
Inhibitors of Deubiquitinating Enzymes Block HIV-1 Replication and Augment the Presentation of Gag-Derived MHC-I Epitopes

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Transfection
2.2. Expression Plasmids
2.3. Viruses
2.4. Inhibitors
2.5. Cultivation and Preparation of Primary Cells
2.6. Ethical Statement
2.7. Investigation of Gag Processing and Virus Release by Steady State Analysis
2.8. In Vitro Processing of Gag Polyproteins
2.9. SDS-PAGE and Western Blotting
2.10. Detection of Ubiquitinated Gag
2.11. Flow Cytometry
2.12. Single Round Infection Assay
2.13. Infection of HLA Cultures
2.14. Determination of the Replication Capacity
2.15. Assessment of Cell Viability
3. Results
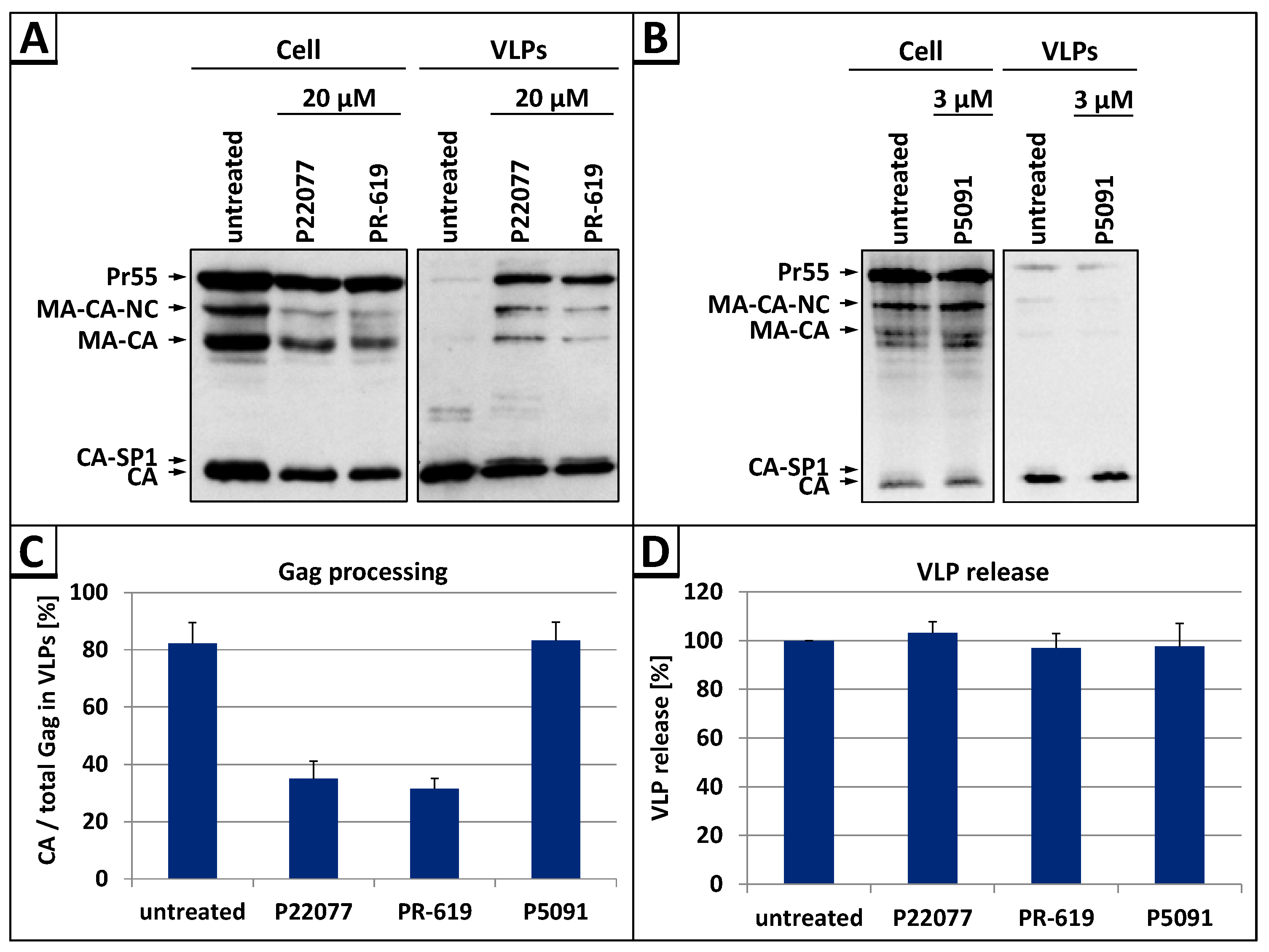
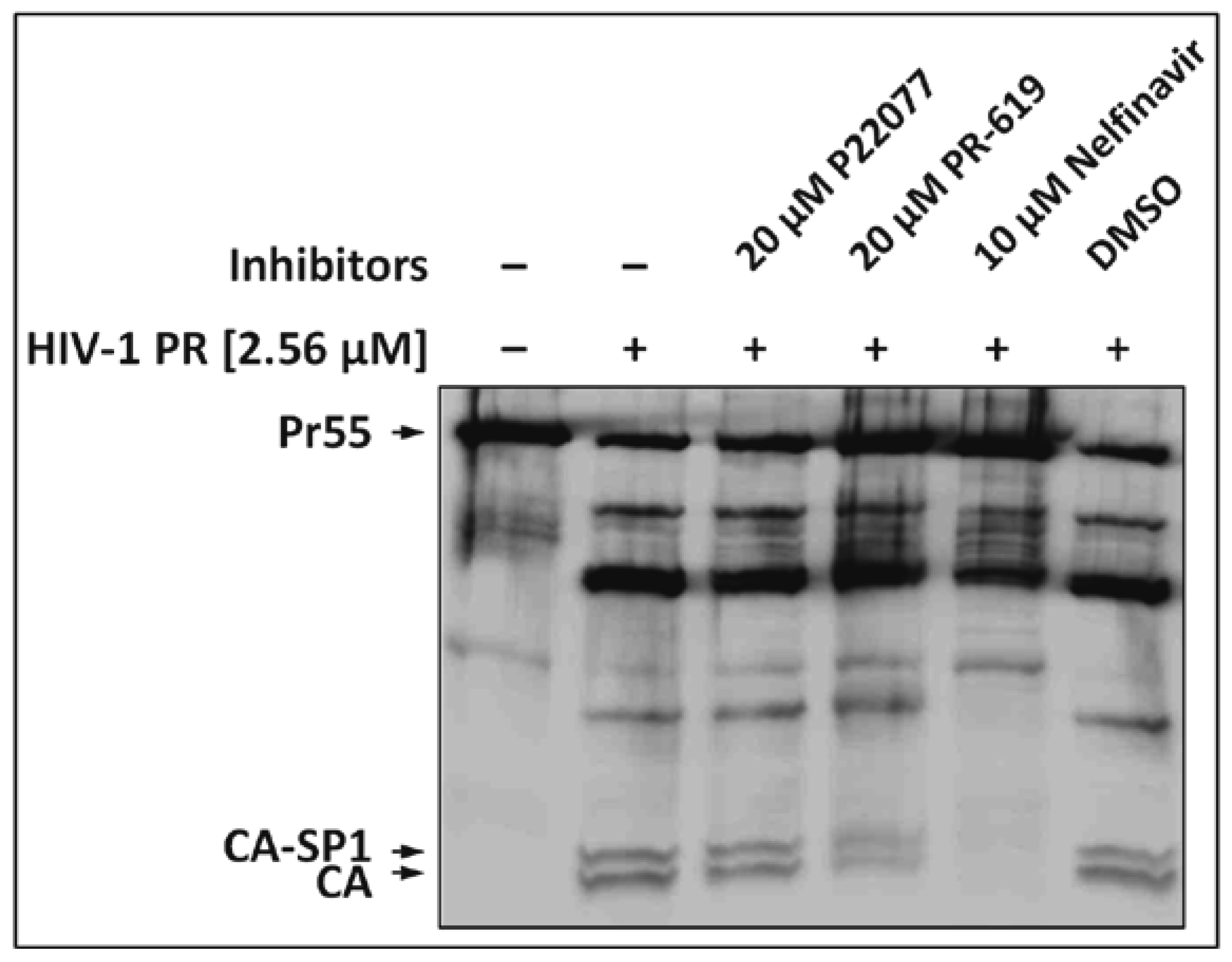
3.1. The DUB-Inhibitors P22077 and PR-619 Impair Gag Processing without Affecting Protease Activity
3.2. The DUB-Inhibitors P22077 and PR-619 Reduce Virus Infectivity
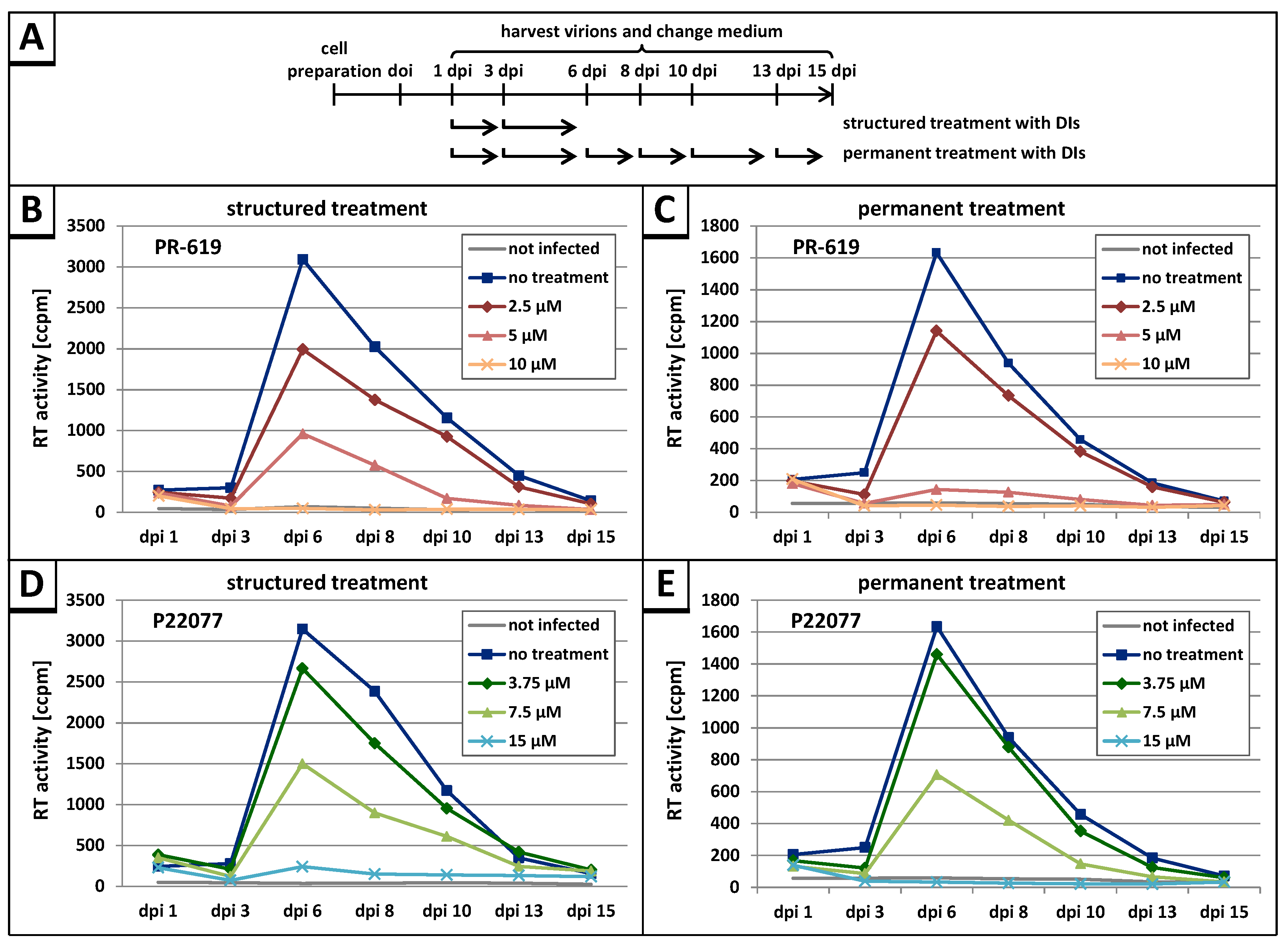
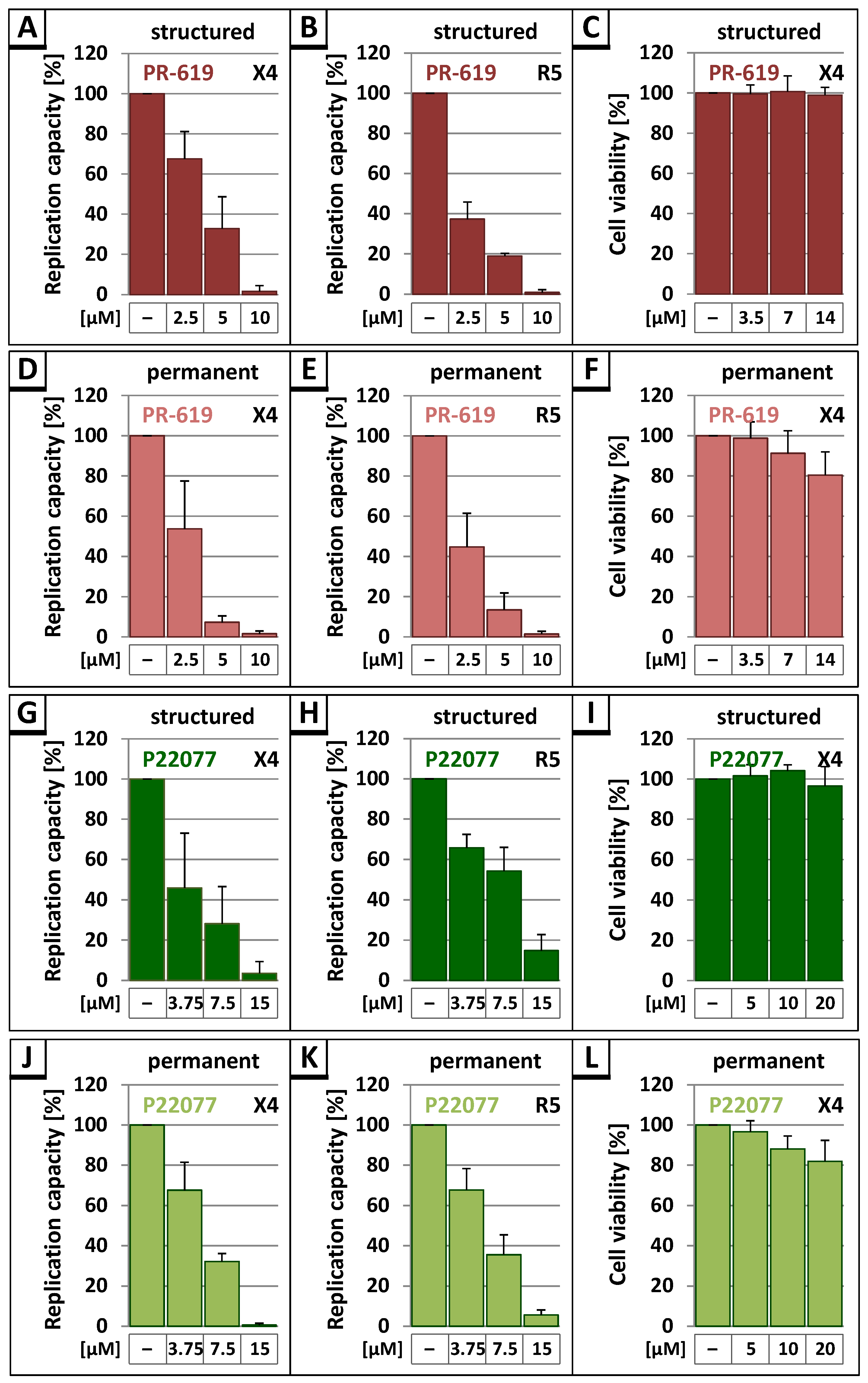
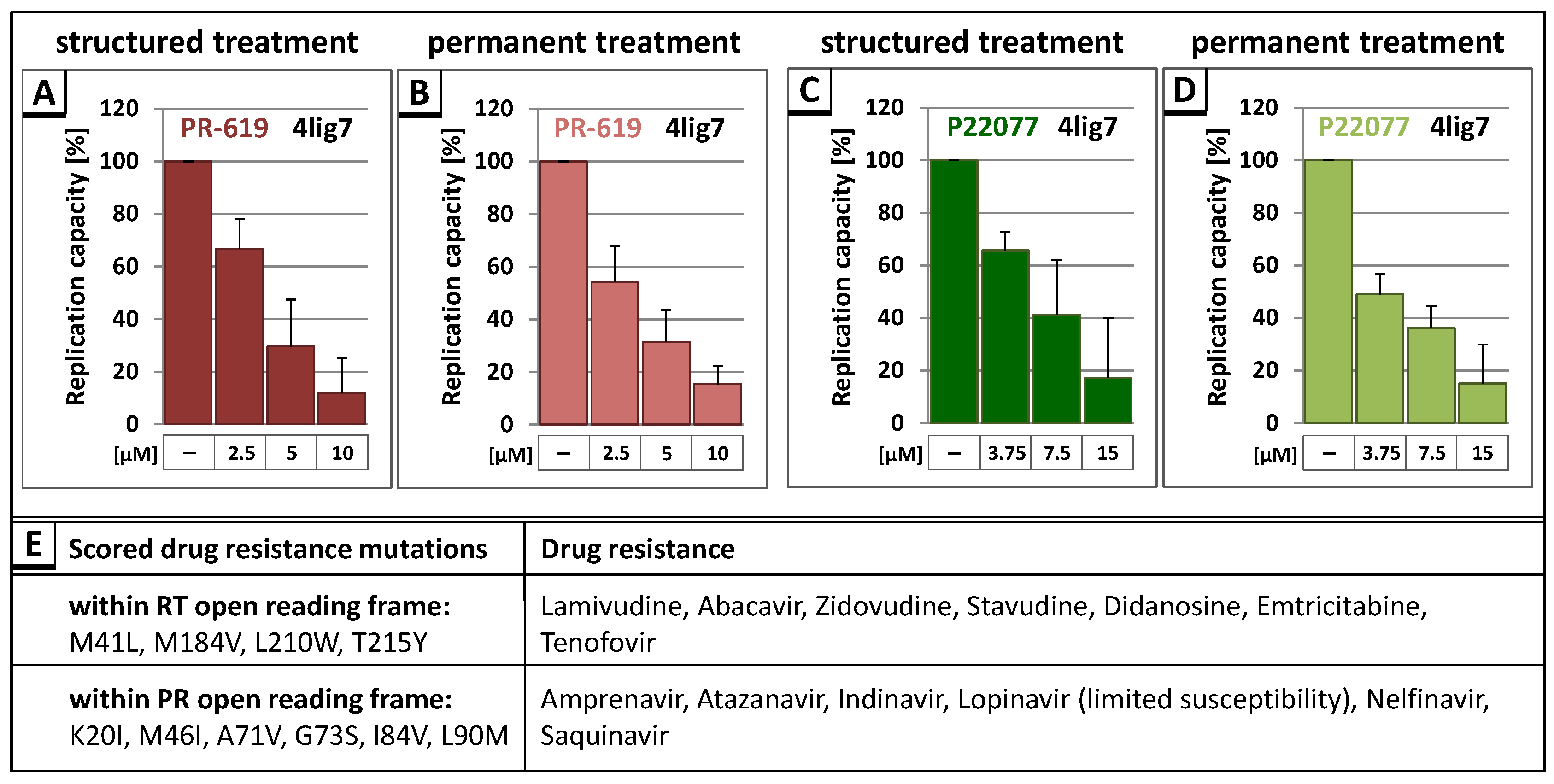
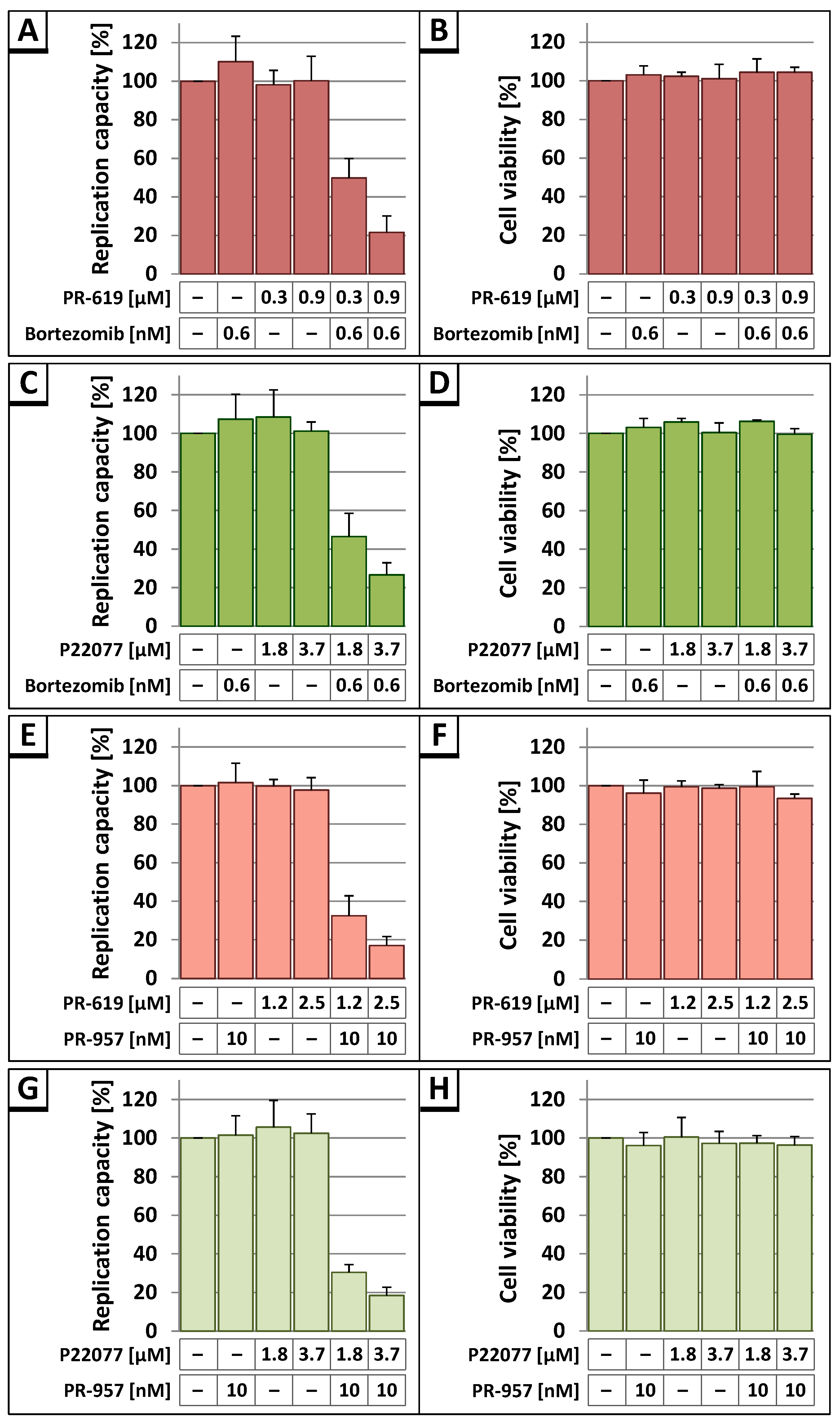
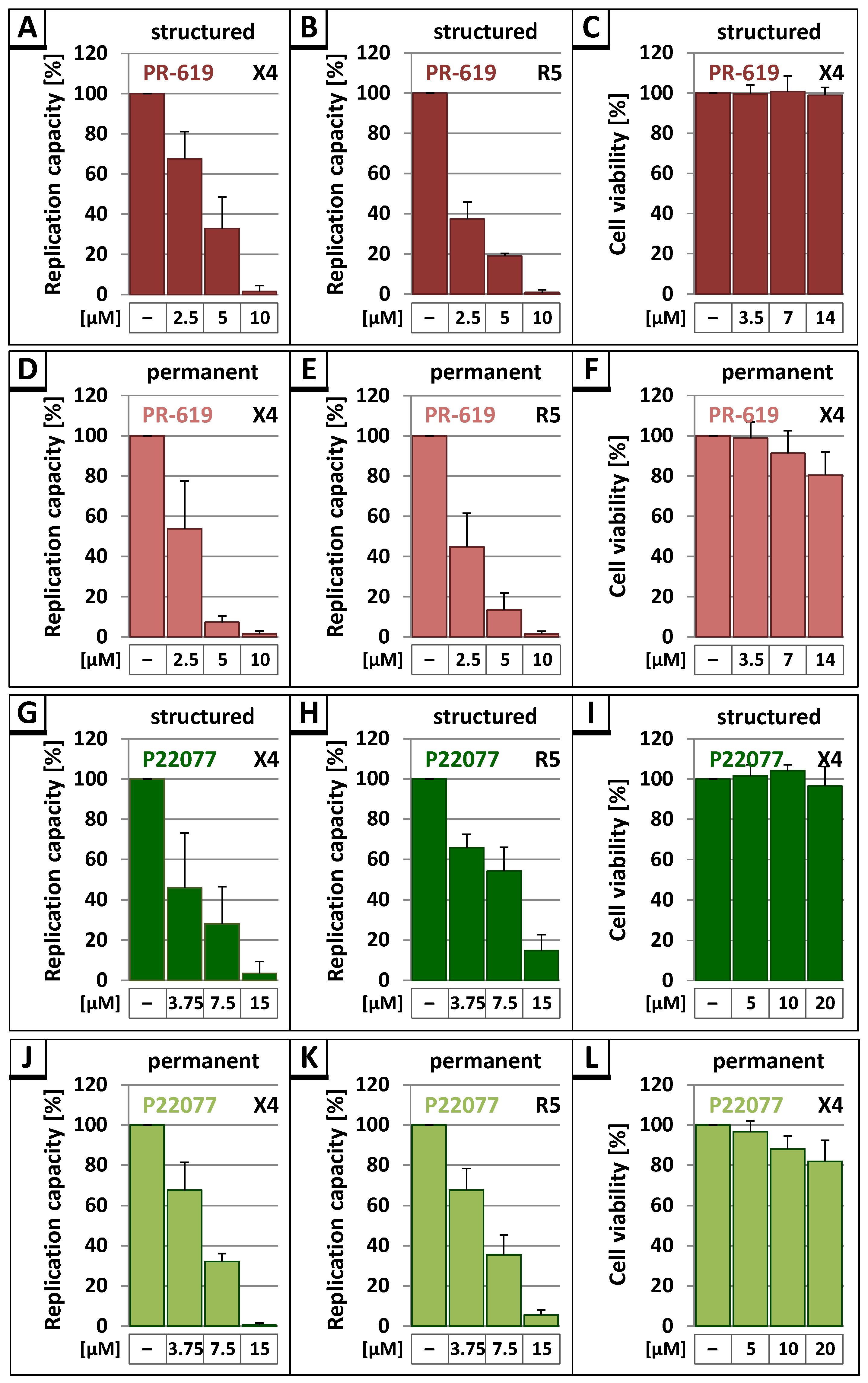
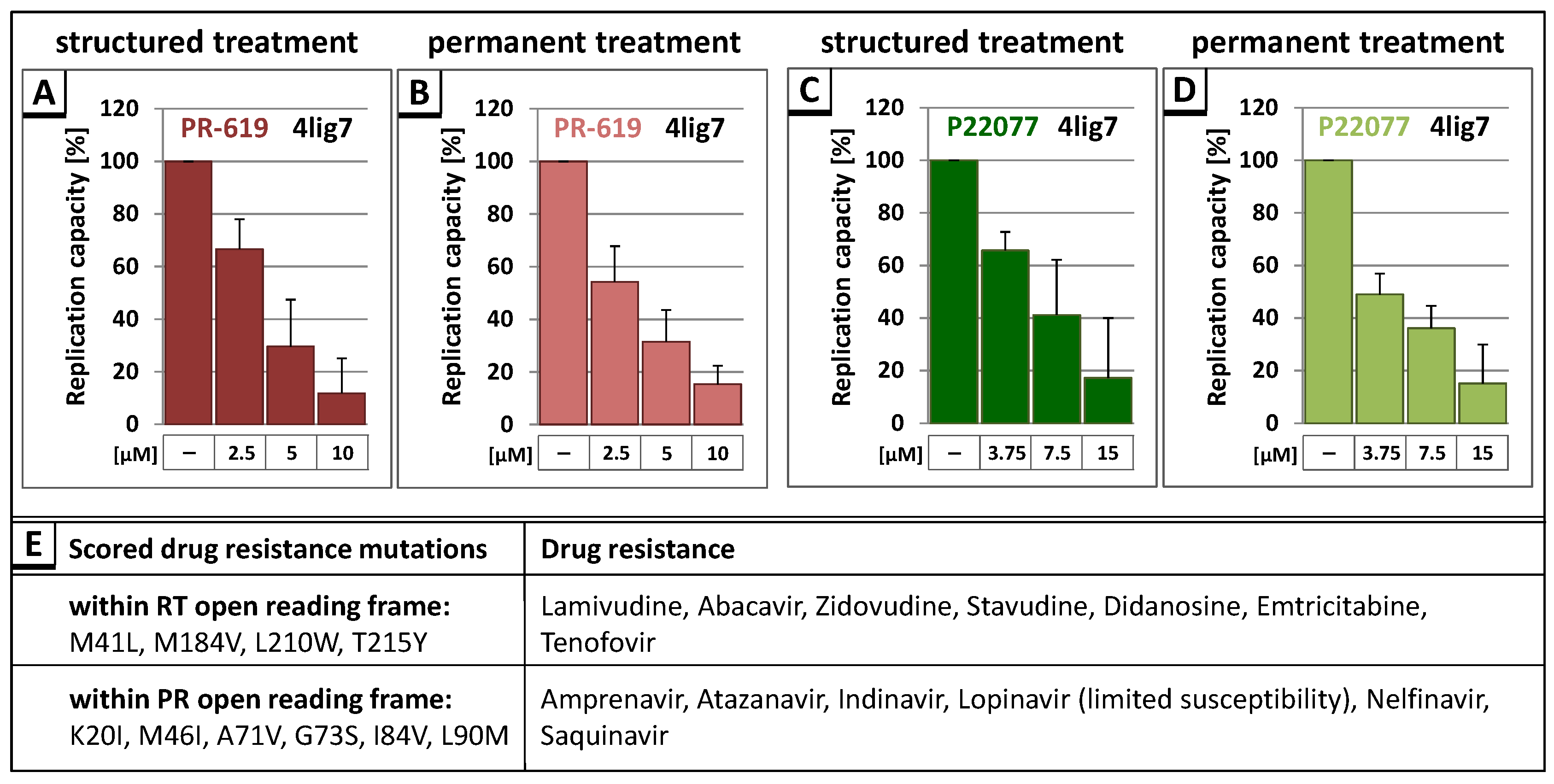
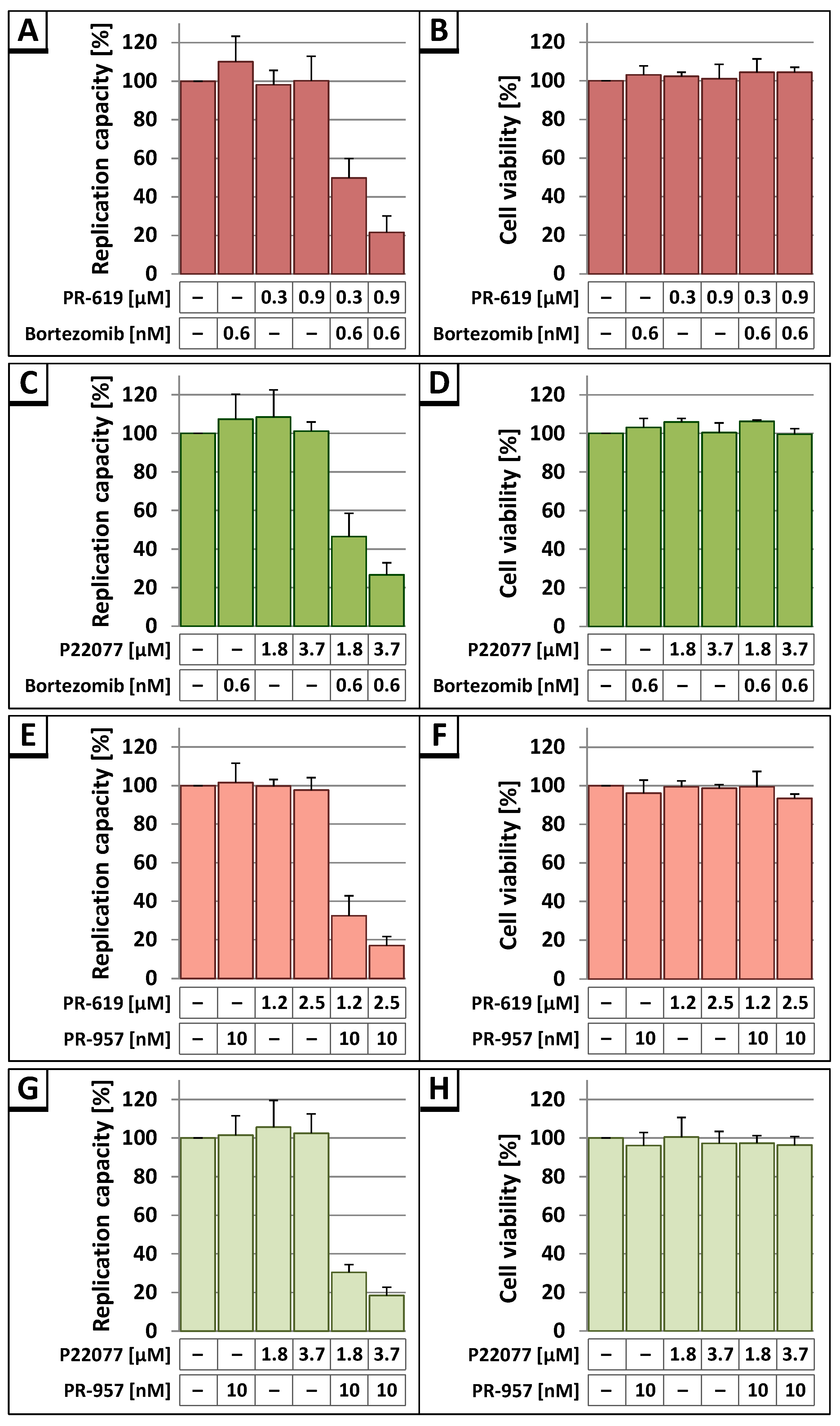
3.3. The DUB-Inhibitors P22077 and PR-619 Reduce HIV-1 Replication in a Dose-Dependent Manner
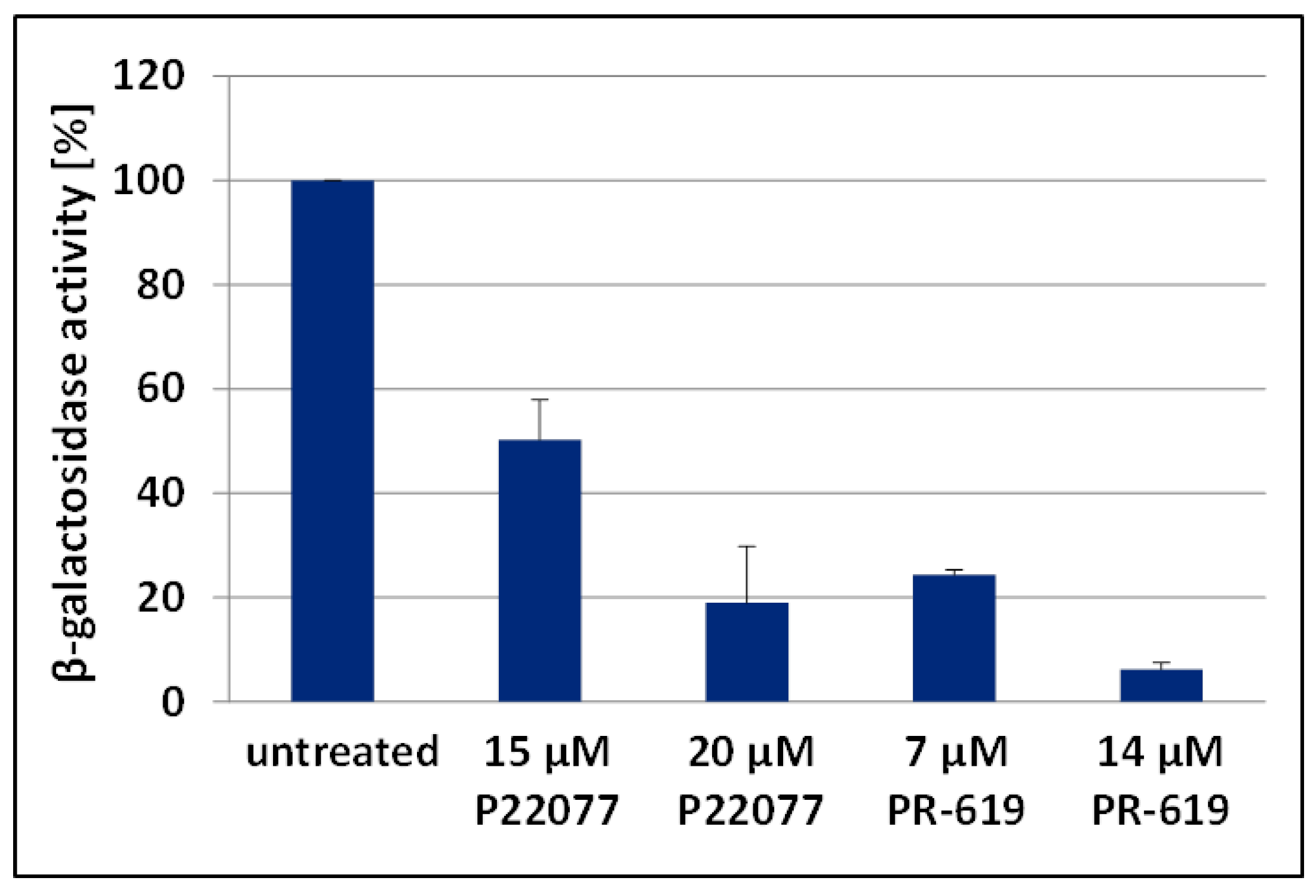
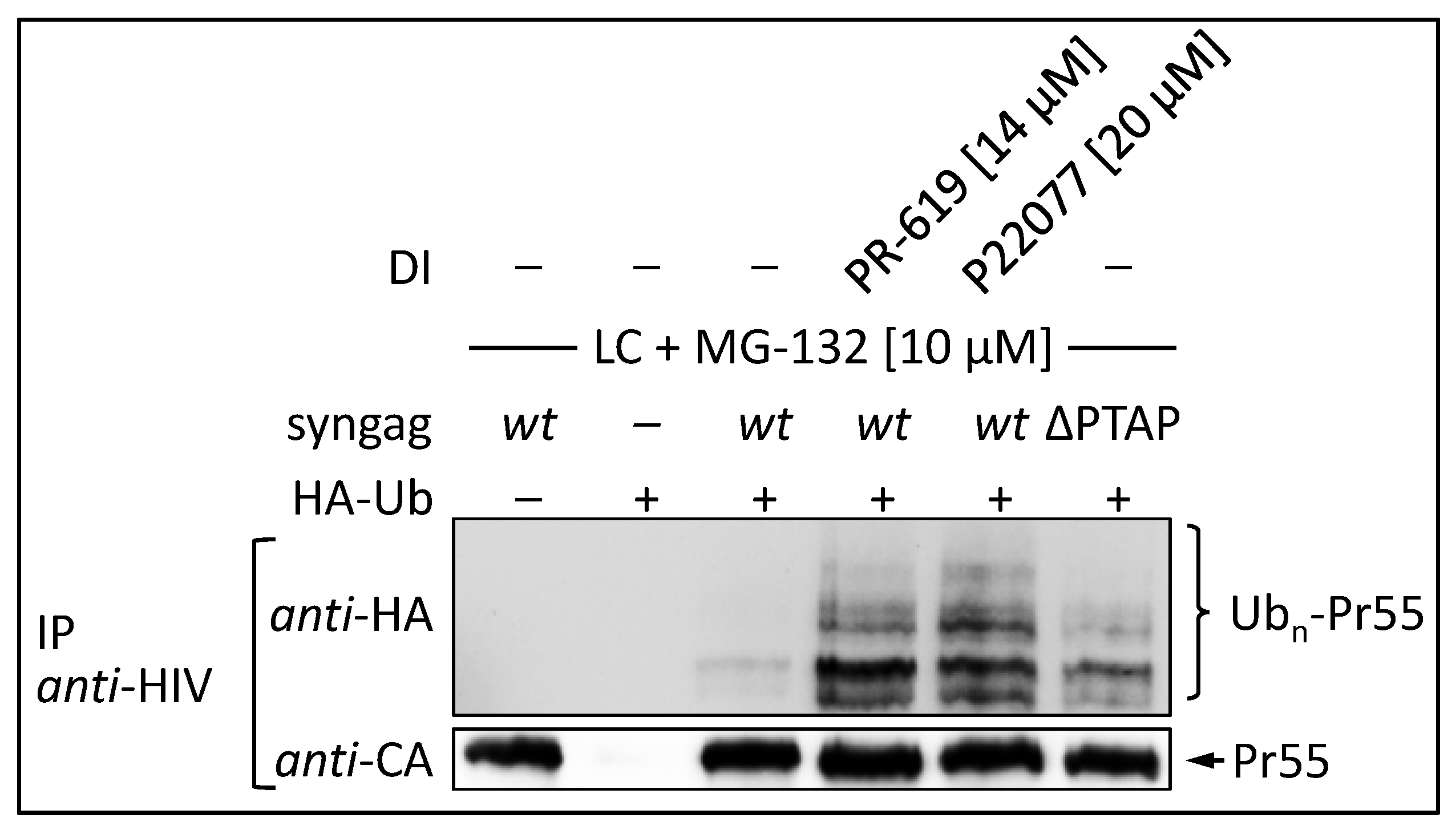
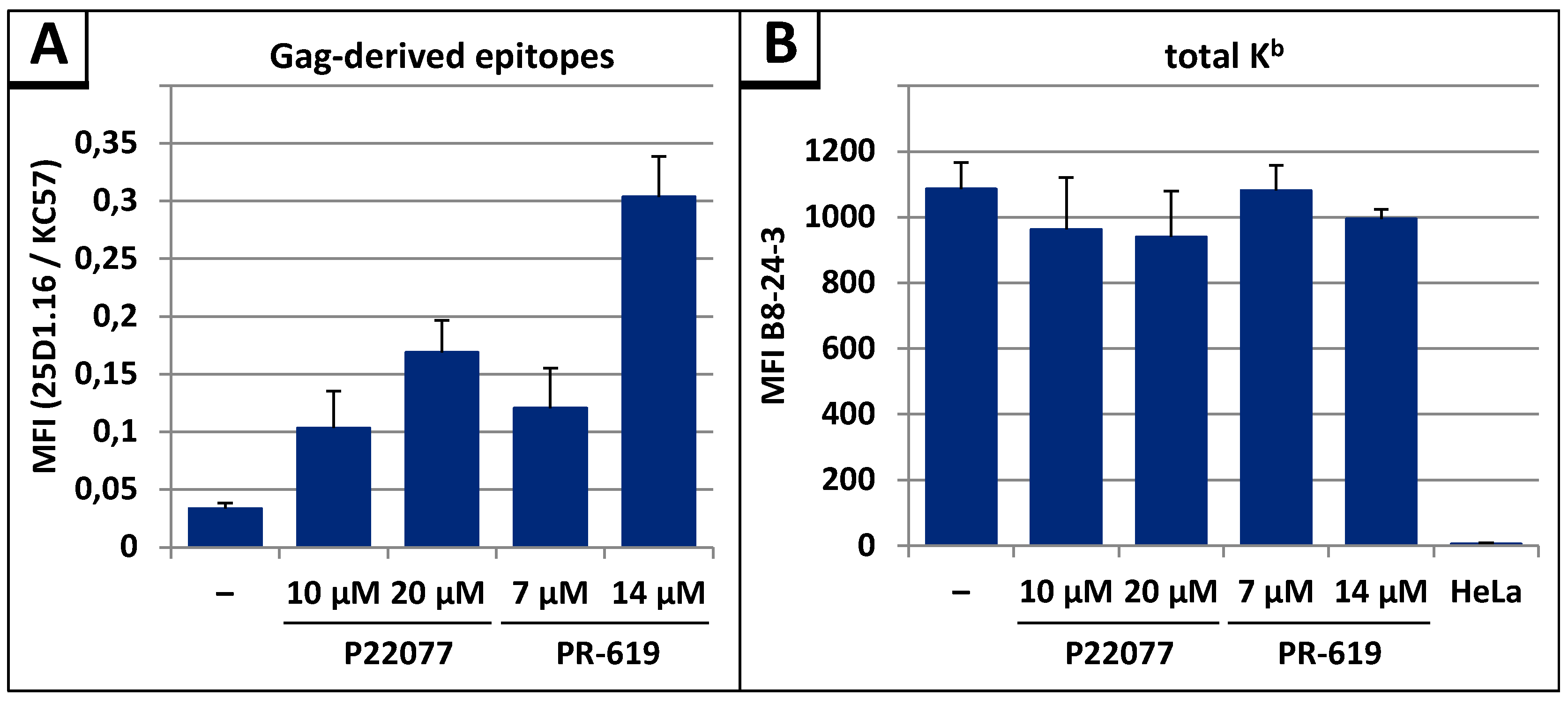
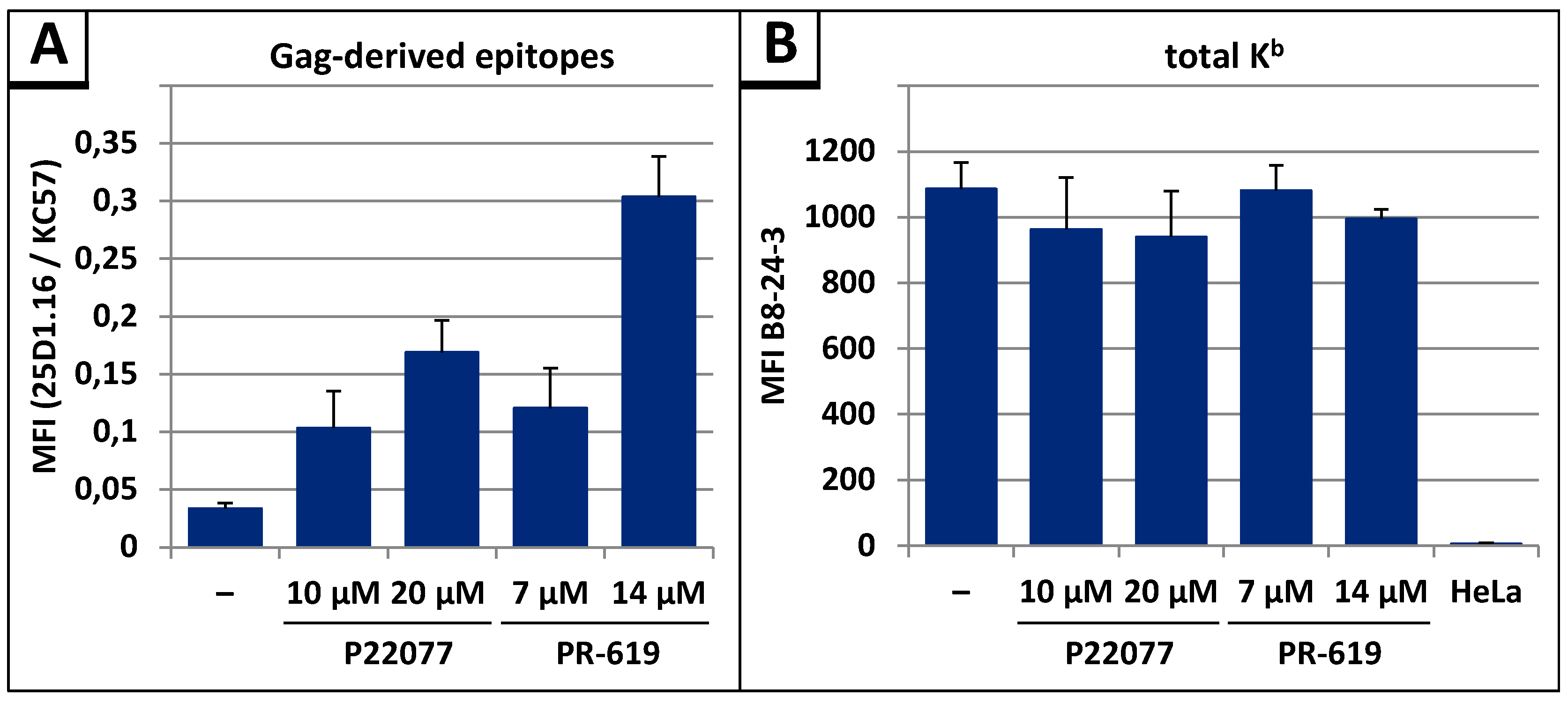
3.4. The DUB-inhibitors P22077 and PR-619 Cause an Enhanced Entry of Gag into the UPS and thus into the MHC-I Antigen Presentation Pathway
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pickart, C.M. Mechanisms underlying ubiquitination. Annu. Rev. BioChem. 2001, 70, 503–533. [Google Scholar] [CrossRef]
- Komander, D. The emerging complexity of protein ubiquitination. BioChem. Soc. Trans. 2009, 37, 937–953. [Google Scholar] [CrossRef]
- Reyes-Turcu, F.E.; Ventii, K.H.; Wilkinson, K.D. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 2009, 78, 363–397. [Google Scholar] [CrossRef]
- Hanpude, P.; Bhattacharya, S.; Dey, A.K.; Maiti, T.K. Deubiquitinating enzymes in cellular signaling and disease regulation. IUBMB Life 2015, 67, 544–555. [Google Scholar] [CrossRef]
- Clague, M.J.; Coulson, J.M.; Urbe, S. Cellular functions of the dubs. J. Cell. Sci. 2012, 125, 277–286. [Google Scholar] [CrossRef]
- Bailey-Elkin, B.A.; Knaap, R.C.M.; Kikkert, M.; Mark, B.L. Structure and function of viral deubiquitinating enzymes. J. Mol. Biol. 2017. [Google Scholar] [CrossRef]
- Dong, X.; Guan, J.; Zheng, C.; Zheng, X. The herpes simplex virus 1 UL36USP deubiquitinase suppresses DNA repair in host cells via deubiquitination of proliferating cell nuclear antigen. J. Biol. Chem. 2017, 292, 8472–8483. [Google Scholar] [CrossRef]
- Ye, R.; Su, C.; Xu, H.; Zheng, C. Herpes simplex virus 1 ubiquitin-specific protease UL36 abrogates NF-κb activation in DNA sensing signal pathway. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Kim, Y.E.; Oh, S.E.; Kwon, K.M.; Lee, C.H.; Ahn, J.H. Involvement of the N-terminal deubiquitinating protease domain of human cytomegalovirus UL48 tegument protein in autoubiquitination, virion stability, and virus entry. J. Virol. 2016, 90, 3229–3242. [Google Scholar] [CrossRef]
- van Gent, M.; Braem, S.G.; de Jong, A.; Delagic, N.; Peeters, J.G.; Boer, I.G.; Moynagh, P.N.; Kremmer, E.; Wiertz, E.J.; Ovaa, H.; et al. Epstein-barr virus large tegument protein BPLF1 contributes to innate immune evasion through interference with Toll-like receptor signaling. PLoS Pathog. 2014, 10, e1003960. [Google Scholar] [CrossRef] [Green Version]
- Whitehurst, C.B.; Ning, S.; Bentz, G.L.; Dufour, F.; Gershburg, E.; Shackelford, J.; Langelier, Y.; Pagano, J.S. The epstein-barr virus (EBV) deubiquitinating enzyme BPLF1 reduces ebv ribonucleotide reductase activity. J. Virol. 2009, 83, 4345–4353. [Google Scholar] [CrossRef]
- Mattern, M.R.; Wu, J.; Nicholson, B. Ubiquitin-based anticancer therapy: Carpet bombing with proteasome inhibitors vs surgical strikes with E1, E2, E3, or DUB inhibitors. Biochim. Biophys. Acta 2012, 1823, 2014–2021. [Google Scholar] [CrossRef]
- D’Arcy, P.; Brnjic, S.; Olofsson, M.H.; Fryknas, M.; Lindsten, K.; De Cesare, M.; Perego, P.; Sadeghi, B.; Hassan, M.; Larsson, R.; et al. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat. Med. 2011, 17, 1636–1640. [Google Scholar]
- Pal, A.; Young, M.A.; Donato, N.J. Emerging potential of therapeutic targeting of ubiquitin-specific proteases in the treatment of cancer. Cancer Res. 2014, 74, 4955–4966. [Google Scholar] [CrossRef]
- Lopez-Castejon, G.; Edelmann, M.J. Deubiquitinases: Novel therapeutic targets in immune surveillance? Mediators Inflamm. 2016, 2016, 3481371. [Google Scholar] [CrossRef]
- Isaacson, M.K.; Ploegh, H.L. Ubiquitination, ubiquitin-like modifiers, and deubiquitination in viral infection. Cell Host Microbe 2009, 5, 559–570. [Google Scholar] [CrossRef]
- Perry, J.W.; Ahmed, M.; Chang, K.O.; Donato, N.J.; Showalter, H.D.; Wobus, C.E. Antiviral activity of a small molecule deubiquitinase inhibitor occurs via induction of the unfolded protein response. PLoS Pathog. 2012, 8, e1002783. [Google Scholar] [CrossRef]
- Nag, D.K.; Finley, D. A small-molecule inhibitor of deubiquitinating enzyme USP14 inhibits dengue virus replication. Virus Res. 2012, 165, 103–106. [Google Scholar] [CrossRef]
- Ching, W.; Koyuncu, E.; Singh, S.; Arbelo-Roman, C.; Hartl, B.; Kremmer, E.; Speiseder, T.; Meier, C.; Dobner, T. A ubiquitin-specific protease possesses a decisive role for adenovirus replication and oncogene-mediated transformation. PLoS Pathog. 2013, 9, e1003273. [Google Scholar] [CrossRef]
- Gonzalez-Hernandez, M.J.; Pal, A.; Gyan, K.E.; Charbonneau, M.E.; Showalter, H.D.; Donato, N.J.; O’Riordan, M.; Wobus, C.E. Chemical derivatives of a small molecule deubiquitinase inhibitor have antiviral activity against several RNA viruses. PLoS ONE 2014, 9, e94491. [Google Scholar] [CrossRef]
- Ratia, K.; Pegan, S.; Takayama, J.; Sleeman, K.; Coughlin, M.; Baliji, S.; Chaudhuri, R.; Fu, W.; Prabhakar, B.S.; Johnson, M.E.; et al. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl. Acad. Sci. USA 2008, 105, 16119–16124. [Google Scholar] [CrossRef]
- Putterman, D.; Pepinsky, R.B.; Vogt, V.M. Ubiquitin in avian leukosis virus particles. Virology 1990, 176, 633–637. [Google Scholar] [CrossRef]
- Martin-Serrano, J. The role of ubiquitin in retroviral egress. Traffic 2007, 8, 1297–1303. [Google Scholar] [CrossRef]
- Vogt, V.M. Ubiquitin in retrovirus assembly: Actor or bystander? Proc. Natl. Acad. Sci. USA 2000, 97, 12945–12947. [Google Scholar] [CrossRef]
- Jager, S.; Gottwein, E.; Krausslich, H.G. Ubiquitination of human immunodeficiency virus type 1 Gag is highly dependent on gag membrane association. J. Virol. 2007, 81, 9193–9201. [Google Scholar] [CrossRef]
- Hahn, S.; Setz, C.; Wild, J.; Schubert, U. The PTAP sequence within the p6 domain of human immunodeficiency virus type 1 Gag regulates its ubiquitination and MHC class I antigen presentation. J. Immunol. 2011, 186, 5706–5718. [Google Scholar] [CrossRef]
- Ott, D.E.; Coren, L.V.; Copeland, T.D.; Kane, B.P.; Johnson, D.G.; Sowder, R.C., II; Yoshinaka, Y.; Oroszlan, S.; Arthur, L.O.; Henderson, L.E. Ubiquitin is covalently attached to the p6Gag proteins of human immunodeficiency virus type 1 and simian immunodeficiency virus and to the p12Gag protein of moloney murine leukemia virus. J. Virol. 1998, 72, 2962–2968. [Google Scholar]
- Gurer, C.; Berthoux, L.; Luban, J. Covalent modification of human immunodeficiency virus type 1 p6 by SUMO-1. J. Virol. 2005, 79, 910–917. [Google Scholar] [CrossRef]
- Ott, D.E.; Coren, L.V.; Chertova, E.N.; Gagliardi, T.D.; Schubert, U. Ubiquitination of HIV-1 and MuLV Gag. Virology 2000, 278, 111–121. [Google Scholar] [CrossRef]
- Gottwein, E.; Krausslich, H.G. Analysis of human immunodeficiency virus type 1 Gag ubiquitination. J. Virol. 2005, 79, 9134–9144. [Google Scholar] [CrossRef]
- Gottwein, E.; Jager, S.; Habermann, A.; Krausslich, H.G. Cumulative mutations of ubiquitin acceptor sites in human immunodeficiency virus type 1 Gag cause a late budding defect. J. Virol. 2006, 80, 6267–6275. [Google Scholar] [CrossRef]
- Hahn, F.; Setz, C.; Friedrich, M.; Rauch, P.; Solbak, S.M.; Froystein, N.A.; Henklein, P.; Votteler, J.; Fossen, T.; Schubert, U. Mutation of the highly conserved SER-40 of the HIV-1 p6 Gag protein to phe causes the formation of a hydrophobic patch, enhances membrane association, and polyubiquitination of Gag. Viruses 2014, 6, 3738–3765. [Google Scholar] [CrossRef]
- Friedrich, M.; Setz, C.; Hahn, F.; Matthaei, A.; Fraedrich, K.; Rauch, P.; Henklein, P.; Traxdorf, M.; Fossen, T.; Schubert, U. Glutamic acid residues in HIV-1 p6 regulate virus budding and membrane association of Gag. Viruses 2016, 8, 117. [Google Scholar] [CrossRef]
- Wei, M.; Zhao, X.; Liu, M.; Huang, Z.; Xiao, Y.; Niu, M.; Shao, Y.; Kleiman, L. Inhibition of HIV-1 assembly by coiled-coil domain containing protein 8 in human cells. Sci. Rep. 2015, 5, 14724. [Google Scholar] [CrossRef]
- Amit, I.; Yakir, L.; Katz, M.; Zwang, Y.; Marmor, M.D.; Citri, A.; Shtiegman, K.; Alroy, I.; Tuvia, S.; Reiss, Y.; et al. Tal, a TSG101-specific E3 ubiquitin ligase, regulates receptor endocytosis and retrovirus budding. Genes Dev. 2004, 18, 1737–1752. [Google Scholar] [CrossRef]
- Kim, B.Y.; Olzmann, J.A.; Barsh, G.S.; Chin, L.S.; Li, L. Spongiform neurodegeneration-associated E3 ligase mahogunin ubiquitylates TSG101 and regulates endosomal trafficking. Mol. Biol. Cell. 2007, 18, 1129–1142. [Google Scholar] [CrossRef]
- Alroy, I.; Tuvia, S.; Greener, T.; Gordon, D.; Barr, H.M.; Taglicht, D.; Mandil-Levin, R.; Ben-Avraham, D.; Konforty, D.; Nir, A.; et al. The trans-golgi network-associated human ubiquitin-protein ligase posh is essential for HIV type 1 production. Proc. Natl. Acad. Sci. USA 2005, 102, 1478–1483. [Google Scholar] [CrossRef]
- Schubert, U.; Ott, D.E.; Chertova, E.N.; Welker, R.; Tessmer, U.; Princiotta, M.F.; Bennink, J.R.; Krausslich, H.G.; Yewdell, J.W. Proteasome inhibition interferes with Gag polyprotein processing, release, and maturation of HIV-1 and HIV-2. Proc. Natl. Acad. Sci. USA 2000, 97, 13057–13062. [Google Scholar] [CrossRef]
- Sundquist, W.I.; Krausslich, H.G. HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2012, 2, a006924. [Google Scholar] [CrossRef]
- Freed, E.O. HIV-1 assembly, release and maturation. Nat. Rev. Microbiol. 2015, 13, 484–496. [Google Scholar] [CrossRef]
- Wills, J.W.; Craven, R.C. Form, function, and use of retroviral Gag proteins. AIDS 1991, 5, 639–654. [Google Scholar] [CrossRef]
- Adachi, A.; Gendelman, H.E.; Koenig, S.; Folks, T.; Willey, R.; Rabson, A.; Martin, M.A. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 1986, 59, 284–291. [Google Scholar]
- Khan, M.A.; Aberham, C.; Kao, S.; Akari, H.; Gorelick, R.; Bour, S.; Strebel, K. Human immunodeficiency virus type 1 VIF protein is packaged into the nucleoprotein complex through an interaction with viral genomic RNA. J. Virol. 2001, 75, 7252–7265. [Google Scholar] [CrossRef]
- Papkalla, A.; Munch, J.; Otto, C.; Kirchhoff, F. NEF enhances human immunodeficiency virus type 1 infectivity and replication independently of viral coreceptor tropism. J. Virol. 2002, 76, 8455–8459. [Google Scholar] [CrossRef]
- Ratner, L.; Fisher, A.; Jagodzinski, L.L.; Mitsuya, H.; Liou, R.S.; Gallo, R.C.; Wong-Staal, F. Complete nucleotide sequences of functional clones of the AIDS virus. AIDS Res. Hum. Retroviruses 1987, 3, 57–69. [Google Scholar] [CrossRef]
- Graf, M.; Bojak, A.; Deml, L.; Bieler, K.; Wolf, H.; Wagner, R. Concerted action of multiple cis-acting sequences is required for REV dependence of late human immunodeficiency virus type 1 gene expression. J. Virol. 2000, 74, 10822–10826. [Google Scholar] [CrossRef]
- Deml, L.; Bojak, A.; Steck, S.; Graf, M.; Wild, J.; Schirmbeck, R.; Wolf, H.; Wagner, R. Multiple effects of codon usage optimization on expression and immunogenicity of DNA candidate vaccines encoding the human immunodeficiency virus type 1 Gag protein. J. Virol. 2001, 75, 10991–11001. [Google Scholar] [CrossRef]
- Goldwich, A.; Hahn, S.S.; Schreiber, S.; Meier, S.; Kampgen, E.; Wagner, R.; Lutz, M.B.; Schubert, U. Targeting HIV-1 Gag into the defective ribosomal product pathway enhances MHC class I antigen presentation and CD8+ T cell activation. J. Immunol. 2008, 180, 372–382. [Google Scholar] [CrossRef]
- Rotzschke, O.; Falk, K.; Stevanovic, S.; Jung, G.; Walden, P.; Rammensee, H.G. Exact prediction of a natural T cell epitope. Eur. J. Immunol. 1991, 21, 2891–2894. [Google Scholar] [CrossRef]
- Tschochner, M.; Schwingel, E.; Thein, C.; Wittmann, S.; Paatz, C.; Walter, H. Superiority of infectivity-based over particle-based methods for quantitation of drug resistant HIV-1 as inocula for cell cultures. J. Virol. Methods 2007, 141, 87–96. [Google Scholar] [CrossRef]
- Walter, H.; Low, P.; Harrer, T.; Schmitt, M.; Schwingel, E.; Tschochner, M.; Helm, M.; Korn, K.; Uberla, K.; Schmidt, B. No evidence for persistence of multidrug-resistant viral strains after a 7-month treatment interruption in an HIV-1-infected individual. J. Acquir. Immune Defic. Syndr. 2002, 31, 137–146. [Google Scholar] [CrossRef]
- Glushakova, S.; Baibakov, B.; Margolis, L.B.; Zimmerberg, J. Infection of human tonsil histocultures: A model for HIV pathogenesis. Nat. Med. 1995, 1, 1320–1322. [Google Scholar] [CrossRef]
- Glushakova, S.; Baibakov, B.; Zimmerberg, J.; Margolis, L.B. Experimental HIV infection of human lymphoid tissue: Correlation of CD4+ T cell depletion and virus syncytium-inducing/non-syncytium-inducing phenotype in histocultures inoculated with laboratory strains and patient isolates of HIV type 1. AIDS Res. Hum. Retroviruses 1997, 13, 461–471. [Google Scholar] [CrossRef]
- Aida; Version 4.22.034; Raytest: Straubenhardt, Germany, 2008.
- Konvalinka, J.; Litterst, M.A.; Welker, R.; Kottler, H.; Rippmann, F.; Heuser, A.M.; Krausslich, H.G. An active-site mutation in the human immunodeficiency virus type 1 proteinase (PR) causes reduced pr activity and loss of pr-mediated cytotoxicity without apparent effect on virus maturation and infectivity. J. Virol. 1995, 69, 7180–7186. [Google Scholar]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage t4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Köhler, G.; Lindahl, K.F.; Heusser, C. Characterization of a monoclonal anti-H-2Kb antibody. In The Immune System; Karger Publishers: Basel, Switzerland, 1981; pp. 202–208. [Google Scholar]
- Willey, R.L.; Smith, D.H.; Lasky, L.A.; Theodore, T.S.; Earl, P.L.; Moss, B.; Capon, D.J.; Martin, M.A. In vitro mutagenesis identifies a region within the envelope gene of the human immunodeficiency virus that is critical for infectivity. J. Virol. 1988, 62, 139–147. [Google Scholar]
- Altun, M.; Kramer, H.B.; Willems, L.I.; McDermott, J.L.; Leach, C.A.; Goldenberg, S.J.; Kumar, K.G.; Konietzny, R.; Fischer, R.; Kogan, E.; et al. Activity-based chemical proteomics accelerates inhibitor development for deubiquitylating enzymes. Chem. Biol. 2011, 18, 1401–1412. [Google Scholar] [CrossRef]
- Chauhan, D.; Tian, Z.; Nicholson, B.; Kumar, K.G.; Zhou, B.; Carrasco, R.; McDermott, J.L.; Leach, C.A.; Fulcinniti, M.; Kodrasov, M.P.; et al. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell 2012, 22, 345–358. [Google Scholar] [CrossRef]
- Muller, B.; Anders, M.; Akiyama, H.; Welsch, S.; Glass, B.; Nikovics, K.; Clavel, F.; Tervo, H.M.; Keppler, O.T.; Krausslich, H.G. HIV-1 Gag processing intermediates trans-dominantly interfere with HIV-1 infectivity. J. Biol. Chem. 2009, 284, 29692–29703. [Google Scholar] [CrossRef]
- Eckstein, D.A.; Penn, M.L.; Korin, Y.D.; Scripture-Adams, D.D.; Zack, J.A.; Kreisberg, J.F.; Roederer, M.; Sherman, M.P.; Chin, P.S.; Goldsmith, M.A. HIV-1 actively replicates in naive CD4+ T cells residing within human lymphoid tissues. Immunity 2001, 15, 671–682. [Google Scholar] [CrossRef]
- Obermeier, M.; Pironti, A.; Berg, T.; Braun, P.; Daumer, M.; Eberle, J.; Ehret, R.; Kaiser, R.; Kleinkauf, N.; Korn, K.; et al. HIV-grade: A publicly available, rules-based drug resistance interpretation algorithm integrating bioinformatic knowledge. Intervirology 2012, 55, 102–107. [Google Scholar] [CrossRef]
- Yu, L.; Mohanram, V.; Simonson, O.E.; Smith, C.I.; Spetz, A.L.; Mohamed, A.J. Proteasome inhibitors block HIV-1 replication by affecting both cellular and viral targets. Biochem. Biophys. Res. Commun. 2009, 385, 100–105. [Google Scholar] [CrossRef]
- Adams, J.; Kauffman, M. Development of the proteasome inhibitor Velcade (bortezomib). Cancer Investig. 2004, 22, 304–311. [Google Scholar] [CrossRef]
- Muchamuel, T.; Basler, M.; Aujay, M.A.; Suzuki, E.; Kalim, K.W.; Lauer, C.; Sylvain, C.; Ring, E.R.; Shields, J.; Jiang, J.; et al. A selective inhibitor of the immunoproteasome subunit lmp7 blocks cytokine production and attenuates progression of experimental arthritis. Nat. Med. 2009, 15, 781–787. [Google Scholar] [CrossRef]
- Porgador, A.; Yewdell, J.W.; Deng, Y.; Bennink, J.R.; Germain, R.N. Localization, quantitation, and in situ detection of specific peptide-MHC class I complexes using a monoclonal antibody. Immunity 1997, 6, 715–726. [Google Scholar] [CrossRef]
- York, I.A.; Chang, S.C.; Saric, T.; Keys, J.A.; Favreau, J.M.; Goldberg, A.L.; Rock, K.L. The ER aminopeptidase ERAP1 enhances or limits antigen presentation by trimming epitopes to 8–9 residues. Nat. Immunol. 2002, 3, 1177–1184. [Google Scholar] [CrossRef]
- Strack, B.; Calistri, A.; Accola, M.A.; Palu, G.; Gottlinger, H.G. A role for ubiquitin ligase recruitment in retrovirus release. Proc. Natl. Acad. Sci. USA 2000, 97, 13063–13068. [Google Scholar] [CrossRef]
- Patnaik, A.; Chau, V.; Wills, J.W. Ubiquitin is part of the retrovirus budding machinery. Proc. Natl. Acad. Sci. USA 2000, 97, 13069–13074. [Google Scholar] [CrossRef]
- Garrus, J.E.; von Schwedler, U.K.; Pornillos, O.W.; Morham, S.G.; Zavitz, K.H.; Wang, H.E.; Wettstein, D.A.; Stray, K.M.; Cote, M.; Rich, R.L.; et al. TSG101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 2001, 107, 55–65. [Google Scholar] [CrossRef]
- Sette, P.; Nagashima, K.; Piper, R.C.; Bouamr, F. Ubiquitin conjugation to Gag is essential for ESCRT-mediated HIV-1 budding. Retrovirology 2013, 10, 79. [Google Scholar] [CrossRef]
- Zhadina, M.; McClure, M.O.; Johnson, M.C.; Bieniasz, P.D. Ubiquitin-dependent virus particle budding without viral protein ubiquitination. Proc. Natl. Acad. Sci. USA 2007, 104, 20031–20036. [Google Scholar] [CrossRef]
- Zhadina, M.; Bieniasz, P.D. Functional interchangeability of late domains, late domain cofactors and ubiquitin in viral budding. PLoS Pathog. 2010, 6, e1001153. [Google Scholar] [CrossRef]
- Morita, E.; Sundquist, W.I. Retrovirus budding. Annu. Rev. Cell. Dev. Biol. 2004, 20, 395–425. [Google Scholar] [CrossRef]
- Shackelford, J.; Pagano, J.S. Tumor viruses and cell signaling pathways: Deubiquitination versus ubiquitination. Mol. Cell. Biol. 2004, 24, 5089–5093. [Google Scholar] [CrossRef]
- Furman, M.H.; Ploegh, H.L. Lessons from viral manipulation of protein disposal pathways. J. Clin. Investig. 2002, 110, 875–879. [Google Scholar] [CrossRef]
- Calistri, A.; Munegato, D.; Carli, I.; Parolin, C.; Palu, G. The ubiquitin-conjugating system: Multiple roles in viral replication and infection. Cells 2014, 3, 386–417. [Google Scholar] [CrossRef]
- Agromayor, M.; Martin-Serrano, J. Interaction of amsh with ESCRT-III and deubiquitination of endosomal cargo. J. Biol. Chem. 2006, 281, 23083–23091. [Google Scholar] [CrossRef]
- Zamborlini, A.; Usami, Y.; Radoshitzky, S.R.; Popova, E.; Palu, G.; Gottlinger, H. Release of autoinhibition converts ESCRT-III components into potent inhibitors of HIV-1 budding. Proc. Natl. Acad. Sci. USA 2006, 103, 19140–19145. [Google Scholar] [CrossRef]
- D’Arcy, P.; Linder, S. Proteasome deubiquitinases as novel targets for cancer therapy. Int. J. Biochem. Cell Biol. 2012, 44, 1729–1738. [Google Scholar] [CrossRef]
- Daviet, L.; Colland, F. Targeting ubiquitin specific proteases for drug discovery. Biochimie 2008, 90, 270–283. [Google Scholar] [CrossRef]
- Liu, J.; Shaik, S.; Dai, X.; Wu, Q.; Zhou, X.; Wang, Z.; Wei, W. Targeting the ubiquitin pathway for cancer treatment. Biochim. Biophys. Acta 2015, 1855, 50–60. [Google Scholar] [CrossRef]
- Nicholson, B.; Marblestone, J.G.; Butt, T.R.; Mattern, M.R. Deubiquitinating enzymes as novel anticancer targets. Future Oncol. 2007, 3, 191–199. [Google Scholar] [CrossRef]
- Maartens, G.; Celum, C.; Lewin, S.R. HIV infection: Epidemiology, pathogenesis, treatment, and prevention. Lancet 2014, 384, 258–271. [Google Scholar] [CrossRef]
- Cuevas, J.M.; Geller, R.; Garijo, R.; Lopez-Aldeguer, J.; Sanjuan, R. Extremely high mutation rate of HIV-1 in vivo. PLoS Biol. 2015, 13, e1002251. [Google Scholar] [CrossRef]
- Drake, J.W.; Charlesworth, B.; Charlesworth, D.; Crow, J.F. Rates of spontaneous mutation. Genetics 1998, 148, 1667–1686. [Google Scholar]
- Piao, J.; Tashiro, A.; Nishikawa, M.; Aoki, Y.; Moriyoshi, E.; Hattori, A.; Kakeya, H. Expression, purification and enzymatic characterization of a recombinant human ubiquitin-specific protease 47. J. Biochem. 2015, 158, 477–484. [Google Scholar] [CrossRef]
- Si, X.; Gao, G.; Wong, J.; Wang, Y.; Zhang, J.; Luo, H. Ubiquitination is required for effective replication of coxsackievirus B3. PLoS ONE 2008, 3, e2585. [Google Scholar] [CrossRef]
- Shields, S.B.; Piper, R.C. How ubiquitin functions with ESCRTS. Traffic 2011, 12, 1306–1317. [Google Scholar] [CrossRef]
- Joshi, A.; Munshi, U.; Ablan, S.D.; Nagashima, K.; Freed, E.O. Functional replacement of a retroviral late domain by ubiquitin fusion. Traffic 2008, 9, 1972–1983. [Google Scholar] [CrossRef]
- Keren-Kaplan, T.; Attali, I.; Estrin, M.; Kuo, L.S.; Farkash, E.; Jerabek-Willemsen, M.; Blutraich, N.; Artzi, S.; Peri, A.; Freed, E.O.; et al. Structure-based in silico identification of ubiquitin-binding domains provides insights into the ALIX-V:Ubiquitin complex and retrovirus budding. EMBO J. 2013, 32, 538–551. [Google Scholar] [CrossRef]
- Dowlatshahi, D.P.; Sandrin, V.; Vivona, S.; Shaler, T.A.; Kaiser, S.E.; Melandri, F.; Sundquist, W.I.; Kopito, R.R. ALIX is a Lys63-specific polyubiquitin binding protein that functions in retrovirus budding. Dev. Cell 2012, 23, 1247–1254. [Google Scholar] [CrossRef]
- Schubert, U.; Anton, L.C.; Gibbs, J.; Norbury, C.C.; Yewdell, J.W.; Bennink, J.R. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 2000, 404, 770–774. [Google Scholar]
- Ciechanover, A. Proteolysis: From the lysosome to ubiquitin and the proteasome. Nat. Rev. Mol. Cell. Biol. 2005, 6, 79–87. [Google Scholar] [CrossRef]
- Yewdell, J.W. Plumbing the sources of endogenous MHC class I peptide ligands. Curr. Opin. Immunol. 2007, 19, 79–86. [Google Scholar] [CrossRef]
- Qian, S.B.; Reits, E.; Neefjes, J.; Deslich, J.M.; Bennink, J.R.; Yewdell, J.W. Tight linkage between translation and MHC class I peptide ligand generation implies specialized antigen processing for defective ribosomal products. J. Immunol. 2006, 177, 227–233. [Google Scholar] [CrossRef]
- Grant, E.P.; Michalek, M.T.; Goldberg, A.L.; Rock, K.L. Rate of antigen degradation by the ubiquitin-proteasome pathway influences MHC class I presentation. J. Immunol. 1995, 155, 3750–3758. [Google Scholar]
- Princiotta, M.F.; Finzi, D.; Qian, S.B.; Gibbs, J.; Schuchmann, S.; Buttgereit, F.; Bennink, J.R.; Yewdell, J.W. Quantitating protein synthesis, degradation, and endogenous antigen processing. Immunity 2003, 18, 343–354. [Google Scholar] [CrossRef]
- Sijts, A.; Zaiss, D.; Kloetzel, P.M. The role of the ubiquitin-proteasome pathway in MHC class I antigen processing: Implications for vaccine design. Curr. Mol. Med. 2001, 1, 665–676. [Google Scholar] [CrossRef]
- Tobery, T.; Siliciano, R.F. Cutting edge: Induction of enhanced CTL-dependent protective immunity in vivo by N-end rule targeting of a model tumor antigen. J. Immunol. 1999, 162, 639–642. [Google Scholar]
- Townsend, A.; Bastin, J.; Gould, K.; Brownlee, G.; Andrew, M.; Coupar, B.; Boyle, D.; Chan, S.; Smith, G. Defective presentation to class I-restricted cytotoxic T lymphocytes in vaccinia-infected cells is overcome by enhanced degradation of antigen. J. Exp. Med. 1988, 168, 1211–1224. [Google Scholar] [CrossRef]
- Wong, S.B.; Buck, C.B.; Shen, X.; Siliciano, R.F. An evaluation of enforced rapid proteasomal degradation as a means of enhancing vaccine-induced ctl responses. J. Immunol. 2004, 173, 3073–3083. [Google Scholar] [CrossRef]
- Starodubova, E.; Boberg, A.; Kashuba, E.V.; Wahren, B.; Karpov, V.; Isaguliants, M. HIV-1 reverse transcriptase targeted for proteasomal degradation as a prototype vaccine against drug-resistant HIV-1. Vaccine 2006, 24, 4541–4547. [Google Scholar] [CrossRef]
- Tellam, J.; Connolly, G.; Webb, N.; Duraiswamy, J.; Khanna, R. Proteasomal targeting of a viral oncogene abrogates oncogenic phenotype and enhances immunogenicity. Blood 2003, 102, 4535–4540. [Google Scholar] [CrossRef]
- Rodriguez, F.; Zhang, J.; Whitton, J.L. DNA immunization: Ubiquitination of a viral protein enhances cytotoxic T-lymphocyte induction and antiviral protection but abrogates antibody induction. J. Virol. 1997, 71, 8497–8503. [Google Scholar]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Setz, C.; Friedrich, M.; Rauch, P.; Fraedrich, K.; Matthaei, A.; Traxdorf, M.; Schubert, U. Inhibitors of Deubiquitinating Enzymes Block HIV-1 Replication and Augment the Presentation of Gag-Derived MHC-I Epitopes. Viruses 2017, 9, 222. https://doi.org/10.3390/v9080222
Setz C, Friedrich M, Rauch P, Fraedrich K, Matthaei A, Traxdorf M, Schubert U. Inhibitors of Deubiquitinating Enzymes Block HIV-1 Replication and Augment the Presentation of Gag-Derived MHC-I Epitopes. Viruses. 2017; 9(8):222. https://doi.org/10.3390/v9080222
Chicago/Turabian StyleSetz, Christian, Melanie Friedrich, Pia Rauch, Kirsten Fraedrich, Alina Matthaei, Maximilian Traxdorf, and Ulrich Schubert. 2017. "Inhibitors of Deubiquitinating Enzymes Block HIV-1 Replication and Augment the Presentation of Gag-Derived MHC-I Epitopes" Viruses 9, no. 8: 222. https://doi.org/10.3390/v9080222