
What Can We Learn from a Metagenomic Analysis of a Georgian Bacteriophage Cocktail?

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. The Intesti Phage Cocktail
2.2. Host-Amplified Samples

{kind=link}
{kind=link}
{kind=link}
| Host Bacterial Strain | Number of Reads |
|---|---|
| Escherichia coli ATCC 25922 | 358,914 |
| Enterococcus faecalis ATCC 29212 | 134,966 |
| Pseudomonasaeruginosa 0407431-2 | 184,790 |
| Pseudomonasaeruginosa PAO1_seq | 265,772 |
| Proteus vulgaris CCUG 36761 (ATCC 13315) | 64,852 |
| Salmonella typhimurium ATCC 14028 | 133,980 |
| Shigellaflexneri iran_1s | 225,664 |
| Shigellasonnei iran_2s | 401,722 |
2.3. Sample Preparation
2.4. Sequencing and Genome Assembly
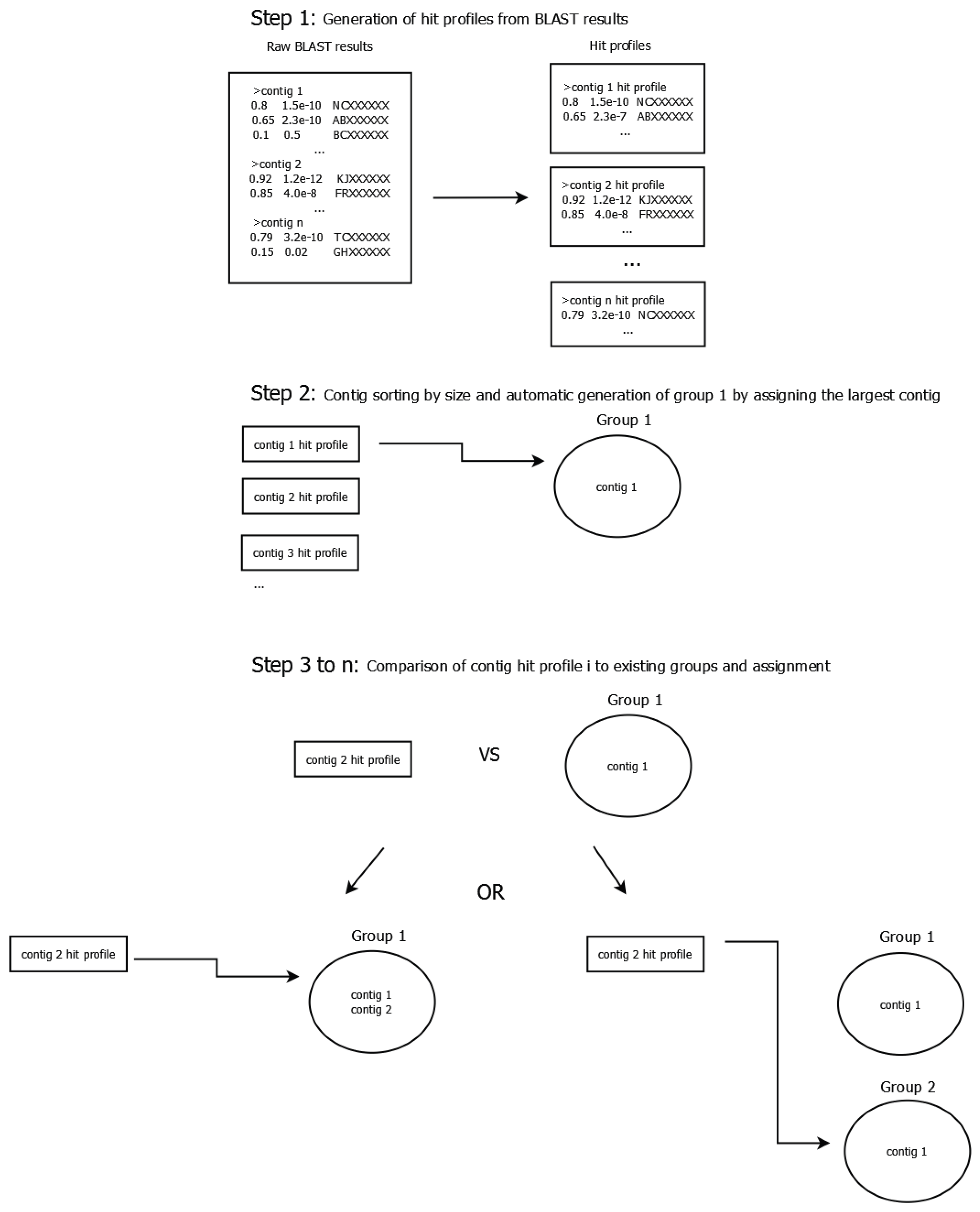
2.5. Construction of Phage Clusters

- If the hit is only present in one of the profiles, its distance is 1.0.
- If the hit is present in both profiles, the hit’s distance is the absolute value of the difference between the query coverage values, as defined below:
2.6. Analysis of the Depth of Coverage
2.7. Gene Prediction and Functional Annotation
2.8. Host Range Estimation
3. Results
3.1. Sequencing Statistics
| Phage Cluster | Cluster Size in bp | Reference Accession | Average Coverage of Phage Cluster | Average Percent Identity | Reference Phage Description Line | Phage Family | Subfamily | Genus | Size RatioCluster/Reference |
|---|---|---|---|---|---|---|---|---|---|
| D1 | 142,025 | KC012913.1 | 99.97 | 99.80 | Staphylococcus phage Team1, complete genome | Myoviridae | 1.01 | ||
| AY954969.1 | 97.98 | 99.74 | Bacteriophage G1, complete genome * | Spounavirinae | Twortlikevirus | 1.02 | |||
| D2 | 76,960 | JX415536.1 | 87.89 | 87.60 | Escherichia phage KBNP135, complete genome | Podoviridae | 1.00 | ||
| D3 | 87,828 | KC862301.1 | 98.97 | 96.16 | Pseudomonas phage PAK_P5, complete genome | Myoviridae | 1.00 | ||
| D4 | 69,023 | KF562340.1 | 87.20 | 94.02 | Escherichia phage vB_EcoP_PhAPEC7, complete genome | Podoviridae | 0.96 | ||
| D5 | 150,530 | FR775895.2 | 92.41 | 98.16 | Enterobacteria phage phi92, complete genome | Myoviridae | 1.01 | ||
| D6 | 81,563 | AB609718.1 | 35.55 | 77.46 | Enterococcus phage phiEF24C-P2 , complete genome | Myoviridae | 0.57 | ||
| D7 | 58,193 | KJ094032.2 | 77.23 | 88.35 | Enterococcus phage VD13, complete genome | Siphoviridae | - | Sap6likevirus | 1.06 |
| D8 | 50,277 | HM035024.1 | 98.16 | 90.67 | Shigella phage Shfl1, complete genome | Siphoviridae | - | Tunalikevirus | 0.99 |
| D9 | 39,912 | EU734172.1 | 88.25 | 93.45 | Enterobacteria phage EcoDS1, complete genome | Podoviridae | 1.02 | ||
| D10 | 145,982 | KJ190158.1 | 93.95 | 93.00 | Escherichia phage vB_EcoM_FFH2, complete genome | Myoviridae | 1.05 | ||
| DQ832317.1 | 93.72 | 92.62 | Escherichia coli bacteriophage rv5, complete sequence | - | “rv5-like virus” * | 1.06 | |||
| D11 | 61,791 | JX094499.1 | 96.33 | 92.95 | Enterobacteria phage Chi, complete genome | Siphoviridae | 1.04 | ||
| KC139512.1 | 95.15 | 93.86 | Salmonella phage FSL SP-088, complete genome | - | Chilikevirus | 1.04 | |||
| D12 | 60,451 | KJ010489.1 | 54.57 | 87.35 | Enterococcus phage IME-EFm1, complete genome | Siphoviridae | 1.42 | ||
| D13 | 188,630 | GU070616.1 | 88.67 | 94.90 | Salmonella phage PVP-SE1, complete genome | Myoviridae | “rv5-like virus” * | 1.29 | |
| D14 | 133,015 | JX128259.1 | 94.55 | 96.24 | Escherichia phage ECML-134, complete genome | Myoviridae | 0.80 | ||
| DQ904452.1 | 93.42 | 96.00 | Bacteriophage RB32, complete genome | Tevenvirinae | T4likevirus | 0.80 | |||
| D15 | 43,967 | GQ468526.1 | 87.06 | 91.27 | Enterobacteria phage 285P, complete genome | Podoviridae | 1.12 | ||
| FJ194439.1 | 87.13 | 90.61 | Kluyvera phage Kvp1, complete sequence | Autographivirinae | T7likevirus | 1.11 | |||
| D16 | 46,882 | KM233151.1 | 93.68 | 91.47 | Enterobacteria phage EK99P-1, complete genome | Siphoviridae | 1.06 | ||
| JX865427.2 | 91.64 | 91.03 | Enterobacteria phage JL1, complete genome | Hk578likevirus | 1.08 | ||||
| D17 | 41,098 | AY370674.1 | 88.68 | 94.28 | Enterobacteria phage K1-5, complete genome | Podoviridae | Autographivirinae | Sp6likevirus | 0.93 |
| D18 | 41,016 | HE775250.1 | 94.95 | 91.57 | Salmonella phage vB_SenS-Ent1 complete genome | Siphoviridae | 0.97 | ||
| JX202565.1 | 92.76 | 91.41 | Salmonella phage wksl3, complete genome | Jerseylikevirus | 0.96 | ||||
| F1 | 13,855 | HG518155.1 | 99.97 | 99.02 | Pseudomonas phage TL complete genome | Podoviridae | 0.30 | ||
| AM910650.1 | 91.92 | 97.11 | Pseudomonas phage LUZ24, complete genome | - | Luz24likevirus | 0.30 | |||
| F2 | 11,476 | EU877232.1 | 99.94 | 91.42 | Enterobacteria phage WV8, complete sequence | Myoviridae | - | Felixounalikevirus | 0.13 |
| F3 | 5706 | HQ665011.1 | 83.42 | 86.09 | Escherichia phage bV_EcoS_AKFV33, complete genome | Siphoviridae | 0.05 | ||
| AY543070.1 | 82.09 | 87.59 | Bacteriophage T5, complete genome | - | T5likevirus | 0.05 | |||
| F4 | 2624 | EF437941.1 | 98.59 | 97.76 | Enterobacteria phage Phi1, complete genome | Myoviridae | Tevenvirinae | T4likevirus | 0.02 |
| Proteus phage | 104,213 | - | - | - | - | Siphoviridae | - |
3.2. Recovered Phage Clusters
3.2.1. Similarity to Known Phages
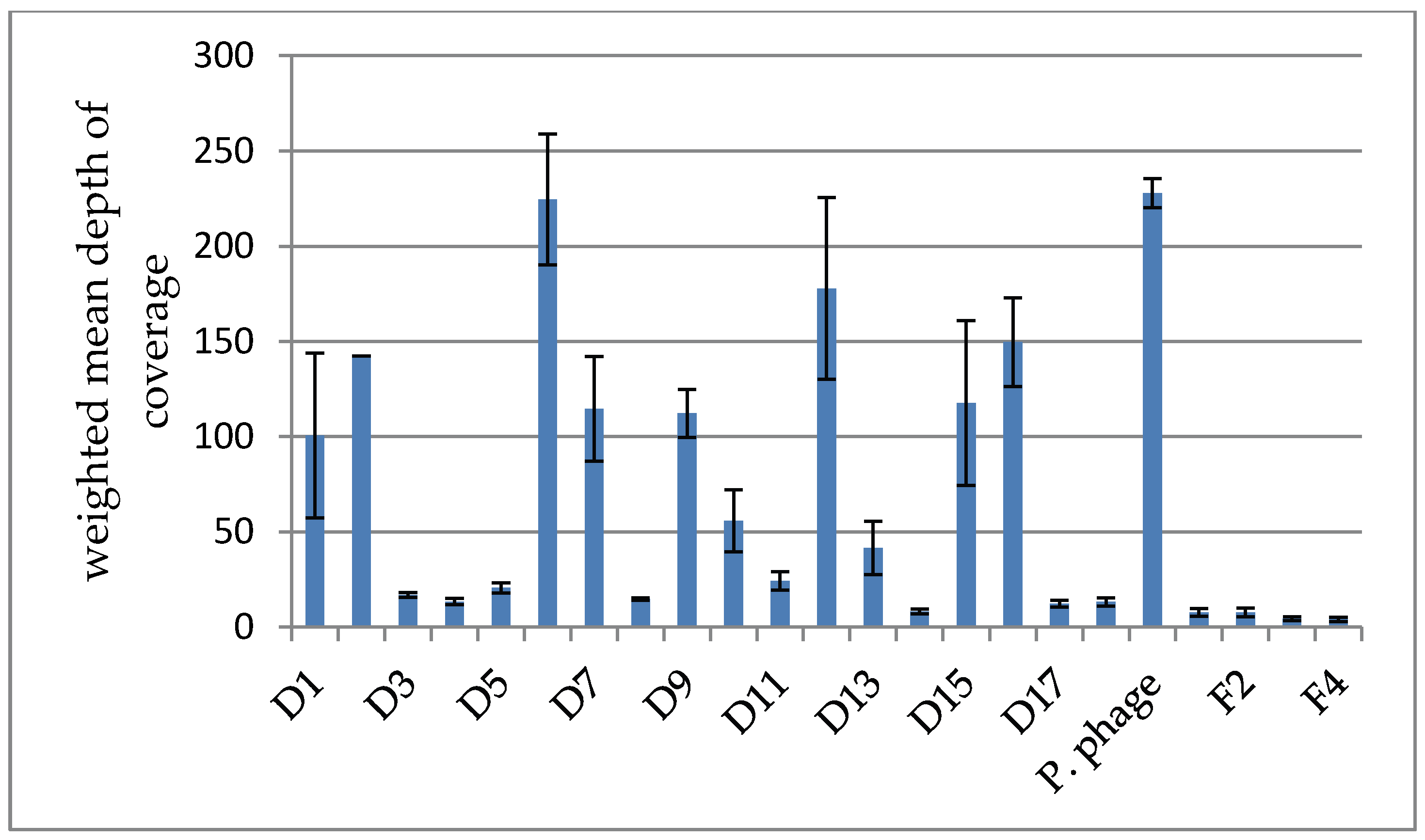
3.2.2. Depth of Coverage in the Intesti Clusters

3.2.3. Depth of Coverage in the Host-Amplified Samples and Comparison of Phage Clusters between Samples
| Cluster | E. coli | Enterococcus | P. aeruginosa PAO1 | P. aeruginosa PA0407 | Shigella flexneri | Shigella sonnei | Proteus |
|---|---|---|---|---|---|---|---|
| D1 | 0.03 | 0.00 | 0.00 | 0.00 | 0.02 | 0.00 | 0.00 |
| D2 | 0.02 | 0.00 | 0.00 | 0.00 | 0.02 | 0.02 | 0.00 |
| D3 | 0.30 | 0.00 | 0.00 | 22.29 | 0.10 | 0.00 | 0.00 |
| D4 | 0.09 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| D5 | 0.11 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| D6 | 0.06 | 0.00 | 0.02 | 0.00 | 0.01 | 0.01 | 0.00 |
| D7 | 0.05 | 2.57 | 0.00 | 0.00 | 0.02 | 0.00 | 0.00 |
| D8 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| D9 | 0.04 | 0.00 | 0.00 | 0.00 | 0.02 | 0.00 | 0.00 |
| D10 | 0.08 | 0.00 | 0.06 | 0.00 | 0.02 | 0.00 | 0.00 |
| D11 | 0.13 | 0.13 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| D12 | 0.04 | 0.00 | 0.01 | 0.00 | 0.02 | 0.00 | 0.00 |
| D13 | 0.05 | 0.05 | 0.00 | 0.00 | 0.04 | 0.00 | 0.00 |
| D14 | 4.74 | 0.00 | 0.00 | 0.00 | 2.82 | 2.06 | 0.00 |
| D15 | 0.04 | 0.00 | 0.00 | 0.00 | 4.97 | 9.84 | 0.00 |
| D16 | 0.04 | 0.00 | 0.00 | 0.00 | 0.02 | 0.02 | 0.00 |
| D17 | 47.17 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 10.01 |
| D18 | 0.37 | - | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| F1 | 0.00 | 0.00 | 1.47 | 0.00 | 0.00 | 0.00 | 0.00 |
| F2 | 0.00 | - | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| F3 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| F4 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Proteus | 0.04 | 0.00 | 0.02 | 0.00 | 0.00 | 0.01 | 0.12 |
| * | 0.00 | 0.00 | 1044.20 | 0.00 | 0.00 | 0.00 | 0.00 |
3.2.4. Gene Prediction and Functional Annotation in the Intesti Clusters
| Text Mining Term | Description Line | Part of Cluster | Annotation by | Top BLAST Hit Description Line | Query Coverage | Percent Positives |
|---|---|---|---|---|---|---|
| “virulence” | Phage virulence-associated protein | D1 | RAST | ORF002 (Staphylococcus phage G1) | 100% | 100% |
| Phage virulence-associated protein | D6 | RAST | putative adsorption associated tail protein (Enterococcus phage phiEF24C) | 100% | 95% | |
| “resistance” | Acridine resistance | D14 | RAST | - | - | - |
| Acriflavin resistance protein | D3 | RAST | hypothetical protein PAK_P500103 (Pseudomonas phage PAK_P5) | 100% | 100% | |
| Tellurium resistance protein TerD | D5 | RAST | Phi92_gp172 (Enterobacteria phage phi92) | 100% | 100% | |
| Tellurium resistance protein TerD | D5 | RAST | Phi92_gp173 (Enterobacteria phage phi92) | 100% | 100% | |
| Tellurite resistance protein | D5 | RAST | Phi92_gp178 (Enterobacteria phage phi92) | 100% | 100% | |
| “methyltransferase” or “methylase” | DNA methylase | D7 | RAST/BLAST | See “Description line” | 100% | 99% |
| DNA N-6-adenine-methyltransferase | D8 | RAST/BLAST | See “Description line” | 94% | 90% | |
| putative site specific DNA methylase | D8 | BLAST | See “Description line“ | 100% | 99% | |
| DNA methyltransferase | D13 | RAST/BLAST | See “Description line” | 100% | 99% | |
| putative DNA N-6-adenine methyltransferase | D10 | RAST/BLAST | See “Description line” | 100% | 99% | |
| Dam methylase | D8 | BLAST | See “Description line” | 100% | 100% | |
| putative DNA adenine methylase | D11 | BLAST | See “Description line” | 100% | 100% | |
| putative DNA methyltransferase | unassigned | BLAST | See “Description line” | 100% | 100% | |
| DNA adenine methyltransferase | D14 | BLAST | See “Description line” | 100% | 99% | |
| putative DNA adenine methylase | D11 | RAST/BLAST | See “Description line” | 100% | 97% | |
| dCMPhydroxymethylase | D14 | RAST/BLAST | See “Description line” | 100% | 100% | |
| putative adenine methyltransferase | D10 | RAST/BLAST | See “Description line” | 100% | 98% | |
| DNA-cytosine methyltransferase | D5 | RAST | Phi92_gp043 (Enterobacteria phage phi92) | 100% | 99% | |
| Adenine-specific methyltransferase | D5 | RAST | Phi92_gp155 (Enterobacteria phage phi92) | 100% | 99% | |
| “integrase” | Phage integrase | D2 | RAST/BLAST | putative integrase (Escherichia phage KBNP1711) | 100% | 98% |
| Phage integrase | D4 | RAST/BLAST | integrase (Enterobacter phage IME11) | 100% | 99% |
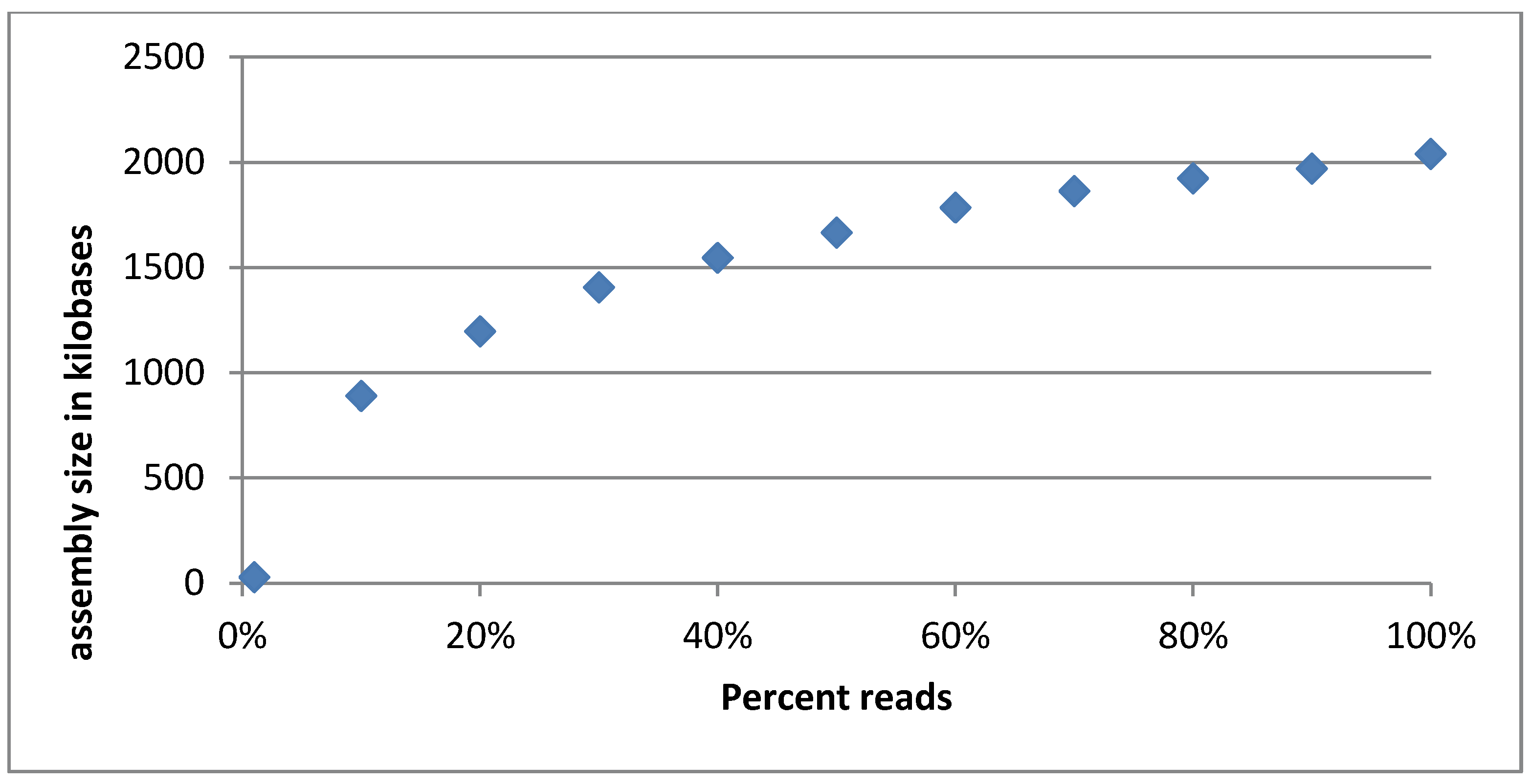
3.2.5. Evaluation of Sequencing Depth of the Cocktail

3.3. Host Range Estimation
| Pathogen | Susceptible Strains |
|---|---|
| Salmonella Enterica | 10/10 |
| Staphylococcus | 5/10 |
| Shigella | 5/5 |
| Pseudomonas Aeruginosa | 5/7 |
| E. coli | 2/6 |
| Proteus | 3/5 |
| Enterococcus | 2/5 |
4. Discussion
4.1. Completeness and Accuracy of the Analysis
4.2. Concerning the Synonymous Clusters and Amplification by Bacterial Hosts
4.3. Comparison to Other Phage Cocktail Studies Employing Metagenomics
4.4. Future Perspectives
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization. Antimicrobial Resistance Global Report on Surveillance 2014; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Lu, T.K.; Koeris, M.S. The next generation of bacteriophage therapy. Curr. Opin. Microbiol. 2011, 14, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.K.; Abedon, S.T.; Loc-Carrillo, C. Phage cocktails and the future of phage therapy. Future Microbiol. 2013, 8, 769–783. [Google Scholar] [CrossRef] [PubMed]
- Hatfull, G.F. Bacteriophage genomics. Curr. Opin. Microbiol. 2008, 11, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Reardon, S. Phage therapy gets revitalized. Nature 2014, 510, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Harper, D.R.; Anderson, J.; Enright, M.C. Phage therapy: Delivering on the promise. Ther. Deliv. 2011, 2, 935–947. [Google Scholar] [CrossRef] [PubMed]
- Kutateladze, M.; Adamia, R. Bacteriophages as potential new therapeutics to replace or supplement antibiotics. Trends Biotechnol. 2010, 28, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Kutter, E.; Borysowski, J.; Międzybrodzki, R.; Górski, A.; Weber-Dąbrowska, B.; Kutateladze, M. Clinical phage therapy. In Phage Therapy: Current Research and Applications, 1st ed.; Borysowski, J., Międzybrodzki, R., Górski, A., Eds.; Caister Academic Press: Poole, UK, 2014; pp. 253–284. [Google Scholar]
- Kutter, E.; de Vos, D.; Gvasalia, G.; Alavidze, Z.; Gogokhia, L.; Kuhl, S.; Abedon, S.T. Phage therapy in clinical practice: Treatment of human infections. Curr. Pharm. Biotechnol. 2010, 11, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Abedon, S.T.; Kuhl, S.J.; Blasdel, B.G.; Kutter, E.M. Phage treatment of human infections. Bacteriophage 2011, 1, 66–85. [Google Scholar] [CrossRef] [PubMed]
- Sulakvelidze, A.; Alavidze, Z.; Morris, J.G. Bacteriophage therapy. Antimicrob. Agents Chemother. 2011, 45, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Bruttin, A.; Brüssow, H. Human volunteers receiving Escherichia coli phage T4 orally: A safety test of phage therapy. Antimicrob. Agents Chemother. 2005, 49, 2874–2878. [Google Scholar] [CrossRef] [PubMed]
- Sarker, S.A.; McCallin, S.; Barretto, C.; Berger, B.; Pittet, A.-C.; Sultana, S.; Krause, L.; Huq, S.; Bibiloni, R.; Bruttin, A.; et al. Oral T4-like phage cocktail application to healthy adult volunteers from Bangladesh. Virology 2012, 434, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.; Hawkins, C.H.; Anggård, E.E.; Harper, D.R. A controlled clinical trial of a therapeutic bacteriophage preparation in chronic otitis due to antibiotic-resistant Pseudomonas aeruginosa; a preliminary report of efficacy. Clin. Otolaryngol. 2009, 34, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, D.D.; Wolcott, R.D.; Kuskowski, M.A.; Wolcott, B.M.; Ward, L.S.; Sulakvelidze, A. Bacteriophage therapy of venous leg ulcers in humans: Results of a phase I safety trial. J. Wound Care 2009, 18, 237–243. [Google Scholar] [CrossRef] [PubMed]
- McCallin, S.; Sarker, S.A.; Barretto, C.; Sultana, S.; Berger, B.; Huq, S.; Krause, L.; Bibiloni, R.; Schmitt, B.; Reuteler, G.; et al. Safety analysis of a Russian phage cocktail: From metagenomic analysis to oral application in healthy human subjects. Virology 2013, 443, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Brüssow, H. What is needed for phage therapy to become a reality in Western medicine? Virology 2012, 434, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Salamon, P.; Andresen, B.; Mahaffy, J.M.; Segall, A.M.; Mead, D.; Azam, F.; Rohwer, F. Genomic analysis of uncultured marine viral communities. Proc. Natl. Acad. Sci. USA 2002, 99, 14250–14255. [Google Scholar] [CrossRef] [PubMed]
- D’Herelle, F. The Bacteriophage; Williams & Wilkins Company: Baltimore, MD, USA, 1922. [Google Scholar]
- Mikeladze, C.; Nemsadze, E.; Alexidze, N.; Assanichvili, T. Sur le traitement de la fievre typhoide et des colites aigues par le bacteriophage de d'Herelle. La Médecine 1936, 17, 33–38. (In French) [Google Scholar]
- Chanishvili, N. A Literature Review of the Practical Application of Bacteriophage Research; Nova Science Publishers: New York, NY, USA, 2012. [Google Scholar]
- Kuhl, S.J.; Mazure, H. d’Hérelle. Preparation of Therapeutic Bacteriophages, Appendix 1 from: Le Phénomène de la Guérison dans les maladies infectieuses: Masson et Cie, 1938, Paris—OCLC 5784382. Bacteriophage 2011, 1, 55–65. [Google Scholar] [CrossRef]
- Andrews, S. FastQC—A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc/ (accessed on 1 February 2015).
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed]
- Laserson, J.; Jojic, V.; Koller, D. Genovo: De novo assembly for metagenomes. J. Comput. Biol. 2011, 18, 429–443. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Deurenberg, R.H.; Stobberingh, E.E. The evolution of Staphylococcus aureus. Infect. Genet. Evol. 2008, 8, 747–763. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9. [Google Scholar] [CrossRef] [PubMed]
- Besemer, J. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef] [PubMed]
- Lobocka, M.; Hejnowicz, M.S.; Gagala, U.; Weber-Dąbrowska, B.; Wegrzyn, G.; Dadlez, M. The first step to bacteriophage therapy: How to choose the correct phage. In Phage Therapy: Current Research and Applications; Borysowski, J., Międzybrodzki, R., Gorski, A., Eds.; Caister Academic Press: Norfolk, UK, 2014; pp. 23–67. [Google Scholar]
- Samson, J.E.; Magadán, A.H.; Sabri, M.; Moineau, S. Revenge of the phages: Defeating bacterial defences. Nat. Rev. Microbiol. 2013, 11, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef] [PubMed]
- Joensen, K.G.; Scheutz, F.; Lund, O.; Hasman, H.; Kaas, R.S.; Nielsen, E.M.; Aarestrup, F.M. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 2014, 52, 1501–1510. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.B.; Kropinski, A.M.; Ceyssens, P.-J.; Ackermann, H.-W.; Villegas, A.; Lavigne, R.; Krylov, V.N.; Carvalho, C.M.; Ferreira, E.C.; Azeredo, J. Genomic and proteomic characterization of the broad-host-range Salmonella phage PVP-SE1: Creation of a new phage genus. J. Virol. 2011, 85, 11265–11273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarzer, D.; Buettner, F.F.R.; Browning, C.; Nazarov, S.; Rabsch, W.; Bethe, A.; Oberbeck, A.; Bowman, V.D.; Stummeyer, K.; Mühlenhoff, M.; et al. A multivalent adsorption apparatus explains the broad host range of phage phi92: A comprehensive genomic and structural analysis. J. Virol. 2012, 86, 10384–10398. [Google Scholar] [CrossRef] [PubMed]
- Zuo, G.; Xu, Z.; Hao, B. Shigella strains are not clones of Escherichia coli but sister species in the genus Escherichia. Genom. Proteom. Bioinform. 2013, 11, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, A.T. Antiseptic “resistance”: Real or perceived threat? Clin. Infect. Dis. 2005, 40, 1650–1656. [Google Scholar] [CrossRef] [PubMed]
- Piechowski, M.M.; Susman, M. Acridine-resistance in phage T4D. Genetics 1967, 56, 133–148. [Google Scholar] [PubMed]
- Kawai, M.; Yamada, S.; Ishidoshiro, A.; Oyamada, Y.; Ito, H.; Yamagishi, J.-I. Cell-wall thickness: Possible mechanism of acriflavine resistance in meticillin-resistant Staphylococcus aureus. J. Med. Microbiol. 2009, 58, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.J.; Ripley, L.S. The spectrum of acridine resistant mutants of bacteriophage T4 reveals cryptic effects of the tsL141 DNA polymerase allele on spontaneous mutagenesis. Genetics 1998, 148, 1655–1665. [Google Scholar] [PubMed]
- Deghorain, M.; van Melderen, L. The Staphylococci phages family: An overview. Viruses 2012, 4, 3316–3335. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zschach, H.; Joensen, K.G.; Lindhard, B.; Lund, O.; Goderdzishvili, M.; Chkonia, I.; Jgenti, G.; Kvatadze, N.; Alavidze, Z.; Kutter, E.M.; et al. What Can We Learn from a Metagenomic Analysis of a Georgian Bacteriophage Cocktail? Viruses 2015, 7, 6570-6589. https://doi.org/10.3390/v7122958
Zschach H, Joensen KG, Lindhard B, Lund O, Goderdzishvili M, Chkonia I, Jgenti G, Kvatadze N, Alavidze Z, Kutter EM, et al. What Can We Learn from a Metagenomic Analysis of a Georgian Bacteriophage Cocktail? Viruses. 2015; 7(12):6570-6589. https://doi.org/10.3390/v7122958
Chicago/Turabian StyleZschach, Henrike, Katrine G. Joensen, Barbara Lindhard, Ole Lund, Marina Goderdzishvili, Irina Chkonia, Guliko Jgenti, Nino Kvatadze, Zemphira Alavidze, Elizabeth M. Kutter, and et al. 2015. "What Can We Learn from a Metagenomic Analysis of a Georgian Bacteriophage Cocktail?" Viruses 7, no. 12: 6570-6589. https://doi.org/10.3390/v7122958