
Impacts of Humanized Mouse Models on the Investigation of HIV-1 Infection: Illuminating the Roles of Viral Accessory Proteins in Vivo

Abstract
:1. Introduction
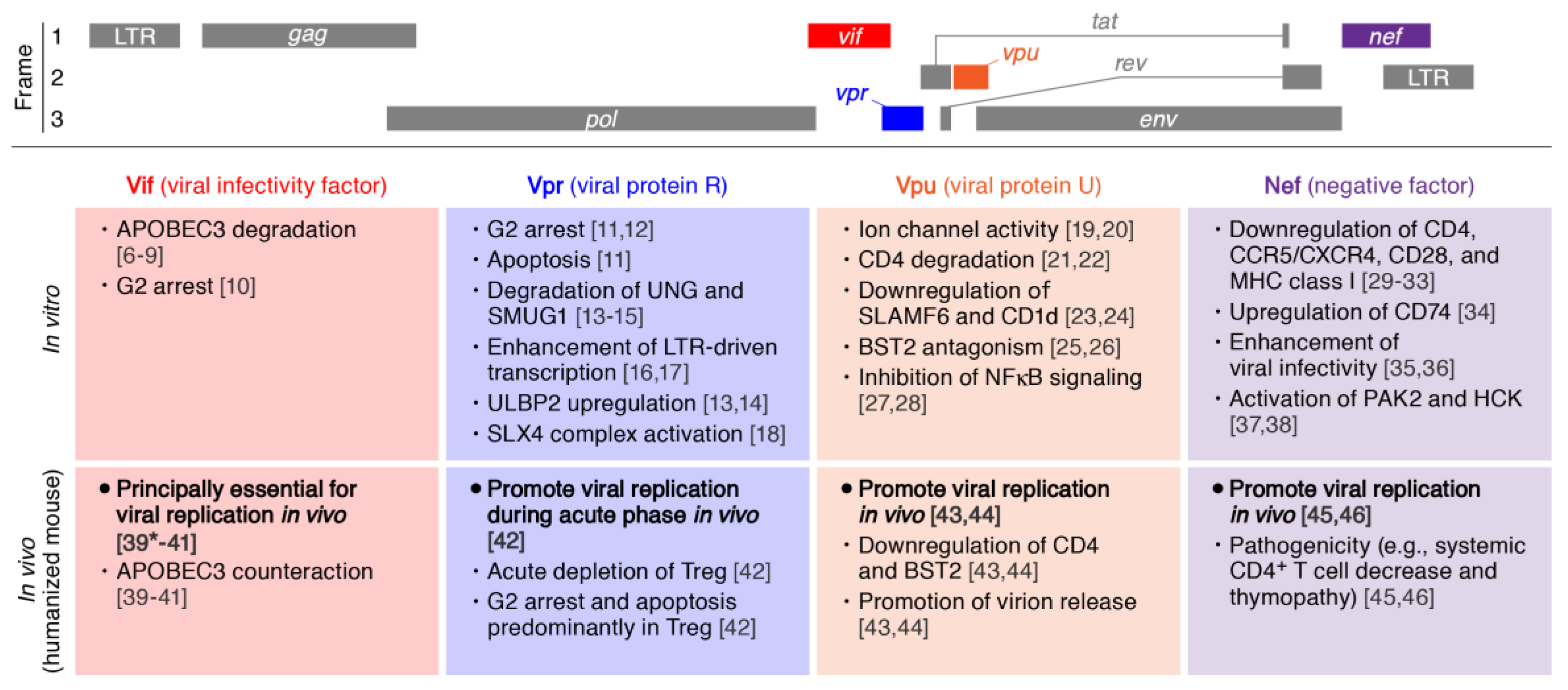
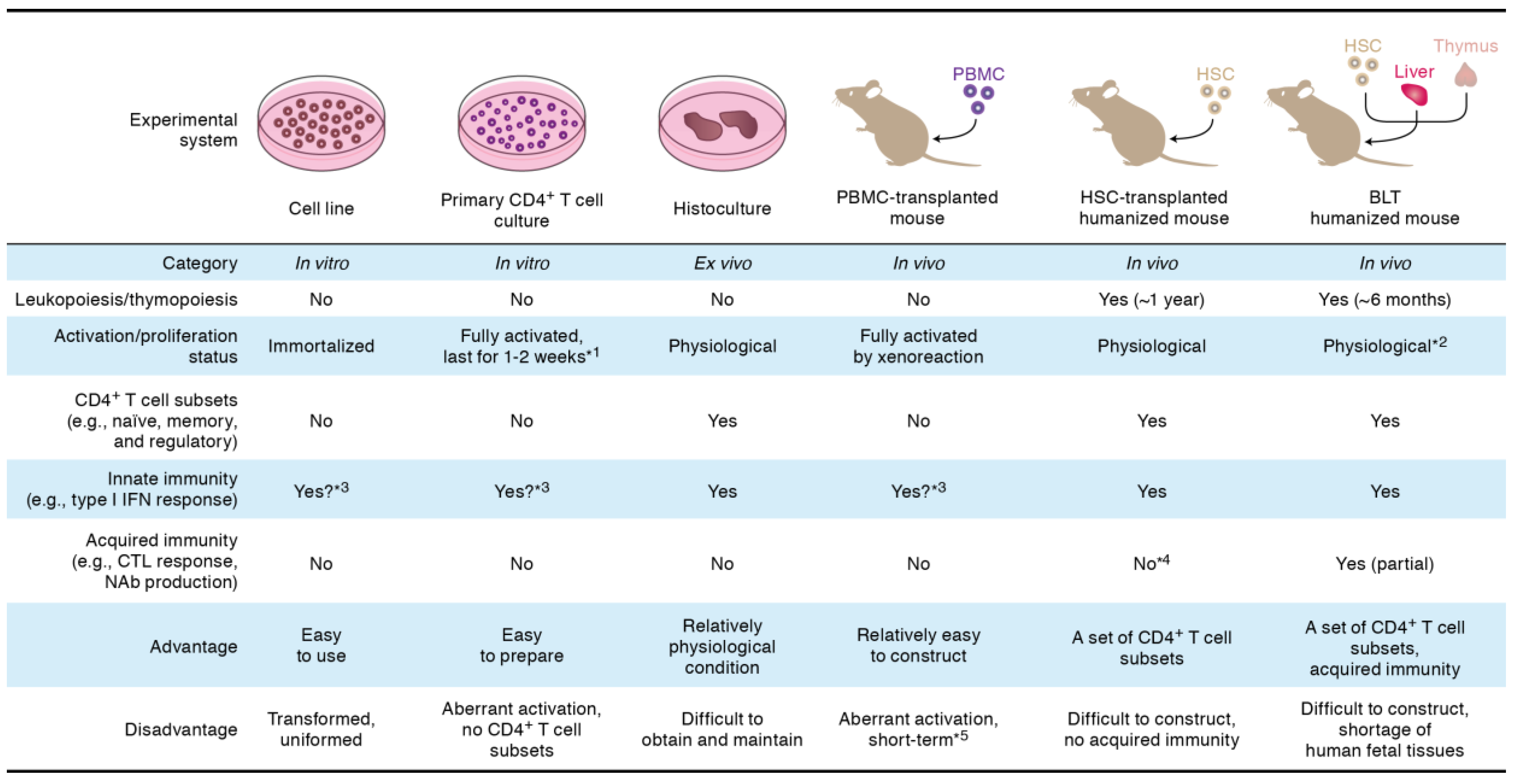
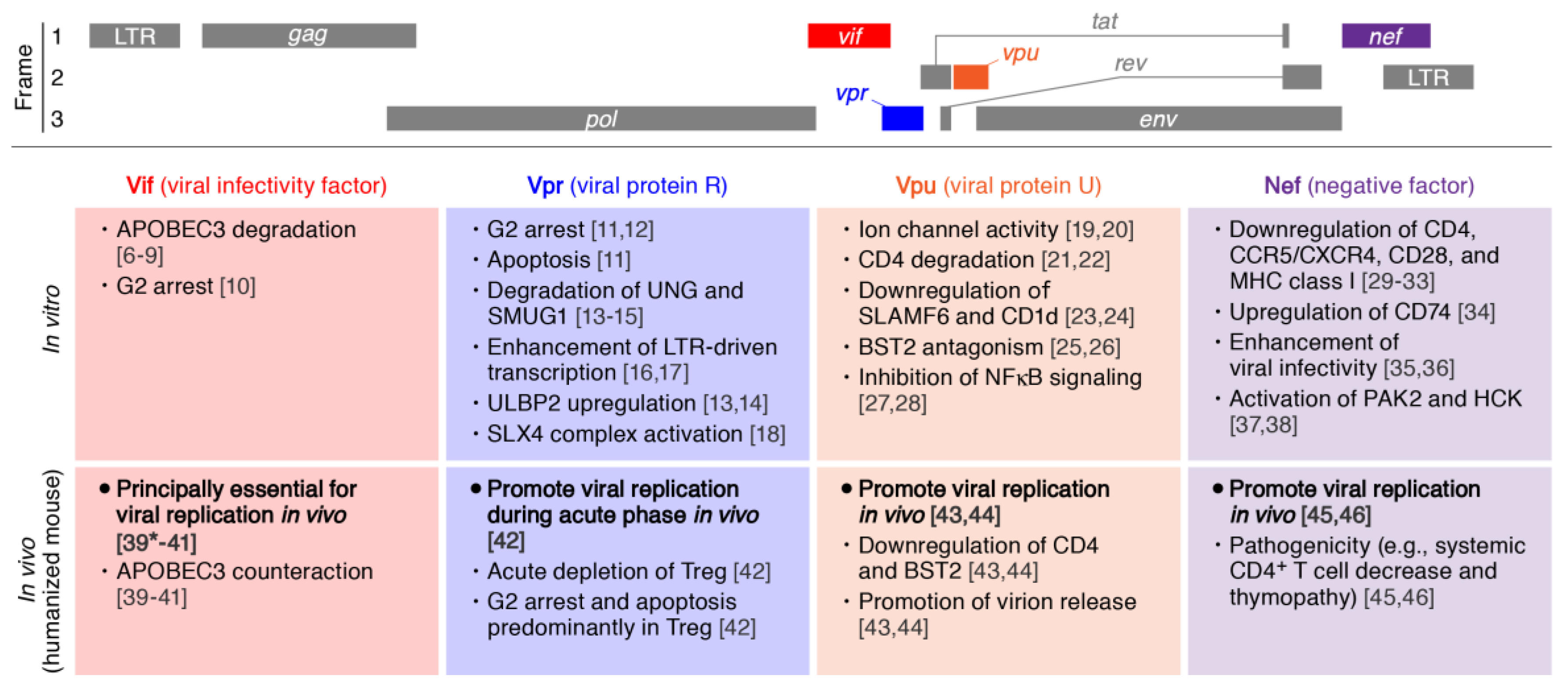
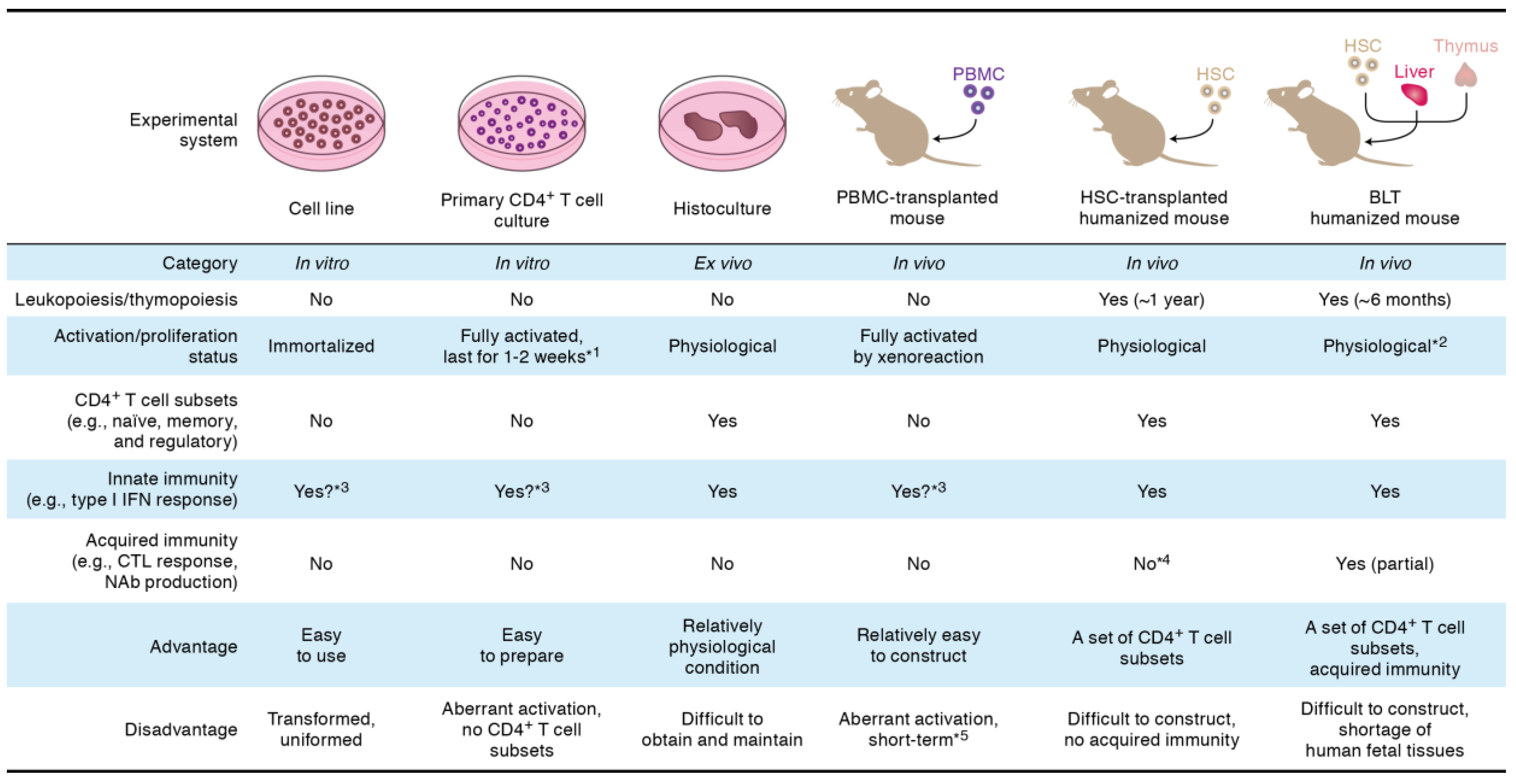
2. Viral Infectivity Factor (Vif)

{kind=link}
{kind=link}
| Gene | Strain | Coreceptor Usage | Mutation Type | Reference a |
|---|---|---|---|---|
| vif | JRCSF | CCR5 | Deletion | [40] |
| JRCSF | CCR5 | Deletion | [39] | |
| JRCSF | CCR5 | Frame shift | [39] | |
| LAI | CXCR4 | Deletion | [39] | |
| NLCSFV3 | CCR5 | DRMR/AAAA substitution (4A) | [41] | |
| NLCSFV3 | CCR5 | YRHHY/AAAAA substitution (5A) | [41] | |
| NLCSFV3 | CCR5 | Both of above (4A5A) | [41] | |
| vpr | JRCSF | CCR5 | Deletion | [42] |
| NL4-3 | CXCR4 | Deletion | [42] | |
| vpu | AD8 | CCR5 | Deletion | [44] |
| NLADA b | CCR5 | Deletion | [43] | |
| NLADA b | CCR5 | S52D, S56D substitution | [43] | |
| nef | LAI | CXCR4 | Deletion | [46] |
| LAI | CXCR4 | Frame shift | [45] | |
| LAI | CXCR4 | fsΔ-1 c | [45] | |
| LAI | CXCR4 | fsΔ-13 c | [45] | |
| LAI | CXCR4 | P72A, P75A substitution | [45] |
3. Viral Protein R (Vpr)
4. Viral Protein U (Vpu)
5. Negative Factor (Nef)
6. Future Perspective
Acknowledgments
Conflicts of Interest
References
- Barre-Sinoussi, F.; Chermann, J.C.; Rey, F.; Nugeyre, M.T.; Chamaret, S.; Gruest, J.; Dauguet, C.; Axler-Blin, C.; Vezinet-Brun, F.; Rouzioux, C.; Rozenbaum, W.; Montagnier, L. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 1983, 220, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Gallo, R.C.; Sarin, P.S.; Gelmann, E.P.; Robert-Guroff, M.; Richardson, E.; Kalyanaraman, V.S.; Mann, D.; Sidhu, G.D.; Stahl, R.E.; Zolla-Pazner, S.; Leibowitch, J.; Popovic, M. Isolation of human T-cell leukemia virus in acquired immune deficiency syndrome (AIDS). Science 1983, 220, 865–867. [Google Scholar] [CrossRef] [PubMed]
- Freed, E.O.; Martin, M.A. HIVs and Their Replication. In Fields Virology, 5th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, 2007; Volume 2, pp. 2107–2185. [Google Scholar]
- Kirchhoff, F. Immune evasion and counteraction of restriction factors by HIV-1 and other primate lentiviruses. Cell Host Microbe 2010, 8, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Malim, M.H.; Bieniasz, P.D. HIV Restriction Factors and Mechanisms of Evasion. Cold Spring Harb. Perspect. Med. 2012, 2, a006940. [Google Scholar] [CrossRef] [PubMed]
- Albin, J.S.; Harris, R.S. Interactions of host APOBEC3 restriction factors with HIV-1 in vivo: implications for therapeutics. Expert Rev. Mol. Med. 2010, 12, e4. [Google Scholar] [CrossRef] [PubMed]
- Desimmie, B.A.; Delviks-Frankenberrry, K.A.; Burdick, R.C.; Qi, D.; Izumi, T.; Pathak, V.K. Multiple APOBEC3 restriction factors for HIV-1 and one Vif to rule them all. J. Mol. Biol. 2014, 426, 1220–1245. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, S.; Ode, H.; Iwatani, Y. Structural features of antiviral APOBEC3 proteins are linked to their functional activities. Front Microbiol. 2011, 2, 258. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Izumi, T.; Io, K.; Matsui, M.; Shirakawa, K.; Shinohara, M.; Nagai, Y.; Kawahara, M.; Kobayashi, M.; Kondoh, H.; Misawa, N.; Koyanagi, Y.; Uchiyama, T.; Takaori-Kondo, A. HIV-1 viral infectivity factor interacts with TP53 to induce G2 cell cycle arrest and positively regulate viral replication. Proc. Natl. Acad. Sci. U S A 2010, 107, 20798–20803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, J.L.; Le Rouzic, E.; Planelles, V. HIV-1 Vpr: mechanisms of G2 arrest and apoptosis. Exp. Mol. Pathol. 2008, 85, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Goh, W.C.; Rogel, M.E.; Kinsey, C.M.; Michael, S.F.; Fultz, P.N.; Nowak, M.A.; Hahn, B.H.; Emerman, M. HIV-1 Vpr increases viral expression by manipulation of the cell cycle: a mechanism for selection of Vpr in vivo. Nat. Med. 1998, 4, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Hrecka, K.; Gierszewska, M.; Srivastava, S.; Kozaczkiewicz, L.; Swanson, S.K.; Florens, L.; Washburn, M.P.; Skowronski, J. Lentiviral Vpr usurps Cul4-DDB1[VprBP] E3 ubiquitin ligase to modulate cell cycle. Proc. Natl. Acad. Sci. U S A 2007, 104, 11778–11783. [Google Scholar] [CrossRef] [PubMed]
- Schrofelbauer, B.; Hakata, Y.; Landau, N.R. HIV-1 Vpr function is mediated by interaction with the damage-specific DNA-binding protein DDB1. Proc. Natl. Acad. Sci. U S A 2007, 104, 4130–4135. [Google Scholar] [CrossRef] [PubMed]
- Schrofelbauer, B.; Yu, Q.; Zeitlin, S.G.; Landau, N.R. Human immunodeficiency virus type 1 Vpr induces the degradation of the UNG and SMUG uracil-DNA glycosylases. J. Virol. 2005, 79, 10978–10987. [Google Scholar] [CrossRef] [PubMed]
- Forget, J.; Yao, X.J.; Mercier, J.; Cohen, E.A. Human immunodeficiency virus type 1 vpr protein transactivation function: mechanism and identification of domains involved. J. Mol. Biol. 1998, 284, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Sawaya, B.E.; Khalili, K.; Mercer, W.E.; Denisova, L.; Amini, S. Cooperative actions of HIV-1 Vpr and p53 modulate viral gene transcription. J. Biol. Chem. 1998, 273, 20052–20057. [Google Scholar] [CrossRef] [PubMed]
- Laguette, N.; Bregnard, C.; Hue, P.; Basbous, J.; Yatim, A.; Larroque, M.; Kirchhoff, F.; Constantinou, A.; Sobhian, B.; Benkirane, M. Premature activation of the SLX4 complex by Vpr promotes G2/M arrest and escape from innate immune sensing. Cell 2014, 156, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Ewart, G.D.; Sutherland, T.; Gage, P.W.; Cox, G.B. The Vpu protein of human immunodeficiency virus type 1 forms cation-selective ion channels. J. Virol. 1996, 70, 7108–7115. [Google Scholar] [PubMed]
- Gonzalez, M.E.; Carrasco, L. Viroporins. FEBS Lett 2003, 552, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Gottlinger, H.G.; Dorfman, T.; Cohen, E.A.; Haseltine, W.A. Vpu protein of human immunodeficiency virus type 1 enhances the release of capsids produced by gag gene constructs of widely divergent retroviruses. Proc. Natl. Acad. Sci. U S A 1993, 90, 7381–7385. [Google Scholar] [CrossRef] [PubMed]
- Schubert, U.; Clouse, K.A.; Strebel, K. Augmentation of virus secretion by the human immunodeficiency virus type 1 Vpu protein is cell type independent and occurs in cultured human primary macrophages and lymphocytes. J. Virol. 1995, 69, 7699–7711. [Google Scholar] [PubMed]
- Moll, M.; Andersson, S.K.; Smed-Sorensen, A.; Sandberg, J.K. Inhibition of lipid antigen presentation in dendritic cells by HIV-1 Vpu interference with CD1d recycling from endosomal compartments. Blood 2010, 116, 1876–1884. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.H.; Sowrirajan, B.; Davis, Z.B.; Ward, J.P.; Campbell, E.M.; Planelles, V.; Barker, E. Degranulation of natural killer cells following interaction with HIV-1-infected cells is hindered by downmodulation of NTB-A by Vpu. Cell Host Microbe 2010, 8, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.J.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Galao, R.P.; Le Tortorec, A.; Pickering, S.; Kueck, T.; Neil, S.J. Innate sensing of HIV-1 assembly by Tetherin induces NFκB-dependent proinflammatory responses. Cell Host Microbe 2012, 12, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Sauter, D.; Hotter, D.; Van Driessche, B.; Sturzel, C.M.; Kluge, S.F.; Wildum, S.; Yu, H.; Baumann, B.; Wirth, T.; Plantier, J.C.; Leoz, M.; Hahn, B.H.; Van Lint, C.; Kirchhoff, F. Differential regulation of NF-κB-mediated proviral and antiviral host gene expression by primate lentiviral Nef and Vpu proteins. Cell Rep. 2015, 10, 586–599. [Google Scholar] [CrossRef] [PubMed]
- Hrecka, K.; Swigut, T.; Schindler, M.; Kirchhoff, F.; Skowronski, J. Nef proteins from diverse groups of primate lentiviruses downmodulate CXCR4 to inhibit migration to the chemokine stromal derived factor 1. J. Virol. 2005, 79, 10650–10659. [Google Scholar] [CrossRef] [PubMed]
- Landi, A.; Iannucci, V.; Nuffel, A.V.; Meuwissen, P.; Verhasselt, B. One protein to rule them all: modulation of cell surface receptors and molecules by HIV Nef. Curr. HIV Res. 2011, 9, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Michel, N.; Allespach, I.; Venzke, S.; Fackler, O.T.; Keppler, O.T. The Nef protein of human immunodeficiency virus establishes superinfection immunity by a dual strategy to downregulate cell-surface CCR5 and CD4. Curr. Biol. 2005, 15, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, O.; Marechal, V.; Le Gall, S.; Lemonnier, F.; Heard, J.M. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat. Med. 1996, 2, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Swigut, T.; Shohdy, N.; Skowronski, J. Mechanism for down-regulation of CD28 by Nef. EMBO J 2001, 20, 1593–1604. [Google Scholar] [CrossRef] [PubMed]
- Keppler, O.T.; Tibroni, N.; Venzke, S.; Rauch, S.; Fackler, O.T. Modulation of specific surface receptors and activation sensitization in primary resting CD4+ T lymphocytes by the Nef protein of HIV-1. J. Leukoc. Biol. 2006, 79, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, M.A.; Warmerdam, M.T.; Atchison, R.E.; Miller, M.D.; Greene, W.C. Dissociation of the CD4 downregulation and viral infectivity enhancement functions of human immunodeficiency virus type 1 Nef. J. Virol. 1995, 69, 4112–4121. [Google Scholar] [PubMed]
- Pizzato, M.; Helander, A.; Popova, E.; Calistri, A.; Zamborlini, A.; Palu, G.; Gottlinger, H.G. Dynamin 2 is required for the enhancement of HIV-1 infectivity by Nef. Proc. Natl. Acad. Sci. U S A 2007, 104, 6812–6817. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, K.C.; Mukerji, J.; Gabuzda, D. Nef-mediated enhancement of cellular activation and human immunodeficiency virus type 1 replication in primary T cells is dependent on association with p21-activated kinase 2. Retrovirology 2011, 8, 64. [Google Scholar] [CrossRef] [PubMed]
- Trible, R.P.; Emert-Sedlak, L.; Smithgall, T.E. HIV-1 Nef selectively activates Src family kinases Hck, Lyn, and c-Src through direct SH3 domain interaction. J. Biol. Chem. 2006, 281, 27029–27038. [Google Scholar] [CrossRef] [PubMed]
- Krisko, J.F.; Martinez-Torres, F.; Foster, J.L.; Garcia, J.V. HIV restriction by APOBEC3 in humanized mice. PLoS Pathog. 2013, 9, e1003242. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Izumi, T.; Misawa, N.; Kobayashi, T.; Yamashita, Y.; Ohmichi, M.; Ito, M.; Takaori-Kondo, A.; Koyanagi, Y. Remarkable lethal G-to-A mutations in vif-proficient HIV-1 provirus by individual APOBEC3 proteins in humanized mice. J. Virol. 2010, 84, 9546–9556. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Takeuchi, J. S.; Misawa, N.; Izumi, T.; Kobayashi, T.; Kimura, Y.; Iwami, S.; Takaori-Kondo, A.; Hu, W.S.; Aihara, K.; Ito, M.; An, D.S.; Pathak, V.K.; Koyanagi, Y. APOBEC3D and APOBEC3F potently promote HIV-1 diversification and evolution in humanized mouse model. PLoS Pathog. 2014, 10, e1004453. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Misawa, N.; Iwami, S.; Satou, Y.; Matsuoka, M.; Ishizaka, Y.; Ito, M.; Aihara, K.; An, D.S.; Koyanagi, Y. HIV-1 Vpr accelerates viral replication during acute infection by exploitation of proliferating CD4+ T cells in vivo. PLoS Pathog. 2013, 9, e1003812. [Google Scholar] [CrossRef] [PubMed]
- Dave, V.P.; Hajjar, F.; Dieng, M.M.; Haddad, E.; Cohen, E.A. Efficient BST2 antagonism by Vpu is critical for early HIV-1 dissemination in humanized mice. Retrovirology 2013, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Misawa, N.; Fukuhara, M.; Iwami, S.; An, D.S.; Ito, M.; Koyanagi, Y. Vpu augments the initial burst phase of HIV-1 propagation and downregulates BST2 and CD4 in humanized mice. J. Virol. 2012, 86, 5000–5013. [Google Scholar] [CrossRef] [PubMed]
- Watkins, R.L.; Zou, W.; Denton, P.W.; Krisko, J.F.; Foster, J.L.; Garcia, J.V. In vivo analysis of highly conserved Nef activities in HIV-1 replication and pathogenesis. Retrovirology 2013, 10, e125. [Google Scholar] [CrossRef]
- Zou, W.; Denton, P.W.; Watkins, R.L.; Krisko, J.F.; Nochi, T.; Foster, J.L.; Garcia, J.V. Nef functions in BLT mice to enhance HIV-1 replication and deplete CD4+CD8+ thymocytes. Retrovirology 2012, 9, 44. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A.; Travers, P.; Walport, M.; Shlomchik, M.J. T cell-mediated immunity. In Immunobiology: The Immune System in Health and Disease, 6th ed.; Garland Science: New York, USA, 2005; pp. 25–51. [Google Scholar]
- Glushakova, S.; Baibakov, B.; Margolis, L.B.; Zimmerberg, J. Infection of human tonsil histocultures: a model for HIV pathogenesis. Nat. Med. 1995, 1, 1320–1322. [Google Scholar] [CrossRef] [PubMed]
- Introini, A.; Vanpouille, C.; Lisco, A.; Grivel, J.C.; Margolis, L. Interleukin-7 facilitates HIV-1 transmission to cervico-vaginal tissue ex vivo. PLoS Pathog. 2013, 9, e1003148. [Google Scholar] [CrossRef] [PubMed]
- Merbah, M.; Arakelyan, A.; Edmonds, T.; Ochsenbauer, C.; Kappes, J.C.; Shattock, R.J.; Grivel, J.C.; Margolis, L.B. HIV-1 expressing the envelopes of transmitted/founder or control/reference viruses have similar infection patterns of CD4 T-cells in human cervical tissue ex vivo. PLoS One 2012, 7, e50839. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.J.; Wang, Y.; Huo, Z.; Giemza, R.; Babaahmady, K.; Rahman, D.; Shattock, R.J.; Singh, M.; Lehner, T. Effect of vaginal immunization with HIVgp140 and HSP70 on HIV-1 replication and innate and T cell adaptive immunity in women. J. Virol. 2014, 88, 11648–11657. [Google Scholar] [CrossRef] [PubMed]
- Adjali, O.; Montel-Hagen, A.; Swainson, L.; Marty, S.; Vicente, R.; Mongellaz, C.; Jacquet, C.; Zimmermann, V.; Taylor, N. In vivo and ex vivo gene transfer in thymocytes and thymocyte precursors. Methods Mol. Biol. 2009, 506, 171–190. [Google Scholar] [PubMed]
- Mosier, D. E.; Gulizia, R.J.; Baird, S.M.; Wilson, D.B.; Spector, D.H.; Spector, S.A. Human immunodeficiency virus infection of human-PBL-SCID mice. Science 1991, 251, 791–794. [Google Scholar] [CrossRef] [PubMed]
- Gauduin, M.C.; Parren, P.W.; Weir, R.; Barbas, C.F.; Burton, D.R.; Koup, R.A. Passive immunization with a human monoclonal antibody protects hu-PBL-SCID mice against challenge by primary isolates of HIV-1. Nat. Med. 1997, 3, 1389–1393. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, J.; Shpitz, B.; Gallinger, S.; Hozumi, N. Human primary immune response in SCID mice engrafted with human peripheral blood lymphocytes. J. Immunol. 1994, 152, 3806–3813. [Google Scholar] [PubMed]
- Shultz, L.D.; Ishikawa, F.; Greiner, D.L. Humanized mice in translational biomedical research. Nat. Rev. Immunol. 2007, 7, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Traggiai, E.; Chicha, L.; Mazzucchelli, L.; Bronz, L.; Piffaretti, J.C.; Lanzavecchia, A.; Manz, M.G. Development of a human adaptive immune system in cord blood cell-transplanted mice. Science 2004, 304, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Baenziger, S.; Tussiwand, R.; Schlaepfer, E.; Mazzucchelli, L.; Heikenwalder, M.; Kurrer, M.O.; Behnke, S.; Frey, J.; Oxenius, A.; Joller, H.; Aguzzi, A.; Manz, M.G.; Speck, R.F. Disseminated and sustained HIV infection in CD34+ cord blood cell-transplanted Rag2-/- γc-/- mice. Proc. Natl. Acad. Sci. U S A 2006, 103, 15951–15956. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Hiramatsu, H.; Kobayashi, K.; Suzue, K.; Kawahata, M.; Hioki, K.; Ueyama, Y.; Koyanagi, Y.; Sugamura, K.; Tsuji, K.; Heike, T.; Nakahata, T. NOD/SCID/γcnull mouse: an excellent recipient mouse model for engraftment of human cells. Blood 2002, 100, 3175–3182. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Brehm, M.A.; Garcia-Martinez, J.V.; Greiner, D.L. Humanized mice for immune system investigation: progress, promise and challenges. Nat. Rev. Immunol. 2012, 12, 786–798. [Google Scholar] [CrossRef] [PubMed]
- Hatziioannou, T.; Evans, D.T. Animal models for HIV/AIDS research. Nat. Rev. Microbiol. 2012, 10, 852–867. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Koyanagi, Y. The mouse is out of the bag: insights and perspectives on HIV-1-infected humanized mouse models. Exp. Biol. Med. 2011, 236, 977–985. [Google Scholar] [CrossRef]
- Ito, R.; Takahashi, T.; Katano, I.; Ito, M. Current advances in humanized mouse models. Cell Mol. Immunol. 2012, 9, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Melkus, M.W.; Estes, J.D.; Padgett-Thomas, A.; Gatlin, J.; Denton, P.W.; Othieno, F.A.; Wege, A.K.; Haase, A.T.; Garcia, J.V. Humanized mice mount specific adaptive and innate immune responses to EBV and TSST-1. Nat. Med. 2006, 12, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Tonomura, N.; Habiro, K.; Shimizu, A.; Sykes, M.; Yang, Y.G. Antigen-specific human T-cell responses and T cell-dependent production of human antibodies in a humanized mouse model. Blood 2008, 111, 4293–4296. [Google Scholar] [CrossRef] [PubMed]
- Pearson, T.; Greiner, D.L.; Shultz, L.D. Humanized SCID mouse models for biomedical research. In Humanized mice; Nomura, T., Watanabe, T., Habu, S., Eds.; Springer: Heidelberg, Germany, 2008; pp. 25–51. [Google Scholar]
- Willinger, T.; Rongvaux, A.; Strowig, T.; Manz, M.G.; Flavell, R.A. Improving human hemato-lymphoid-system mice by cytokine knock-in gene replacement. Trends Immunol. 2011, 32, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Rongvaux, A.; Willinger, T.; Martinek, J.; Strowig, T.; Gearty, S.V.; Teichmann, L.L.; Saito, Y.; Marches, F.; Halene, S.; Palucka, A.K.; Manz, M.G.; Flavell, R.A. Development and function of human innate immune cells in a humanized mouse model. Nat. Biotechnol. 2014, 32, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Takagi, S.; Saito, Y.; Hijikata, A.; Tanaka, S.; Watanabe, T.; Hasegawa, T.; Mochizuki, S.; Kunisawa, J.; Kiyono, H.; Koseki, H.; Ohara, O.; Saito, T.; Taniguchi, S.; Shultz, L.D.; Ishikawa, F. Membrane-bound human SCF/KL promotes in vivo human hematopoietic engraftment and myeloid differentiation. Blood 2012, 119, 2768–2777. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Saito, Y.; Najima, Y.; Tanaka, S.; Ochi, T.; Tomizawa, M.; Doi, T.; Sone, A.; Suzuki, N.; Fujiwara, H.; Yasukawa, M.; Ishikawa, F. Generation of functional human T-cell subsets with HLA-restricted immune responses in HLA class I expressing NOD/SCID/IL2rγnull humanized mice. Proc. Natl. Acad. Sci. U S A 2010, 107, 13022–13027. [Google Scholar] [CrossRef] [PubMed]
- Berkowitz, R.D.; Beckerman, K.P.; Schall, T.J.; McCune, J.M. CXCR4 and CCR5 expression delineates targets for HIV-1 disruption of T cell differentiation. J. Immunol. 1998, 161, 3702–3710. [Google Scholar] [PubMed]
- Kitchen, S.G.; Zack, J.A. Distribution of the human immunodeficiency virus coreceptors CXCR4 and CCR5 in fetal lymphoid organs: implications for pathogenesis in utero. AIDS Res. Hum. Retroviruses 1999, 15, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Zamarchi, R.; Allavena, P.; Borsetti, A.; Stievano, L.; Tosello, V.; Marcato, N.; Esposito, G.; Roni, V.; Paganin, C.; Bianchi, G.; Titti, F.; Verani, P.; Gerosa, G.; Amadori, A. Expression and functional activity of CXCR-4 and CCR-5 chemokine receptors in human thymocytes. Clin. Exp. Immunol. 2002, 127, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Connor, R.I.; Sheridan, K.E.; Ceradini, D.; Choe, S.; Landau, N.R. Change in coreceptor use correlates with disease progression in HIV-1-infected individuals. J. Exp. Med. 1997, 185, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Scarlatti, G.; Tresoldi, E.; Bjorndal, A.; Fredriksson, R.; Colognesi, C.; Deng, H.K.; Malnati, M.S.; Plebani, A.; Siccardi, A.G.; Littman, D.R.; Fenyo, E.M.; Lusso, P. In vivo evolution of HIV-1 co-receptor usage and sensitivity to chemokine-mediated suppression. Nat. Med. 1997, 3, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.A.; Pathak, V.K. Identification of two distinct human immunodeficiency virus type 1 Vif determinants critical for interactions with human APOBEC3G and APOBEC3F. J. Virol. 2007, 81, 8201–8210. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.L.; Pathak, V.K. Identification of specific determinants of human APOBEC3F, APOBEC3C, and APOBEC3DE and African green monkey APOBEC3F that interact with HIV-1 Vif. J. Virol. 2010, 84, 12599–12608. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.; Davis, Z.; DeHart, J.; Zimmerman, E.; Bosque, A.; Brunetta, E.; Mavilio, D.; Planelles, V.; Barker, E. HIV-1 Vpr triggers natural killer cell-mediated lysis of infected cells through activation of the ATR-mediated DNA damage response. PLoS Pathog. 2009, 5, e1000613. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.; Sindhu, S.; Pham, T.N.; Belzile, J.P.; Cohen, E.A. HIV-1 Vpr up-regulates expression of ligands for the activating NKG2D receptor and promotes NK cell-mediated killing. Blood 2010, 115, 1354–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyara, M.; Yoshioka, Y.; Kitoh, A.; Shima, T.; Wing, K.; Niwa, A.; Parizot, C.; Taflin, C.; Heike, T.; Valeyre, D.; Mathian, A.; Nakahata, T.; Yamaguchi, T.; Nomura, T.; Ono, M.; Amoura, Z.; Gorochov, G.; Sakaguchi, S. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity 2009, 30, 899–911. [Google Scholar] [CrossRef] [PubMed]
- Nie, C.; Sato, K.; Misawa, N.; Kitayama, H.; Fujino, H.; Hiramatsu, H.; Heike, T.; Nakahata, T.; Tanaka, Y.; Ito, M.; Koyanagi, Y. Selective infection of CD4+ effector memory T lymphocytes leads to preferential depletion of memory T lymphocytes in R5 HIV-1-infected humanized NOD/SCID/IL-2Rγnull mice. Virology 2009, 394, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Miyara, M.; Costantino, C.M.; Hafler, D.A. FOXP3+ regulatory T cells in the human immune system. Nat. Rev. Immunol. 2010, 10, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J. M.; Silvestri, G.; Douek, D.C. Nonprogressive and progressive primate immunodeficiency lentivirus infections. Immunity 2010, 32, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Levesque, K.; Finzi, A.; Binette, J.; Cohen, E.A. Role of CD4 receptor down-regulation during HIV-1 infection. Curr. HIV Res. 2004, 2, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Blasius, A.L.; Giurisato, E.; Cella, M.; Schreiber, R.D.; Shaw, A.S.; Colonna, M. Bone marrow stromal cell antigen 2 is a specific marker of type I IFN-producing cells in the naive mouse, but a promiscuous cell surface antigen following IFN stimulation. J. Immunol. 2006, 177, 3260–3265. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.T.; Serra-Moreno, R.; Singh, R.K.; Guatelli, J.C. BST-2/tetherin: a new component of the innate immune response to enveloped viruses. Trends Microbiol. 2010, 18, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Douglas, J.L.; Gustin, J.K.; Viswanathan, K.; Mansouri, M.; Moses, A.V.; Fruh, K. The great escape: viral strategies to counter BST-2/tetherin. PLoS Pathog. 2010, 6, e1000913. [Google Scholar] [CrossRef] [PubMed]
- Goffinet, C.; Allespach, I.; Homann, S.; Tervo, H.M.; Habermann, A.; Rupp, D.; Oberbremer, L.; Kern, C.; Tibroni, N.; Welsch, S.; Krijnse-Locker, J.; Banting, G.; Krausslich, H.G.; Fackler, O.T.; Keppler, O.T. HIV-1 antagonism of CD317 is species specific and involves Vpu-mediated proteasomal degradation of the restriction factor. Cell Host Microbe 2009, 5, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.S.; Katsura, C.; Skasko, M.A.; Fitzpatrick, K.; Lau, D.; Ruiz, A.; Stephens, E.B.; Margottin-Goguet, F.; Benarous, R.; Guatelli, J.C. Vpu antagonizes BST-2-mediated restriction of HIV-1 release via β-TrCP and endo-lysosomal trafficking. PLoS Pathog. 2009, 5, e1000450. [Google Scholar] [CrossRef] [PubMed]
- Willey, R.L.; Maldarelli, F.; Martin, M.A.; Strebel, K. Human immunodeficiency virus type 1 Vpu protein induces rapid degradation of CD4. J. Virol. 1992, 66, 7193–7200. [Google Scholar] [PubMed]
- Homann, S.; Smith, D.; Little, S.; Richman, D.; Guatelli, J. Upregulation of BST-2/tetherin by HIV infection in vivo. J. Virol. 2011, 85, 10659–10668. [Google Scholar] [CrossRef] [PubMed]
- Schubert, U.; Strebel, K. Differential activities of the human immunodeficiency virus type 1-encoded Vpu protein are regulated by phosphorylation and occur in different cellular compartments. J. Virol. 1994, 68, 2260–2271. [Google Scholar] [PubMed]
- Sauter, D.; Schindler, M.; Specht, A.; Landford, W.N.; Munch, J.; Kim, K. A.; Votteler, J.; Schubert, U.; Bibollet-Ruche, F.; Keele, B.F.; Takehisa, J.; Ogando, Y.; Ochsenbauer, C.; Kappes, J.C.; Ayouba, A.; Peeters, M.; Learn, G.H.; Shaw, G.; Sharp, P.M.; Bieniasz, P.; Hahn, B.H.; Hatziioannou, T.; Kirchhoff, F. Tetherin-driven adaptation of Vpu and Nef function and the evolution of pandemic and nonpandemic HIV-1 strains. Cell Host Microbe 2009, 6, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Sauter, D.; Hue, S.; Petit, S.J.; Plantier, J.C.; Towers, G.J.; Kirchhoff, F.; Gupta, R.K. HIV-1 Group P is unable to antagonize human tetherin by Vpu, Env or Nef. Retrovirology 2011, 8, 103. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.J.; Lopez, L.A.; Exline, C.M.; Haworth, K.G.; Cannon, P.M. Lack of adaptation to human tetherin in HIV-1 group O and P. Retrovirology 2011, 8, 78. [Google Scholar] [CrossRef] [PubMed]
- Fackler, O.T.; Moris, A.; Tibroni, N.; Giese, S.I.; Glass, B.; Schwartz, O.; Krausslich, H.G. Functional characterization of HIV-1 Nef mutants in the context of viral infection. Virology 2006, 351, 322–339. [Google Scholar] [CrossRef] [PubMed]
- Glushakova, S.; Munch, J.; Carl, S.; Greenough, T.C.; Sullivan, J.L.; Margolis, L.; Kirchhoff, F. CD4 down-modulation by human immunodeficiency virus type 1 Nef correlates with the efficiency of viral replication and with CD4+ T-cell depletion in human lymphoid tissue ex vivo. J. Virol. 2001, 75, 10113–10117. [Google Scholar] [CrossRef] [PubMed]
- Lundquist, C.A.; Tobiume, M.; Zhou, J.; Unutmaz, D.; Aiken, C. Nef-mediated downregulation of CD4 enhances human immunodeficiency virus type 1 replication in primary T lymphocytes. J. Virol. 2002, 76, 4625–4633. [Google Scholar] [CrossRef] [PubMed]
- Churchill, M. J.; Rhodes, D.I.; Learmont, J.C.; Sullivan, J.S.; Wesselingh, S.L.; Cooke, I.R.; Deacon, N.J.; Gorry, P.R. Longitudinal analysis of human immunodeficiency virus type 1 nef/long terminal repeat sequences in a cohort of long-term survivors infected from a single source. J. Virol. 2006, 80, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Gorry, P.R.; McPhee, D.A.; Verity, E.; Dyer, W.B.; Wesselingh, S.L.; Learmont, J.; Sullivan, J.S.; Roche, M.; Zaunders, J.J.; Gabuzda, D.; Crowe, S.M.; Mills, J.; Lewin, S.R.; Brew, B.J.; Cunningham, A.L.; Churchill, M.J. Pathogenicity and immunogenicity of attenuated, nef-deleted HIV-1 strains in vivo. Retrovirology 2007, 4, e66. [Google Scholar] [CrossRef]
- Greenough, T.C.; Sullivan, J.L.; Desrosiers, R.C. Declining CD4 T-cell counts in a person infected with nef-deleted HIV-1. N. Engl. J. Med. 1999, 340, 236–237. [Google Scholar] [CrossRef] [PubMed]
- Kondo, M.; Shima, T.; Nishizawa, M.; Sudo, K.; Iwamuro, S.; Okabe, T.; Takebe, Y.; Imai, M. Identification of attenuated variants of HIV-1 circulating recombinant form 01_AE that are associated with slow disease progression due to gross genetic alterations in the nef/long terminal repeat sequences. J. Infect. Dis. 2005, 192, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, D.I.; Ashton, L.; Solomon, A.; Carr, A.; Cooper, D.; Kaldor, J.; Deacon, N. Characterization of three nef-defective human immunodeficiency virus type 1 strains associated with long-term nonprogression. Australian Long-Term Nonprogressor Study Group. J. Virol. 2000, 74, 10581–10588. [Google Scholar]
- Salvi, R.; Garbuglia, A.R.; Di Caro, A.; Pulciani, S.; Montella, F.; Benedetto, A. Grossly defective nef gene sequences in a human immunodeficiency virus type 1-seropositive long-term nonprogressor. J. Virol. 1998, 72, 3646–3657. [Google Scholar] [PubMed]
- Braibant, M.; Xie, J.; Samri, A.; Agut, H.; Autran, B.; Barin, F. Disease progression due to dual infection in an HLA-B57-positive asymptomatic long-term nonprogressor infected with a nef-defective HIV-1 strain. Virology 2010, 405, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.S.; Baugh, L.L.; Denial, S.J.; Watkins, R.L.; Liu, M.; Garcia, J.V.; Foster, J.L. Overlapping effector interfaces define the multiple functions of the HIV-1 Nef polyproline helix. Retrovirology 2012, 9, e47. [Google Scholar] [CrossRef]
- Manninen, A.; Hiipakka, M.; Vihinen, M.; Lu, W.; Mayer, B.J.; Saksela, K. SH3-Domain binding function of HIV-1 Nef is required for association with a PAK-related kinase. Virology 1998, 250, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.L.; Denial, S.J.; Temple, B.R.; Garcia, J.V. Mechanisms of HIV-1 Nef function and intracellular signaling. J. Neuroimmune Pharmacol. 2011, 6, 230–246. [Google Scholar] [CrossRef] [PubMed]
- Balazs, A. B.; Ouyang, Y.; Hong, C.M.; Chen, J.; Nguyen, S.M.; Rao, D.S.; An, D.S.; Baltimore, D. Vectored immunoprophylaxis protects humanized mice from mucosal HIV transmission. Nat. Med. 2014, 20, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Holt, N.; Wang, J.; Kim, K.; Friedman, G.; Wang, X.; Taupin, V.; Crooks, G.M.; Kohn, D.B.; Gregory, P.D.; Holmes, M.C.; Cannon, P.M. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat. Biotechnol. 2010, 28, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Pickering, S.; Hue, S.; Kim, E.Y.; Reddy, S.; Wolinsky, S.M.; Neil, S.J. Preservation of tetherin and CD4 counter-activities in circulating Vpu alleles despite extensive sequence variation within HIV-1 infected individuals. PLoS Pathog. 2014, 10, e1003895. [Google Scholar] [CrossRef] [PubMed]
- Iwabu, Y.; Kinomoto, M.; Tatsumi, M.; Fujita, H.; Shimura, M.; Tanaka, Y.; Ishizaka, Y.; Nolan, D.; Mallal, S.; Sata, T.; Tokunaga, K. Differential anti-APOBEC3G activity of HIV-1 Vif proteins derived from different subtypes. J. Biol. Chem. 2010, 285, 35350–35358. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamada, E.; Yoshikawa, R.; Nakano, Y.; Misawa, N.; Koyanagi, Y.; Sato, K. Impacts of Humanized Mouse Models on the Investigation of HIV-1 Infection: Illuminating the Roles of Viral Accessory Proteins in Vivo. Viruses 2015, 7, 1373-1390. https://doi.org/10.3390/v7031373
Yamada E, Yoshikawa R, Nakano Y, Misawa N, Koyanagi Y, Sato K. Impacts of Humanized Mouse Models on the Investigation of HIV-1 Infection: Illuminating the Roles of Viral Accessory Proteins in Vivo. Viruses. 2015; 7(3):1373-1390. https://doi.org/10.3390/v7031373
Chicago/Turabian StyleYamada, Eri, Rokusuke Yoshikawa, Yusuke Nakano, Naoko Misawa, Yoshio Koyanagi, and Kei Sato. 2015. "Impacts of Humanized Mouse Models on the Investigation of HIV-1 Infection: Illuminating the Roles of Viral Accessory Proteins in Vivo" Viruses 7, no. 3: 1373-1390. https://doi.org/10.3390/v7031373