Identification of Alpha and Beta Coronavirus in Wildlife Species in France: Bats, Rodents, Rabbits, and Hedgehogs

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
- -
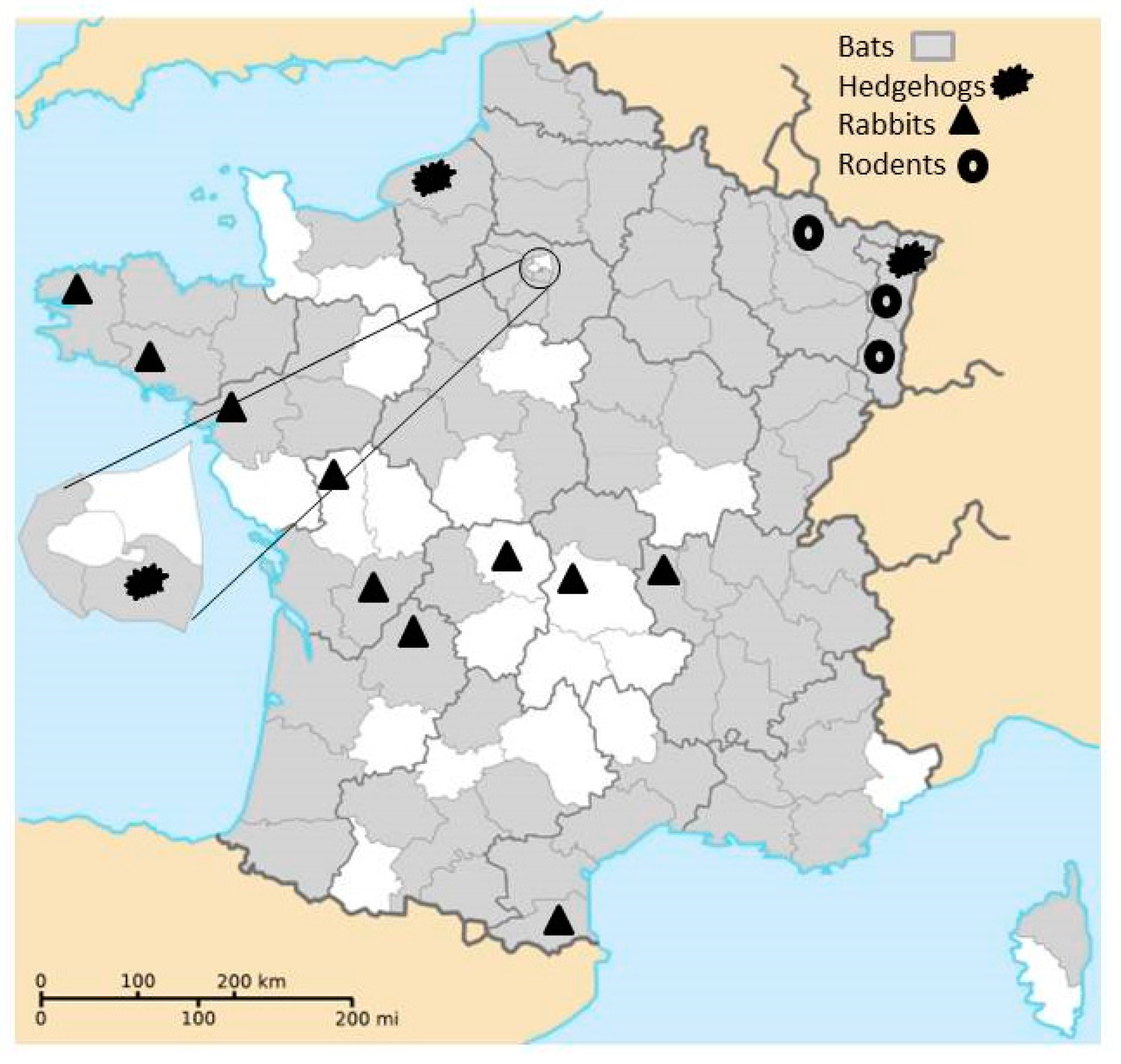
- From 2013 to 2015, intestinal samples from carcasses of bats were collected within the lyssaviruses surveillance in France and originated from 74 of the 101 French administrative department (with only one overseas French department concerned, French Guyana, Figure 1).
- -
- From 2014 to 2016, rodents were trapped in five areas of eastern France (Figure 1) in the context of other studies. Rodents were captured using pieces of carrot and sunflower seeds as bait in different kinds of traps adapted to species. Captured animals were euthanized in accordance with the French Animal Protection Law and Directive 2010/63/EU of the European Parliament and of the Council on the protection of animals used for scientific purposes (identification code of the approval (29 September 2016) project AP AFIS#2939-20160106142231, name of the ethics committee: Cometh).
- -
- From 2007 to 2009, intestinal samples from rabbits (Oryctolagus cuniculus) collected in 10 departments by the French National Hunting and Wildlife Agency for another research project were analyzed (Figure 1). Intestinal samples were taken from animals killed by hunters during rabbit hunting. Therefore, no wild animal was killed specifically for the purpose of this study.
2.2. RNA Extraction and RT-PCR of the Partial RNA-Dependent RNA Polymerase Protein (RdRp) Gene
2.3. Phylogenetic Analysis
2.4. Statistical Analysis
3. Results
3.1. Bats
3.2. Hedgehogs
3.3. Rodents
3.4. Rabbits
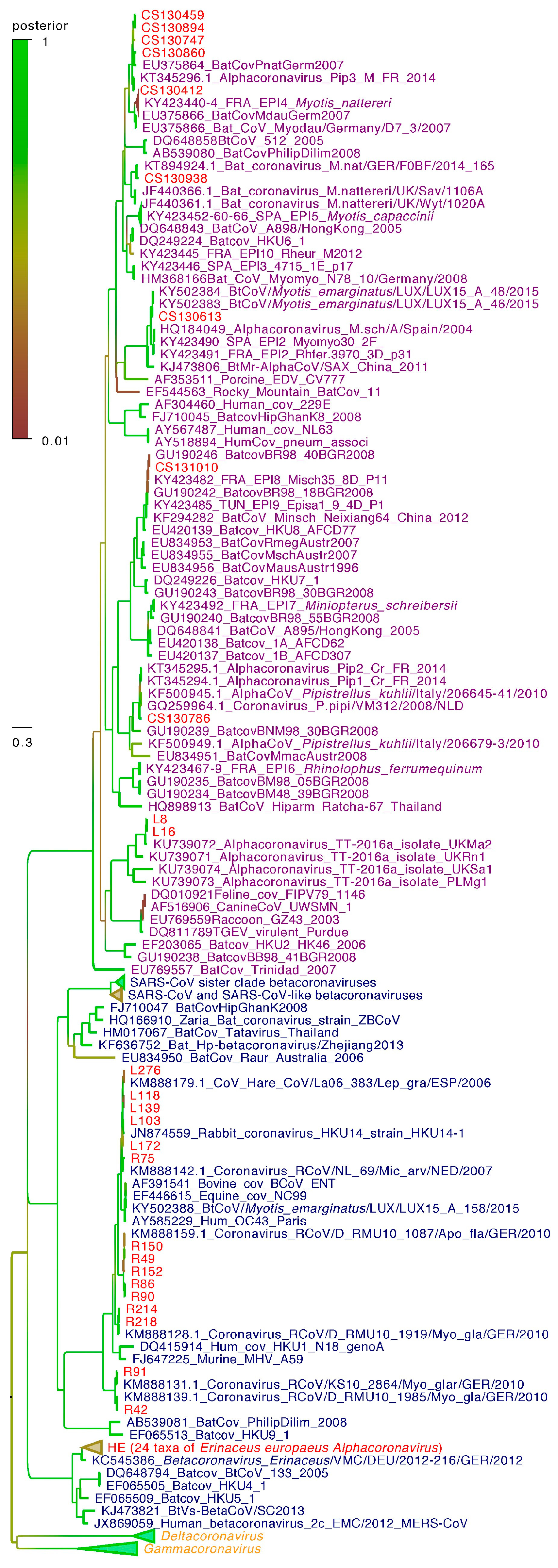
3.5. Phylogenetic Diversity
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Annan, A.; Baldwin, H.J.; Corman, V.M.; Klose, S.M.; Owusu, M.; Nkrumah, E.E.; Badu, E.K.; Anti, P.; Agbenyega, O.; Meyer, B.; et al. Human betacoronavirus 2c EMC/2012-related viruses in bats, Ghana and Europe. Emerg. Infect. Dis. 2013, 19, 456–459. [Google Scholar] [CrossRef] [PubMed]
- Goffard, A.; Demanche, C.; Arthur, L.; Pinçon, C.; Michaux, J.; Dubuisson, J. Alphacoronaviruses detected in french bats are phylogeographically linked to coronaviruses of european bats. Viruses 2015, 7, 6279–6290. [Google Scholar] [CrossRef] [PubMed]
- Corman, V.M.; Kallies, R.; Philipps, H.; Göpner, G.; Müller, M.A.; Eckerle, I.; Brünink, S.; Drosten, C.; Drexler, J.F. Characterization of a novel betacoronavirus related to middle east respiratory syndrome coronavirus in european hedgehogs. J. Virol. 2014, 88, 717–724. [Google Scholar] [CrossRef] [PubMed]
- Tsoleridis, T.; Onianwa, O.; Horncastle, E.; Dayman, E.; Zhu, M.; Danjittrong, T.; Wachtl, M.; Behnke, J.M.; Chapman, S.; Strong, V.; et al. Discovery of novel alphacoronaviruses in European rodents and shrews. Viruses 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.P.; Woo, P.C.Y.; Yip, C.C.Y.; Fan, R.Y.Y.; Huang, Y.; Wang, M.; Guo, R.; Lam, C.S.F.; Tsang, A.K.L.; Lai, K.K.Y.; et al. Isolation and characterization of a novel Betacoronavirus subgroup a coronavirus, rabbit coronavirus HKU14, from domestic rabbits. J. Virol. 2012, 86, 5481–5496. [Google Scholar] [CrossRef] [PubMed]
- Lelli, D.; Papetti, A.; Sabelli, C.; Moreno, A.; Boniotti, M.B. Detection of coronaviruses in bats of various species in Italy. Viruses 2013, 5, 2679–2689. [Google Scholar] [CrossRef] [PubMed]
- Gouilh, M.A.; Puechmaille, S.J.; Gonzalez, J.P.; Teeling, E.; Kittayapong, P.; Manuguerra, J.C. SARS-Coronavirus ancestor’s foot-prints in South-East Asian bat colonies and the refuge theory. Infect. Genet. Evol. 2011, 11, 1690–1702. [Google Scholar] [CrossRef] [PubMed]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.D.; Cai, T.T.; DasGupta, A. Interval estimation for a binomial proportion. Stat. Sci. 2001, 16, 101–133. [Google Scholar]
- Van Gucht, S.; Nazé, F.; Kadaani, K.E.; Bauwens, D.; Francart, A.; Brochier, B.; Wuillaume, F.; Thomas, I. No evidence of coronavirus infection by reverse transcriptase-PCR in bats in Belgium. J. Wildl. Dis. 2014, 50, 969–971. [Google Scholar] [CrossRef] [PubMed]
- August, T.A.; Fensome, A.G.; Nunn, M.A. Alphacoronavirus detected in bats in the United Kingdom. Vector Borne Zoonotic Dis. 2012, 12, 530–533. [Google Scholar] [CrossRef] [PubMed]
- Pauly, M.; Pir, J.B.; Loesch, C.; Sausy, A.; Snoeck, C.J.; Hubschen, J.M.; Muller, C.P. Novel Alphacoronaviruses and Paramyxoviruses co-circulate with Type 1 and SARS-related Betacoronaviruses in synanthropic bats in Luxembourg. Appl. Environ. Microbial. 2017, 83. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Gloza-Rausch, F.; Glende, J.; Corman, V.M.; Muth, D.; Goettsche, M.; Seebens, A.; Niedrig, M.; Pfefferle, S.; Yordanov, S.; et al. Genomic characterization of severe acute respiratory syndrome-related coronavirus in European bats and classification of coronaviruses based on partial RNA-dependent RNA polymerase gene sequences. J. Virol. 2010, 84, 11336–11349. [Google Scholar] [CrossRef] [PubMed]
- Falcón, A.; Vázquez-Morón, S.; Casas, I.; Aznar, C.; Ruiz, G.; Pozo, F.; Perez-Breña, P.; Juste, J.; Ibáñez, C.; Garin, I.; et al. Detection of alpha and betacoronaviruses in multiple Iberian bat species. Arch. Virol. 2011, 156, 1883–1890. [Google Scholar] [CrossRef] [PubMed]
- Kemenesi, G.; Dallos, B.; Gorfol, T.; Boldogh, S.; Estok, P.; Kurucz, K.; Kutas, A.; Földes, F.; Oldal, M.; Németh, V.; et al. Molecular survey of RNA viruses in Hungarian bats: Discovering novel astroviruses, coronaviruses, and caliciviruses. Vector Borne Zoonotic Dis. 2014, 14, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Reusken, C.B.; Lina, P.H.; Pielaat, A.; de Vries, A.; Dam-Deisz, C.; Adema, J.; Drexler, J.F.; Drosten, C.; Kool, E.A. Circulation of group 2 coronaviruses in a bat species common to urban areas in Western Europe. Vector Borne Zoonotic Dis. 2010, 10, 785–791. [Google Scholar] [CrossRef] [PubMed]
- De Benedictis, P.; Marciano, S.; Scaravelli, D.; Priori, P.; Zecchin, B.; Capua, I.; Monne, I.; Cattoli, G. Alpha and lineage C betaCoV infections in Italian bats. Virus Genes 2014, 48, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.F.; Corman, V.M.; Wegner, T.; Tateno, A.F.; Zerbinati, R.M.; Gloza-Rausch, F.; Seebens, A.; Müller, M.A.; Drosten, C. Amplification of emerging viruses in a bat colony. Emerg. Infect. Dis. 2011, 17, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; Zeus, V.; Kwasnitschka, L.; Kerth, G.; Haase, M.; Groschup, M.H.; Balkema-Buschmann, A. Insectivorous bats carry host specific astroviruses and coronaviruses across different regions in Germany. Infect. Genet. Evol. 2016, 37, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Gloza-Rausch, F.; Ipsen, A.; Seebens, A.; Göttsche, M.; Panning, M.; Drexler, J.F.; Petersen, N.; Annan, A.; Grywna, K.; Müller, M.; et al. Detection and prevalence petterns of group I coronaviruses in bats, northern Germany. Emerg. Infect. Dis. 2008, 14, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Vijgen, L.; Keyaerts, E.; Moës, E.; Thoelen, I.; Wollants, E.; Lemey, P.; Vandamme, A.M.; Van Ranst, M. Complet genomic sequence of human coronavirus OC43: Molecular clock analysis suggests a relatively recent zoonotic coronavirus transmission event. J. Virol. 2005, 79, 1595–1604. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, M.; Bloch, J.I.; Flynn, J.J.; Gaudin, T.J.; Giallombardo, A.; Giannini, N.P.; Goldberg, S.L.; Kraatz, B.P.; Luo, Z.X.; Meng, J.; et al. The placental mammal ancestor and the post-K-Pg radiation of placentals. Science 2013, 339, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Anthony, S.J.; Johnson, C.K.; Greig, D.J.; Kramer, S.; Che, X.; Wells, H.; Hicks, A.L.; Joly, D.O.; Wolfe, N.D.; Daszak, P.; et al. Global patterns in coronavirus diversity. Virus Evol. 2017, 3. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.; Godeke, G.J.; Raamsman, M.J.; Masters, P.S.; Rottier, P.J. Retargeting of coronavirus by substitution of the spike glycoprotein ectodomain: Crossing the host cell species barrier. J. Virol. 2000, 74, 1393–1406. [Google Scholar] [CrossRef] [PubMed]
- Van Doremalen, N.; Miazgowicz, K.L.; Milne-Price, S.; Bushmaker, T.; Robertson, S.; Scott, D.; Kinne, J.; McLellan, J.S.; Zhu, J.; Munster, V.J. Host species restriction of Middle East respiratory syndrome coronavirus through its receptor, dipeptidyl peptidase 4. J. Virol. 2014, 88, 9220–9232. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Species | Number of Carcasses Total (Positive Samples) | |||
|---|---|---|---|---|
| 2013 | 2014 | 2015 | total | |
| Non-identified bats | 2 | 4 | 3 | 9 |
| Rhinolophus ferrumequinum | 1 | 0 | 1 | 2 |
| Rhinolophus hipposideros | 4 | 2 | 2 | 8 |
| Rhinolophus euryale | 0 | 0 | 3 | 3 |
| Barbastella barbatellus | 1 | 3 | 0 | 4 |
| Myotis myotis | 0 | 1 | 0 | 1 |
| Myotis blythii | 0 | 0 | 1 | 1 |
| Myotis mystacinus | 0 | 1 | 4 | 5 |
| Myotis emarginatus | 1 | 11 (2) | 1 | 13 |
| Myotis bechsteinii | 3 | 0 | 0 | 3 |
| Myotis daubentonii | 1 | 1 | 0 | 2 |
| Myotis nattereri | 0 | 0 | 1 (1) | 1 |
| Myotis sp. | 0 | 0 | 1 | 1 |
| Nyctalus noctula | 2 | 0 | 0 | 2 |
| Nyctalus leisleri | 2 | 9 | 4 | 15 |
| Plecotus austriacus | 4 | 4 | 2 | 10 |
| Plecotus auritus | 3 | 4 | 8 | 15 |
| Pipistrellus pipistrellus | 42 | 127 (4) | 78 (1) | 247 |
| Pipistrellus kuhlii | 9 | 20 | 11 | 40 |
| Pipistrellus nathusii | 2 | 20 | 5 | 27 |
| Pipistrellus pygmaeus | 17 | 3 | 3 | 23 |
| Pipistrellus sp. | 11 | 20 (2) | 7 | 38 |
| Vespertilio murinus | 0 | 1 | 1 | 2 |
| Eptesicus serotinus | 7 | 11 | 8 | 26 |
| Eptesicus nilssonii | 1 | 0 | 0 | 1 |
| Miniopterus schreibersii | 0 | 1 (1) | 4 (1) | 5 |
| Total | 113 | 243 (9) | 148 (3) | 504 (12) |
| Year | Species | Bitche Infected/Total | Forbach Infected/Total | La Petite Pierre Infected/Total | Murbach Infected/Total | Wissembourg Infected/Total | Prevalence by Species |
|---|---|---|---|---|---|---|---|
| 2014 | Apodemus sp. | NC | 6/82 | 0/4 | 0/0 | 0/0 | (6/86) 6.98 [3.24; 14.40] |
| Myodes glareolus | NC | 3/32 | 0/4 | 0/2 | 0/0 | (3/38) 7.89 [2.72; 20.80] | |
| Arvicola terrestris | NC | 0/0 | 0/0 | 0/0 | 0/16 | (0/16) 0 [0.00; 19.36] | |
| Microtus sp. | NC | 0/0 | 0/0 | 0/0 | 0/3 | (0/3) 0 [0.00; 56.15] | |
| Total | NC | 9/114 | 0/8 | 0/2 | 0/19 | ||
| 2015 | Apodemus sp. | 8/64 | NC | 1/38 | 0/2 | 0/0 | (9/104) 8.65 [4.62; 15.63] |
| Myodes glareolus | 1/7 | NC | 0/17 | 1/15 | 0/0 | (2/39) 5.13 [1.42; 16.89] | |
| Arvicola terrestris | 0/0 | NC | 0/0 | 0/0 | 0/18 | (0/18) 0 [0.00; 17.59] | |
| Microtus sp. | 0/6 | NC | 0/0 | 0/0 | 0/0 | (0/6) 0 [0.00; 39.30] | |
| Total | 9/77 | NC | 1/55 | 1/17 | 0/18 | ||
| 2016 | Apodemus sp. | 1/4 | NC | 0/11 | 0/1 | NC | (1/16) 6.25 [1.11; 28.33] |
| Myodes glareolus | 0/2 | NC | 0/0 | 0/1 | NC | (0/3) 0 [0.00; 56.15] | |
| Arvicola terrestris | 0/0 | NC | 0/1 | 0/0 | NC | (0/1) 0 [0.00; 79.35] | |
| Microtus sp. | 0/0 | NC | 0/0 | 0/0 | NC | NE | |
| Total | 1/6 | NC | 0/12 | 0/2 | NC | ||
| Total from 2014 to 2016 | Apodemus sp. | 9/68 | 6/82 | 1/53 | 0/3 | 0/0 | (16/206) 7.77 [4.84; 12.24] |
| Myodes glareolus | 1/9 | 3/32 | 0/21 | 1/18 | 0/0 | (5/80) 6.25 [2.70; 13.81] | |
| Arvicola terrestris | 0/0 | 0/0 | 0/1 | 0/0 | 0/34 | (0/35) 0 [0.00; 9.89] | |
| Microtus sp. | 0/6 | 0/0 | 0/0 | 0/0 | 0/3 | (0/9) 0 [0.00; 29.91] | |
| Total | 10/83 | 9/114 | 1/75 | 1/21 | 0/37 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monchatre-Leroy, E.; Boué, F.; Boucher, J.-M.; Renault, C.; Moutou, F.; Ar Gouilh, M.; Umhang, G. Identification of Alpha and Beta Coronavirus in Wildlife Species in France: Bats, Rodents, Rabbits, and Hedgehogs. Viruses 2017, 9, 364. https://doi.org/10.3390/v9120364
Monchatre-Leroy E, Boué F, Boucher J-M, Renault C, Moutou F, Ar Gouilh M, Umhang G. Identification of Alpha and Beta Coronavirus in Wildlife Species in France: Bats, Rodents, Rabbits, and Hedgehogs. Viruses. 2017; 9(12):364. https://doi.org/10.3390/v9120364
Chicago/Turabian StyleMonchatre-Leroy, Elodie, Franck Boué, Jean-Marc Boucher, Camille Renault, François Moutou, Meriadeg Ar Gouilh, and Gérald Umhang. 2017. "Identification of Alpha and Beta Coronavirus in Wildlife Species in France: Bats, Rodents, Rabbits, and Hedgehogs" Viruses 9, no. 12: 364. https://doi.org/10.3390/v9120364
APA StyleMonchatre-Leroy, E., Boué, F., Boucher, J.-M., Renault, C., Moutou, F., Ar Gouilh, M., & Umhang, G. (2017). Identification of Alpha and Beta Coronavirus in Wildlife Species in France: Bats, Rodents, Rabbits, and Hedgehogs. Viruses, 9(12), 364. https://doi.org/10.3390/v9120364