Alterations in Expression and Function of ABC Family Transporters at Blood-Brain Barrier under Liver Failure and Their Clinical Significances

Center of Drug Metabolism and Pharmacokinetics, China Pharmaceutical University, Nanjing 210009, China
*
Author to whom correspondence should be addressed.
Pharmaceutics 2018, 10(3), 102; https://doi.org/10.3390/pharmaceutics10030102
Submission received: 27 June 2018
/
Revised: 20 July 2018
/
Accepted: 21 July 2018
/
Published: 23 July 2018
(This article belongs to the Special Issue ABC Transporter-Mediated Drug Disposition)
Abstract
:Liver failure is often associated with hepatic encephalopathy, due to dyshomeostasis of the central nervous system (CNS). Under physiological conditions, the CNS homeostasis is precisely regulated by the blood-brain barrier (BBB). The BBB consists of brain microvessel endothelial cells connected with a junctional complex by the adherens junctions and tight junctions. Its main function is to maintain brain homoeostasis via limiting the entry of drugs/toxins to brain. The brain microvessel endothelial cells are characterized by minimal pinocytotic activity, absent fenestrations, and highly expressions of ATP-binding cassette (ABC) family transporters (such as P-glycoprotein, breast cancer resistance protein and multidrug resistance-associated proteins). These ABC transporters prevent brain from toxin accumulation by pumping toxins out of brain. Accumulating evidences demonstrates that liver failure diseases altered the expression and function of ABC transporters at The BBB, indicating that the alterations subsequently affect drugs’ brain distribution and CNS activity/neurotoxicity. ABC transporters also mediate the transport of endogenous substrates across the BBB, inferring that ABC transporters are also implicated in some physiological processes and the development of hepatic encephalopathy. This paper focuses on the alteration in the BBB permeability, the expression and function of ABC transporters at the BBB under liver failure status and their clinical significances.
1. Introduction
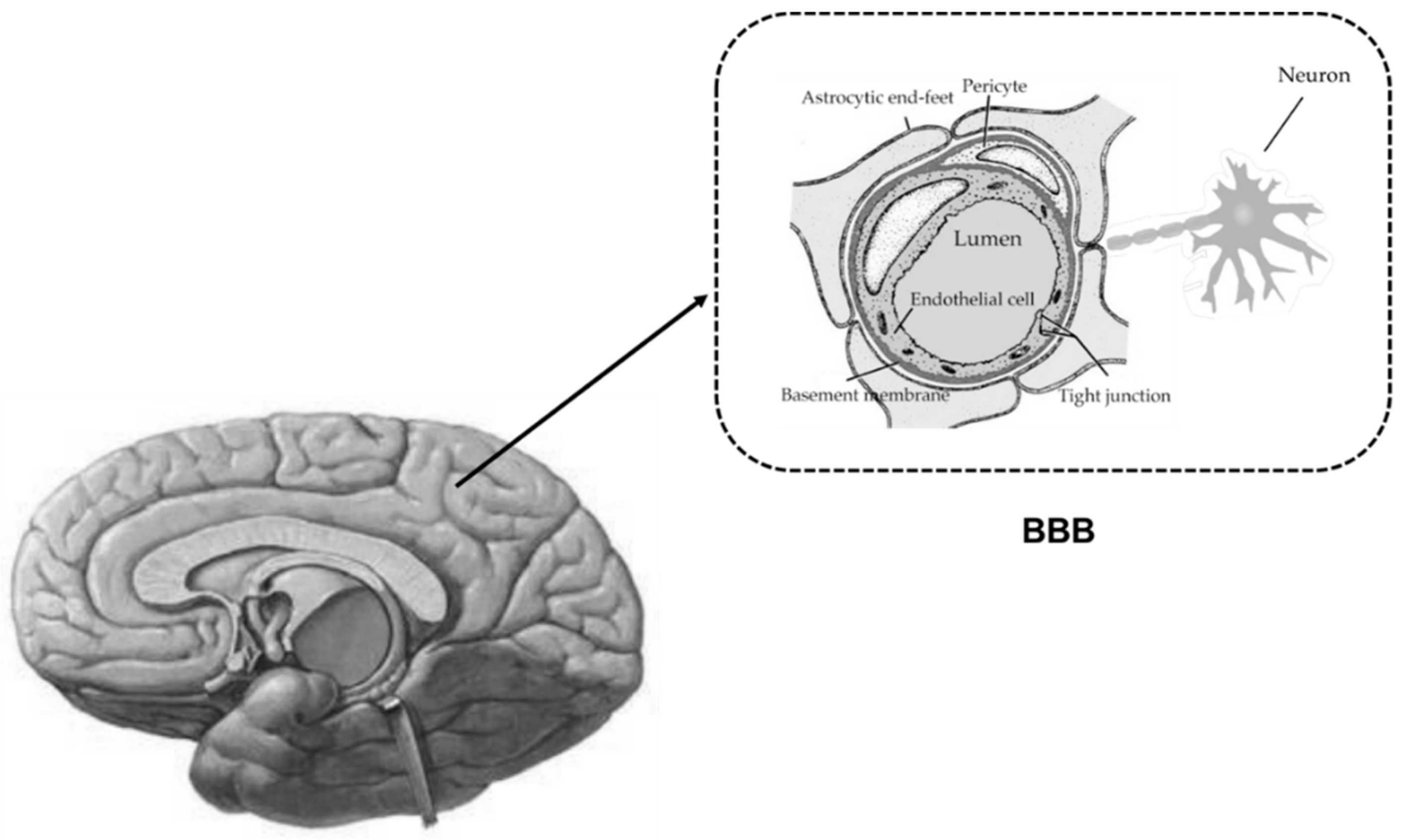
Liver failure is often associated with hepatic encephalopathy, due to accumulation of neurotoxins in brain. Under physiological conditions, the homeostasis of central nervous system (CNS) is precisely regulated by the blood–brain barrier (BBB). Serving as an interface for communication between CNS and blood circulation, the BBB is crucial in limiting free diffusion between brain fluids and blood. This provides transport processes for essential nutrients, ions, signaling molecules and metabolic waste products. The BBB consists of brain microvessel endothelial cells connected with a junctional complex by the adherens junctions and tight junctions. In addition, multiple transporters, receptors and enzymes are highly expressed at the BBB. Thus, the BBB is not only an anatomical barrier, but also a dynamic tissue. The primary anatomic units of the BBB are brain capillaries, where the endothelial cells of brain capillaries and the closely apposed pericytes are completely unsheathed by overlapping astrocytic end-feet, microglia and neuronal terminals, termed as a “neurovascular unit” (Figure 1) [1].
Compared to peripheral vasculature, endothelial cells of brain capillaries are characterized by minimal pinocytotic activity, absent fenestrations and high expression of tight junction proteins at cell-cell contacts [1,2]. Tight junctions consist of multiple proteins, and include junctional adhesion molecules(JAMs), occludin, claudins (such as claudin-1, claudin-3, and claudin-5), and zonulla occluden proteins (such as ZO-1, ZO-2 and ZO-3) [2]. They form the primary physical barrier component of the BBB, and their function is to restrict paracellular entry of various endogenous and exogenous substances—leading to higher transendothelial electrical resistance (1500~2000 Ωcm2) across the BBB compared with that (i.e., 3–33 Ωcm2) in other vascular tissues [1,2].
About 99% of cerebral capillaries are covered by astrocyte end-feets. The critical cell-cell interactions directly modulate the BBB’s characteristics by secreting some factors, such as transforming growth factor-β (TGF-β), glial cell line-derived neurotrophic factor (GDNF), and basic fibroblast growth factor (BFGF). Pericytes are attached to the abluminal side of brain capillary endothelial cells and to the luminal side of the astrocyte end feet, which contribute not only to vascular contractility and immune responses but also to BBB functional integrity. It was reported that the percentage of cerebral capillaries covered by pericytes positively correlated with “tightness” of the junctions between endothelial cells and inversely correlated with BBB permeability [2,3]. Pericytes are considered to be essential for proper localization of endogenous BBB proteins and induction of BBB properties [4,5]. In addition, the extracellular matrix regulates the expression of tight junction proteins and BBB integrity. Disruption of the extracellular matrix was reported to be associated with the loss of the BBB function and the increases in BBB permeability [2].
Both brain microvessel endothelial cells and associated astrocyte processes are distinctly connected with noradrenergic, serotonergic, cholinergic and GABAergic neurons, demonstrating that BBB function is probably regulated by these neurons. Loss of direct noradrenergic input from the locus coeruleus was reported to increase BBB susceptibility to acute hypertension, with a significant increase in BBB permeability to [125I]-albumin [6]. Furthermore, stimulation of the postganglionic parasympathetic fibers of the sphenopalatine ganglion also increased vascular leakage as well as the levels of anti-HER2 monoclonal antibody and etoposide in brain [7].
In general, hepatic encephalopathy is considered to be the main cause of accumulation of brain neurotoxins due to BBB dysfunctions (i.e., liver-brain axis) [8,9,10]. A report showed that efflux of phenylalanine from brain to blood significantly decreased in patients with hepatic encephalopathy, leading to a significant increase in brain interstitial fluid phenylalanine concentration [9]. The extent of alterations was positively correlated to coma grade [9]. Quinolinic acid is also an endogenous neuroexcitant derived from tryptophan. It was found that quinolinic acid levels in brain of patients who died of acute liver failure were significantly elevated [10]. Rat experiment showed that elevated levels of brain quinolinic acid were strongly correlated with fulminant hepatic failure [11]. These results indicate that quinolinic acid is involved in pathogenesis of hepatic encephalopathy.
2. ABC Drug Transporters Expressed at BBB and Their Functions
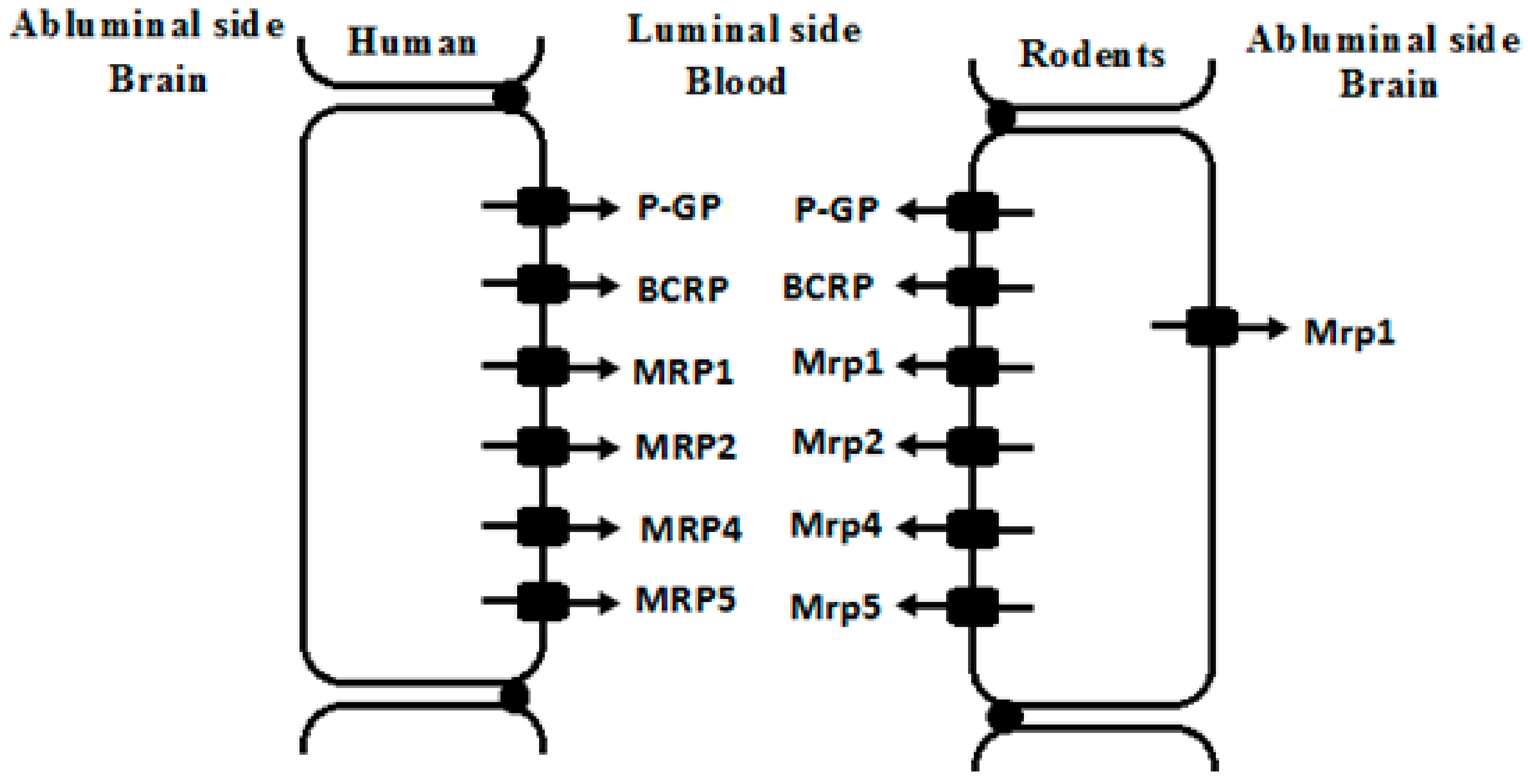
Endothelial cells of brain capillaries highly express ATP-binding cassette (ABC) transporters and solute carriers (SLC), constructing BBB components. The SLC transporters expressed at the BBB include organic anion transporting polypeptides (OATPs), organic anion transporters (OATs) and organic cation transporters (OCTs). These SLC transporters are usually located in the luminal or abluminal membranes of the endothelial cells and facilitate drug transport across BBB. Here, we focus on the alterations in the expression and function of brain ABC transporters under liver failure conditions. ABC transporters expressed at brain include P-glycoprotein (P-GP/ABCB1), breast cancer resistance protein (BCRP/ABCG2), and multidrug associated resistance proteins (MRPs/ABCCs). These ABC transporters are mainly located in the luminal membranes of the endothelial cells (Figure 2) and efflux drugs out of CNS [2,3].
P-GP, coded by ABCB1 gene, is mainly located in the luminal membranes of brain microvessel endothelial cells, playing the function of protecting CNS from exposure to potential neurotoxins and maintaining the homeostasis [12,13]. The importance of P-GP in CNS protection has been highlighted in Abcb1-/- mice [14,15,16,17]. It was found that Abcb1b-/- mice showed a 100-fold increase in brain uptake of ivermectin, a neurotoxic pesticide, leading to a significant increase in ivermectin toxicity compared with wild-type mice [17]. Abcb1-/- mice showed a nine-fold higher brain concentration of asimadoline, and an eight-fold more sensitivity to the sedative effect of asimadoline compared with wild-type mice [15]. The relationship between increases in brain penetration and Abcb1 deficiency was also demonstrated in other drugs such as amiodarone, loperamide, quinidine, verapamil and digoxin [14,16].
P-GP possesses a broad spectrum of substrates, such as: Antibiotics, calcium channel blockers, cardiac glycosides, chemotherapeutics, immunosuppressant, anti-epileptics, HIV-1 protease inhibitors, and some toxins. In addition, P-GP is also involved in the transport of some drug-delivery vectors (i.e., carbon allotropes) [18]. Thus P-GP expressed at the BBB becomes a formidable obstacle to CNS drug delivery?limiting the ability of effective treat for CNS disorders. P-GP also transports some endogenous substrates, including cytokines, lipids, steroid hormones, and peptides [12,13], which shows its physiological roles. Some diseases alter the expression of P-GP at BBB. Clinical trials and animal experiments have demonstrated that Alzheimer’s disease (AD) significantly downregulated the expression and function of P-GP at brain [19,20,21,22]. Some peripheral diseases, such as diabetes [23,24,25] and liver failure [26,27,28], also downregulated brain P-GP expression and function—enhancing pharmacological effect on CNS. Conversely, overexpression of P-GP also occurred in capillary endothelial cells of epilepsia patients [29] or epilepsia animals [30]. Some antiepileptic drugs are P-GP substrates, indicating that P-GP overexpression at the BBB contributes to the pharmacoresistance observed in epilepsia patients [30,31].
BCRP, another important efflux transporter coded by ABCG2 gene, is predominantly expressed at the luminal membrane of microvessel endothelial cells, and protects brain from the toxicity of xenobiotics. Tyrosine kinase inhibitors (such as gefitinib, imatinib, nilotinib, erlotinib, lapatinib, and sunitinib), methotrexate, other drugs (such as prazosin, elacridar, and mitoxantrone), or some endogenous compounds (such as dehydroepiandrosterone sulfate and urate) are BCRP substrates. The BCRP expression at the BBB also impedes these drugs to reach their intracerebral targets for treating CNS disorder, such as brain tumors.
It is noteworthy that in vitro BCRP functional analysis using cell lines overexpressing BCRP may not often accurately reflect in vivo BCRP function. For example, in vitro data demonstrated that some compounds (cimetidine, alfuzosin, dipyridamole, LY2228820, dehydroepiandrosterone sulfate, and mitoxantrone) were substrates of BCRP, but Abcg2 deficiency slightly affected brain uptakes of these compounds in mice [32,33]. At the BBB, BCRP is co-located with P-GP, and also share substrates with P-GP: Indicating that the two transporters work together to limit xenobiotics entering into the brain. In vitro study demonstrated that BCRP contributed more efficient transport of vemurafenib than P-GP, but Abcg2 deficiency did not increase brain distribution of vemurafenib. However, Abcb1 deficiency still significantly increased brain distribution of vemurafenib (2.9-fold). Importantly, both Abcb1 and Abcg2 knockout resulted in remarkable 80-fold increase in brain distribution of vemurafenib [34], demonstrating the “synergistic” role of the two transporters at the BBB. The “synergistic” effect was also found in other drugs such as dasatinib [35], elacridar [36],erlotinib [37], gefitinib [38], rucaparib [39], sorafenib [40], and topotecan [41]. Cooperation of P-GP with BCRP implies that the absence of either P-GP or BCRP alone does not appreciably increase brain penetration of these dual substrates, and the greatest enhancement in brain penetration of dual substrates is always seen when both P-GP and BCRP are absent. Accordantly, dual P-GP/BCRP inhibitor elacridar showed the strongest enhancement of brain distribution of gefitinib although P-GP inhibitor (LY335979) or BCRP inhibitor (Ko143) alone mildly altered brain distribution of gefitinib [38].
MRPs, coded by ABCC gene, also extrude xenobiotics from brain. Substrates of MRPs are generally organic anions and their glucuronidated, sulfated, and glutathione-conjugated metabolites. However, the expression and distribution of MRPs at the BBB often show controversial reports. For example, MRP1, MRP2, MRP3, MRP4 and MRP5 mRNA were detected in brain samples of human, among which expression of MRP5 mRNA showed the highest expression. However, MRP1, MRP2, MRP3, and MRP4 mRNA showed considerable variation. Immunofluorescence analysis showed that MRP1, MRP4 and MRP5 were primarily localized in the luminal membrane of capillary endothelial cells, but no reactivity for the MRP2 or MRP3 proteins [42]. Human brain microvascular endothelial cells exhibited the strongest expression of MRP1 mRNA, followed by MRP4, MRP5 and MRP6, but MRP2 and MRP3 showed very weak expression [43].
Expression of MRPs also shows large species variations. A report on five species (human, rat, mouse, pig and cow) showed that mRNA levels of MRP1/Mrp1 were detected to be high in brain microvessel endothelial cells of human, rat and cow, low in mouse, and negligible in pig. MRP3/Mrp3 expressions in mouse and pig were higher than other species. The highest expression of MRP4/Mrp4 was found in rodents, followed by pig and human. MRP5/Mrp5 was highly expressed in brain endothelial cells of human, rat, pig, and cow, with lower expression in mice. No significant expression of MRP2/Mrp2 was detected in the brain of indicated species (Table 1) [44]. Mrp2 expression in brain of mice is dependent on strain. For example, FVB mice lacked Mrp2 in the brain vascular and choroid plexus endothelium although Mrp2 was still present in liver and kidney of the strain, indicating that FVB mice represented a spontaneous, brain-specific “knockout” of Mrp2 [45].
Cystic fibrosis transmembrane conductance regulator (CFTR), belonging to ABCC family (ABCC7), is also exclusively expressed in neurons of adult and neonatal human brains [46,47] as well as rat brains [48]. It was found that CFTR expression in developing cerebral cortex was increased progressively during development although faint expression was observed at early stage. Moreover, cystic fibrosis patient (p.F508del mutation) showed also very weak expression of brain CFTR, which was consistent with a slight delay in maturation, especially at the level of the neuroepithelium [46]. These results indicate that CFTR is involved in neuronal maturation and differentiation.
3. Alterations in BBB Permeability under Liver Failure
Liver failure, especially acute liver failure (ALF), is often accompanied by hepatic encephalopathy, brain edema and hepatic coma. Brain edema and hepatic coma are lethal. The incidence of brain edema in patients with hepatic failure was reported to reach 51.4% [49]. The mechanisms of brain edema are generally categorized as cytotoxic and vasogenic. Accumulation of water and plasma constituents in the extracellular region was attributed to alteration of structural BBB [49], although ultrastructural alterations in the BBB were slightly changed. It was found that in cerebral cortex obtained immediately after death from nine patients with acute liver failure, the intercellular tight junctions were intact except for being slightly widened in two patients. However, the endothelial cells and perivascular astroglial foot were swollen, accompanied by increased vesicles and vacuoles [50]. ALF rabbits also demonstrated the swelling of perivascular astroglial foot and the intact tight junctions [51]. In ALF mice induced by d-galactosamine and liposaccharide, it was found that increased BBB permeability by ALF was partly attributed to disruption of tight junctions and loss of the tight junction-associated protein occluding, which might be involved in both vasogenic and cytotoxic mechanisms [52]. Our previous study also demonstrated intercellular tight junctions were intact in thioacetamide-induced ALF rats, without leakage of Evans blue into brain and alteration of the expression of tight junction proteins (claudin-5 and occluding) [26,53]. These results indicate that a vasogenic brain edema results from subtle modifications of tight junctional proteins without an obvious disruption of the BBB. However, chronic liver failure obviously disrupted BBB, leading to a significant increase of Evans blue extravasation into brain [27].
Liver failure is often associated with hyperammonemia, which is attributed to hepatic encephalopathy and brain edema. Several reports showed that arterial ammonia level directly correlated to the risk of impending brain herniation [54,55]. Ammmonia was reported to increase paracellular permeability of BBB partly via encompassing oxidative/nitrosative stress and activating matrix metalloproteinases [56]. Clinical trials [57] and animal experiments [53,58] have showed that liver failure also significantly increased some inflammatory molecules including cytokines interleukin-1 (IL-1), interleukin-6 (IL-6), and tumor necrosis factor-alpha (TNF-α) in plasma. Levels of TNF-α in plasma were reported to positively correlate with the severity of hepatic encephalopathy [59] or with the intracranial pressure [60]. Selective deletion of cellular receptors of IL-1 and TNF-α attenuated the development of encephalopathy and brain edema in experimental ALF [61]. Contrastingly, an injection of TNF-α into CNS increased BBB permeability in a dose-dependent manner [62]. Furthermore, the anti-TNF-α antibody could inhibit the increase of BBB permeability and block the entering of bacterium to CNS [62,63]. Similarly, TNF-α antibody or TNF-α-R1 antibody increased the expression of tight junction protein (occludin and ZO-1), and decreased the concentration of Evans Blue in brains of ALF mice induced by D-galactosamine and lipopolysaccharide [62,63]. In acetaminophen-induced ALF mice, it was found that increases in BBB permeability positively correlated with elevated serum TNF-α levels, which could be prevented by administering anti-TNFα-IgG [63].
Matrix Metalloproteinases (MMPs), especially, MMP-9, play an important role in numerous physiologic and pathologic processes, via digesting capillary endothelial constituents and tight junction proteins of the BBB. Brain edema and tight junction protein degradation in brain of azoxymethane-induced ALF mice were associated with increase in circulating MMP-9 levels. Blocking MMP-9 with either GM6001 or MMP-9 monoclonal antibody significantly attenuated brain extravasation, astrocytic end foot swelling, and brain edema [64]. Moreover, liver failure also increased circulating transforming growth factor β1 (TGFβ1), further promoting endothelial cell permeability. In azoxymethane-treated mice, it was reported that an administration of neutralizing antibodies against TGFβ significantly reduced BBB permeability. In vitro data also demonstrated that treatment of bEnd.3 cells with recombinant transforming growth factor β1(rTGFβ1) dose-dependently increased MMP9 expression and suppressed claudin-5 expression [65]. Furthermore, MMP-9 downregulated occludin expression via inducing transactivation of epidermal growth factor receptor (EGFR) and p38 mitogen-activated protein kinase/nuclear factor-kappa B (MAPK/NFκB) signals [66].
The increase in BBB permeability was also associated with vascular remodeling due to up-regulation of vascular endothelial growth factor (VEGF) and its receptors [67]. VEGF is known to play a main role in vascular permeability by activating MMPs. The activations of MMPs, especially MMP-9, increased BBB permeability via breaking vessel walls and degrading tight junctions [68,69,70]. Systemic and cerebral VEGF levels were significantly elevated under experimental ALF, commonly contributing to the chronic impairment of the BBB [71].
4. Alterations in Expression and Function of ABC Transporters at BBB by Liver Failure
Accumulating evidence [26,27,28,53,72,73,74,75] has demonstrated that liver failure altered the expression and function of ABC transporters at the BBB, and that the alterations were dependent on types of the developed liver failure and the species of ABC transporters. ALF rats were developed using intraperitoneal injection of thioacetamide (TAA) (300 mg/kg) for two days with a 24-h interval [26,53]. The functions of P-GP, BCRP and Mrp2 at rat brain were represented as brain-to-plasma ratios of their substrate (rhodamine 123 and vincristine for P-GP, prazosin and methotrexate for BCRP, dinitrophenyl-S-glutathione and sulfobromophthalein for MRP2) levels, respectively. Expressions of these transporter proteins were measured using Western blotting. The results showed that ALF significantly increased brain-to-plasma ratios of both P-GP and BCRP substrates. Concordantly, expressions of P-GP and BCRP protein in brain of ALF rats were significantly suppressed. Data from the leakage of Evans blue and expression of tight junctional protein (occludin and claudin-5) demonstrated that BBB integrity of ALF rats was intact. These results indicated that the increased distributions of the P-GP and BCRP substrates in brain of ALF rats were mainly attributed to the reduction of P-GP and BCRP function and expression [26,53]. Being different from P-GP and BCRP, the expression and function of Mrp2 at brain of ALF rats were significantly upregulated, leading to lower brain-to-plasma ratios of its substrates [26]. These findings were further confirmed in TAA-induced ALF mice [28]. Moreover, TAA-induced chronic liver failure also downregulated the expression and function of P-GP and upregulated the expression and function of Mrp2 at the BBB of rats, although BBB integrity of chronic liver failure rats was impaired [27]. However, CCl4-induced ALF was reported to suppress the function of P-GP in brain of rats without affecting the expression of P-GP [74]. Similarly, partial hepatectomy increased BBB permeability to cyclosporine A in mice due to suppression of P-GP function, although expressions of Mdr1a/1b mRNA in brain were not altered [75].
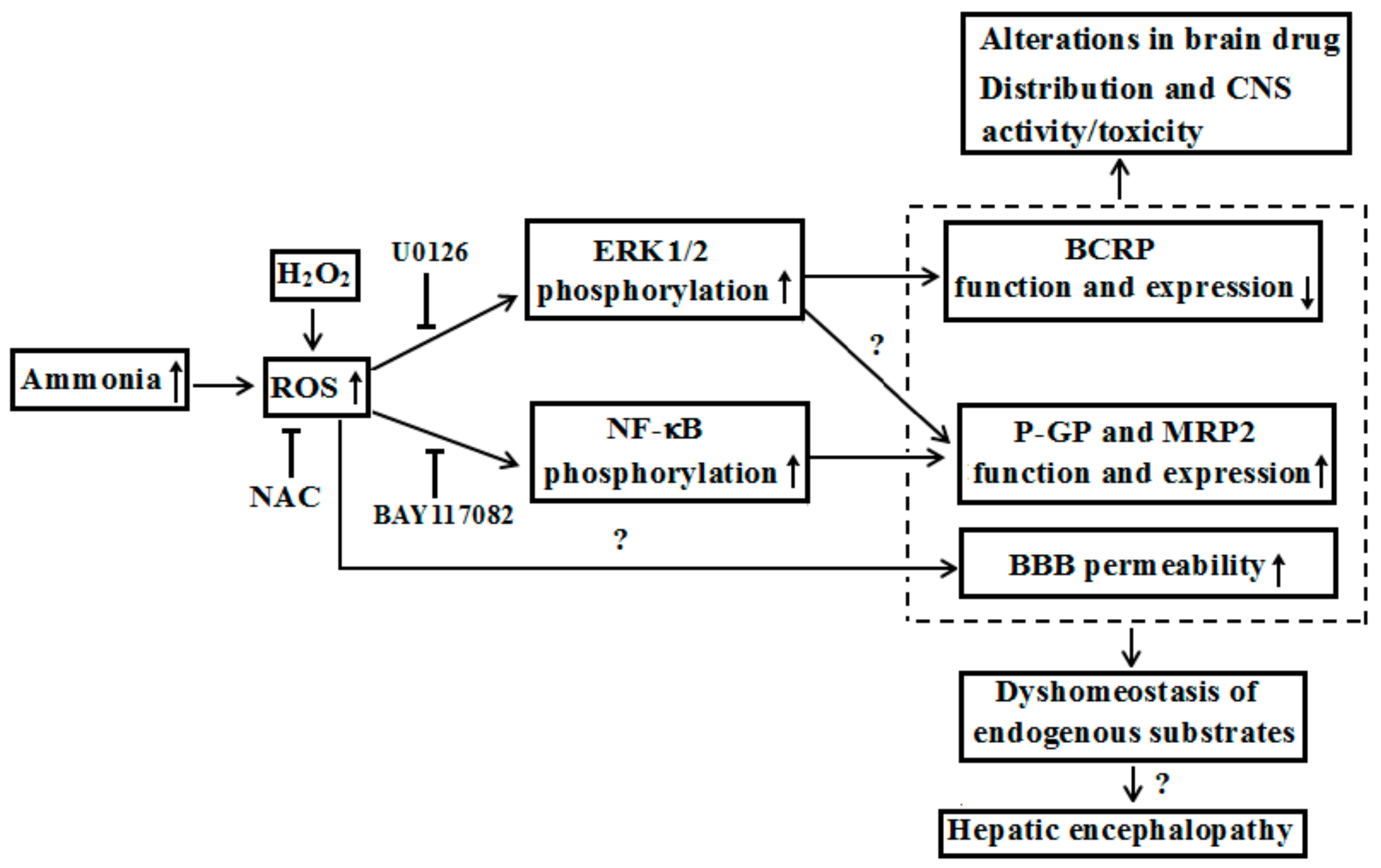
In general, liver failure is accompanied by increases in levels of plasma ammonia, bilirubin or bile acid, which seems to become predisposing factors for alterations in the expression and function of these ABC transporters in brain. Although liver failure is often characterized hyperammonemia, the acute hyperammonemia by ammonium acetate was reported to induce rather than decrease the expression and function of P-GP and Mrp2 in brain of rats, accompanied by activation of nuclear factor-κB (NF-κB) p65 in brain of rats [73]. In vitro data, from primarily cultured rat brain microvessel endothelial cells (rBMECs), demonstrated that the expression and function of P-GP and Mrp2 in rBMECs were significantly increased after incubation with NH4Cl (5 mM) for 24 h, with NF-κB pathway activation. The alterations induced by ammonia were reversed by NF-κB inhibitor BAY117082, indicating that hyperammonemia increased the expression and function of P-GP and Mrp2 at BBB via activating NF-κB pathway [73]. Chronic hyperammonemia induced by feeding with diet containing ammonium acetate plus partial portal vein ligation also significantly increased P-GP function, accompanied by increases in levels of extracellular-regulated protein kinase 1/2 (ERK1/2) phosphorylation, and reactive oxygen species (ROS) at brain of rats [unpublished]. Western blotting analysis demonstrated that chronic hyperammonemia plus partial portal vein ligation enhanced the translocation of P-GP from cytoplasm to plasma membrane. Moreover, 72-h incubation with 1 mM ammonia or H2O2 increased membrane expression and function of P-GP, ROS generation and phosphorylated ERK1/2 in HCMEC/D3 cells, which were reversed by ROS scavenger N-acetylcysteine and ERK1/2 inhibitor U0126, but not NF-κB inhibitor BAY117082[unpublised]. However, these results did not explain the downregulated expression and function of P-GP at the BBB under ALF, indicating the existence of other mechanisms. It was reported that the function of P-GP was suppressed in brain of CCl4-induced ALF rats without change of P-GP protein level [74]. In line, the plasma from the CCl4-induced ALF rats also suppressed the function of P-GP in Caco-2 cells [76]. Pre-incubation with plasma obtained from hepatectomized mice also enhanced the accumulation of rhodamine 123 in mouse brain endothelial cells (MBEC4), compared with sham-operated mice; even though the levels of mdr1a/mdr1b mRNA in cerebral capillaries from partial hepatectomized and sham-operated mice were similar [75]. We once reported that 10% serum from TAA-induced ALF rat slightly affected the uptakes of rhodamine 123 and sulfobromophthalein in rBMECs, and Caco-2 cells [26]. Concentrations of rat serum higher than 10% were not used because of their damage to cells. These results indicated the differences of expression and function of P-GP under various ALF models due to different mechanisms. Effects of several bile acids (cholic acid, deoxycholic acid, lithocholic acid, chenodeoxycholic acid, and ursodeoxycholic acid) and unconjugated bilirubin (UCB) on P-GP function and expression in HCMEC/D3 and MDCK-BCRP cells were investigated. The results showed that only chenodeoxycholic acid downregulated P-GP expression, and function in both HCMEC/D3 and MDCK-BCRP cells in a concentration-dependent manner [28]. These results indicated that increased chenodeoxycholic acid might be partly attributed to the downregulation of P-GP function and expression in brain of TAA-induced ALF rats and mice. Further investigations are needed to elucidate the contributions of other factors such as proinflammatory cytokines.
Unlike P-GP, both TAA-induced ALF and acute hyperammonemia induced by intraperitoneally injected ammonium acetate significantly downregulated the expression and function of BCRP, accompanied by marked increases of ERK1/2 phosphorylation and ROS at BBB of rats [53]. In MDCK-BCRP cells and rBMECs, it was found that NH4Cl (5 mM) and H2O2 strikingly downregulated the expression and function of BCRP. This is accompanied by remarkably increases in the levels of phosphorylated ERK1/2 and ROS. ROS scavenger N-acetylcysteine and ERK1/2 inhibitor U0126 restored the alterations by ammonia and H2O2. These results indicated that ALF down-regulated the expression and function of brain that BCRP is partly via ammonia-ROS-ERK1/2 pathway [53].
Hyperbilirubinemia is also a feature of ALF. In liver failure rats induced by bile duct ligation (BDL), it was found that BDL rats exhibited progressive decline in liver function and hyperbilirubinemia from day 3 to day 14, following BDL. In the brain tissues of BDL rats, both the function and protein levels of BCRP were progressively decreased along with time, but BBB integrity was intact. Furthermore, incubation with BDL rat serum significantly decreased BCRP function and protein levels in both HCMEC/D3 and MDCK-BCRP cells, indicating that some abnormal components in BDL rat serum might impair BCRP function at the BBB. BDL rats exhibited significant elevations of UCB and bile acids [72]. In vitro data showed that only incubation with 25 μmol/L UCB significantly increased the uptake of prazosin, and downregulated the expression of BCRP protein in HCMEC/D3 and MDCK-BCRP cells. Although bile acids (chenodeoxycholic acid, ursodeoxycholic acid and deoxycholic acid) concentration-dependently increased the uptake of prazosin in HCMEC/D3 cells, the expression of BCRP protein was unaltered. Moreover, the alterations in function of BCRP by bile acids in HCMEC/D3 cells were not confirmed in MDCK-BCRP cells. Hyperbilirubinemia rats were developed by intravenous injection of UCB. The results demonstrated that hyperbilirubinemia downregulated BCRP expression and function at BBB of rats. Correlation analysis revealed that the level of UCB negatively correlated with BCRP expression in the brains of BDL rats and hyperbilirubinemia rats. Two types of cells tested in vitro further demonstrated that UCB elevation in plasma of BDL rats contributed to the impaired function, and expression of BCRP at the BBB of BDL rats [72].
Transport of phenobarbital across the BBB is mediated by P-GP. TAA-induced ALF was reported to significantly increase brain concentration of phenobarbital, and brain-to-plasma concentration ratios of phenobarbital in mice—which was in consistence with the reduction of P-GP function and expression [28]. The ALF mice showed a significantly larger duration and shorter latency in the loss of righting reflex by phenobarbital, which was in line with increases in phenobarbital distribution of phenobarbital [28]. Similarly, cyclosporin A, a P-GP substrate, was reported to markedly aggravate harmine-induced tremors in sham-operated mice and partially hepatectomized mice at postoperative day 1 and 3, but partial hepatectomy showed stronger effect. In particular, at postoperative day 1, partial hepatectomy significantly augmented harmine-induced tremors by 53% compared with sham operation in cyclosporin A-treated mice. Further study showed that the increased effects of cyclosporine A were in line with the increased brain uptake of cyclosporine A and decreased P-GP function in brain [75], indicating the possibility that cyclosporine A increases the risk of neurotoxicity at an early phase of liver transplantation.
5. Future Perspectives
Based on current knowledge, we proposed that liver failure not only altered systemic pharmacokinetics of drugs, but also affected their brain distribution due to BBB alterations: Including changes of permeability as well as function and expression of ABC transporters. Those alterations may further affect activities/toxicities of those drugs towards CNS (Figure 3). It was reported that hepatitis A, B, and E viruses accounted for most ALF cases in the developing world [77]. Overall incidence of seizures in ALF patients was up to 33% [76]. Moreover, many drugs, such as antiepileptics (lamotrigine), antivirals (lamivudine, zidovudine and abacavir), antibiotic (levofloxacin), and immunomodulators (mycophenolate mofetil, sirolimus, tacrolimus and cyclosporine A) are substrates of BCRP or P-GP [78,79,80], some of which are also MRP2 substrates. These drugs may be administrated to liver failure patients [81,82,83,84] or liver transplant patients [85], indicating that the altered expression and function of ABC transporters at the BBB probably affect their pharmacological/toxic effects on CNS—although data cannot be directly extrapolated to human. It should be mentioned that the effects of liver failure on the expression and function of ABC transporters at BBB are often dependent on species of ABC transporters, brain regions, duration of liver failure and types of liver failure. Moreover, P-GP and BCRP are also highly expressed in intestine and liver and mediate excretion of their substrate drugs via intestinal wall and bile duct, demonstrating important roles in pharmacokinetics and pharmacology/toxicity of drugs [86,87]. Several reports demonstrated that liver failure downregulated the function and expression of intestinal P-GP [88], but increased the expression of hepatic BCRP and P-GP [89,90]. Thus, better understanding of alterations in ABC transporter functions and expressions under liver failure is of major pharmacological importance to the development and optimization of therapeutics.
ABC transporters also transport endogenous substrates. For instance, transport of steroid hormones (such as cortisol, corticosterone, aldosterone, and progesterone) is mediated by P-GP [91]. Some metabolites (dehydroepiandrosterone sulfate and dihydrotestosterone) of neurosteroids are BCRP substrates [92]. These neurosteroids are considered to contribute to the pathogenesis of hepatic encephalopathy by affecting γ-aminobutyric acid (GABA) ergic tone [93,94,95,96]. It was reported that GABA receptor positive allosteric modulator allopregnanolone, a metabolite of progesterone, was significantly increased in brain tissue from hepatic coma patients [95], On the contrast, GABA receptor negative allosteric modulator dehydroepiandrosterone sulfate was significantly decreased [96]. The reduction of dehydroepiandrosterone sulfate, together with concomitantly increased levels of allopregnanolone in brain would contribute to the phenomenon of “increased GABAergic tone” in hepatic encephalopathy. These findings indicate that the alterations in function and expression of brain ABC transporters under liver failure status will be implicated in the development of hepatic encephalopathy, which needs further investigation.
Author Contributions
Both authors shared in the searching for publication, preparation and editing of the manuscript. Both authors have approved the final manuscript.
Funding
This research was funded by National Science Foundation of China [No. 81473273, 81373482 and 81573490].
Conflicts of Interest
The authors declare no conflict of interest.
References
- Redzic, Z. Molecular biology of the blood-brain and the blood-cerebrospinal fluid barriers: Similarities and differences. Fluids Barriers CNS 2011, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Covarrubias, L.; Slosky, L.M.; Thompson, B.J.; Davis, T.P.; Ronaldson, P.T. Transporters at CNS barrier sites: Obstacles or opportunities for drug delivery? Curr. Pharm. Des. 2014, 20, 1422–1449. [Google Scholar] [CrossRef] [PubMed]
- Strazielle, N.; Ghersi-Egea, J.F. Physiology of blood-brain interfaces in relation to brain disposition of small compounds and macromolecules. Mol. Pharm. 2013, 10, 1473–1491. [Google Scholar] [CrossRef] [PubMed]
- Al Ahmad, A.; Taboada, C.B.; Gassmann, M.; Ogunshola, O.O. Astrocytes and pericytes differentially modulate blood-brain barrier characteristics during development and hypoxic insult. J. Cereb. Blood Flow Metab. 2011, 31, 693–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 2010, 468, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Ben-Menachem, E.; Johansson, B.B.; Svensson, T.H. Increased vulnerability of the blood-brain barrier to acute hypertension following depletion of brain noradrenaline. J. Neural Transm. 1982, 53, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Yarnitsky, D.; Gross, Y.; Lorian, A.; Shalev, A.; Lamensdorf, I.; Bornstein, R.; Shorer, S.; Mayevsky, A.; Patel, K.P.; Abbott, N.J.; et al. Blood-brain barrier opened by stimulation of the parasympathetic sphenopalatine ganglion: A new method for macromolecule delivery to the brain. J. Neurosurg. 2004, 101, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, R.F. The liver-brain axis in liver failure: Neuroinflammation and encephalopathy. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, G.M.; Schmidt, J.; Almdal, T.; Paulson, O.B.; Vilstrup, H. Passage of amino acids and glucose across the blood-brain barrier in patients with hepatic encephalopathy. Hepatology 1993, 17, 987–992. [Google Scholar] [CrossRef] [PubMed]
- Basile, A.S.; Saito, K.; al-Mardini, H.; Record, C.O.; Hughes, R.D.; Harrison, P.; Williams, R.; Li, Y.; Heyes, M.P. The relationship between plasma and brain quinolinic acid levels and the severity of hepatic encephalopathy. Gastroenterology 1995, 108, 818–823. [Google Scholar] [CrossRef]
- Basile, A.S.; Saito, K.; Li, Y.; Heyes, M.P. The relationship between plasma and brain quinolinic acid levels and the severity of hepatic encephalopathy in animal models of fulminant hepatic failure. J. Neurochem. 1995, 64, 2607–2614. [Google Scholar] [CrossRef] [PubMed]
- Abdullahi, W.; Davis, T.P.; Ronaldson, P.T. Functional expression of P-glycoprotein and organic anion transporting polypeptides at the blood-brain barrier: Understanding transport mechanisms for improved CNS drug delivery? AAPS J. 2017, 19, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, A.H.; Jonker, J.W. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: An overview. Adv. Drug Deliv. Rev. 2003, 55, 3–29. [Google Scholar] [CrossRef]
- Doran, A.; Obach, R.S.; Smith, B.J.; Hosea, N.A.; Becker, S.; Callegari, E.; Chen, C.; Chen, X.; Choo, E.; Cianfrogna, J.; et al. The impact of P-glycoprotein on the disposition of drugs targeted for indications of the central nervous system: Evaluation using the mdr1a/1b knockout mouse model. Drug Metab. Dispos. 2005, 33, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Jonker, J.W.; Wagenaar, E.; van Deemter, L.; Gottschlich, R.; Bender, H.M.; Dasenbrock, J.; Schinkel, A.H. Role of blood-brain barrier P-glycoprotein in limiting brain accumulation and sedative side-effects of asimadoline, a peripherally acting analgaesic drug. Br. J. Pharmacol. 1999, 127, 43–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, U.; Wagenaar, E.; Dorobek, B.; Beijnen, J.H.; Borst, P.; Schinkel, A.H. Full blockade of intestinal P-glycoprotein and extensive inhibition of blood-brain barrier P-glycoprotein by oral treatment of mice with PSC833. J. Clin. Investig. 1997, 100, 2430–2436. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, A.H.; Smit, J.J.; van Tellingen, O.; Beijnen, J.H.; Wagenaar, E.; van Deemter, L.; Mol, C.A.; van der Valk, M.A.; Robanus-Maandag, E.C.; te Riele, H.P.; et al. Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell 1994, 77, 491–502. [Google Scholar] [CrossRef]
- Shityakov, S.; Forster, C. Multidrug resistance protein P-gp interaction with nanoparticles (fullerenes and carbon nanotube) to assess their drug delivery potential: A theoretical molecular docking study. Int. J. Comput. Biol. Drug Des. 2013, 6, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.; Miller, M.C.; Monahan, R.; Osgood, D.P.; Stopa, E.G.; Silverberg, G.D. P-glycoprotein expression and amyloid accumulation in human aging and alzheimer’s disease: Preliminary observations. Neurobiol. Aging 2015, 36, 2475–2482. [Google Scholar] [CrossRef] [PubMed]
- Wijesuriya, H.C.; Bullock, J.Y.; Faull, R.L.; Hladky, S.B.; Barrand, M.A. ABC efflux transporters in brain vasculature of alzheimer’s subjects. Brain Res. 2010, 1358, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Deo, A.K.; Borson, S.; Link, J.M.; Domino, K.; Eary, J.F.; Ke, B.; Richards, T.L.; Mankoff, D.A.; Minoshima, S.; O’Sullivan, F.; et al. Activity of P-glycoprotein, a beta-amyloid transporter at the blood-brain barrier, is compromised in patients with mild alzheimer disease. J. Nucl. Med. 2014, 55, 1106–1111. [Google Scholar] [CrossRef] [PubMed]
- Hartz, A.M.; Miller, D.S.; Bauer, B. Restoring blood-brain barrier P-glycoprotein reduces brain amyloid-beta in a mouse model of alzheimer’s disease. Mol. Pharmacol. 2010, 77, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Xu, X.; Yang, Z.; Deng, Y.; Liu, X.; Xie, L. Impaired function and expression of P-glycoprotein in blood-brain barrier of streptozotocin-induced diabetic rats. Brain Res. 2006, 1123, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liu, X.; Jia, L.; Liu, Y.; Yang, H.; Wang, G.; Xie, L. Insulin therapy restores impaired function and expression of P-glycoprotein in blood-brain barrier of experimental diabetes. Biochem. Pharmacol. 2008, 75, 1649–1658. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, D.; Xu, X.; Liu, X.; Wang, G.; Xie, L.; Pang, X.; Liu, L. Attenuated function and expression of P-glycoprotein at blood-brain barrier and increased brain distribution of phenobarbital in streptozotocin-induced diabetic mice. Eur. J. Pharmacol. 2007, 561, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Wang, X.T.; Liu, L.; Yao, D.; Liu, C.; Zhang, M.; Guo, H.F.; Liu, X.D. P-glycoprotein and multidrug resistance-associated protein 2 are oppositely altered in brain of rats with thioacetamide-induced acute liver failure. Liver Int. 2013, 33, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.Y.D.; Liu, C.; Wang, X.T.; Zhang, L.L.; Liu, X.D. The effect of chronic liver failure on the function and expression of P-gp and Mrp2 in rat brain. J. China Pharm. Univ. 2012, 43, 64–69. [Google Scholar]
- Liu, L.; Miao, M.; Chen, Y.; Wang, Z.; Sun, B.; Liu, X. Altered function and expression of ABC transporters at the blood-brain barrier and increased brain distribution of phenobarbital in acute liver failure mice. Front. Pharmacol. 2018, 9, 190. [Google Scholar] [CrossRef] [PubMed]
- Loscher, W.; Potschka, H. Role of multidrug transporters in pharmacoresistance to antiepileptic drugs. J. Pharmacol. Exp. Ther. 2002, 301, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, Z.; Yang, J.; Yang, H. Increased P-glycoprotein expression and decreased phenobarbital distribution in the brain of pentylenetetrazole-kindled rats. Neuropharmacology 2007, 53, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Liu, X.; Wen, T.; Xie, S.; Yao, D.; Liu, X.; Wang, G.; Xie, L. Combined effects of epileptic seizure and phenobarbital induced overexpression of P-glycoprotein in brain of chemically kindled rats. Br. J. Pharmacol. 2010, 159, 1511–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.J.; Kusuhara, H.; Jonker, J.W.; Schinkel, A.H.; Sugiyama, Y. Investigation of efflux transport of dehydroepiandrosterone sulfate and mitoxantrone at the mouse blood-brain barrier: A minor role of breast cancer resistance protein. J. Pharmacol. Exp. Ther. 2005, 312, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Raub, T.J.; Sawada, G.A.; Kasper, S.C.; Bacon, J.A.; Bridges, A.S.; Pollack, G.M. Breast cancer resistance protein interacts with various compounds in vitro, but plays a minor role in substrate efflux at the blood-brain barrier. Drug Metab. Dispos. 2009, 37, 1251–1258. [Google Scholar] [CrossRef] [PubMed]
- Mittapalli, R.K.; Vaidhyanathan, S.; Sane, R.; Elmquist, W.F. Impact of P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) on the brain distribution of a novel braf inhibitor: Vemurafenib (PLX4032). J. Pharmacol. Exp. Ther. 2012, 342, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Agarwal, S.; Shaik, N.M.; Chen, C.; Yang, Z.; Elmquist, W.F. P-glycoprotein and breast cancer resistance protein influence brain distribution of dasatinib. J. Pharmacol. Exp. Ther. 2009, 330, 956–963. [Google Scholar] [CrossRef] [PubMed]
- Sane, R.; Agarwal, S.; Mittapalli, R.K.; Elmquist, W.F. Saturable active efflux by P-glycoprotein and breast cancer resistance protein at the blood-brain barrier leads to nonlinear distribution of elacridar to the central nervous system. J. Pharmacol. Exp. Ther. 2013, 345, 111–124. [Google Scholar] [CrossRef] [PubMed]
- De Vries, N.A.; Buckle, T.; Zhao, J.; Beijnen, J.H.; Schellens, J.H.; van Tellingen, O. Restricted brain penetration of the tyrosine kinase inhibitor erlotinib due to the drug transporters P-gp and Bcrp. Investig. New Drugs 2012, 30, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Sane, R.; Gallardo, J.L.; Ohlfest, J.R.; Elmquist, W.F. Distribution of gefitinib to the brain is limited by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2)-mediated active efflux. J. Pharmacol. Exp. Ther. 2010, 334, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Durmus, S.; Sparidans, R.W.; van Esch, A.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. Breast cancer resistance protein (BCRP/ABCG2) and P-glycoprotein (P-GP/ABCB1) restrict oral availability and brain accumulation of the parp inhibitor rucaparib (AG-014699). Pharm. Res. 2015, 32, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Sane, R.; Ohlfest, J.R.; Elmquist, W.F. The role of the breast cancer resistance protein (abcg2) in the distribution of sorafenib to the brain. J. Pharmacol. Exp. Ther. 2011, 336, 223–233. [Google Scholar] [CrossRef] [PubMed]
- De Vries, N.A.; Zhao, J.; Kroon, E.; Buckle, T.; Beijnen, J.H.; van Tellingen, O. P-glycoprotein and breast cancer resistance protein: Two dominant transporters working together in limiting the brain penetration of topotecan. Clin. Cancer Res. 2007, 13, 6440–6449. [Google Scholar] [CrossRef] [PubMed]
- Nies, A.T.; Jedlitschky, G.; Konig, J.; Herold-Mende, C.; Steiner, H.H.; Schmitt, H.P.; Keppler, D. Expression and immunolocalization of the multidrug resistance proteins, Mrp1-Mrp6 (ABCC1-ABCC6), in human brain. Neuroscience 2004, 129, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Kubota, H.; Ishihara, H.; Langmann, T.; Schmitz, G.; Stieger, B.; Wieser, H.G.; Yonekawa, Y.; Frei, K. Distribution and functional activity of P-glycoprotein and multidrug resistance-associated proteins in human brain microvascular endothelial cells in hippocampal sclerosis. Epilepsy Res. 2006, 68, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Warren, M.S.; Zerangue, N.; Woodford, K.; Roberts, L.M.; Tate, E.H.; Feng, B.; Li, C.; Feuerstein, T.J.; Gibbs, J.; Smith, B.; et al. Comparative gene expression profiles of ABC transporters in brain microvessel endothelial cells and brain in five species including human. Pharmacol. Res. 2009, 59, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Soontornmalai, A.; Vlaming, M.L.; Fritschy, J.M. Differential, strain-specific cellular and subcellular distribution of multidrug transporters in murine choroid plexus and blood-brain barrier. Neuroscience 2006, 138, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Marcorelles, P.; Friocourt, G.; Uguen, A.; Lede, F.; Ferec, C.; Laquerriere, A. Cystic fibrosis transmembrane conductance regulator protein (CFTR) expression in the developing human brain: Comparative immunohistochemical study between patients with normal and mutated CFTR. J. Histochem. Cytochem. 2014, 62, 791–801. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Su, M.; McNutt, M.A.; Gu, J. Expression and distribution of cystic fibrosis transmembrane conductance regulator in neurons of the human brain. J. Histochem. Cytochem. 2009, 57, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Mulberg, A.E.; Resta, L.P.; Wiedner, E.B.; Altschuler, S.M.; Jefferson, D.M.; Broussard, D.L. Expression and localization of the cystic fibrosis transmembrane conductance regulator mRNA and its protein in rat brain. J. Clin. Investig. 1995, 96, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Sun, C.M.; Liu, P. Alterations of blood-brain barrier and associated factors in acute liver failure. Gastroenterol. Res. Pract. 2013, 2013, 841707. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Hughes, R.D.; Keays, R.T.; Williams, R. Electron microscopic study of brain capillaries in cerebral edema from fulminant hepatic failure. Hepatology 1992, 15, 1060–1066. [Google Scholar] [CrossRef] [PubMed]
- Traber, P.G.; Dal Canto, M.; Ganger, D.R.; Blei, A.T. Electron microscopic evaluation of brain edema in rabbits with galactosamine-induced fulminant hepatic failure: Ultrastructure and integrity of the blood-brain barrier. Hepatology 1987, 7, 1272–1277. [Google Scholar] [CrossRef] [PubMed]
- Lv, S.; Song, H.L.; Zhou, Y.; Li, L.X.; Cui, W.; Wang, W.; Liu, P. Tumour necrosis factor-alpha affects blood-brain barrier permeability and tight junction-associated occludin in acute liver failure. Liver Int. 2010, 30, 1198–1210. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, J.; Xu, P.; Sun, B.; Zhong, Z.; Liu, C.; Ling, Z.; Chen, Y.; Shu, N.; Zhao, K.; et al. Acute liver failure impairs function and expression of breast cancer-resistant protein (BCRP) at rat blood-brain barrier partly via ammonia-ROS-ERK1/2 activation. J. Neurochem. 2016, 138, 282–294. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, V.; Singh, R.; Acharya, S.K. Predictive value of arterial ammonia for complications and outcome in acute liver failure. Gut 2006, 55, 98–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zwirner, K.; Thiel, C.; Thiel, K.; Morgalla, M.H.; Konigsrainer, A.; Schenk, M. Extracellular brain ammonia levels in association with arterial ammonia, intracranial pressure and the use of albumin dialysis devices in pigs with acute liver failure. Metab. Brain Dis. 2010, 25, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Skowronska, M.; Zielinska, M.; Wojcik-Stanaszek, L.; Ruszkiewicz, J.; Milatovic, D.; Aschner, M.; Albrecht, J. Ammonia increases paracellular permeability of rat brain endothelial cells by a mechanism encompassing oxidative/nitrosative stress and activation of matrix metalloproteinases. J. Neurochem. 2012, 121, 125–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, G.; Shawcross, D.; Olde Damink, S.W.; Jalan, R. Brain cytokine flux in acute liver failure and its relationship with intracranial hypertension. Metab. Brain Dis. 2007, 22, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Desjardins, P.; Butterworth, R.F. Direct evidence for central proinflammatory mechanisms in rats with experimental acute liver failure: Protective effect of hypothermia. J. Cereb. Blood Flow Metab. 2009, 29, 944–952. [Google Scholar] [CrossRef] [PubMed]
- Odeh, M.; Sabo, E.; Srugo, I.; Oliven, A. Relationship between tumor necrosis factor-alpha and ammonia in patients with hepatic encephalopathy due to chronic liver failure. Ann. Med. 2005, 37, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Jalan, R.; Olde Damink, S.W.; Hayes, P.C.; Deutz, N.E.; Lee, A. Pathogenesis of intracranial hypertension in acute liver failure: Inflammation, ammonia and cerebral blood flow. J. Hepatol. 2004, 41, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Bemeur, C.; Qu, H.; Desjardins, P.; Butterworth, R.F. IL-1 or TNF receptor gene deletion delays onset of encephalopathy and attenuates brain edema in experimental acute liver failure. Neurochem. Int. 2010, 56, 213–215. [Google Scholar] [CrossRef] [PubMed]
- Tsao, N.; Hsu, H.P.; Wu, C.M.; Liu, C.C.; Lei, H.Y. Tumour necrosis factor-alpha causes an increase in blood-brain barrier permeability during sepsis. J. Med. Microbiol. 2001, 50, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lv, S.; Zhou, Y.; Fu, J.; Li, C.; Liu, P. Tumor necrosis factor-α affects blood-brain barrier permeability in acetaminophen-induced acute liver failure. Eur. J. Gastroenterol. Hepatol. 2011, 23, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.H.; Yamamoto, S.; Steers, J.; Sevlever, D.; Lin, W.; Shimojima, N.; Castanedes-Casey, M.; Genco, P.; Golde, T.; Richelson, E.; et al. Matrix metalloproteinase-9 contributes to brain extravasation and edema in fulminant hepatic failure mice. J. Hepatol. 2006, 44, 1105–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMillin, M.A.; Frampton, G.A.; Seiwell, A.P.; Patel, N.S.; Jacobs, A.N.; DeMorrow, S. TGFβ1 exacerbates blood-brain barrier permeability in a mouse model of hepatic encephalopathy via upregulation of MMP9 and downregulation of claudin-5. Lab. Investig. 2015, 95, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Hori, T.; Ohashi, N.; Baine, A.M.; Eckman, C.B.; Nguyen, J.H. Occludin is regulated by epidermal growth factor receptor activation in brain endothelial cells and brains of mice with acute liver failure. Hepatology 2011, 53, 1294–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigau, V.; Morin, M.; Rousset, M.C.; de Bock, F.; Lebrun, A.; Coubes, P.; Picot, M.C.; Baldy-Moulinier, M.; Bockaert, J.; Crespel, A.; et al. Angiogenesis is associated with blood-brain barrier permeability in temporal lobe epilepsy. Brain 2007, 130, 1942–1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; Deguchi, K.; Nagotani, S.; Zhang, H.; Sehara, Y.; Tsuchiya, A.; Abe, K. Cerebral ischemia and angiogenesis. Curr. Neurovasc. Res. 2006, 3, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Wang, Z.H.; Zhang, B.; Zhe, X.; Wang, M.J.; Shi, S.T.; Bai, J.; Lin, T.; Guo, C.J.; Zhang, S.J.; et al. Disruption of the blood-brain barrier after generalized tonic-clonic seizures correlates with cerebrospinal fluid MMP-9 levels. J. Neuroinflamm. 2013, 10, 80. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chopp, M. Vascular endothelial growth factor and angiopoietins in focal cerebral ischemia. Trends Cardiovasc. Med. 2002, 12, 62–66. [Google Scholar] [CrossRef]
- Aspinall, R.J.; Weis, S.M.; Barnes, L.; Lutu-Fuga, K.; Bylund, D.J.; Pockros, P.J.; Cheresh, D.A. A src family kinase inhibitor improves survival in experimental acute liver failure associated with elevated cerebral and circulating vascular endothelial growth factor levels. Liver Int. 2011, 31, 1222–1230. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Ling, Z.L.; Zhang, J.; Li, Y.; Shu, N.; Zhong, Z.Y.; Chen, Y.; Di, X.Y.; Wang, Z.J.; Liu, L.; et al. Unconjugated bilirubin elevation impairs the function and expression of breast cancer resistance protein (BCRP) at the blood-brain barrier in bile duct-ligated rats. Acta Pharmacol. Sin. 2016, 37, 1129–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zhang, M.; Sun, B.; Li, Y.; Xu, P.; Liu, C.; Liu, L.; Liu, X. Hyperammonemia enhances the function and expression of P-glycoprotein and Mrp2 at the blood-brain barrier through NF-κB. J. Neurochem. 2014, 131, 791–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.H.; Murakami, T.; Okochi, A.; Yumoyo, R.; Nagai, J.; Takano, M. Expression and function of P-glycoprotein in rats with carbon tetrachloride-induced acute hepatic failure. J. Pharm. Pharmacol. 2001, 53, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, A.; Dohgu, S.; Takata, F.; Watanabe, T.; Nishioku, T.; Matsumoto, J.; Ohkubo, Y.; Shuto, H.; Kataoka, Y. Partial hepatectomy aggravates cyclosporin A-induced neurotoxicity by lowering the function of the blood-brain barrier in mice. Life Sci. 2011, 88, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Ellis, A.J.; Wendon, J.A.; Williams, R. Subclinical seizure activity and prophylactic phenytoin infusion in acute liver failure: A controlled clinical trial. Hepatology 2000, 32, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Bernal, W.; Auzinger, G.; Dhawan, A.; Wendon, J. Acute liver failure. Lancet 2010, 376, 190–201. [Google Scholar] [CrossRef]
- Robey, R.W.; To, K.K.; Polgar, O.; Dohse, M.; Fetsch, P.; Dean, M.; Bates, S.E. ABCG2: A perspective. Adv. Drug Deliv. Rev. 2009, 61, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Reznicek, J.; Ceckova, M.; Ptackova, Z.; Martinec, O.; Tupova, L.; Cerveny, L.; Staud, F. MDR1 and BCRP transporter-mediated drug-drug interaction between rilpivirine and abacavir and effect on intestinal absorption. Antimicrob. Agents Chemother. 2017, 61, e00837-17. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Shi, T.; Zhang, L.; Zhu, P.; Deng, M.; Huang, C.; Hu, T.; Jiang, L.; Li, J. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family in multidrug resistance: A review of the past decade. Cancer Lett. 2016, 370, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Jalan, R. Acute liver failure: Current management and future prospects. J. Hepatol. 2005, 42, S115–S123. [Google Scholar] [CrossRef] [PubMed]
- Mpabanzi, L.; Jalan, R. Neurological complications of acute liver failure: Pathophysiological basis of current management and emerging therapies. Neurochem. Int. 2012, 60, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Romermann, K.; Helmer, R.; Loscher, W. The antiepileptic drug lamotrigine is a substrate of mouse and human breast cancer resistance protein (ABCG2). Neuropharmacology 2015, 93, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Lok, A.S.; McMahon, B.J.; Brown, R.S., Jr.; Wong, J.B.; Ahmed, A.T.; Farah, W.; Almasri, J.; Alahdab, F.; Benkhadra, K.; Mouchli, M.A.; et al. Antiviral therapy for chronic hepatitis B viral infection in adults: A systematic review and meta-analysis. Hepatology 2016, 63, 284–306. [Google Scholar] [CrossRef] [PubMed]
- Calmet, F.H.; Yarur, A.J.; Pukazhendhi, G.; Ahmad, J.; Bhamidimarri, K.R. Endoscopic and histological features of mycophenolate mofetil colitis in patients after solid organ transplantation. Ann. Gastroenterol. 2015, 28, 366–373. [Google Scholar] [PubMed]
- Yang, X.; Liu, K. P-gp inhibition-based strategies for modulating pharmacokinetics of anticancer drugs: An update. Curr. Drug Metab. 2016, 17, 806–826. [Google Scholar] [CrossRef] [PubMed]
- Matzneller, P.; Kussmann, M.; Eberl, S.; Maier-Salamon, A.; Jager, W.; Bauer, M.; Langer, O.; Zeitlinger, M.; Poeppl, W. Pharmacokinetics of the P-gp inhibitor tariquidar in rats after intravenous, oral, and intraperitoneal administration. Eur. J. Drug Metab. Pharmacokinet. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Miao, M.X.; Sun, B.B.; Wang, Z.J.; Tang, X.G.; Chen, Y.; Zhao, K.J.; Liu, X.D.; Liu, L. Acute liver failure enhances oral plasma exposure of zidovudine in rats by downregulation of hepatic UGT2B7 and intestinal P-gp. Acta Pharmacol. Sin. 2017, 38, 1554–1565. [Google Scholar] [CrossRef] [PubMed]
- Barnes, S.N.; Aleksunes, L.M.; Augustine, L.; Scheffer, G.L.; Goedken, M.J.; Jakowski, A.B.; Pruimboom-Brees, I.M.; Cherrington, N.J.; Manautou, J.E. Induction of hepatobiliary efflux transporters in acetaminophen-induced acute liver failure cases. Drug Metab. Dispos. 2007, 35, 1963–1969. [Google Scholar] [CrossRef] [PubMed]
- Miah, M.K.; Shaik, I.H.; Bickel, U.; Mehvar, R. Effects of hepatic ischemia-reperfusion injury on the P-glycoprotein activity at the liver canalicular membrane and blood-brain barrier determined by in vivo administration of rhodamine 123 in rats. Pharm. Res. 2014, 31, 861–873. [Google Scholar] [CrossRef] [PubMed]
- Uhr, M.; Holsboer, F.; Muller, M.B. Penetration of endogenous steroid hormones corticosterone, cortisol, aldosterone and progesterone into the brain is enhanced in mice deficient for both mdr1a and mdr1b P-glycoproteins. J. Neuroendocrinol. 2002, 14, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Grube, M.; Hagen, P.; Jedlitschky, G. Neurosteroid transport in the brain: Role of ABC and Slc transporters. Front. Pharmacol. 2018, 9, 354. [Google Scholar] [CrossRef] [PubMed]
- Zaman, S.H. Endogenous steroids and pathogenesis of hepatic encephalopathy. Lancet 1990, 336, 573–574. [Google Scholar] [CrossRef]
- Ahboucha, S.; Pomier-Layrargues, G.; Mamer, O.; Butterworth, R.F. Increased levels of pregnenolone and its neuroactive metabolite allopregnanolone in autopsied brain tissue from cirrhotic patients who died in hepatic coma. Neurochem. Int. 2006, 49, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Ahboucha, S.; Butterworth, R.F. The neurosteroid system: Implication in the pathophysiology of hepatic encephalopathy. Neurochem. Int. 2008, 52, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Ahboucha, S.; Talani, G.; Fanutza, T.; Sanna, E.; Biggio, G.; Gamrani, H.; Butterworth, R.F. Reduced brain levels of dheas in hepatic coma patients: Significance for increased gabaergic tone in hepatic encephalopathy. Neurochem. Int. 2012, 61, 48–53. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Cellular interfaces between blood and brain parenchyma (blood-brain barrier, BBB). The endothelial cells and the closely apposed pericytes are completely unsheathed by overlapping astrocytic end-feet and neuronal terminals, forming “neurovascular unit” (NVU).
Figure 1.
Cellular interfaces between blood and brain parenchyma (blood-brain barrier, BBB). The endothelial cells and the closely apposed pericytes are completely unsheathed by overlapping astrocytic end-feet and neuronal terminals, forming “neurovascular unit” (NVU).

Figure 2.
Possible location of some ATP-binding cassette (ABC) drug transporters in brain microvessel endothelial cells of human and rodents. P-GP, glycoprotein; BCRP, breast cancer resistance protein; MRPs/Mrps, multidrug associated resistance proteins.
Figure 2.
Possible location of some ATP-binding cassette (ABC) drug transporters in brain microvessel endothelial cells of human and rodents. P-GP, glycoprotein; BCRP, breast cancer resistance protein; MRPs/Mrps, multidrug associated resistance proteins.
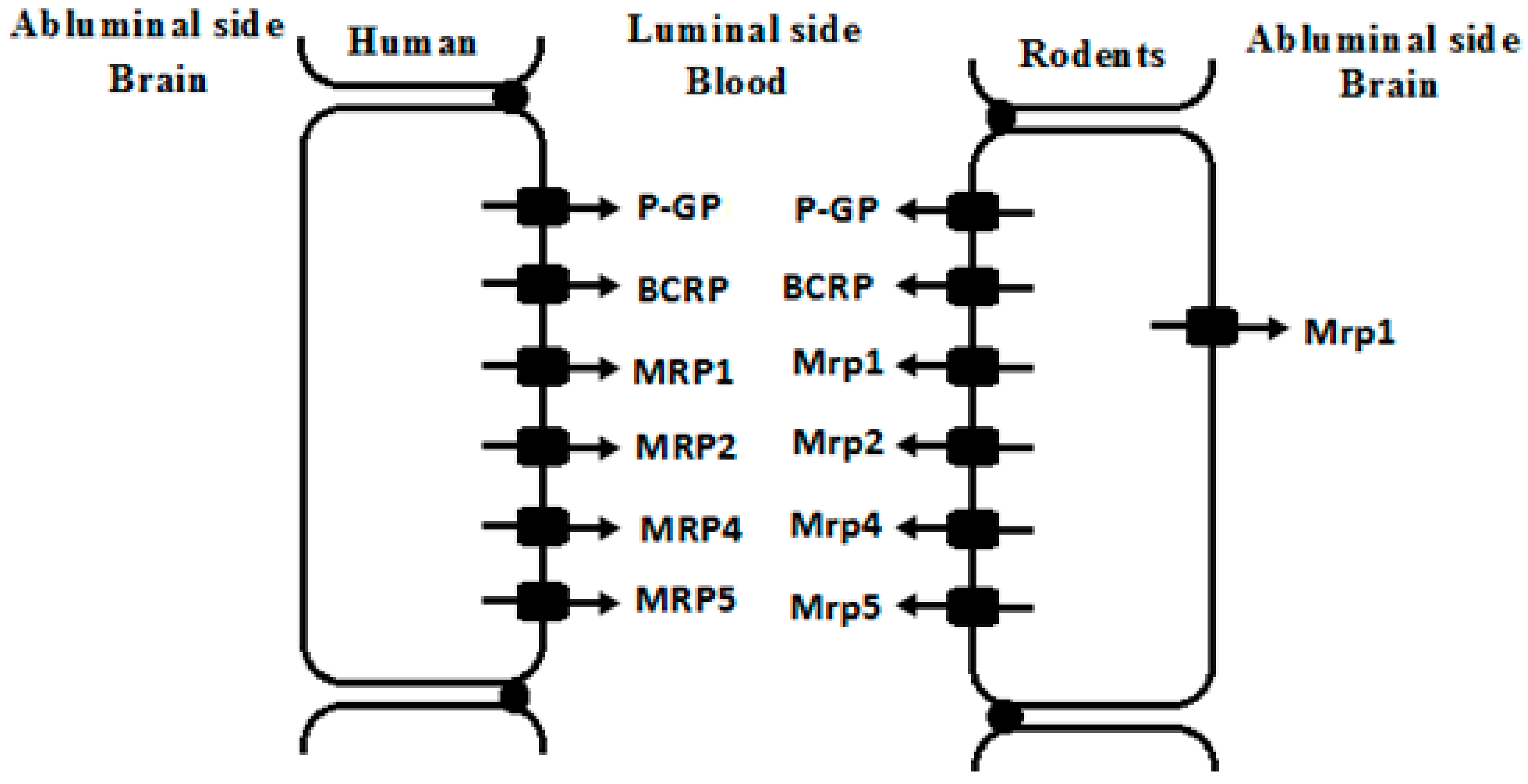
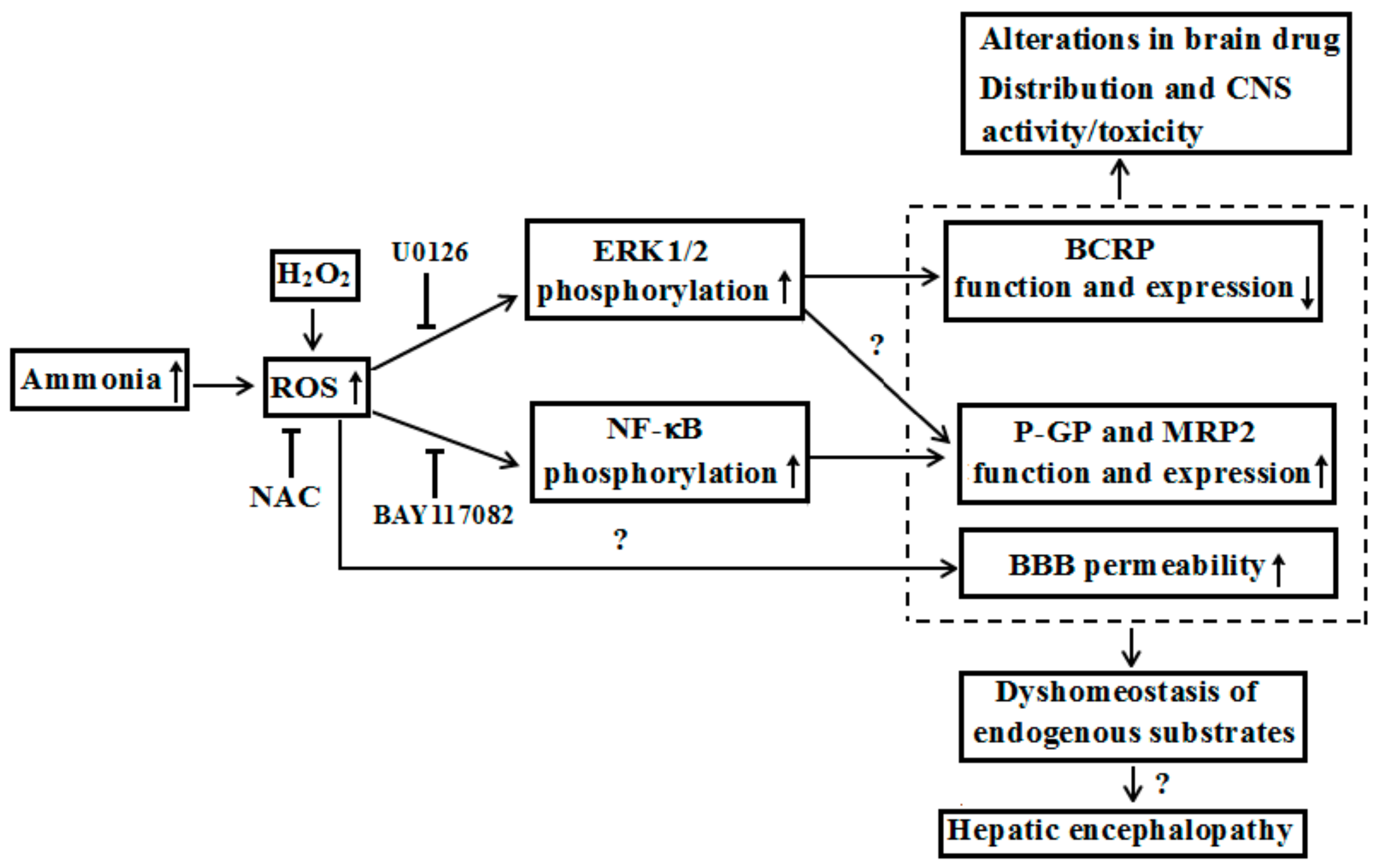
Figure 3.
Possible mechanisms that hyperammonemia altered expression and function of ABC transporters at BBB under liver failure and its contributions to hepatic encephalopathy.
Figure 3.
Possible mechanisms that hyperammonemia altered expression and function of ABC transporters at BBB under liver failure and its contributions to hepatic encephalopathy.

{kind=link}
{kind=link}
{kind=link}
Table 1.
Comparison of MRP/Mrp mRNA levels in brain microvessel endothelial cell from five species. ++++, >10% of GLUT1; +++, >4% of GLUT1; ++, >2% of GLUT1; +, >1% of GLUT1; ±, <10% of GLUT1 and -, no detected, data were derived from reference [44].
Table 1.
Comparison of MRP/Mrp mRNA levels in brain microvessel endothelial cell from five species. ++++, >10% of GLUT1; +++, >4% of GLUT1; ++, >2% of GLUT1; +, >1% of GLUT1; ±, <10% of GLUT1 and -, no detected, data were derived from reference [44].
| Species | MRP1 | MRP2 | MRP3 | MRP4 | MRP5 | MRP6 |
|---|---|---|---|---|---|---|
| Human | ++ | - | ± | + | ++ | + |
| Rat | ++ | ± | ± | ++++ | ++++ | ++ |
| Mouse | ± | - | + | ++++ | + | + |
| Pig | ± | - | ++ | + | +++ | - |
| Cow | ++ | - | - | - | ++++ | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Fan, Y.; Liu, X. Alterations in Expression and Function of ABC Family Transporters at Blood-Brain Barrier under Liver Failure and Their Clinical Significances. Pharmaceutics 2018, 10, 102. https://doi.org/10.3390/pharmaceutics10030102
AMA Style
Fan Y, Liu X. Alterations in Expression and Function of ABC Family Transporters at Blood-Brain Barrier under Liver Failure and Their Clinical Significances. Pharmaceutics. 2018; 10(3):102. https://doi.org/10.3390/pharmaceutics10030102
Chicago/Turabian StyleFan, Yilin, and Xiaodong Liu. 2018. "Alterations in Expression and Function of ABC Family Transporters at Blood-Brain Barrier under Liver Failure and Their Clinical Significances" Pharmaceutics 10, no. 3: 102. https://doi.org/10.3390/pharmaceutics10030102
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.