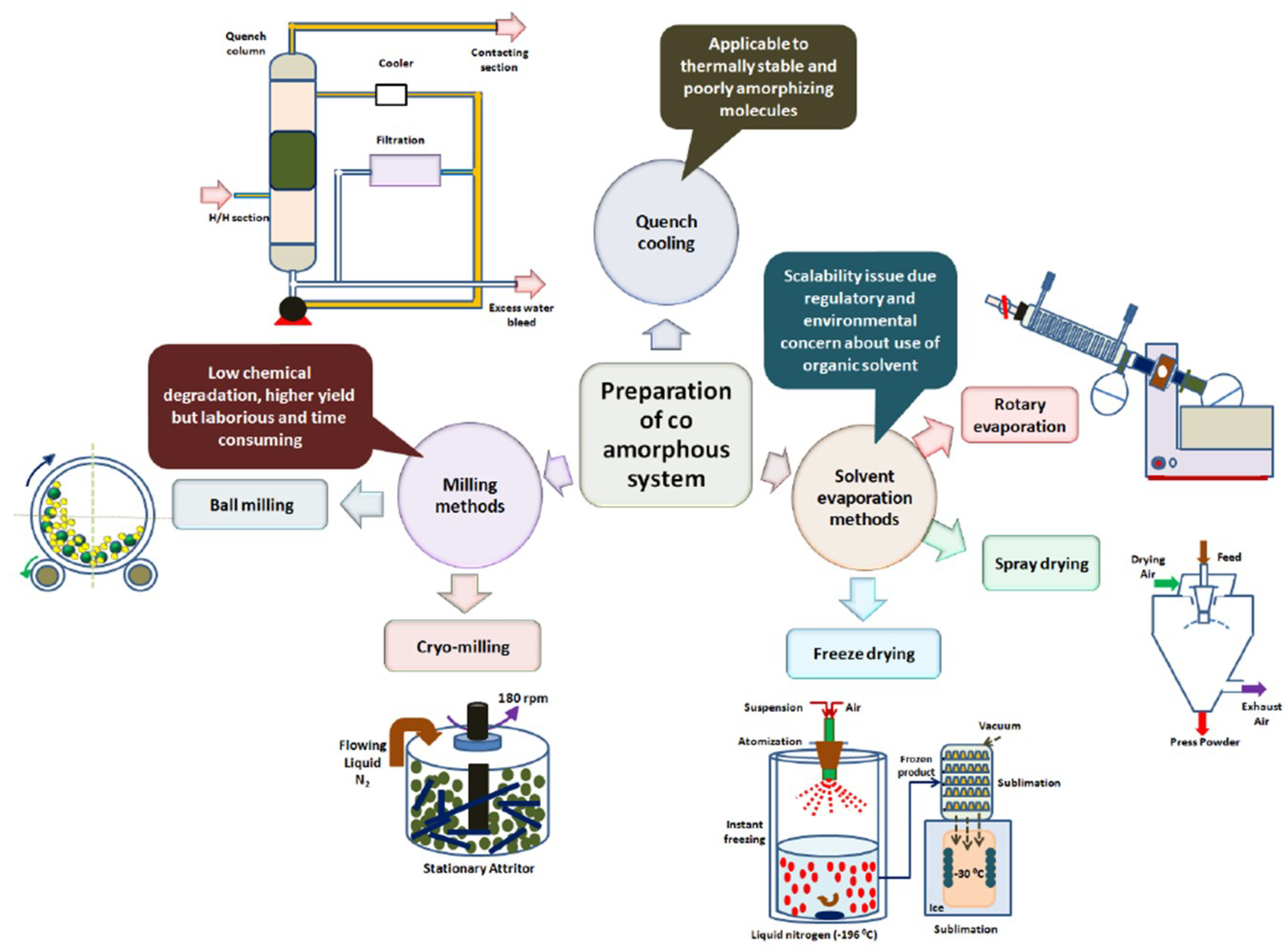
5. Preparation of Co-Amorphous Solid Dispersions
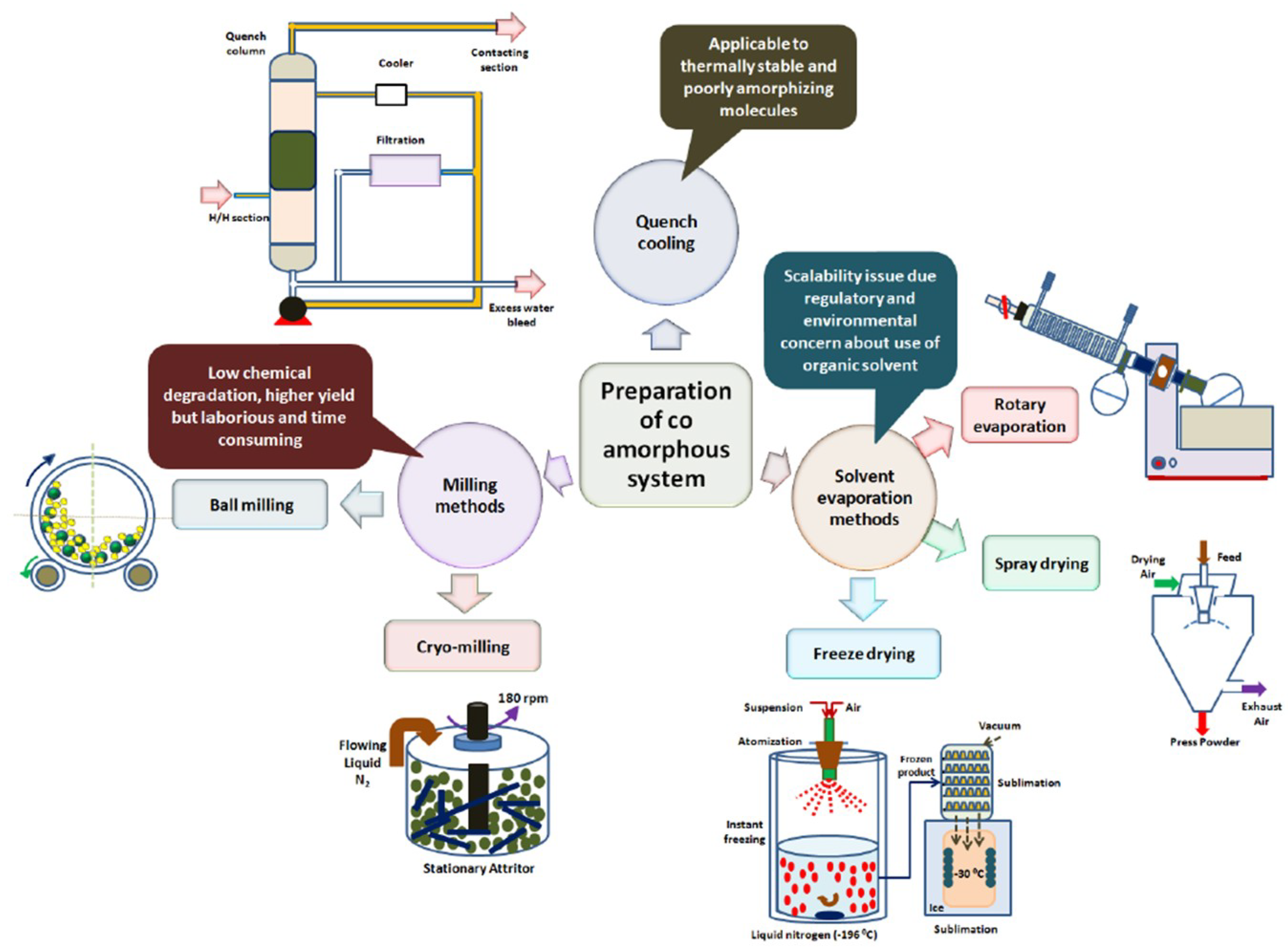
5.1. Quench Cooling
5.2. Vibrational Ball Milling
5.3. Cryomilling
5.4. Solvent Evaporation
5.5. Spray Drying
5.5.1. Comparison of Spray Drying with Milling
5.5.2. Spray Drying Applications with Aqueous Solutions
5.6. Hot-Melt-Extrusion (HME)
5.7. Other Methods
6. Examples of Co-Amorphous Dispersions with Pharmacological Effects
6.1. Drug-Drug Combinations
- ➢
- Cimetidine is sometimes co-administered with naproxen for the treatment of NSAID-induced gastro-intestinal disorders. Allesø et al. [17] prepared co-amorphous naproxen-cimetidine SDs with increased Tg by milling. The IDR of naproxen and cimetidine in the co-amorphous SD was respectively four-fold and two-fold higher than the individual crystalline forms. While the dissolution rate of amorphous cimetidine was found to be identical to the crystalline drug due to recrystallization upon contact with the dissolution medium, the dissolution rate of co-amorphous naproxen SD remained high without recrystallization.
- ➢
- RTV ritonavir, a protease inhibitor, was stabilized with indomethacin in co-amorphous SDs, prepared by solvent evaporation. These were stable for 90 days, while the solubility of both drugs was about three-fold greater compared to the corresponding crystalline analogs. Moreover, co-administration of the two drugs improved the efficiency of antiretroviral therapy and the reduction of ritonavir side effects [66].
- ➢
- Co-amorphous naproxen–indomethacin SD was prepared by spray drying [88]. The influence of the process conditions on the resulting initial sample crystallinity and the recrystallization of the drugs was studied. Recrystallization was affected by the total and individual recrystallization rates of the co-amorphous components, the formation of indomethacin polymorphs and by the inlet temperature and feed rate of the spray dryer.
- ➢
- Combined oral therapy of hypoglycemic agents may assist reduction of side effects and costs. Co-amorphous dispersions of nateglinide (NT) and metformin hydrochloride were formed at therapeutic doses and showed a significantly increased release rate of NT [70]. They also showed synergistic action in patients with type 2 diabetes that could not be controlled with monotherapy. Co-administration did not affect the pharmacokinetics.
- ➢
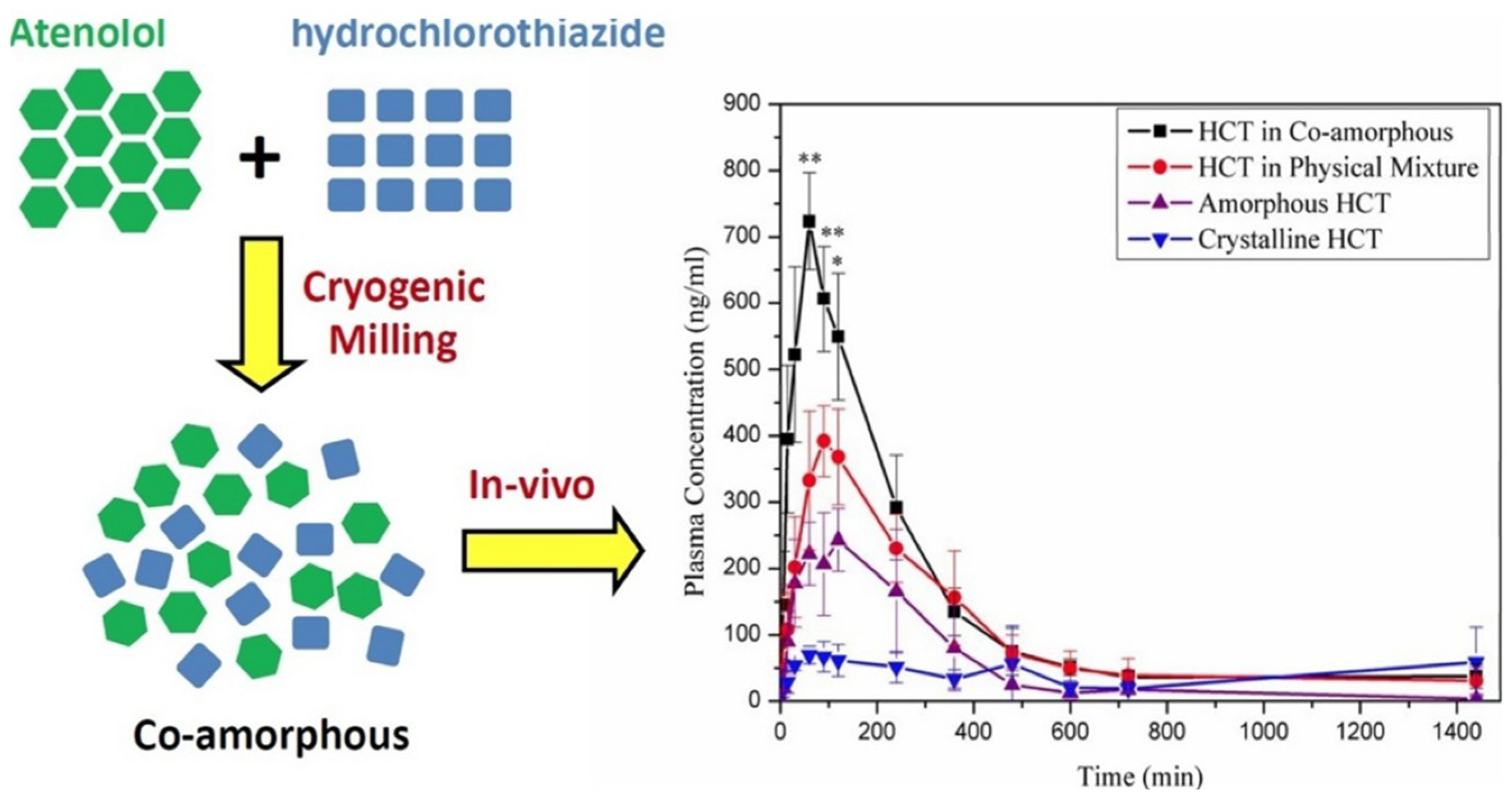
- Recently, an interesting co-amorphous drug-drug system consisting of atenolol (cardio-selective beta blocker) and hydrochlorothiazide (diuretic) was developed by cryomilling for the combined therapy of hypertensive patients. The co-amorphous SD showed better physical stability (30 days) compared to the individual amorphous drugs [38].
6.2. Drug-Carboxylic Acids
- ➢
- Masuda et al. [96] studied solid dispersions of acyclovir with different carboxylic acids prepared by cooling or solvent evaporation of drug solutions in various solvents. Acyclovir formed cocrystal with tartaric acid but co-amorphous mixture with citric acid. The amorphized mixture was characterized by XRPD, TG/DSC IR and HPLC.
- ➢
- Ali et al. [97] prepared stable clozapine (CZ) co-amorphous systems with low MW/low Tg carboxylic acids such as citrate, d-tartrate or oxalate, by rapid solvent evaporation in vacuum via formation of H-bonds. The co-amorphous mixture with tartaric acid had the highest dissolution rates (>95% in 20 min) compared to pure crystalline CZ (56%).
- ➢
- In another study by Maher et al. [71], solvent evaporation was also applied to prepare co-amorphous SDs of olanzapine (OL) using polycarboxylic acids as co-formers. The mixtures were incorporated into rapidly dissolving oral polymer films containing additionally HPMC, Na-CMC, glycerin, PG, PEG 400, citric acid, saccharin, menthol as an easy and simple way of administration. Co-amorphization of OL-ascorbic acid was achieved at 1:2 molar ratio through H-bonding and showed a more than 600-fold increase in OL solubility. The films showed high dissolution rate (97% in 10 min). The co-amorphous dispersion in the films remained stable for over 12 weeks at 40 °C/75% RH. Pharmacokinetic data showed improved OL AUC (144 ng h/mL) and Cmax (14.2 ng/mL) compared to commercial formulations.
6.3. Drug-Amino Acids
- ➢
- Löbmann et al. [41,42] prepared co-amorphous systems of AAs in combination with carbamazepine (CBZ) and indomethacin (IND) by milling. Selection of AA was based on the binding site at the biological receptors arginine (ARG) and tyrosine (TYR) for IND binding to cyclooxygenase 2, and receptors phenylalanine (PHE), tryptophan (TRP) for CBZ binding to neural Na+ channels. Specific hydrogen bonding and π−π interactions were found in all co-amorphous dispersions that contained at least one binding AA from the biological target site of the drug, whereas the blends of IND with the non-receptor binding AAs PHE and TRP showed no interactions [42]. CBZ could only be prepared as co-amorphous with TRP while IND gave co-amorphous binary mixture with ARG but not with TYR. However, binary co-amorphous IND-amino acid mixtures were also obtained with the non-receptors PHE and TRP. The stability of the co-amorphous SDs was over 6 months, whereas for the pure amorphous it was less than 7 days.
- ➢
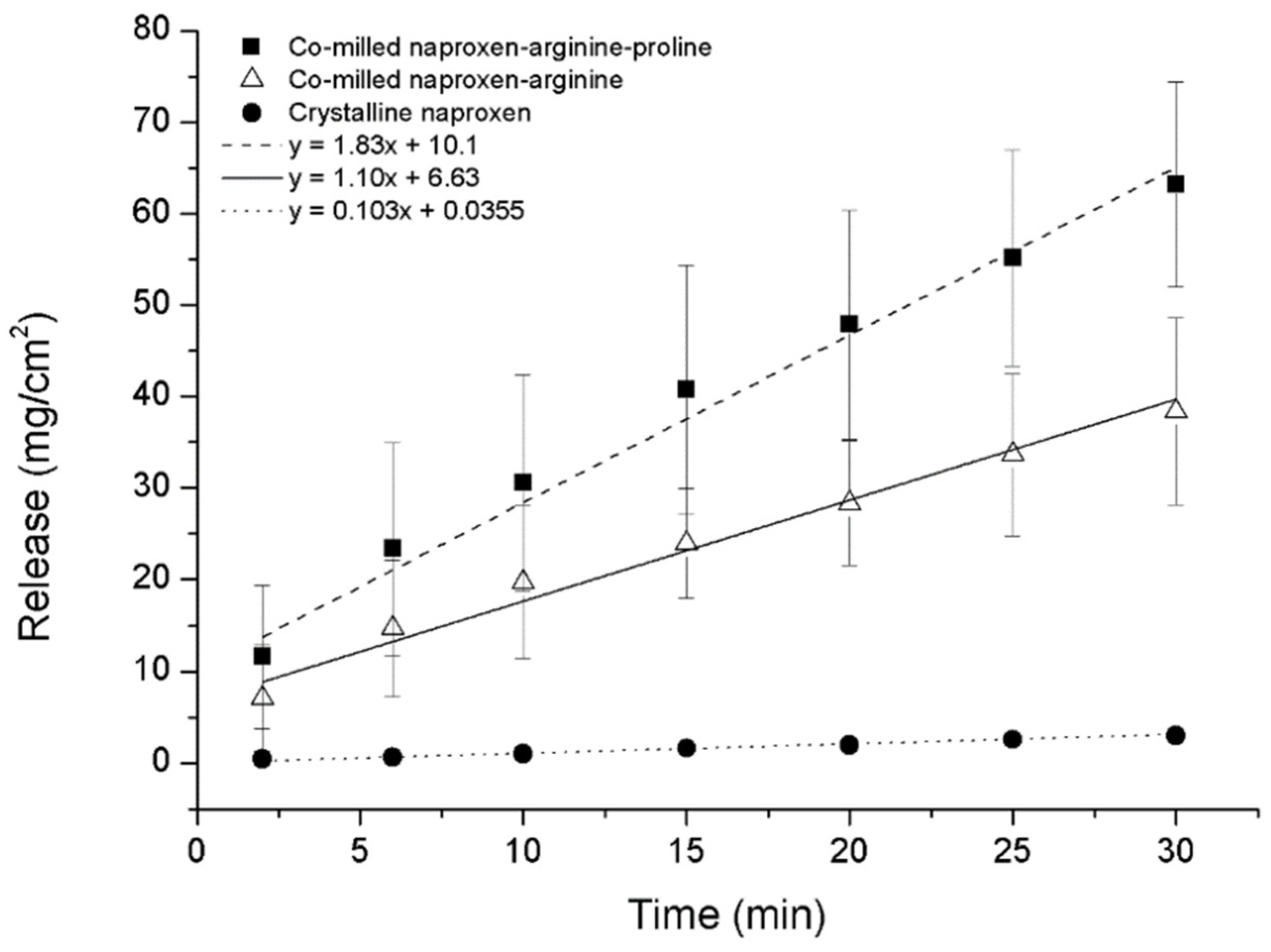
- ARG has been found to form co-amorphous salts with weak acids such as NSAIDs and was used successfully as a co-former for indomethacin [6,42]. Additionally, it can make π−π interactions with molecules having aromatic rings, e.g., naproxen-arginine co-amorphous mixture which showed physical stability and 10-fold faster IDR compared to crystalline drug [42].
- ➢
- On the basis of the ability of ARG for co-amorphization it was thought that the structurally similar citrulline (CTL) could also show similar behavior. However, although its incorporation in mixtures with drugs reduced their crystallinity, fully co-amorphous mixture was obtained only with furosemide (FUR-CTL) and nitrofurantoin (NIT-CTL). Hence, although structural similarity is related to molecular mixing, it cannot predict co-amorphization [98].
- ➢
- In the study by Laitinen et al. [74], from the receptor binding aminoacids of simvastatin (SVS): aspartic acid, lysine and serine, the drug formed co-amorphous only with LYS, whereas glibenclamide (GBC) was able to form co-amorphous with both serine (receptor) and threonine (non-receptor) at s 1:1 molar ratio and with glibenclamide-serine-threonine at 1:1:1. Co-amorphous systems showed improved physical stability, for SVC-LYS 3 months and for GBC-SER for 6 months. A possible reason for the stabilization of GBC was prevention by the AA of the conversion of the amide group into the unstable tautomeric imide form.
6.4. Co-Amorphous Dispersions with Salt Formation
- ➢
- Valsartan VAL, a selective angiotensin antagonist used in hypertension and heart failure, was processed in a vibrational ball mill with l-histidine (HIS), l-lysine (LYS) and l-Arginine (ARG). Co-amorphous drug-AA binary dispersions were obtained and showed increased solubility and IDR at pH 4.6, 6.8. The increase of solubility achieved from co-amorphous ternary mixtures having a second AA was approximately 1000-fold greater, indicating that the second AA had an important role. The reason for the remarkable IDR increase of VAL from the co-amorphous SD was the ionization and salt drug-amino acid formation, as well as the high-water solubility of AAs. The co-amorphous VAL-ARG and VAL-LYS SDs were stable for 3 months [79].
6.5. Examples of Permeability Together with Solubility Improvement
- ➢
- Co-amorphous compositions can address problems of solubility as well as permeability of BCS IV drugs [37]. Apart from the physicochemical properties of the drug molecule (size, polarity), poor permeability may arise if the drugs are substrates to efflux glycoproteins P-gp. These reside in the gastrointestinal epithelium and act as a physiological barrier. In order to overcome these problems, Teja et al. [37] co-amorphisized the poorly water soluble and low permeability drug talinolol, an efflux pump substrate, with naringin, an efflux pump inhibitor. In this way they improved both the dissolution rate and the permeability-absorption of the drug by inhibiting the action of P-gp. The permeability of talinolol was found to increase from 2.48 × 10−5 (control value) to 3.16 × 10−5 cm/s when formulated as a co-amorphous system. The increase of the bioavailability of talinolol in rats was also improved, while the average AUC was found to be 5.4-fold higher than the crystalline drug, which was attributed to a combination of increased solubility and inhibition of P-gp.
7. Formulation of Co-Amorphous Dispersions into Tablets
- ➢
- Processing into a tablet or capsules form has the risk of crystallization due to the influence of environmental factors (moisture, temperature), while mechanical stresses during compression into tablets may alter the amorphous structure, the stability and the dissolution profile [18,23,99]. Other problems arising from the soft and sticky nature of the amorphous SDs may be difficulty in granulation and sieving, poor flowability and compressibility [100]. Wet granulation is not recommended since the added water may cause plasticization and crystallization. Also, during melt granulation, the heat input could increase the temperature above the Tg of the co-amorphous components with the risk of recrystallization.
- ➢
- Lenz et al. [18] were the first to report a successful tablet formulation of the co-amorphous indomethacin-arginine (IND-ARG) dispersion prepared by spray drying and compressed into tablets using mannitol, croscarmellose sodium, colloidal silicon dioxide, magnesium stearate as tableting aids. Molecular interactions were unaffected by compression and no crystallization occurred. Tablets showed immediate release of IND, four-fold supersaturation concentration over crystalline IND and long-term stability. AUC was not affected by tableting compared to spray dried co-amorphous IND-ARG powder. The AUC at 24 h of the IND-ARG tablets was two-fold that of the tablets of physical mixtures of the crystalline drugs (IND-ARG) and three-fold that of the IND. The dissolution plateaus after 24 h were different for spray dried IND-ARG tablets, physical mixtures IND-ARG tablets and neat spray dried IND-ARG powder which was explained due to the formation of different indomethacin polymorphs.
- ➢
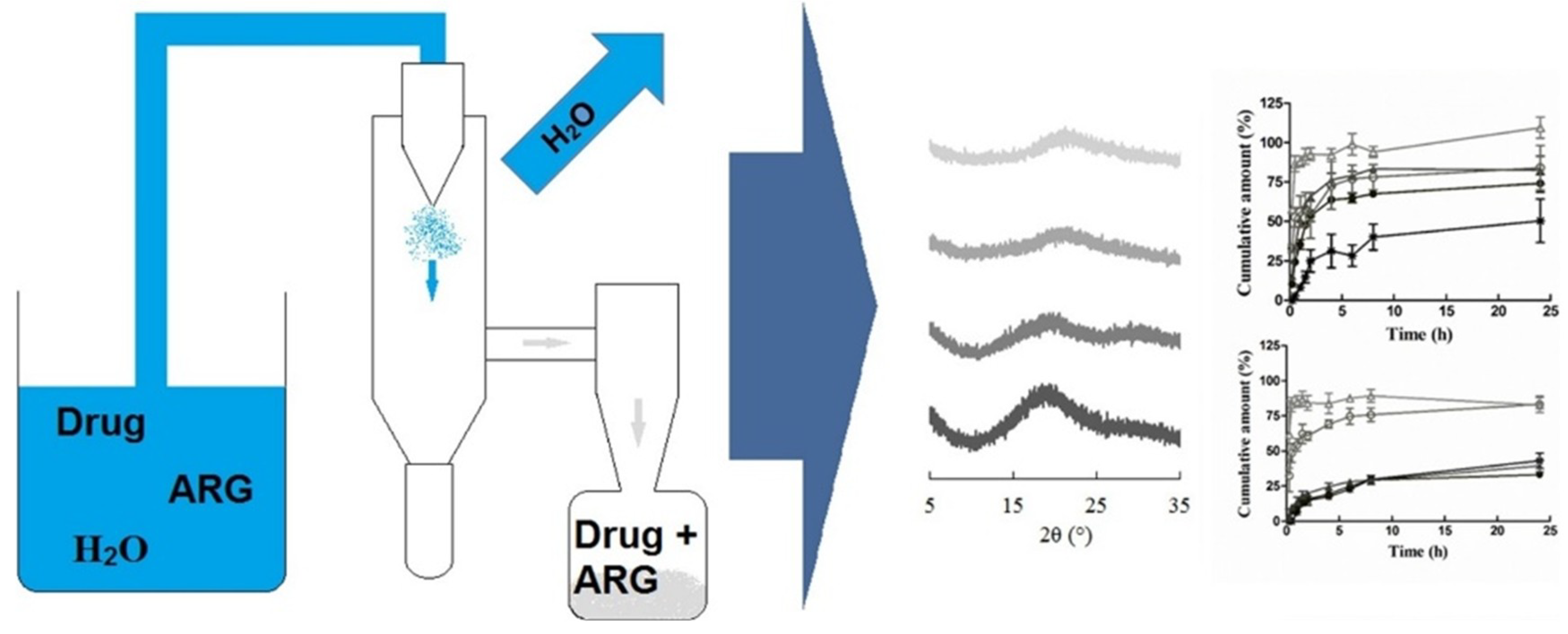
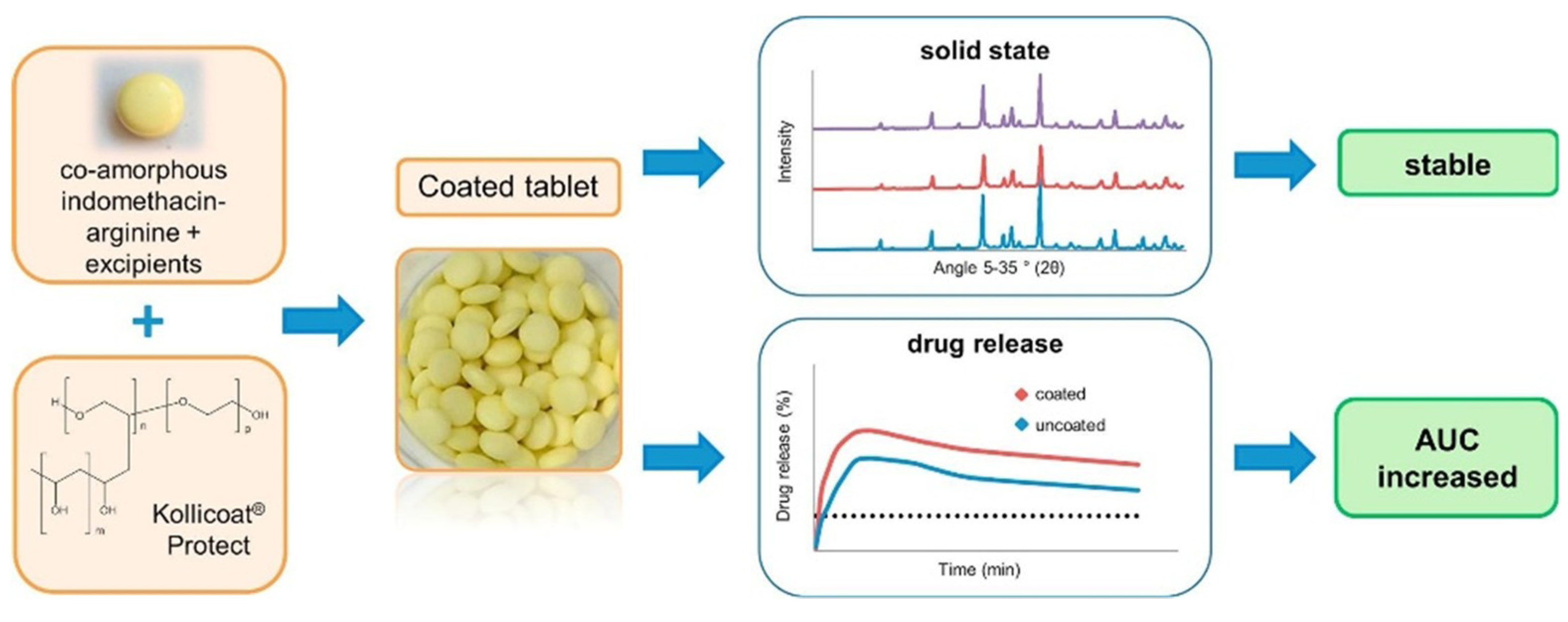
- Long-term stability of co-amorphous systems can be ensured by the application of polymeric coatings. Petry et al. [101] prepared a co-amorphous indomethacin-arginine homogeneous system (SD IND-ARG) by spray drying, which was then processed into tablets and coated with polyvinyl alcohol-polyethylene glycol graft copolymer (Kollicoat® Protect). The steps of the processing are shown in Figure 13. Contact with water, heat and stress during coating did not induce recrystallization of IND-ARG. The coated tablets were stable for at least 94 days, and there was no recrystallization after storage at RT/75% for 91 days, which demonstrated high physical stability even under humid conditions. They also found that the applied coating enhanced dissolution of the co-amorphous tablets by about 30% compared to neat spray dried IND-ARG.
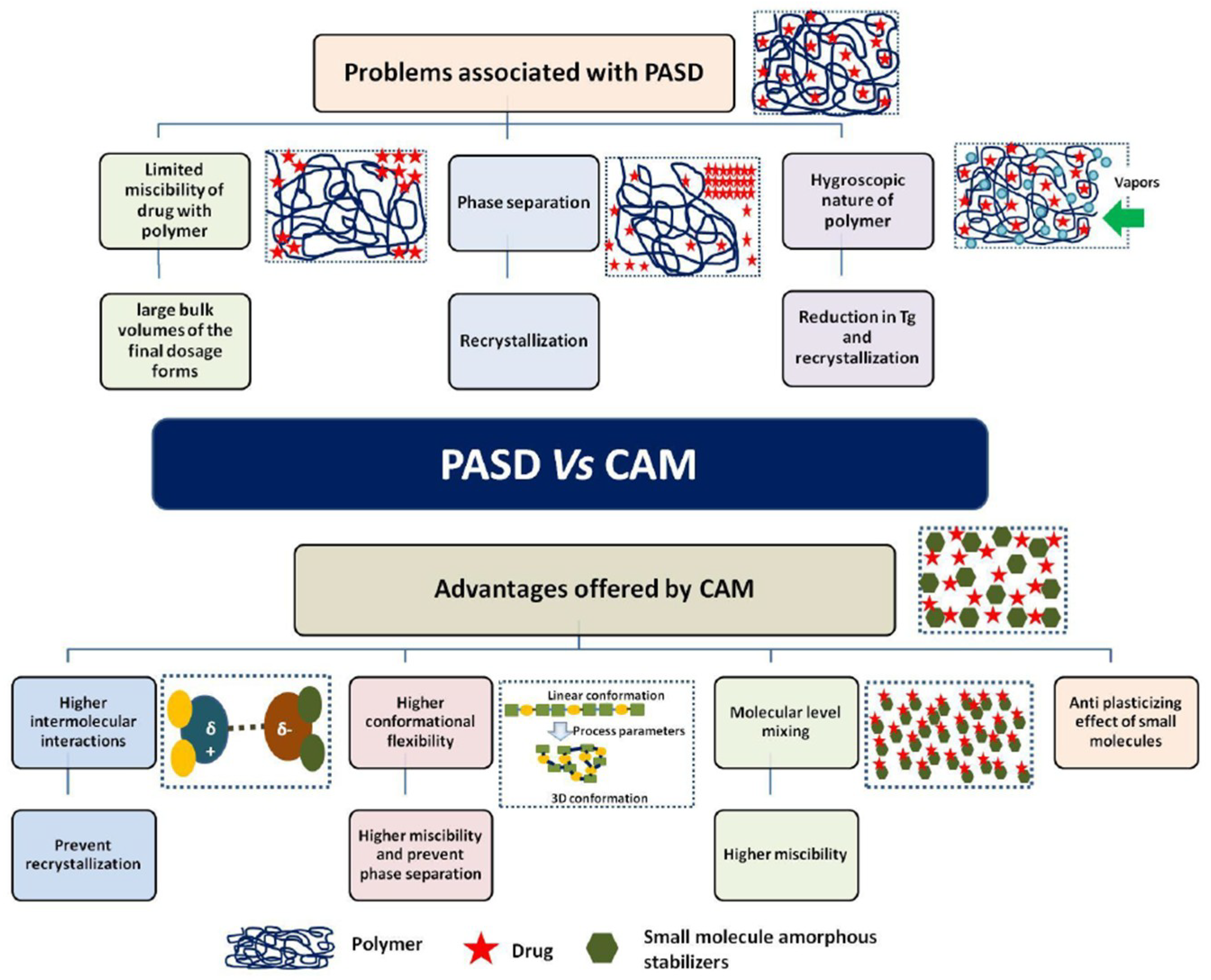
8. Current Issues, Limitations and Potential Solutions
Author Contributions
Funding
Conflicts of Interest
References
- Hauss, D.J. Oral lipid-based formulations. Adv. Drug Deliv. Rev. 1995, 59, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.D.; Trevaskis, N.L.; Charman, S.A.; Shanker, R.M.; Charman, W.N.; Pouton, C.W.; Porter, C.J. Strategies to address low drug solubility in discovery and development. Pharmacol. Rev. 2013, 65, 315–499. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, T.; Sarmento, B.; Costa, P. Solid dispersions as strategy to improve oral bioavailability of poor water-soluble drugs. Drug Discov. Today 2007, 12, 1068–1075. [Google Scholar] [CrossRef] [PubMed]
- Sareen, S.; Mathew, G.; Joseph, L. Improvement in solubility of poor water-soluble drugs by solid dispersion. Int. J. Pharm. Investig. 2012, 2, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Yu, L. Amorphous pharmaceutical solids: Preparation, characterization and stabilization. Adv. Drug Deliv. Rev. 2001, 48, 27–42. [Google Scholar] [CrossRef]
- Ojarinta, R.A.; Heikkinen, T.; Sievänen, E.; Laitinen, R. Dissolution behavior of co-amorphous amino acid-indomethacin mixtures: The ability of amino acids to stabilize the supersaturated state of indomethacin. Eur. J. Pharm. Biopharm. 2017, 112, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiou, W.L.; Riegelman, S. Pharmaceutical applications of solid dispersion systems. Pharmaceutical applications of solid dispersion systems. J. Pharm. Sci. 1971, 60, 1281–1302. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.D.; Lee, P.I. Haste makes waste the interplay between dissolution and precipitation of supersaturating formulations. AAPS J. 2015, 17, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Vaka, S.R.K.; Bommana, M.M.; Desai, D.; Djordjevic, J.; Phuapradit, W.; Shah, N. Excipients for amorphous solid dispersions, Amorphous solid Dispersions. In Amorphous Solid Dispersions; Shah, N., Sandhu, H., Choi, D.S., Chokshi, H., Malick, A.W., Eds.; Springer: New York, NY, USA, 2014; pp. 123–161. [Google Scholar]
- Dengale, S.J.; Grohganz, H.; Rades, T.; Löbmann, K. Recent advances in co-amorphous drug formulations. Adv. Drug Deliv. Rev. 2016, 100, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Janssens, S.; Van den Mooter, G. Physical chemistry of solid dispersions. J. Pharm. Pharmacol. 2009, 61, 1571–1586. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, A.M.; Gupta, P.; Bansal, A.K. Amorphous drug delivery systems: Molecular aspects, design and performance. Crit. Rev. Ther. Drug 2004, 21, 133–193. [Google Scholar] [CrossRef]
- Hancock, B.C.; Shamblin, S.L.; Zografi, G. Molecular mobility of amorphous pharmaceutical solids below their glass transition temperatures. Pharm. Res. 1995, 12, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Serajuddin, A.T.M. Solid dispersion of poorly water-soluble drugs: Early promises, subsequent problems, and recent breakthroughts. J. Pharm. Sci. 1999, 88, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Chieng, N.; Aaltonen, J.; Saville, D.; Rades, T. Physical characterization and stability of amorphous indomethacin and ranitidine hydrochloride binary systems prepared by mechanical activation. Eur. J. Pharm. Biopharm. 2009, 71, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Löbmann, K.; Jensen, K.T.; Laitinen, R.; Rades, T.; Strachan, C.; Grohganz, H. Stabilized amorphous solid dispersions with small molecule excipients. In Amorphous Solid Dispersions; Shah, N., Sandhu, H., Choi, D.S., Chokshi, H., Malick, A.W., Eds.; Springer: New York, NY, USA, 2014; pp. 613–636. [Google Scholar]
- Allesø, M.; Chieng, N.; Rehder, S.; Rantanen, J.; Rades, T.; Aaltonen, J. Enhanced dissolution rate and synchronized release of drugs in binary systems through formulation: Amorphous naproxen–cimetidine mixtures prepared by mechanical activation. J. Controll. Release 2009, 136, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Lenz, E.; Jensen, K.T.; Blaabjerg, L.I.; Knop, K.; Grohganz, H.; Löbmann, K.; Rades, T.; Kleinebudde, P. Solid-state properties and dissolution behaviour of tablets containing co-amorphous indomethacin-arginine. Eur. J. Pharm. Biopharm. 2015, 96, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Löbmann, K.; Laitinen, R.; Grohganz, H.; Gordon, K.C.; Strachan, C.; Rades, T. Coamorphous drug systems: Enhanced physical stability and dissolution rate of indomethacin and naproxen. Mol. Pharm. 2011, 8, 1919–1928. [Google Scholar] [CrossRef] [PubMed]
- Suresh, K.; Mannava, M.K.C.; Nangia, A. A novel curcumin–artemisinin coamorphous solid: Physical properties and pharmacokinetic profile. RSC Adv. 2014, 4, 58357–58361. [Google Scholar] [CrossRef]
- Laitinen, R.; Löbmann, K.; Strachan, C.J.; Grohganz, H.; Rades, T. Emerging trends in the stabilization of amorphous drugs. Int. J. Pharm. 2013, 453, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Gao, H.; Babu, S.; Garad, S. Co-Amorphous Formation of High-Dose Zwitterionic Compounds with Amino Acids to Improve Solubility and Enable Parenteral Delivery. Mol. Pharm. 2018, 15, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Chavan, R.B.; Thipparaboina, R.; Kumar, D.; Shastri, N.R. Co amorphous systems: A product development perspective. Int. J. Pharm. 2016, 515, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Löbmann, K.; Strachan, C.; Grohganz, H.; Rades, T.; Korhonen, O.; Laitinen, R. Co-amorphous simvastatin and glipizide combinations show improved physical stability without evidence of intermolecular interactions. Eur. J. Pharm. Biopharm. 2012, 81, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Schantz, S.; Hoppu, P.; Juppo, A.M. Solid-State NMR Study of Phase Structure, Molecular Interactions, and Mobility in Blends of Citric Acid and Paracetamol. J. Pharm. Sci. 2009, 98, 1862–1870. [Google Scholar] [CrossRef] [PubMed]
- Pajula, K.; Taskinen, M.; Lehto, V.P.; Ketolainen, J.; Korhonen, O. Predicting the formation and stability of amorphous small molecule binary mixtures from computationally determined Flory–Huggins interaction parameter and phase diagram. Mol. Pharm. 2010, 7, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Kojima, T.; Karashima, M.; Ikeda, Y. Physicochemical Evaluation and Developability Assessment of Co-amorphouses of Low Soluble Drugs and Comparison to the Co-crystals. Chem. Pharm. Bull. 2016, 64, 1739–1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sovago, I.; Wang, W.; Qiu, D.; Raijada, D.; Rantanen, J.; Grohganz, H.; Rades, T.; Bond, A.D.; Löbmann, K. Properties of the Sodium Naproxen-Lactose-Tetrahydrate Co-Crystal upon Processing and Storage. Molecules 2016, 21, 509. [Google Scholar] [CrossRef] [PubMed]
- Jayasankar, A.; Good, D.J.; Rodríguez-Hornedo, N. Mechanisms by which moisture generates cocrystals. Mol. Pharm. 2007, 4, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Seefeldt, K.; Miller, J.; Alvarez-Nunez, F.; Rodriquez-Hornedo, N. Crystallization pathways and kinetics of carbamazepine–nicotinamide cocrystals from the amorphous state by in situ thermomicroscopy, spectroscopy and calorimetry studies. J. Pharm. Sci. 2007, 96, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Korhonen, O.; Pajula, K.; Laitinen, R. Rational excipient selection for co-amorphous formulations. Expert Opin. Drug Deliv. 2017, 1, 551–569. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, S.; Momose, Y.; Takahashi, K.; Nagatani, S. Solid-state interaction between cimetidine and naproxen. Drug Stab. 1996, 1, 173–178. [Google Scholar]
- Yamamura, S.; Gotoh, H.; Sakamoto, Y.; Momose, Y. Physicochemical properties of amorphous precipitates of cimetidine–indomethacin binary system. Eur. J. Pharm. Biopharm. 2000, 49, 259–265. [Google Scholar] [CrossRef]
- Yamamura, S.; Gotoh, H.; Sakamoto, Y.; Momose, Y. Physicochemical properties of amorphous salt of cimetidine and diflunisal system. Int. J. Pharm. 2002, 241, 213–221. [Google Scholar] [CrossRef]
- Ueda, H.; Kadota, K.; Imono, M.; Ito, T.; Kunita, A.; Tozuka, Y. Co-amorphous Formation Induced by Combination of Tranilast and Diphenhydramine Hydrochloride. J. Pharm. Sci. 2017, 106, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Heikkinen, A.T.; DeClerck, L.; Löbmann, K.; Grohganz, H.; Rades, T.; Laitinen, R. Dissolution properties of co-amorphous drug-amino acid formulations in buffer and biorelevant media. Pharmazie 2015, 70, 452–457. [Google Scholar] [PubMed]
- Teja, A.; Musmade, P.B.; Khade, A.B.; Dengale, S.J. Simultaneous improvement of solubility and permeability by fabricating binary glassy materials of Talinolol with Naringin: Solid state characterization, in-vivo in-situ evaluation. Eur. J. Pharm. Sci. 2015, 78, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Moinuddin, S.M.; Ruan, S.; Huang, Y.; Gao, Q.; Shi, Q.; Cai, B.; Cai, T. Facile formation of co-amorphous atenolol and hydrochlorothiazide mixtures via cryogenic-milling: Enhanced physical stability, dissolution and pharmacokinetic profile. Int. J. Pharm. 2017, 532, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Liao, J.; Qi, X.; Zhang, J. Coamorphous repaglinide-saccharin with enhanced dissolution. Int. J. Pharm. 2013, 450, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Shayanfar, A.; Ghavimi, H.; Hamishekar, H.; Jouyban, A. Coamorphous atorvastatin calcium to improve its physicochemical and pharmacokinetic properties. J. Pharm. Sci. 2013, 16, 577–587. [Google Scholar]
- Löbmann, K.; Grohganz, H.; Laitinen, R.; Strachan, C.; Rades, T. Amino acids as coamorphous stabilizers for poorly water soluble drugs—Part 1: Preparation, stability and dissolution enhancement. Eur. J. Pharm. Biopharm. 2013, 85, 873–881. [Google Scholar] [PubMed]
- Löbmann, K.; Laitinen, R.; Strachan, C.; Rades, T.; Grohganz, H. Amino acids as coamorphous stabilizers for poorly water-soluble drugs—Part 2: Molecular interactions. Eur. J. Pharm. Biopharm. 2013, 85, 882–888. [Google Scholar] [CrossRef] [PubMed]
- Kasten, G.; Grohganz, H.; Rades, T.; Löbmann, K. Development of a screening method for co-amorphous formulations of drugs and amino acids. Eur. J. Pharm. Sci. 2016, 95, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.T.; Blaabjerg, L.I.; Lenz, E.; Bohr, A.; Grohganz, H.; Kleinebudde, P.; Rades, T.; Löbmann, K. Preparation and characterization of spray dried co-amorphous drug-amino acid salts. J. Pharm. Pharmacol. 2015, 68, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.; Susan, M.; Reutzel-Edens, M.S.; Zografi, G. Coamorphous Active Pharmaceutical Ingredient-Small Molecule Mixtures: Considerations in the Choice of Coformers for Enhancing Dissolution and Oral Bioavailability. J. Pharm. Sci. 2018, 107, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.T.; Löbmann, K.; Rades, T.; Grohganz, H. Improving Co-Amorphous Drug Formulations by the Addition of the Highly Water Soluble Amino Acid, Proline. Pharmaceutics 2014, 6, 416–435. [Google Scholar] [CrossRef] [PubMed]
- Russo, G.M.; Sancho, M.I.; Silva, L.M.A.; Baldoni, H.A.; Venancio, T.; Javier, E.; Narda, G.E. Looking for the interactions between omeprazole and amoxicillin in a disordered phase. An experimental and theoretical study. Spectrochim. Acta Part A 2016, 156, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Kasten, G.; Nouri, K.; Grohganz, H.; Rades, T.; Löbmann, K. Performance comparison between crystalline and co-amorphous salts of indomethacin-lysine. Int. J. Pharm. 2017, 533, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Ueda, H.; Muranushi, N.; Sakuma, S.; Ida, Y.; Endoh, T.; Kadota, K.; Tozuka, Y. A Strategy for Co-former Selection to Design Stable Co-amorphous Formations Based on Physicochemical Properties of Non-steroidal Inflammatory Drugs. Pharm. Res. 2016, 33, 1018–1029. [Google Scholar] [CrossRef] [PubMed]
- Alhalaweh, A.; Alzghoul, A.; Kaialy, W.; Mahlin, D.; Bergstrom, C.A.S. Computational predictions of glass-forming ability and crystallization tendency of drug molecules. Mol. Pharm. 2014, 11, 3123–3132. [Google Scholar] [CrossRef] [PubMed]
- Pajula, K.; Wittoek, L.; Lehto, V.P.; Ketolainen, J.; Korhonen, O. Phase Separation in Coamorphous Systems: In Silico Prediction and the Experimental Challenge of Detection. Mol. Pharm. 2014, 11, 2271–2279. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.T.; Larsen, F.H.; Cornett, C.; Löbmann, K.; Grohganz, H.; Rades, T. Formation Mechanism of Coamorphous Drug-Amino Acid Mixtures. Mol. Pharm. 2015, 12, 2484–2492. [Google Scholar] [CrossRef] [PubMed]
- Marsac, P.J.; Li, T.; Taylor, L.S. Estimation of drug-polymer miscibility and solubility in amorphous solid dispersions using experimentally determined interaction parameters. Pharm. Res. 2009, 26, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.T.; Larsen, F.H.; Löbmann, K.; Rades, T.; Grohganz, H. Influence of variation in molar ratio on co-amorphous drug-amino acid systems. Eur. J. Pharm. Biopharm. 2016, 107, 32–39. [Google Scholar] [CrossRef] [PubMed]
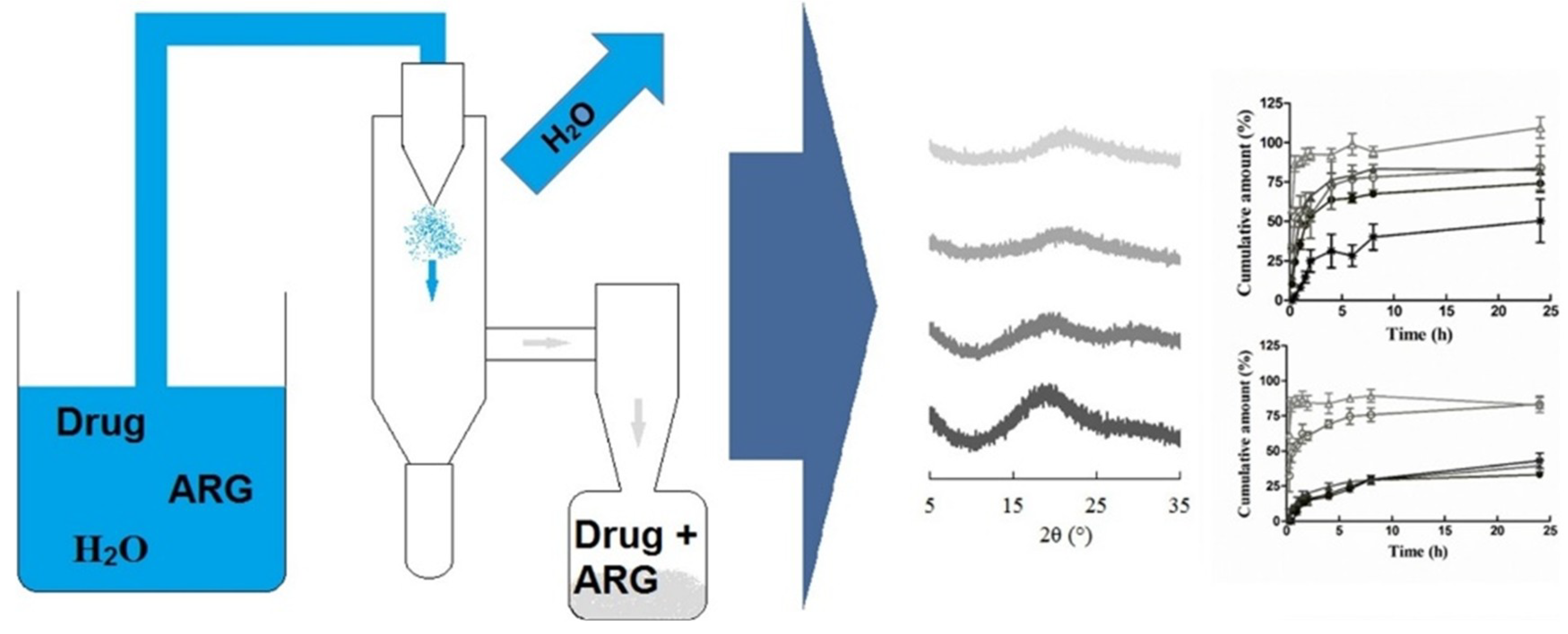
- Ojarinta, R.A.; Lerminiaux, L.; Laitinen, R. Spray drying of poorly soluble drugs from aqueous arginine solution. Int. J. Pharm. 2017, 532, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Beyer, A.; Grohganz, H.; Löbmann, K.; Rades, T.; Leopold, C.S. Influence of the cooling rate and the blend ratio on the physical stability of co-amorphous naproxen/indomethacin. Eur. J. Pharm. Biopharm. 2016, 109, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Beyer, A.; Grohganz, H.; Löbmann, K.; Rades, T.; Leopold, C.S. Multivariate Quantification of the Solid State Phase Composition of Co-Amorphous Naproxen-Indomethacin. Molecules 2015, 20, 19571–19587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Royall, P.G.; Craig, D.Q.M.; Doherty, C. Characterisation of the glass transition of an amorphous drug using modulated DSC. Pharm. Res. 1998, 15, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Paudel, A.; Meeus, J.; Van den Mooter, G. Structural Characterization of Amorphous Solid Dispersions, Amorphous Solid Dispersions; Springer: New York, NY, USA, 2014; pp. 421–485. [Google Scholar]
- Nair, R.; Nyamweya, N.; Gönen, S.; Martinez-Miranda, L.J.; Hoag, S.W. Influence of various drugs on the glass transition temperature of poly(vinylpyrrolidone): A thermodynamic and spectroscopic investigation. Int. J. Pharm. 2001, 225, 83–96. [Google Scholar] [CrossRef]
- Martínez, L.M.; Videa, M.; Sosa, N.G.; Ramírez, J.H.; Castro, S. Long-Term Stability of New Co-Amorphous Drug Binary Systems: Study of Glass Transitions as a Function of Composition and Shelf Time. Molecules 2016, 21, 1712. [Google Scholar] [CrossRef] [PubMed]
- Taylor, L.S.; Zografi, G. Spectroscopic characterization of interactions between PVP and indomethacin in amorphous molecular dispersions. Pharm. Res. 1997, 14, 1691–1698. [Google Scholar] [CrossRef] [PubMed]
- Chieng, N.; Rades, T.; Aaltonen, J. An overview of recent studies on the analysis of pharmaceutical polymorphs. J. Pharm. Biomed. Anal. 2011, 55, 618–644. [Google Scholar] [CrossRef] [PubMed]
- Heinz, A.; Strachan, C.J.; Gordon, K.C.; Rades, T. Analysis of solid-state transformations of pharmaceutical compounds using vibrational spectroscopy. J. Pharm. Pharm. 2009, 61, 971–988. [Google Scholar] [CrossRef] [Green Version]
- Löbmann, K.; Laitinen, R.; Grohganz, H.; Strachan, C.; Rades, T.; Gordon, K.C. A theoretical and spectroscopic study of co-amorphous naproxen and indomethacin. Int. J. Pharm. 2013, 453, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Dengale, S.J.; Ranjan, O.P.; Hussen, S.S.; Krishna, B.S.; Musmade, P.B.; Shenoy, G.G.; Bhat, K. Preparation and characterization of co-amorphous Ritonavir-Indomethacin systems by solvent evaporation technique: Improved dissolution behavior and physical stability without evidence of intermolecular interactions. Eur. J. Pharm. Sci. 2014, 62, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Bounartzi, M.; Panagopoulou, A.; Kantiranis, N.; Malamataris, S.; Nikolakakis, I. Effect of plasticiser type on the hot melt extrusion of venlafaxine hydrochloride. J. Pharm. Pharmacol. 2013, 66, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Baird, J.A.; Taylor, L.S. Evaluation of amorphous solid dispersion properties using thermal analysis techniques. Adv. Drug Deliv. Rev. 2012, 64, 396–421. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Gniado, K.; Erxleben, A.; McArdle, P. Mechanochemical Reaction of Sulfathiazole with Carboxylic Acids: Formation of a Cocrystal, a Salt, and Coamorphous Solids. Cryst. Growth Des. 2014, 14, 803–813. [Google Scholar] [CrossRef]
- Wairkar, S.; Gaud, R. Co-Amorphous Combination of Nateglinide-Metformin Hydrochloride for Dissolution Enhancement. AAPS PharmSciTech 2016, 17, 673–681. [Google Scholar] [CrossRef] [PubMed]
- Maher, E.M.; Ali, A.M.; Salem, H.F.; Abdelrahman, A.A. In vitro/in vivo evaluation of an optimized fast dissolving oral film containing olanzapine co-amorphous dispersion with selected carboxylic acids. Drug Deliv. 2016, 23, 3088–3100. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, P.D. Recent Advances in Nonlinear Optical Analyses of Pharmaceutical Materials in the Solid State. Mol. Pharm. 2017, 14, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Feng, X.; Williams, R.O., III; Zhang, F. Characterization of amorphous solid dispersions. J. Pharm. Investig. 2017, 48, 1–23. [Google Scholar] [CrossRef]
- Laitinen, R.; Löbmann, K.; Grohganz, H.; Strachan, C.; Rades, T. Amino acids as co-amorphous excipients for simvastatin and glibenclamide: Physical properties and stability. Mol. Pharm. 2014, 11, 2381–2389. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Heng, W.; Wei, Y.; Zhang, J.; Gao, Y. Co-amorphous lurasidone hydrochloride– saccharin with charge-assisted hydrogen bonding interaction shows improved physical stability and enhanced dissolution with pH-independent solubility behavior. Cryst. Growth Des. 2015, 15, 2920–2928. [Google Scholar] [CrossRef]
- Lim, A.W.; Löbmann, K.; Grohganz, H.; Rades, T.; Chieng, N. Investigation of physical properties and stability of indomethacin-cimetidine and naproxen-cimetidine co-amorphous systems prepared by quench cooling, coprecipitation and ball milling. J. Pharm. Pharmacol. 2016, 68, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Craye, G.; Löbmann, K.; Grohganz, H.; Rades, T.; Laitinen, R. Characterization of Amorphous and Co-Amorphous Simvastatin Formulations Prepared by Spray Drying. Molecules 2015, 20, 21532–21548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyer, A.; Grohganz, H.; Löbmann, K.; Rades, T.; Leopold, C.S. Improvement of the physicochemical properties of Co-amorphous naproxen-indomethacin by naproxen-sodium. Int. J. Pharm. 2017, 526, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhang, Q.; Wang, J.R.; Lin, K.L.; Mei, X. Amino acids as co-amorphous excipients for tackling the poor aqueous solubility of valsartan. Pharm. Dev. Technol. 2017, 22, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Pang, W.; Lv, J.; Wang, J. Solid state characterization of azelnidipine–oxalic acid co-crystal and co-amorphous complexes: The effect of different azelnidipine polymorphs. J. Pharm. Biomed. Anal. 2017, 138, 302–315. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, T.; Marques, S.; das Neves, J.; Sarmento, B. Amorphous solid dispersions: Rational selection of a manufacturing process. Adv. Drug Deliv. Rev. 2016, 100, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Hancock, B.C.; Zografi, G. Characteristics and significance of the amorphous state in pharmaceutical systems. J. Pharm. Sci. 1997, 86, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Alonzo, D.E.; Zhang, G.G.Z.; Zhou, D.; Gao, Y.; Taylor, L.S. Understanding the behavior of amorphous pharmaceutical systems during dissolution. Pharm. Res. 2010, 27, 608–618. [Google Scholar] [CrossRef] [PubMed]
- Descamps, M.J.; Willart, F.; Dudognon, E.; Caron, V. Transformation of pharmaceutical compounds upon milling and comilling: The role of T(g). J. Pharm. Sci. 2007, 96, 1398–1407. [Google Scholar] [CrossRef] [PubMed]
- Willart, J.F.; Descamps, M. Solid state amorphization of pharmaceuticals. Mol. Pharm. 2008, 5, 905–920. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Park, H.J.; Cho, W.; Cha, K.-H.; Kang, Y.-S.; Hwang, S.-J. Preparation and pharmaceutical characterization of amorphous cefdinir using spray-drying and SAS-process. Int. J. Pharm. 2010, 396, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Baghel, S.; Cathcart, H.; O’Reilly, N.J. Polymeric amorphous solid dispersions: A review of amorphization, crystallization, stabilization, solid-State characterization, and aqueous solubilization of biopharmaceutical classification system class II drugs. J. Pharm. Sci. 2016, 105, 2527–2544. [Google Scholar] [CrossRef] [PubMed]
- Beyer, A.; Radi, L.; Grohganz, H.; Löbmann, K.; Rades, T.; Leopold, C.S. Preparation and recrystallization behavior of spray-dried co-amorphous naproxen-indomethacin. Eur. J. Pharm. Biopharm. 2016, 104, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Nagir, S.; Yamamoto, H.; Kawashima, Y. Solid dispersion particles of tolbutamide with fine silica particles by the spray-drying method. Powder Technol. 2004, 195, 141–187. [Google Scholar] [CrossRef]
- Karmwar, P.; Graeser, K.; Gordon, K.C.; Strachan, C.J.; Rades, T. Investigation of properties and recrystallisation behaviour of amorphous indomethacin samples prepared by different methods. Int. J. Pharm. 2011, 417, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Hitzer, P.; Bäuerle, T.; Drieschner, T.; Ostertag, E.; Paulsen, K.; van Lishaut, H.; Lorenz, G.; Rebner, K. Process analytical techniques for hot-melt extrusion and their application to amorphous solid dispersions. Anal. Bioanal. Chem. 2017, 409, 4321–4333. [Google Scholar] [CrossRef] [PubMed]
- Arnfast, L.; Kamruzzaman, M.; Löbmann, K.; Aho, J.; Baldursdottir, S.; Rades, T.; Rantanen, J. Melt Extrusion of High-Dose Co-Amorphous Drug-Drug Combinations. Pharm. Res. 2017, 34, 2689–2697. [Google Scholar] [CrossRef] [PubMed]
- Lenz, E.; Löbmann, K.; Rades, T.; Knop, K.; Kleinebudde, P. Hot Melt Extrusion and Spray Drying of Co-amorphous Indomethacin-Arginine with Polymers. J. Pharm. Sci. 2017, 106, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Wickstrom, H.; Palo, M.; Rijckaert, K.; Kolakovic, R.; Nyman, J.O.; Määttänen, A.; Ihalainen, P.; Peltonen, J.; Genina, N.; de Beer, T.; et al. Improvement of dissolution rate of indomethacin by inkjet printing. Eur. J. Pharm. Sci. 2015, 75, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Elshaer, A. Amino Acids in Oral Drug Delivery: Salts, Ion-Pairs and Transcriptomics. Ph.D. Thesis, Aston University, Birmingham, UK, 2012. [Google Scholar]
- Masuda, T.; Yoshihashi, Y.; Yonemochi, E.; Fujii, K.; Uekusa, H.; Terada, K. Cocrystallization and amorphization induced by drug-excipient interaction improves the physical properties of acyclovir. Int. J. Pharm. 2012, 422, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.M.; Ali, A.A.; Maghrabi, I.A. Clozapine-carboxylic acid plasticized co-amorphous dispersions: Preparation, characterization and solution stability evaluation. Acta Pharm. 2015, 65, 133–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Löbmann, K.; Rades, T.; Grohganz, H. On the role of salt formation and structural similarity of co-formers in co-amorphous drug delivery systems. Int. J. Pharm. 2017, 535, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Renuka Singh, S.K.; Gulati, M.; Narang, R. Stable amorphous binary systems of glipizide and atorvastatin powders with enhanced dissolution profiles: Formulation and characterization. Pharm. Dev. Technol. 2017, 22, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Lipiäinen, T.; Peltoniemi, M.; Räikkönen, H.; Juppo, A. Spray-dried amorphous isomalt and melibiose, two potential protein-stabilizing excipients. Int. J. Pharm. 2016, 510, 311–332. [Google Scholar] [CrossRef] [PubMed]
- Petry, I.; Löbmann, K.; Grohganz, H.; Rades, T.; Leopold, C.S. Solid state properties and drug release behavior of co-amorphous indomethacin-arginine tablets coated with Kollicoat® Protect. Eur. J. Pharm. Biopharm. 2017, 119, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Wyttenbach, N.; Kuentz, M. Glass-forming ability of compounds in marketed amorphous drug Products. Eur. J. Pharm. Biopharm. 2017, 112, 204–208. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karagianni, A.; Kachrimanis, K.; Nikolakakis, I. Co-Amorphous Solid Dispersions for Solubility and Absorption Improvement of Drugs: Composition, Preparation, Characterization and Formulations for Oral Delivery. Pharmaceutics 2018, 10, 98. https://doi.org/10.3390/pharmaceutics10030098
Karagianni A, Kachrimanis K, Nikolakakis I. Co-Amorphous Solid Dispersions for Solubility and Absorption Improvement of Drugs: Composition, Preparation, Characterization and Formulations for Oral Delivery. Pharmaceutics. 2018; 10(3):98. https://doi.org/10.3390/pharmaceutics10030098
Chicago/Turabian StyleKaragianni, Anna, Kyriakos Kachrimanis, and Ioannis Nikolakakis. 2018. "Co-Amorphous Solid Dispersions for Solubility and Absorption Improvement of Drugs: Composition, Preparation, Characterization and Formulations for Oral Delivery" Pharmaceutics 10, no. 3: 98. https://doi.org/10.3390/pharmaceutics10030098