
Matrix Metalloproteinase-9 Mediates RSV Infection in Vitro and in Vivo

Abstract
:1. Introduction
2. Results
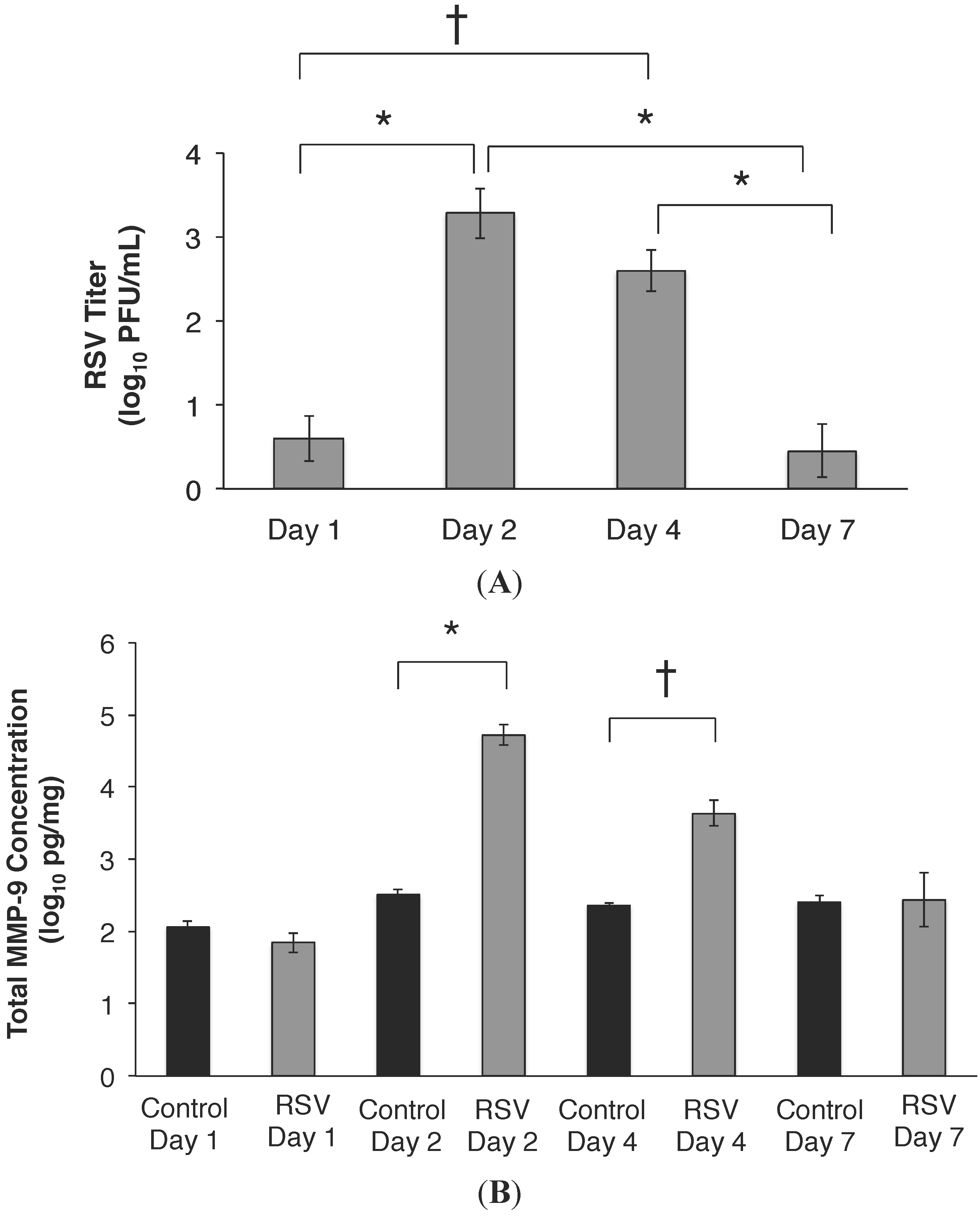
2.1. RSV Infection of 16HBE Cells Led to an Increase in MMP-9 mRNA and Protein Release
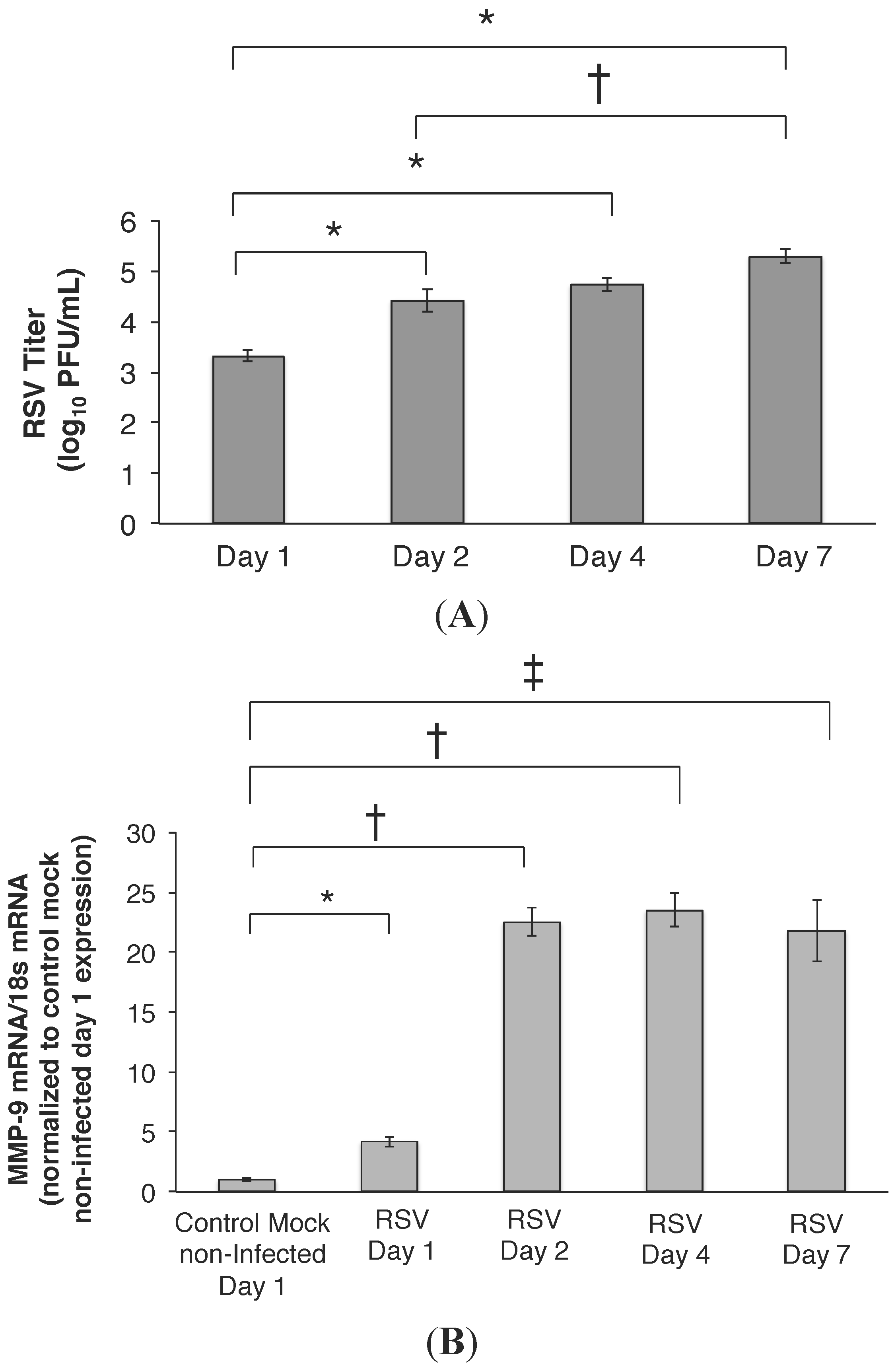
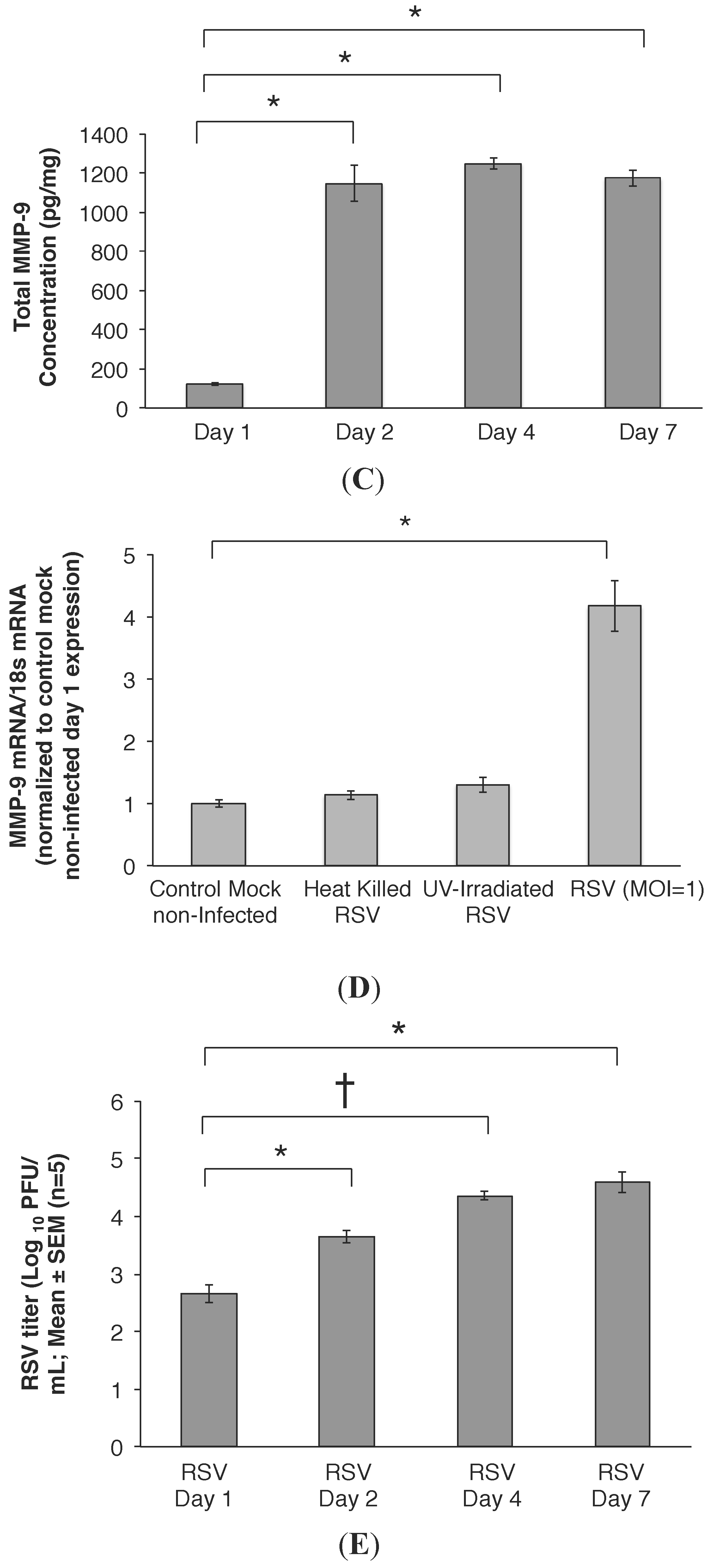

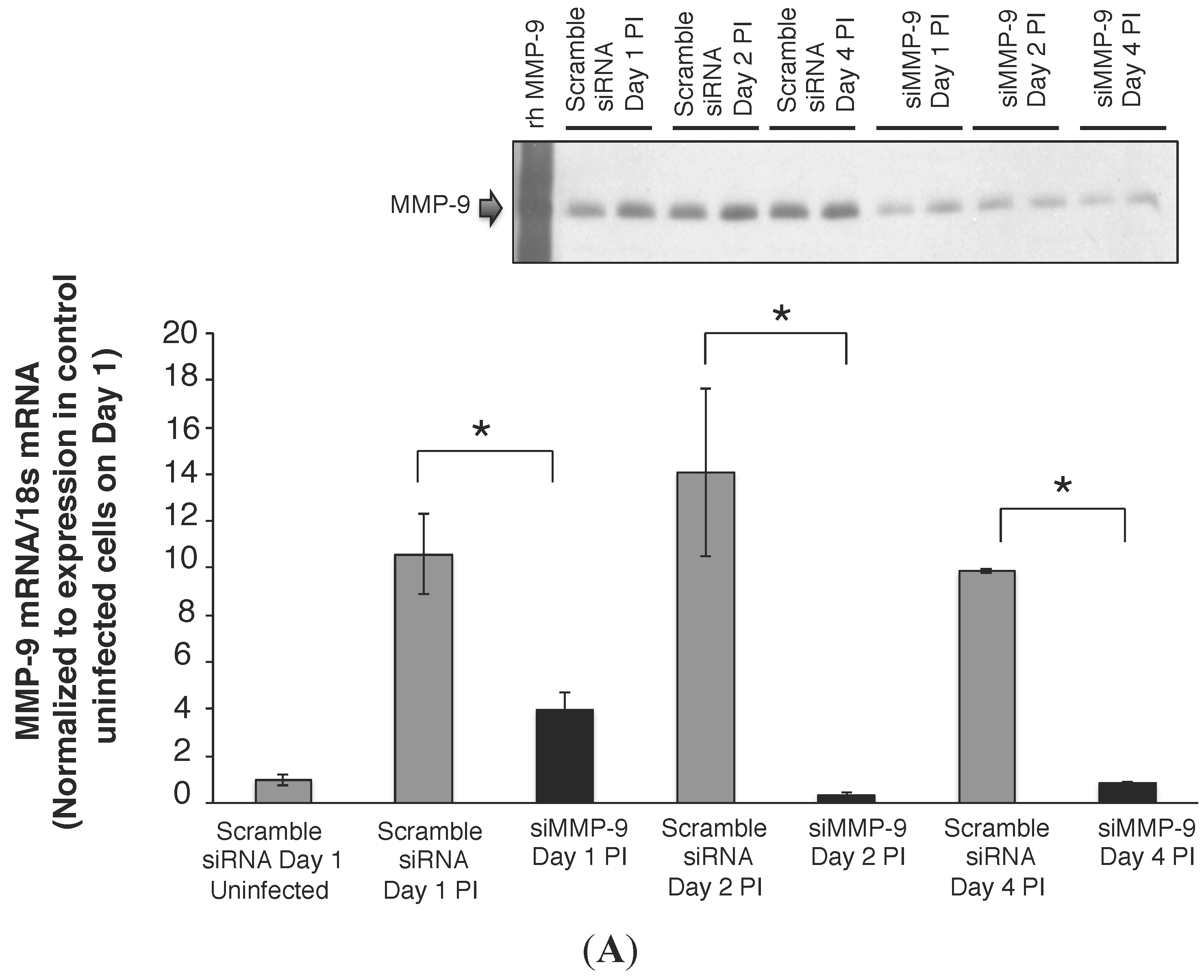
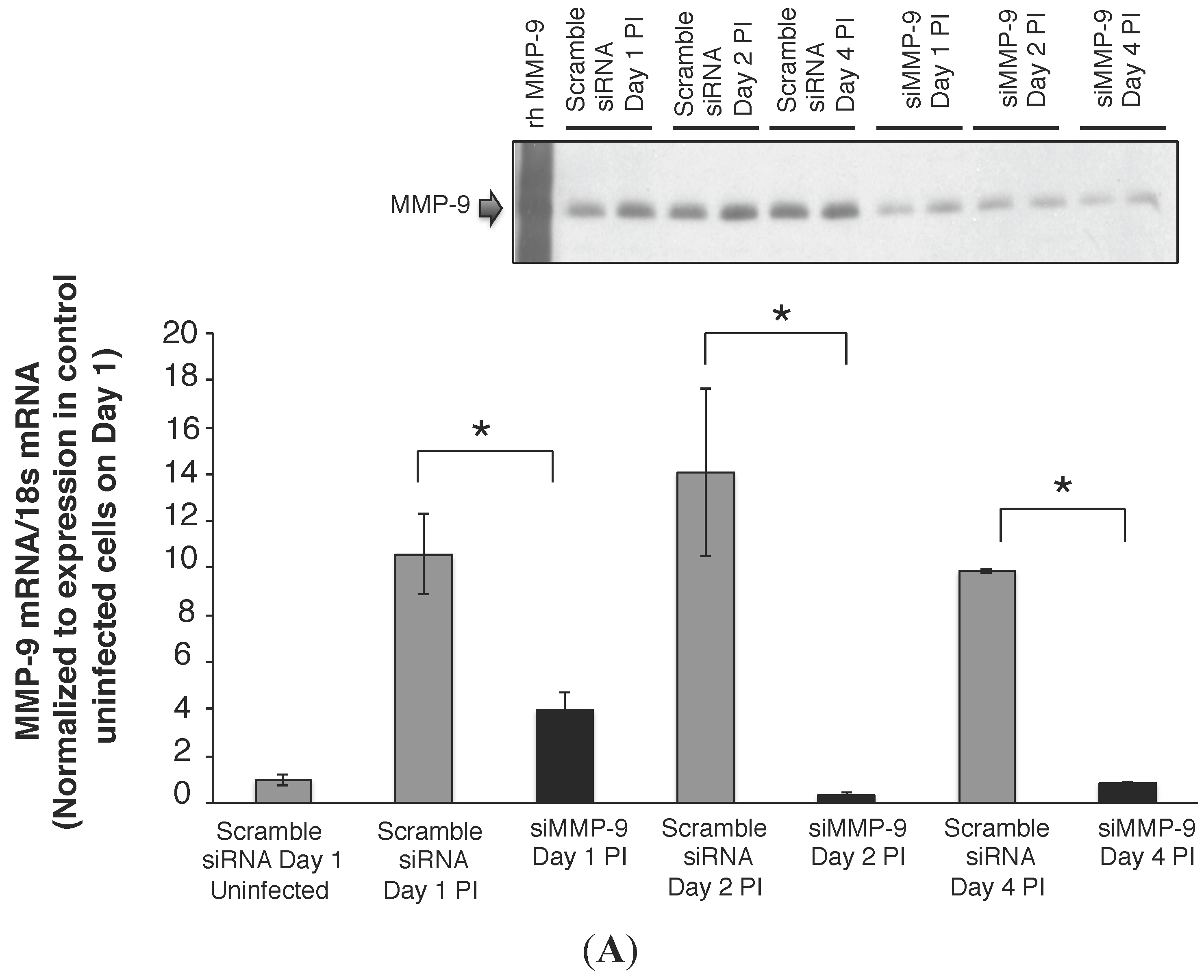
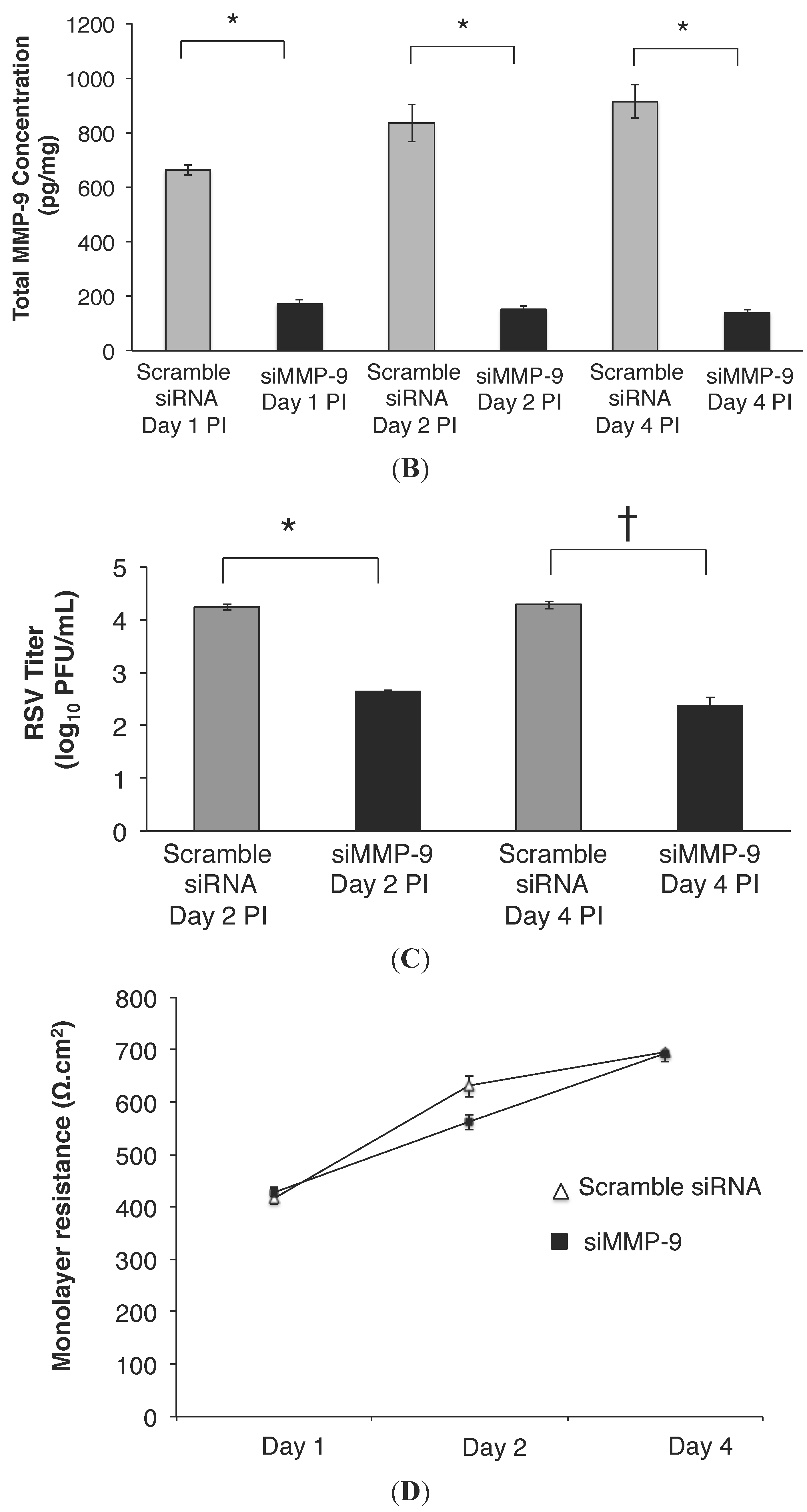
2.2. MMP-9 Knockdown in 16HBE Cells Decreased RSV Titer

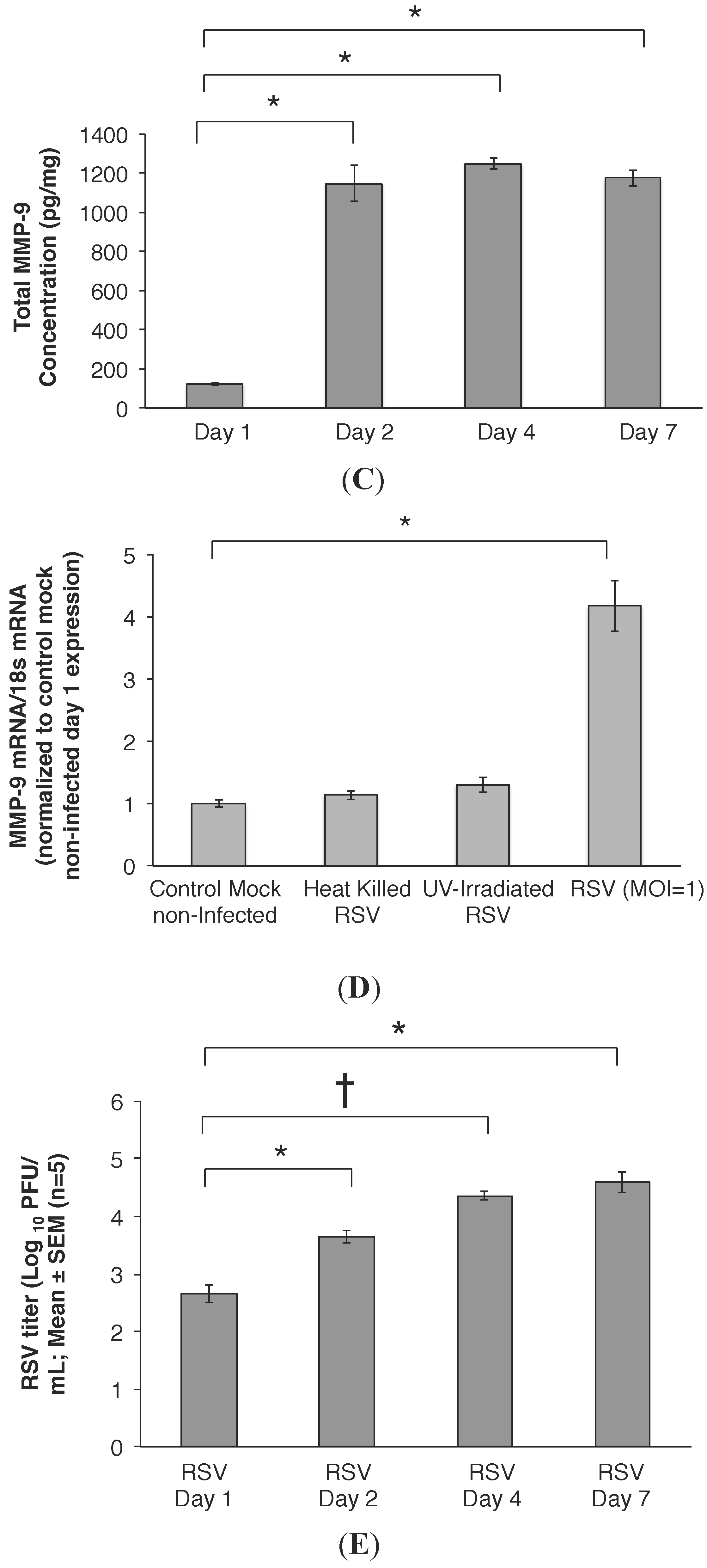
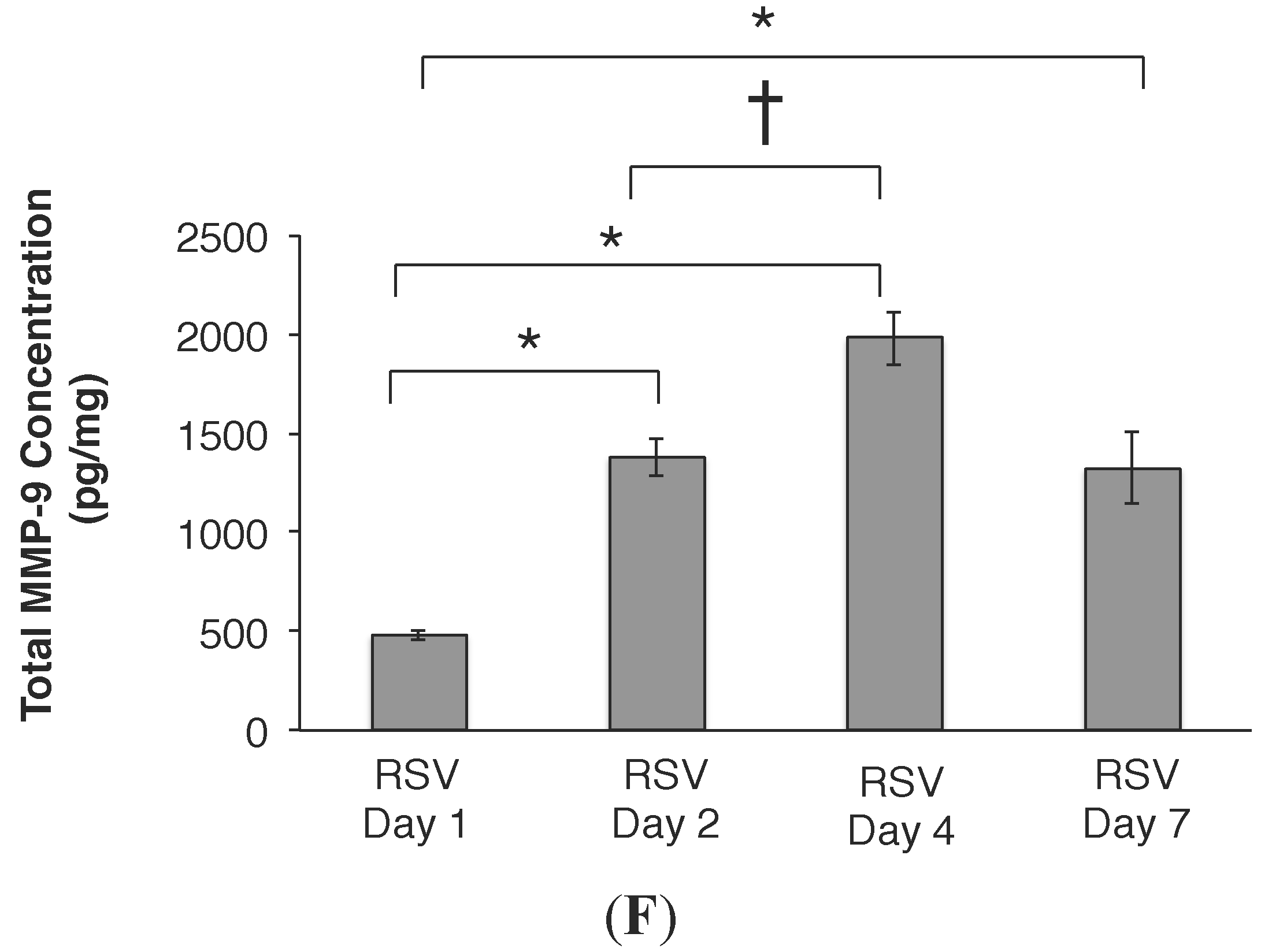
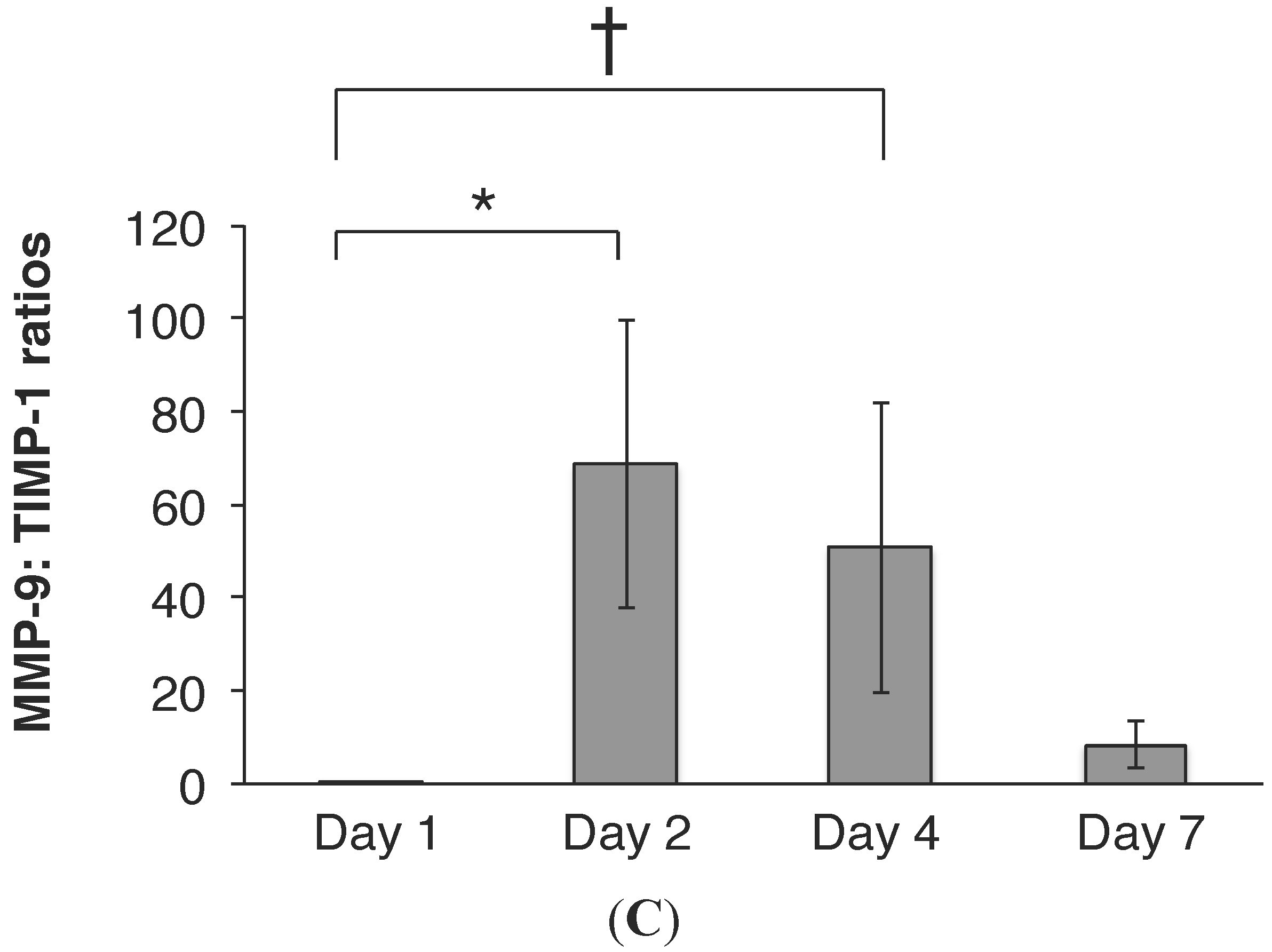
2.3. RSV Infection Led to an Early Increase in MMP-9 and MMP-9:TIMP-1 Ratios in Vivo
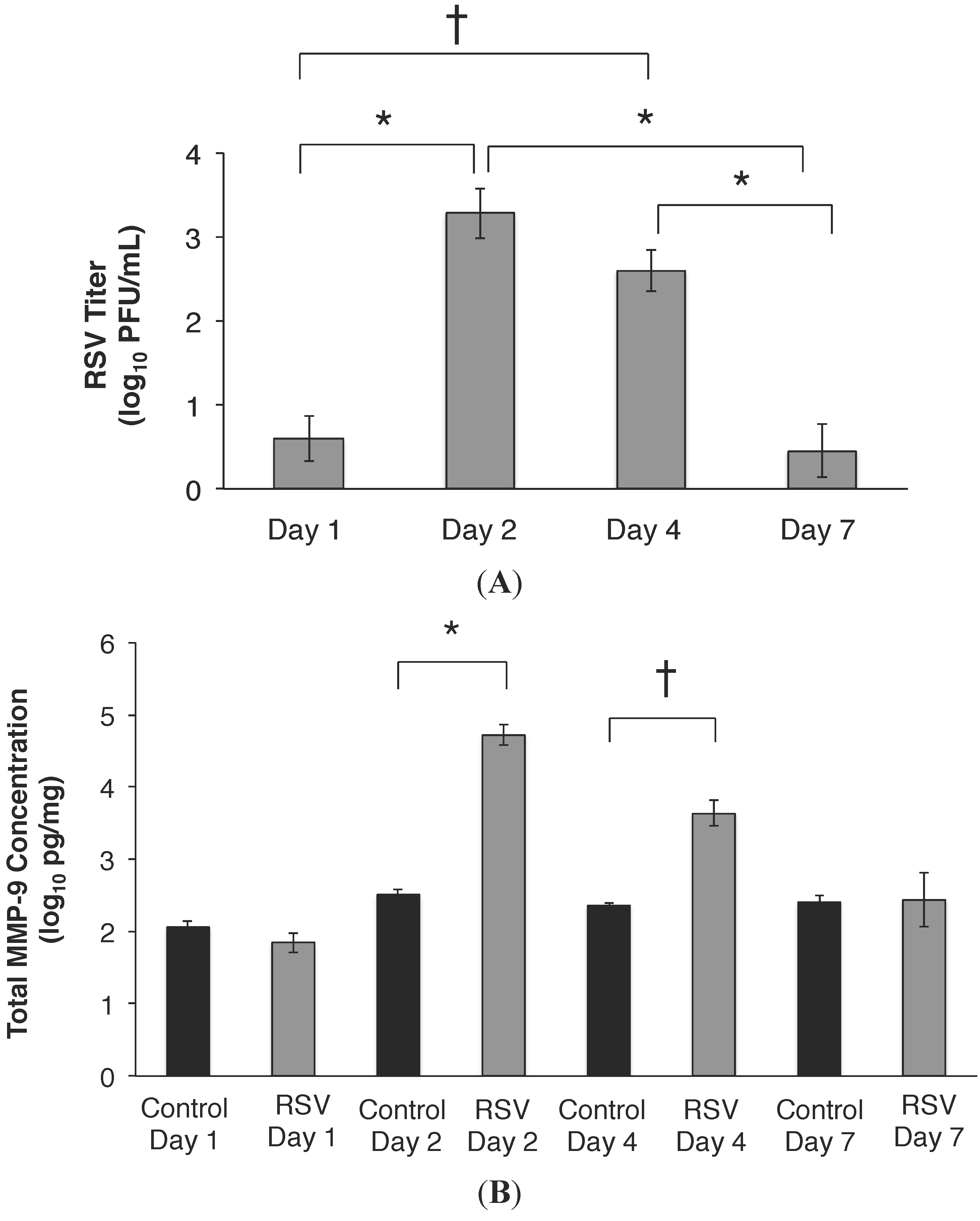

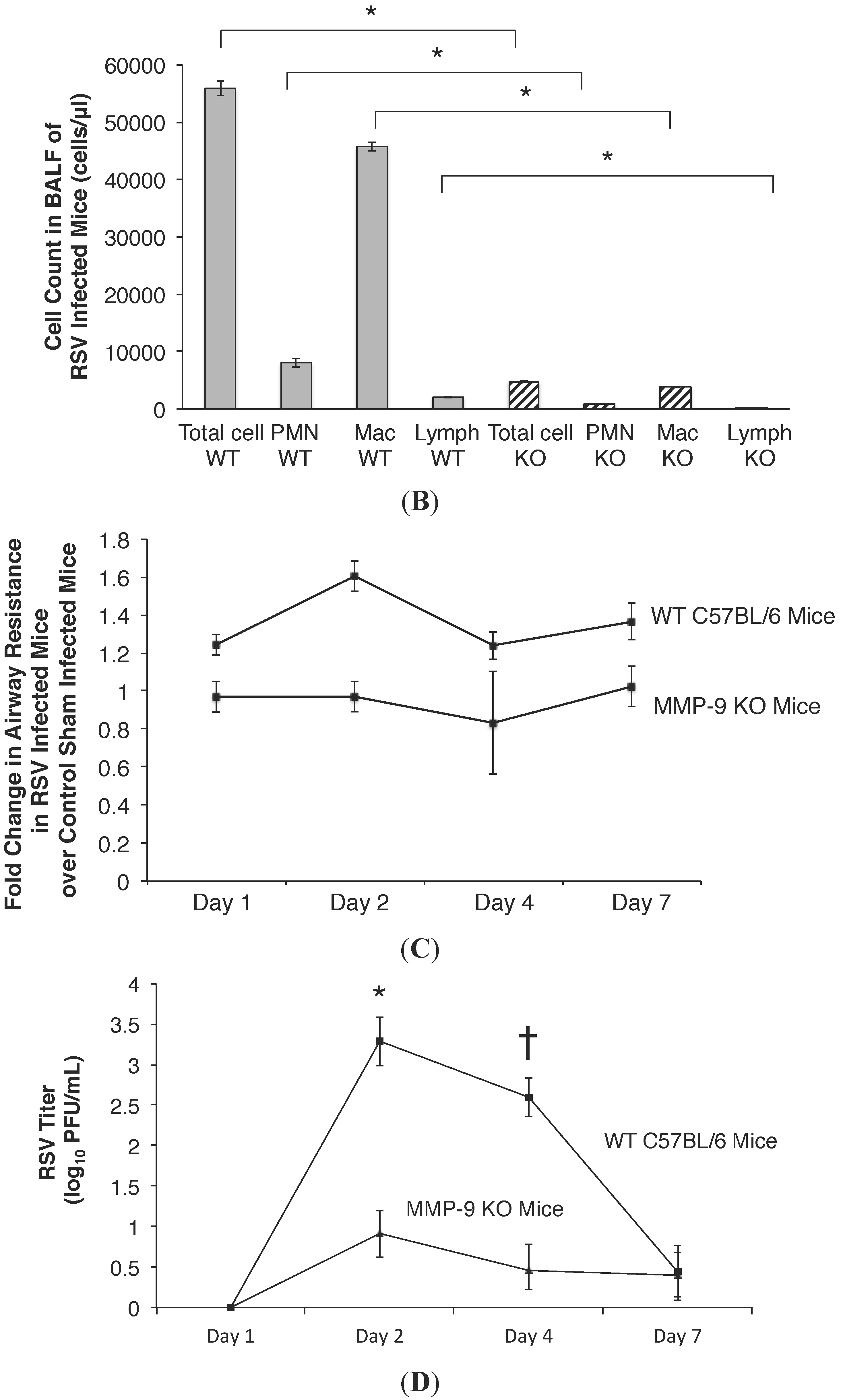
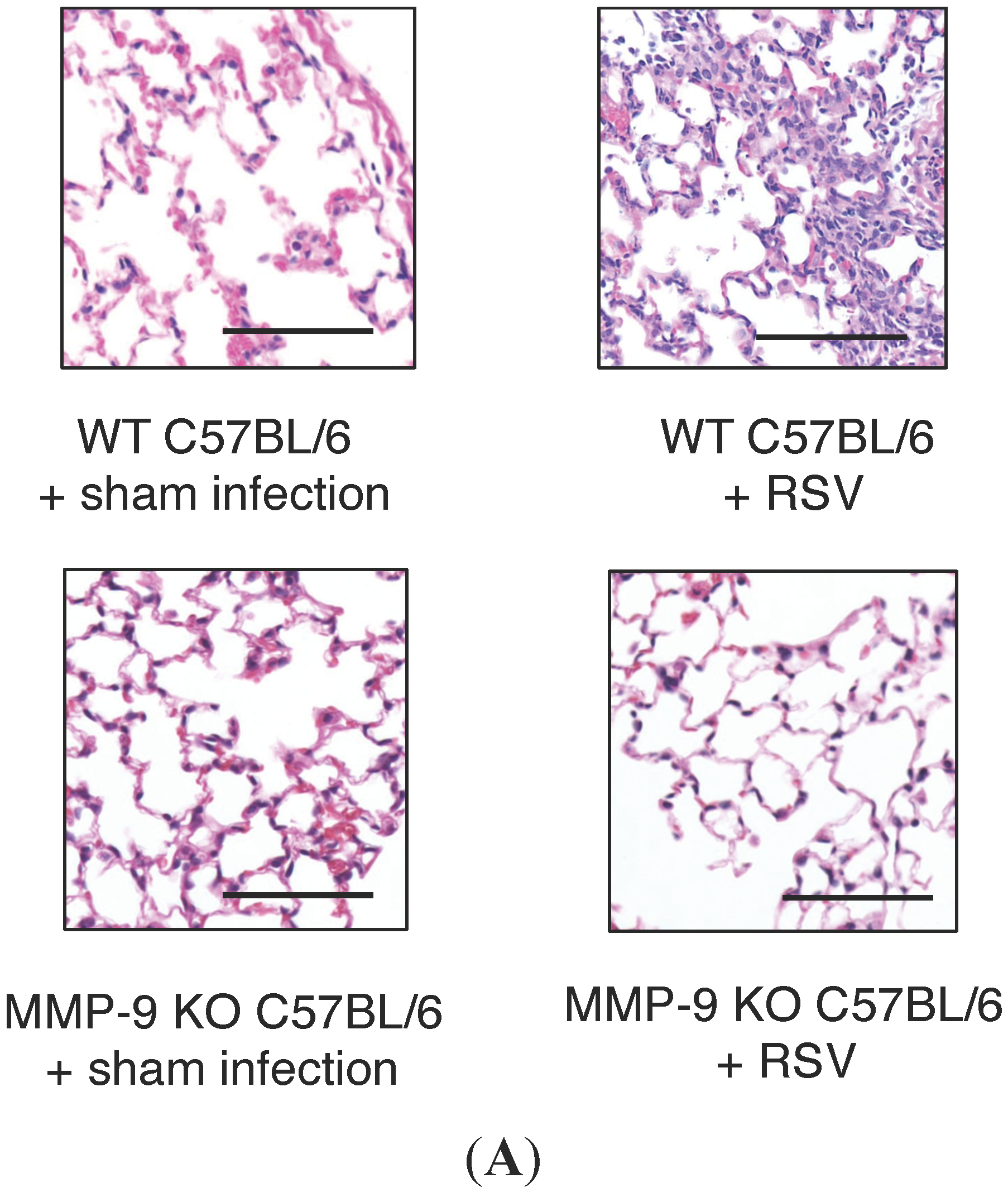
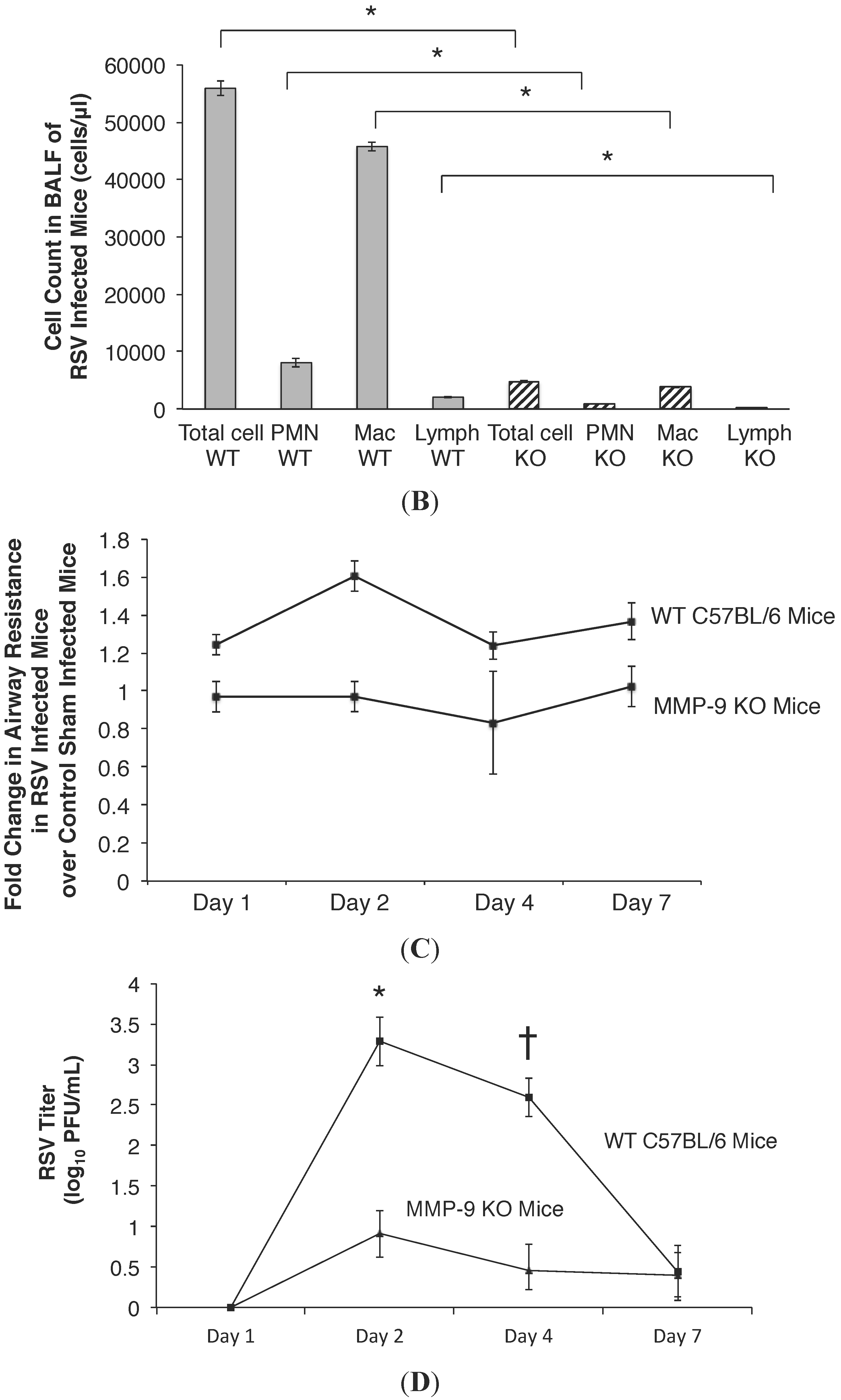
2.4. MMP-9 Knockout Mice have Decreased Inflammation and Viral Load during Acute RSV Infection


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| r | p | |
|---|---|---|
| MMP-9 and RSV Day 2 Post Infection | 0.66 | 0.002 |
| TIMP-1 and RSV Day 2 Post Infection | 0.37 | 0.11 |
| MMP-9:TIMP-1 and RSV Day 2 Post Infection | 0.13 | 0.58 |
| MMP-9 and RSV Day 4 Post Infection | 0.46 | 0.042 |
| TIMP-1 and RSV Day 4 Post Infection | 0.05 | 0.83 |
| MMP-9:TIMP-1 and RSV Day 4 Post Infection | 0.44 | 0.04 |
| MMP-9 and RSV Day 7 Post Infection | 0.64 | 0.04 |
| TIMP-1 and RSV Day 7 Post Infection | 0.09 | 0.83 |
| MMP-9:TIMP-1 and RSV Day 7 Post Infection | 0.66 | 0.07 |
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. RSV Infection of Cell Model System
4.3. Transepithelial Resistance Measurement
4.4. Real Time RT-PCR to Quantify MMP-9 mRNA Expression
4.5. MMP-9 Knockdown in HBE Cells
4.6. Detection of MMP-9 by Western Blot Analysis
4.7. Measurement of MMP-9 and TIMP
4.8. Mice
4.9. Inoculation
4.10. Sample Collection
4.11. Lung Function Analyses
4.12. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Blanken, M.O.; Rovers, M.M.; Molenaar, J.M.; Winkler-Seinstra, P.L.; Meijer, A.; Kimpen, J.L.; Bont, L. Dutch RSV Neonatal Network. Respiratory syncytial virus and recurrent wheeze in healthy preterm infants. N. Engl. J. Med. 2013, 368, 1791–1799. [Google Scholar] [CrossRef] [PubMed]
- Stretton, M.; Ajizian, S.J.; Mitchell, I.; Newth, C.J. Intensive care course and outcome of patients infected with respiratory syncytial virus. Pediatr. Pulmonol. 1992, 13, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, S.C.; Quasney, M.W.; Bush, A.J.; DeVincenzo, J.P. Respiratory syncytial virus infection in the pediatric intensive care unit: Clinical characteristics and risk factors for adverse outcomes. Pediatr. Crit. Care Med. 2001, 2, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Nair, H.; Nokes, D.J.; Gessner, B.D.; Dherani, M.; Madhi, S.A.; Singleton, R.J.; O’Brien, K.L.; Roca, A.; Wright, P.F.; Bruce, N.; et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: A systematic review and meta-analysis. Lancet 2010, 375, 1545–1555. [Google Scholar] [CrossRef]
- Purcell, K.; Fergie, J. Driscoll Children’s Hospital respiratory syncytial virus database: Risk factors, treatment and hospital course in 3308 infants and young children, 1991 to 2002. Pediatr. Infect. Dis. J. 2004, 23, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Yuksel, B.; Greenough, A. Birthweight and hospital readmission in infants born prematurely. Arch. Pediatr. Adolesc. Med. 1994, 148, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.B.; Weinberg, G.A.; Iwane, M.K.; Blumkin, A.K.; Edwards, K.M.; Staat, M.A.; Auinger, P.; Griffin, M.R.; Poehling, K.A.; Erdman, D.; et al. The burden of respiratory syncytial virus infection in young children. N. Engl. J. Med. 2009, 360, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Stevens, T.P.; Sinkin, R.A.; Hall, C.B.; Maniscalco, W.M.; McConnochie, K.M. Respiratory syncytial virus and premature infants born at 32 weeks’ gestation or earlier: Hospitalization and economic implications of prophylaxis. Arch. Pediatr. Adolesc. Med. 2000, 154, 55–61. [Google Scholar] [PubMed]
- McCormick, J.; Tubman, R. Readmission with respiratory syncytial virus (RSV) infection among graduates from a neonatal intensive care unit. Pediatr. Pulmonol. 2002, 34, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.M.; Lennon, D.R.; Broadbent, R.; Byrnes, C.A.; Grimwood, K.; Mildenhall, L.; Richardson, V.; Rowley, S. Palivizumab prophylaxis of respiratory syncytial virus infection in high-risk infants. J. Paediatr. Child Health 2002, 38, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; Oliver, C.; Prince, G.A.; Hemming, V.G.; Pfarr, D.S.; Wang, S.C.; Dormitzer, M.; O’Grady, J.; Koenig, S.; Tamura, J.K.; et al. Development of a humanized monoclonal antibody [MEDI-493] with potent in vitro and in vivo activity against Respiratory Syncytial Virus. J. Infect. Dis. 1997, 176, 1215–1224. [Google Scholar] [CrossRef] [PubMed]
- Prais, D.; Danino, D.; Schonfeld, T.; Amir, J. Impact of palivizumab on admission to the ICU for respiratory syncytial virus bronchiolitis: A national survey. Chest 2005, 128, 2765–2771. [Google Scholar] [CrossRef] [PubMed]
- Homaira, N.; Rawlinson, W.; Snelling, T.L.; Jaffe, A. Effectiveness of Palivizumab in Preventing RSV Hospitalization in High Risk Children: A Real-World Perspective. Int. J. Pediatr. 2014, 2014, 571609. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.W.; Frankel, L.R.; Mathers, L.H.; Tang, A.T.; Ariagno, R.L.; Prober, C.G. A Controlled Trial of Aerosolized Ribavirin in Infants Receiving Mechanical Ventilation for Severe Respiratory Syncytial Virus Infection. N. Engl. J. Med. 1991, 325, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Krilov, L.R. Safety issues related to the administration of ribavirin. Pediatr. Infect. Dis. J. 2002, 21, 479–481. [Google Scholar] [CrossRef] [PubMed]
- Devereux, G.; Steele, S.; Jagelman, T.; Fielding, S.; Muirhead, R.; Brady, J.; Grierson, C.; Brooker, R.; Winter, J.; Fardon, T.; et al. An observational study of matrix metalloproteinase (MMP)-9 in cystic fibrosis. J. Cyst. Fibros. 2014, 13, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.P.; Li, W.; Liu, X.M. Matrix metalloproteinase 9 is involved in airway inflammation in cough variant asthma. Exp. Ther. Med. 2014, 8, 1197–1200. [Google Scholar] [CrossRef] [PubMed]
- Kong, M.; Gaggar, A.; Li, Y.; Winkler, M.; Blalock, J.E.; Clancy, J.P. Matrix metalloproteinase activity in pediatric acute lung injury. Int. J. Med. Sci. 2009, 6, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Kong, M.; Clancy, J.P.; Peng, N.; Li, Y.; Szul, T.J.; Xu, X.; Oster, R.; Sullender, W.; Ambalavanan, N.; Blalock, J.E.; et al. Pulmonary matrix metalloproteinase-9 activity in mechanically ventilated children with respiratory syncytial virus. Eur. Respir. J. 2014, 43, 1086–1096. [Google Scholar] [CrossRef] [PubMed]
- American Academy of Pediatrics Subcommittee on Diagnosis and Management of Bronchiolitis. Diagnosis and management of bronchiolitis. Pediatrics 2006, 118, 1774–1793. [Google Scholar]
- American Academy of Pediatrics Committee on Infectious Diseases; American Academy of Pediatrics Bronchiolitis Guidelines Committee. Updated guidance for palivizumab prophylaxis among infants and young children at increased risk of hospitalization for respiratory syncytial virus infection. Pediatrics 2014, 134, e620–e638. [Google Scholar]
- Ventre, K.; Randolph, A.G. Ribavirin for respiratory syncytial virus infection of the lower respiratory tract in infants and young children. Cochrane Database Syst. Rev. 2007, 1, CD000181. [Google Scholar] [PubMed]
- DeVincenzo, J.P.; Whitley, R.J.; Mackman, R.L.; Scaglioni-Weinlich, C.; Harrison, L.; Farrell, E.; McBride, S.; Lambkin-Williams, R.; Jordan, R.; Xin, Y.; et al. Oral GS-5806 activity in a respiratory syncytial virus challenge study. N. Engl. J. Med. 2014, 371, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, D.D.; Bettiol, J.; Noël, A.; Bartsch, P.; Foidart, J.M.; Louis, R. Matrix metalloproteinase-9, but not tissue inhibitor of matrix metalloproteinase-1, increases in the sputum from allergic asthmatic patients after allergen challenge. Chest 2002, 122, 1553–1559. [Google Scholar] [CrossRef] [PubMed]
- Culpitt, S.V.; Rogers, D.F.; Traves, S.L.; Barnes, P.J.; Donnelly, L.E. Sputum matrix metalloproteinases: Comparison between chronic obstructive pulmonary disease and asthma. Respir. Med. 2005, 99, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Cederqvist, K.; Sorsa, T.; Tervahartiala, T.; Maisi, P.; Reunanen, K.; Lassus, P.; Andersson, S. Matrix metalloproteinases-2, -8, and -9 and TIMP-2 in tracheal aspirates from preterm infants with respiratory distress. Pediatrics 2001, 108, 686–692. [Google Scholar] [CrossRef] [PubMed]
- Gaggar, A.; Li, Y.; Weathington, N.; Kong, M.; Jackson, P.; Blalock, J.E.; Clancy, J.P. Matrix metalloprotease-9 dysregulation in lower airway secretions of cystic fibrosis patients. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Zheng, T.; Zhu, Z.; Wang, Z.; Homer, R.J.; Ma, B.; Riese, R.J.; Chapman, H.A., Jr.; Shapiro, S.D.; Elias, J.A. Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. J. Clin. Investig. 2000, 106, 1081–1093. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.W.; Zindl, C.L.; Lai, J.F.; Weaver, C.T.; Chaplin, D.D. MMP induced by Gr-1+ cells are crucial for recruitment of Th cells into the airways. Eur. J. Immunol. 2009, 39, 2281–2292. [Google Scholar] [CrossRef] [PubMed]
- Malik, M.; Bakshi, C.S.; McCabe, K.; Catlett, S.V.; Shah, A.; Singh, R.; Jackson, P.L.; Gaggar, A.; Metzger, D.W.; Melendez, J.A.; et al. Matrix metalloproteinase 9 activity enhances host susceptibility to pulmonary infection with type A and B strains of Francisella tularensis. J. Immunol. 2007, 178, 1013–1020. [Google Scholar] [CrossRef] [PubMed]
- Churg, A.; Wang, R.; Wang, X.; Onnervik, P.O.; Thim, K.; Wright, J.L. Effect of an MMP-9/MMP-12 inhibitor on smoke-induced emphysema and airway remodelling in guinea pigs. Thorax 2007, 62, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Suk, M.H.; Yoon, D.W.; Lee, S.H.; Hur, G.Y.; Jung, K.H.; Jeong, H.C.; Lee, S.Y.; Lee, S.Y.; Suh, I.B.; et al. Inhibition of matrix metalloproteinase-9 prevents neutrophilic inflammation in ventilator-induced lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L580–L587. [Google Scholar] [CrossRef] [PubMed]
- Sochor, M.; Richter, S.; Schmidt, A.; Hempel, S.; Hopt, U.T.; Keck, T. Inhibition of matrix metalloproteinase-9 with doxycycline reduces pancreatitis-associated lung injury. Digestion 2009, 80, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, D.D.; Tournoy, K.G.; Vermaelen, K.; Munaut, C.; Foidart, J.M.; Louis, R.; Noël, A.; Pauwels, R.A. Matrix metalloproteinase-9 deficiency impairs cellular infiltration and bronchial hyperresponsiveness during allergen-induced airway inflammation. Am. J. Pathol. 2002, 161, 491–498. [Google Scholar] [CrossRef]
- Warner, R.L.; Beltran, L.; Younkin, E.M.; Lewis, C.S.; Weiss, S.J.; Varani, J.; Johnson, K.J. Role of stromelysin 1 and gelatinase B in experimental acute lung injury. Am. J. Respir. Cell Mol. Biol. 2001, 24, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Fernández, F.G.; Campbell, L.G.; Liu, W.; Shipley, J.M.; Itohara, S.; Patterson, G.A.; Senior, R.M.; Mohanakumar, T.; Jaramillo, A. Inhibition of obliterative airway disease development in murine tracheal allografts by matrix metalloproteinase-9 deficiency. Am. J. Transplant. 2005, 5, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Stark, J.M.; McDowell, S.A.; Koenigsknecht, V.; Prows, D.R.; Leikauf, J.E.; le Vine, A.M.; Leikauf, G.D. Genetic susceptibility to respiratory syncytial virus infection in inbred mice. J. Med. Virol. 2002, 67, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Jafri, H.S.; Chavez-Bueno, S.; Mejias, A.; Gomez, A.M.; Rios, A.M.; Nassi, S.S.; Yusuf, M.; Kapur, P.; Hardy, R.D.; Hatfield, J.; et al. Respiratory syncytial virus induces pneumonia, cytokine response, airway obstruction, and chronic inflammatory infiltrates associated with long-term airway hyperresponsiveness in mice. J. Infect. Dis. 2004, 189, 1856–1865. [Google Scholar] [CrossRef] [PubMed]
- Chávez-Bueno, S.; Mejías, A.; Gómez, A.M.; Olsen, K.D.; Ríos, A.M.; Fonseca-Aten, M.; Ramilo, O.; Jafri, H.S. Respiratory syncytial virus-induced acute and chronic airway disease is independent of genetic background: An experimental murine model. Virol. J. 2005, 2, e46. [Google Scholar] [CrossRef] [PubMed]
- Horsfall, F.L.J.; Hahn, R.G. A latent virus in normal mice capable of producing pneumonia in its natural host. J. Exp. Med. 1940, 71, 391–408. [Google Scholar] [CrossRef] [PubMed]
- Domachowske, J.B.; Bonville, C.A.; Dyer, K.D.; Easton, A.J.; Rosenberg, H.F. Pulmonary eosinophilia and production of MIP-1alpha are prominent responses to infection with pneumonia virus of mice. Cell. Immunol. 2000, 200, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Domachowske, J.B.; Bonville, C.A.; Easton, A.J.; Rosenberg, H.F. Differential expression of proiflammatory cytokine genes in vivo in response to pathogenic and nonpathogenic pneumovirus infections. J. Infect. Dis. 2002, 186, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Lukacs, N.W.; Moore, M.L.; Rudd, B.D.; Berlin, A.A.; Collins, R.D.; Olson, S.J.; Ho, S.B.; Peebles, R.S., Jr. Differential immune responses and pulmonary pathophysiology are induced by two different strains of respiratory syncytial virus. Am. J. Pathol. 2006, 169, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.L.; Chi, M.H.; Luongo, C.; Lukacs, N.W.; Polosukhin, V.V.; Huckabee, M.M.; Newcomb, D.C.; Buchholz, U.J.; Crowe, J.E., Jr.; Goleniewska, K.; et al. A chimeric A2 strain of respiratory syncytial virus (RSV) with the fusion protein of RSV strain line 19 exhibits enhanced viral load, mucus, and airway dysfunction. J. Virol. 2009, 83, 4185–4194. [Google Scholar] [CrossRef] [PubMed]
- Dey, N.; Liu, T.; Garofalo, R.P.; Casola, A. TAK1 regulates NF-ΚB and AP-1 activation in airway epithelial cells following RSV infection. Virology 2011, 418, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Benbow, U.; Brinckerhoff, C.E. The AP-1 site and MMP gene regulation: What is all the fuss about? Matrix Biol. 1997, 15, 519–526. [Google Scholar] [CrossRef]
- Cozens, A.L.; Yezzi, M.J.; Kunzelmann, K.; Ohrui, T.; Chin, L.; Eng, K.; Finkbeiner, W.E.; Widdicombe, J.H.; Gruenert, D.C. CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 1994, 10, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Mbiguino, A.; Menezes, J. Purification of human respiratory syncytial virus: Superiority of sucrose gradient over percoll, renografin, and metrizamide gradients. J. Virol. Methods 1991, 31, 161–170. [Google Scholar] [CrossRef]
- Sullender, W.M.; Anderson, K.; Wertz, G.W. The respiratory syncytial virus subgroup B attachment glycoprotein: Analysis of sequence, expression from a recombinant vector, and evaluation as an immunogen against homologous and heterologous subgroup virus challenge. Virology 1990, 178, 195–203. [Google Scholar] [CrossRef]
- Jaovisidha, P.; Peeples, M.E.; Brees, A.A.; Carpenter, L.R.; Moy, J.N. Respiratory syncytial virus stimulates neutrophil degranulation and chemokine release. J. Immunol. 1999, 163, 2816–2820. [Google Scholar] [PubMed]
- Grumbach, Y.; Quynh, N.V.; Chiron, R.; Urbach, V. LXA4 stimulates ZO-1 expression and transepithelial electrical resistance in human airway epithelial (16HBE14o-) cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L101–L108. [Google Scholar] [CrossRef] [PubMed]
- LeSimple, P.; Liao, J.; Robert, R.; Gruenert, D.C.; Hanrahan, J.W. Cystic fibrosis transmembrane conductance regulator trafficking modulates the barrier function of airway epithelial cell monolayers. J. Physiol. 2010, 588, 1195–1209. [Google Scholar] [CrossRef] [PubMed]
- Ambalavanan, N.; Nicola, T.; Li, P.; Bulger, A.; Murphy-Ullrich, J.; Oparil, S.; Chen, Y.F. Role of matrix metalloproteinase-2 in newborn mouse lungs under hypoxic conditions. Pediatr. Res. 2008, 63, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.C.; Sullender, W.M.; Hickman-Davis, J.M.; Lindsey, J.R.; Matalon, S. Nucleotide-mediated inhibition of alveolar fluid clearance in BALB/c mice after respiratory syncytial virus infection. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Nicola, T.; Hagood, J.S.; James, M.L.; Macewen, M.W.; Williams, T.A.; Hewitt, M.M.; Schwiebert, L.; Bulger, A.; Oparil, S.; Chen, Y.F.; et al. Loss of Thy-1 inhibits alveolar development in the newborn mouse lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L738–L750. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kong, M.Y.F.; Whitley, R.J.; Peng, N.; Oster, R.; Schoeb, T.R.; Sullender, W.; Ambalavanan, N.; Clancy, J.P.; Gaggar, A.; Blalock, J.E. Matrix Metalloproteinase-9 Mediates RSV Infection in Vitro and in Vivo. Viruses 2015, 7, 4230-4253. https://doi.org/10.3390/v7082817
Kong MYF, Whitley RJ, Peng N, Oster R, Schoeb TR, Sullender W, Ambalavanan N, Clancy JP, Gaggar A, Blalock JE. Matrix Metalloproteinase-9 Mediates RSV Infection in Vitro and in Vivo. Viruses. 2015; 7(8):4230-4253. https://doi.org/10.3390/v7082817
Chicago/Turabian StyleKong, Michele Y.F., Richard J. Whitley, Ning Peng, Robert Oster, Trenton R. Schoeb, Wayne Sullender, Namasivayam Ambalavanan, John Paul Clancy, Amit Gaggar, and J. Edwin Blalock. 2015. "Matrix Metalloproteinase-9 Mediates RSV Infection in Vitro and in Vivo" Viruses 7, no. 8: 4230-4253. https://doi.org/10.3390/v7082817
APA StyleKong, M. Y. F., Whitley, R. J., Peng, N., Oster, R., Schoeb, T. R., Sullender, W., Ambalavanan, N., Clancy, J. P., Gaggar, A., & Blalock, J. E. (2015). Matrix Metalloproteinase-9 Mediates RSV Infection in Vitro and in Vivo. Viruses, 7(8), 4230-4253. https://doi.org/10.3390/v7082817