
Structure of the Receptor-Binding Carboxy-Terminal Domain of the Bacteriophage T5 L-Shaped Tail Fibre with and without Its Intra-Molecular Chaperone

 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Protein Expression, Purification, Crystallization and Stability Measurement
2.2. Phage Infection Inhibition
2.3. Crystallographic Structure Solution
2.4. Electron Microscopy of pb1(970–1263) and pb1(970–1396)
3. Results
3.1. Protein Expression, Purification and Crystallization
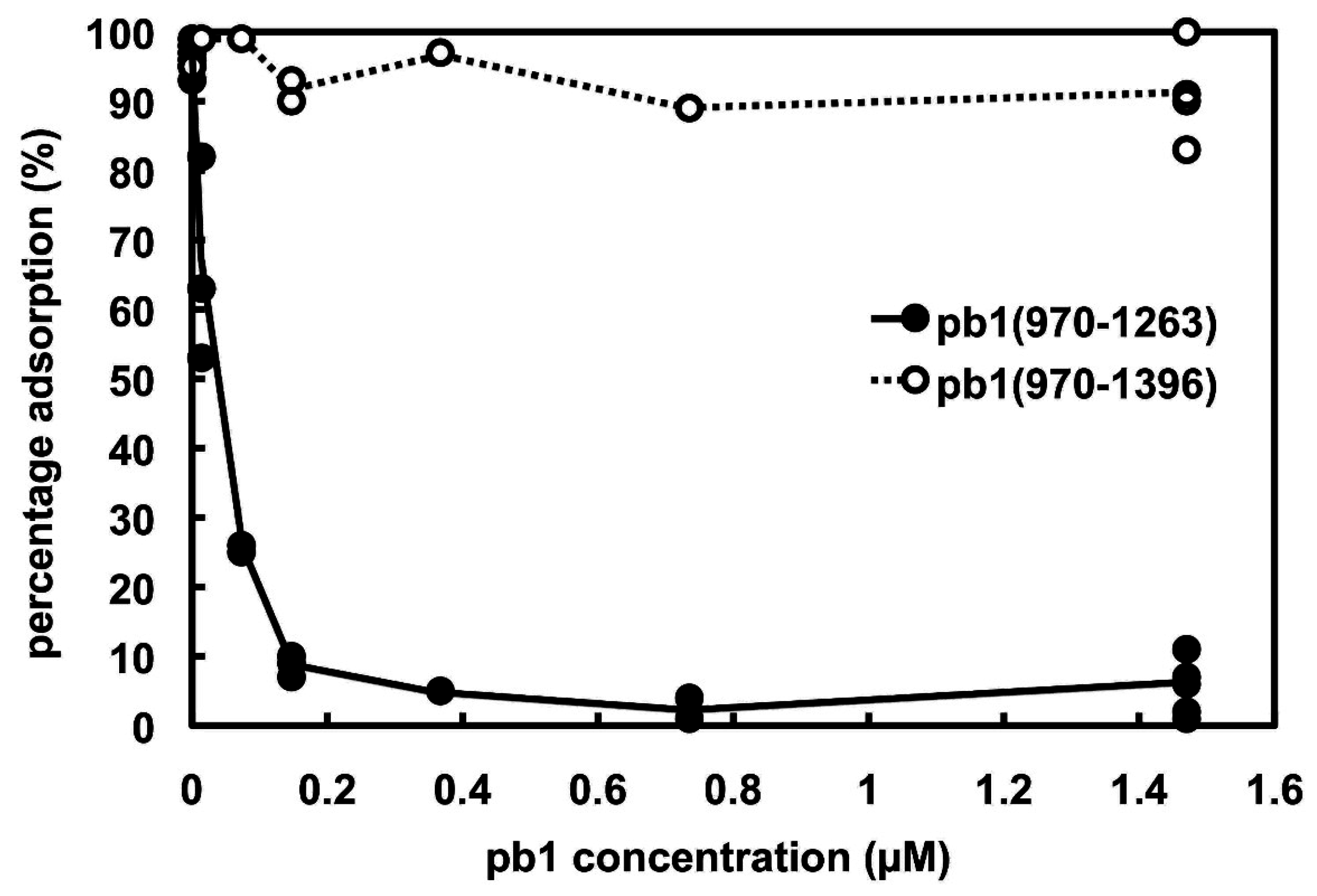
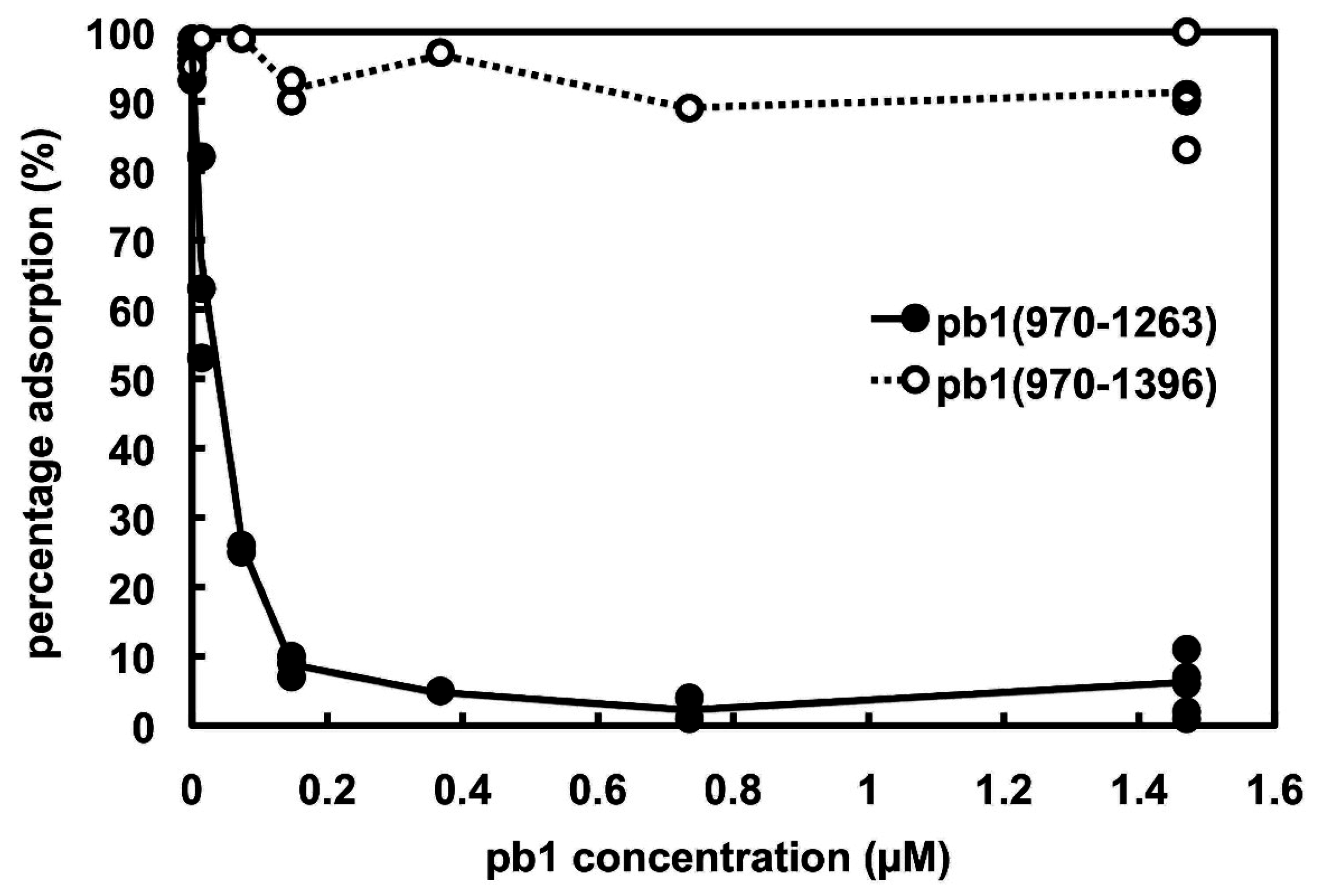
3.2. Phage Infection Inhibition
3.3. Structure Solution


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | SeMet-pb1(970–1263) 1 | pb1(970–1263) | pb1(970–1396) |
|---|---|---|---|
| Data collection | |||
| Wavelength (Å) | 0.9797 | 0.9795 | 1.0972 |
| Space group | C2 | P1 | C2 |
| Cell edges (a, b, c, Å) | 227.4, 58.2, 69.9 | 86.6, 95.0, 127.7 | 160.9, 99.3, 286.2 |
| Cell angles (α, β, γ, °) | 90.0, 98.8, 90.0 | 68.2, 70.2, 83.6 | 90.0, 91.5, 90.0 |
| Resolution range (Å) | 56.2–2.52 (2.59–2.52) 2 | 23.0–2.30 (2.42–2.30) | 95.4–2.52 (2.65–2.52) |
| Unique reflections | 30585 (2237) | 144381 (20958) | 150772 (21539) |
| Multiplicity | 6.3 (6.5) | 2.4 (2.4) | 4.1 (3.8) |
| Completeness | 0.968 (0.958) | 0.913 (0.905) | 0.988 (0.972) |
| I/sigma(I) | 11.4 (2.3) | 7.6 (3.0) | 6.3 (2.5) |
| Rsym 3 | 0.100 (0.644) | 0.080 (0.209) | 0.140 (0.433) |
| CC Imean | 0.996 (0.869) | 0.995 (0.982) | 0.979 (0.784) |
| Phase determination | |||
| Resolution range (Å) | 56.2–2.52 (2.59–2.52) 2 | ||
| Anomalous correlation coefficient | 0.508 (0.059) | ||
| Correlation coefficient (all/weak) 3 | 41.20/24.01 | ||
| Patterson figure of merit 4 | 12.07 | ||
| Correlation coefficient (E) 4 | 0.294 | ||
| R-Cullis 5 | 0.780 | ||
| Phasing power 5 | 1.160 | ||
| Figure of merit 5 | 0.327 | ||
| Solvent flattening 6 | 46.4% solvent | ||
| R-factor before/after density modification 6 | 0.5000/0.1820 | ||
| Correlation on |E|2 before/after dens. mod. 6 | 0.2850/0.6945 | ||
| Hand score (original/inverted) 6,7 | 0.3176/0.4066 | ||
| Refinement | |||
| Resolution range used (Å) | 56.2–2.52 (2.66–2.52) 2 | 23.0–2.30 (2.42–2.30) | 95.4–2.52 (2.65–2.52) |
| Number of reflections used | 28484 (4242) | 140892 (20160) | 148385 (21161) |
| Number of reflections used for Rfree | 2035 (201) | 2906 (467) | 2061 (312) |
| R-factor 8 | 0.213 (0.33) | 0.223 (0.28) | 0.185 (0.28) |
| R-free | 0.266 (0.38) | 0.252 (0.31) | 0.213 (0.29) |
| Atoms (protein/water/other solvent) | 6144/109/18 | 24469/890/0 | 27591/936/117 |
| Ramachandran statistics 9 | 0.931/0.996 | 0.972/1.000 | 0.976/0.999 |
| R.m.s.d. 10 (bonds, Å/angles, °) | 0.012/1.4 | 0.009/1.3 | 0.010/1.4 |
| PDB code | 4UW7 | 5AQ5 | 4UW8 |
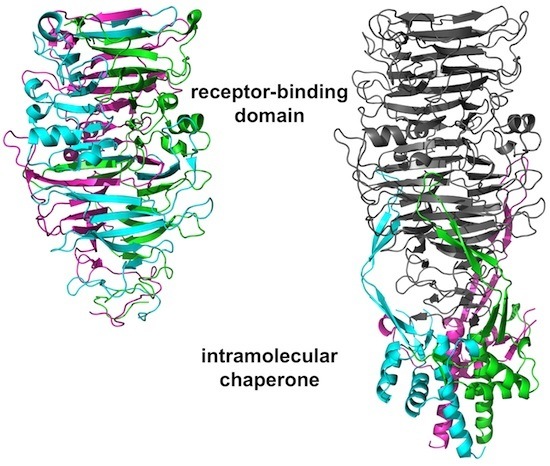
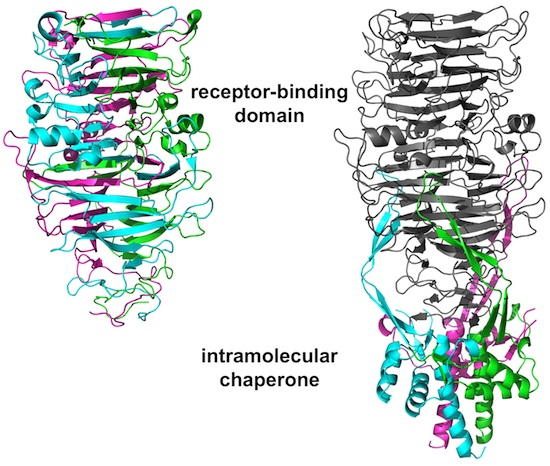
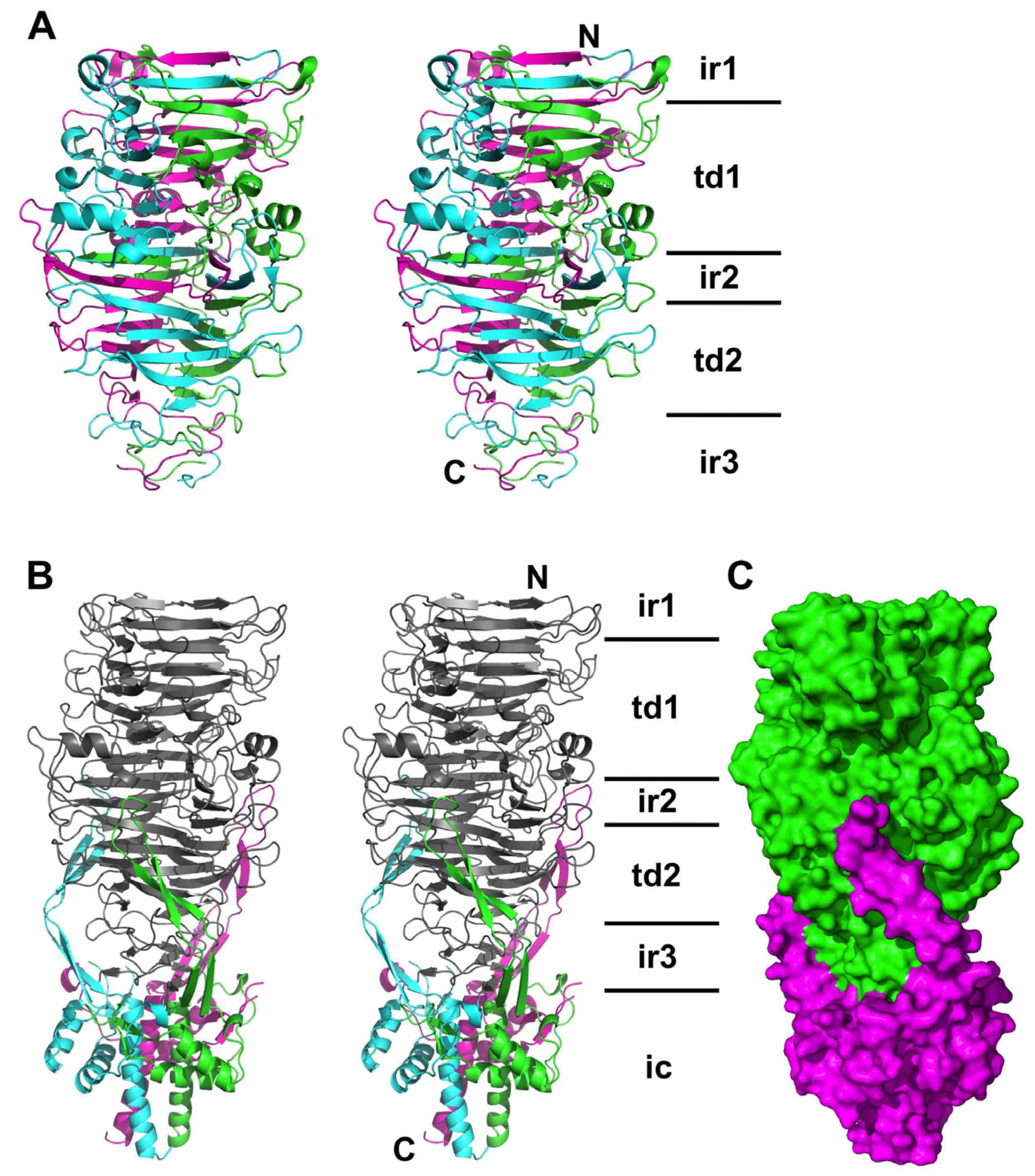
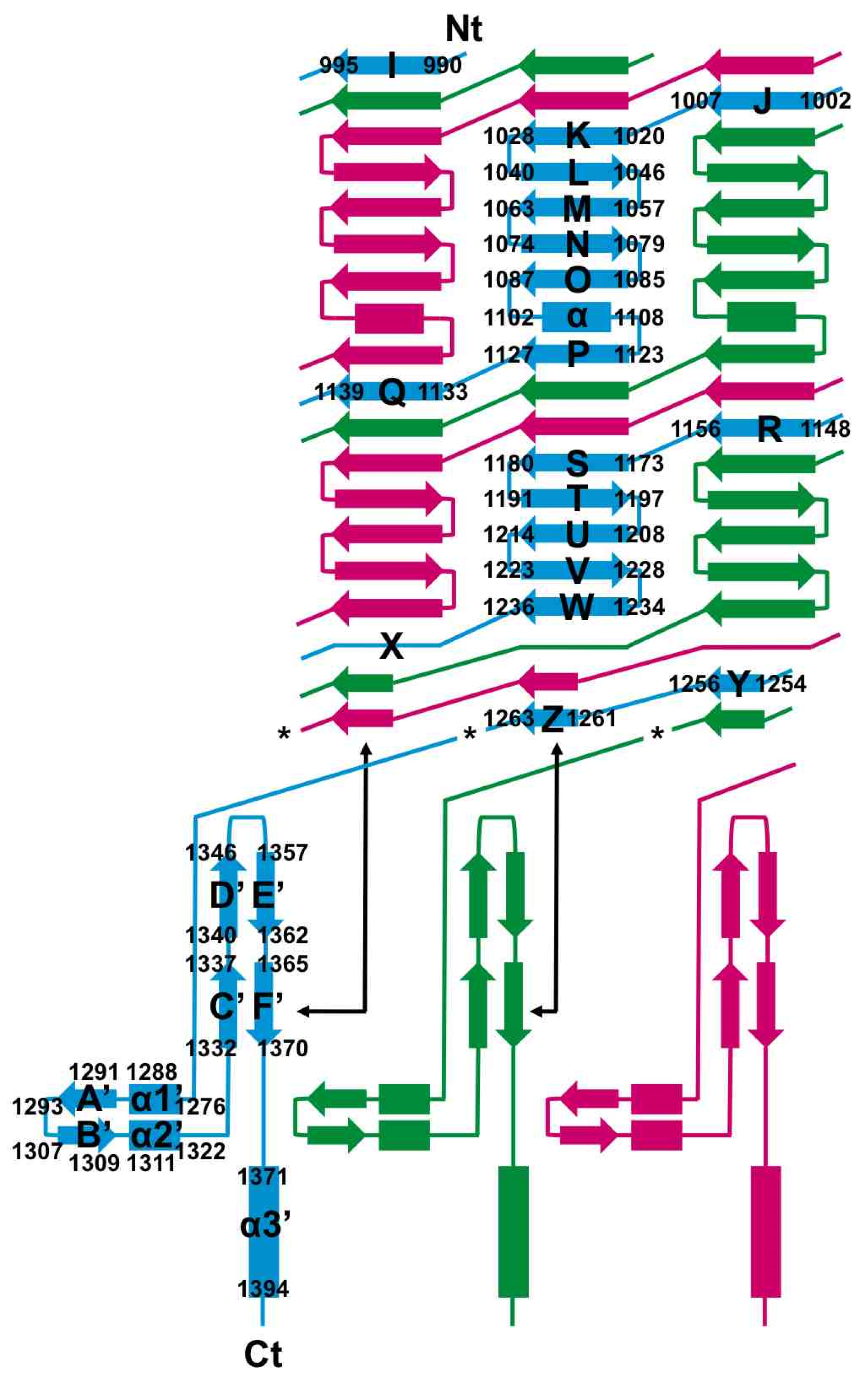
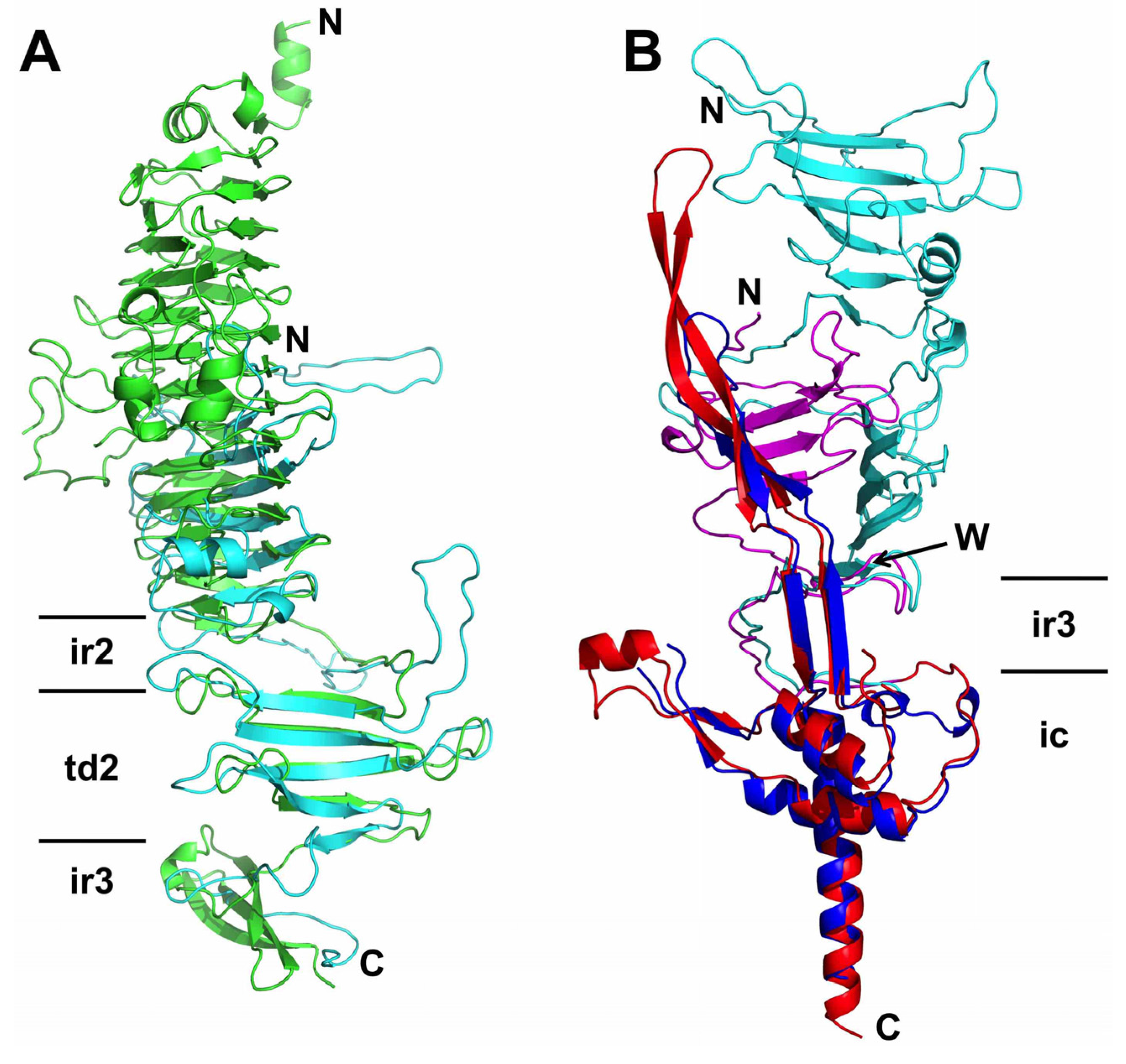
3.4. The Carboxy-Terminal Domain of the Mature Fibre
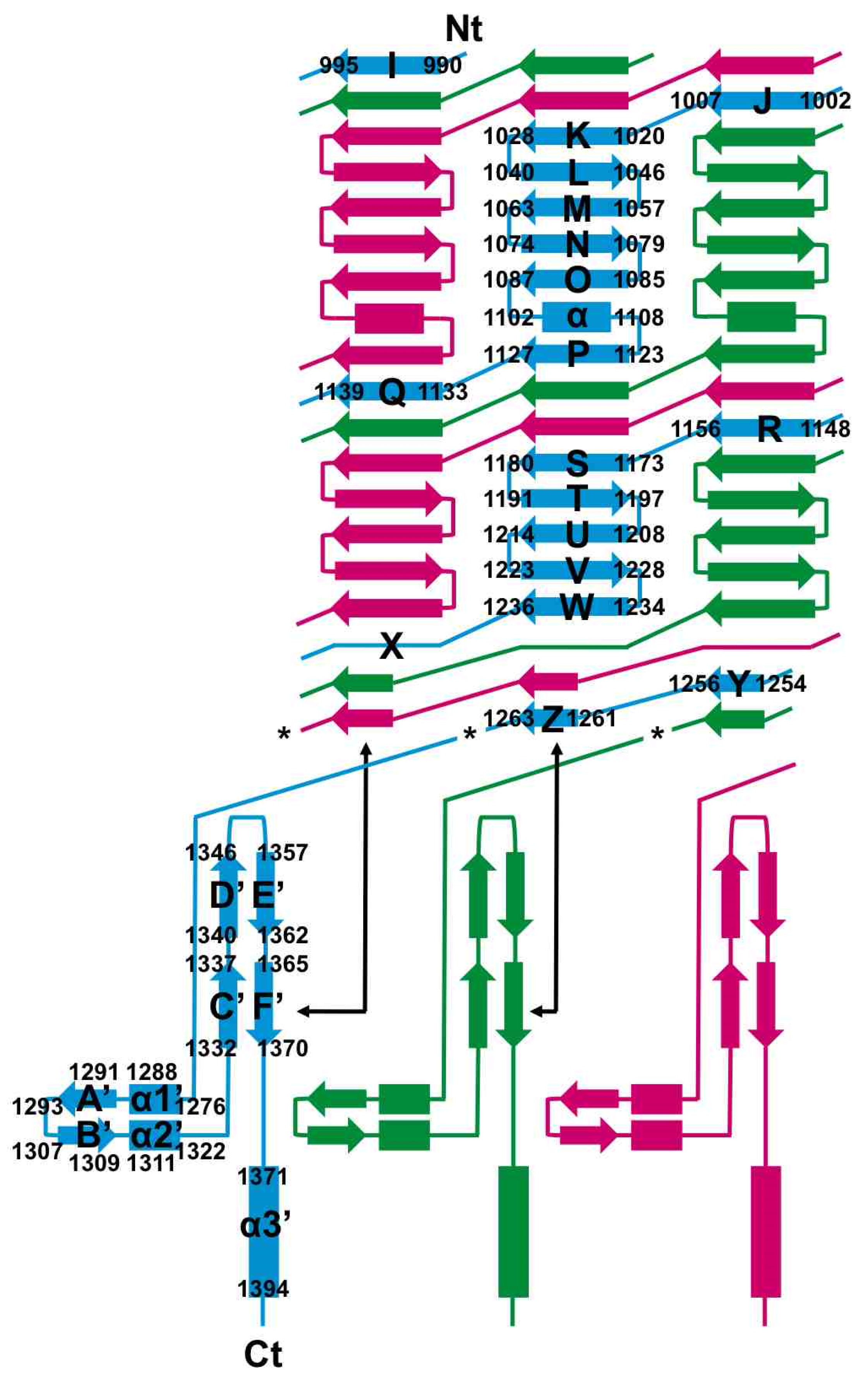
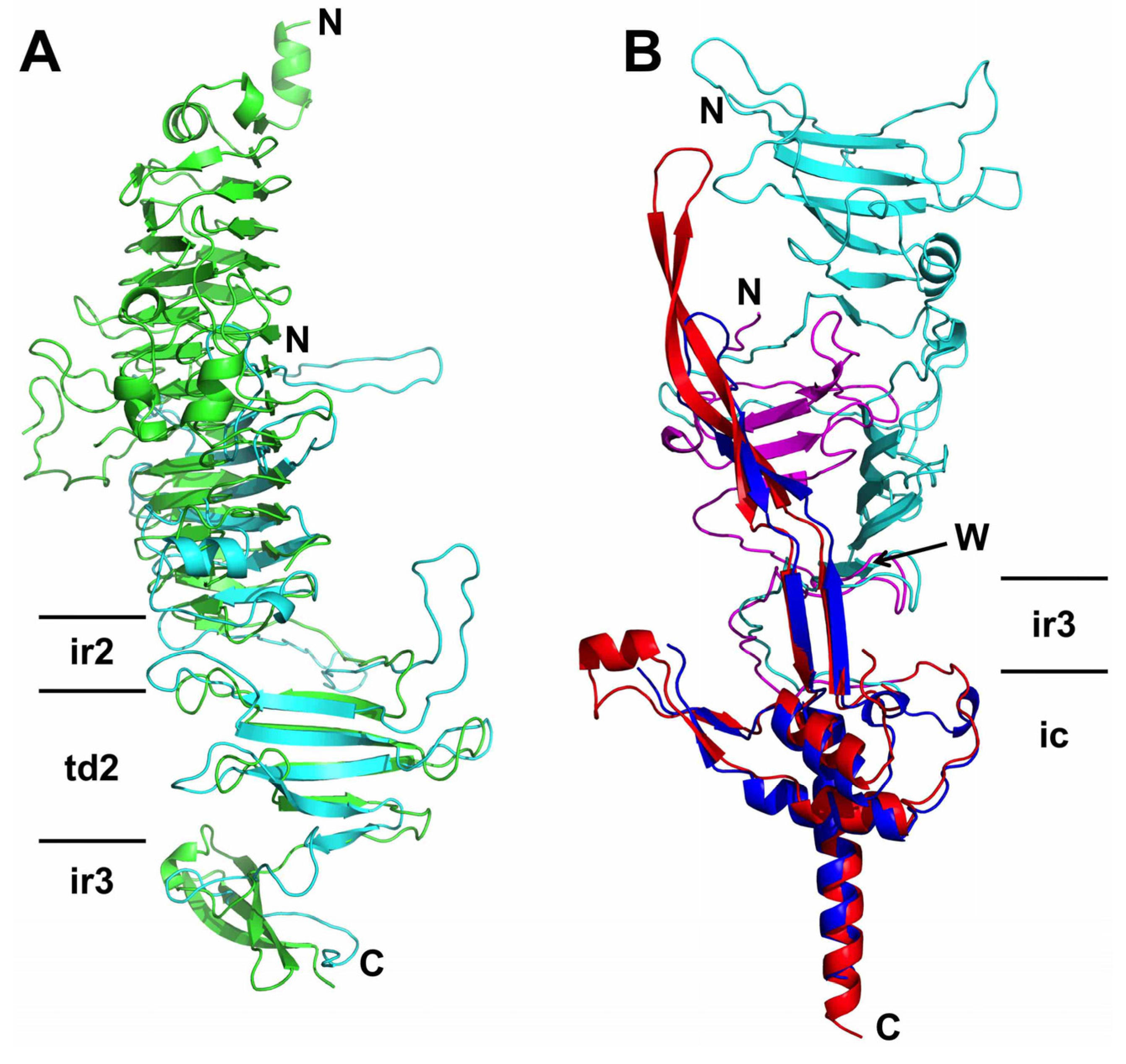
3.5. The Intra-Molecular Chaperone Domain
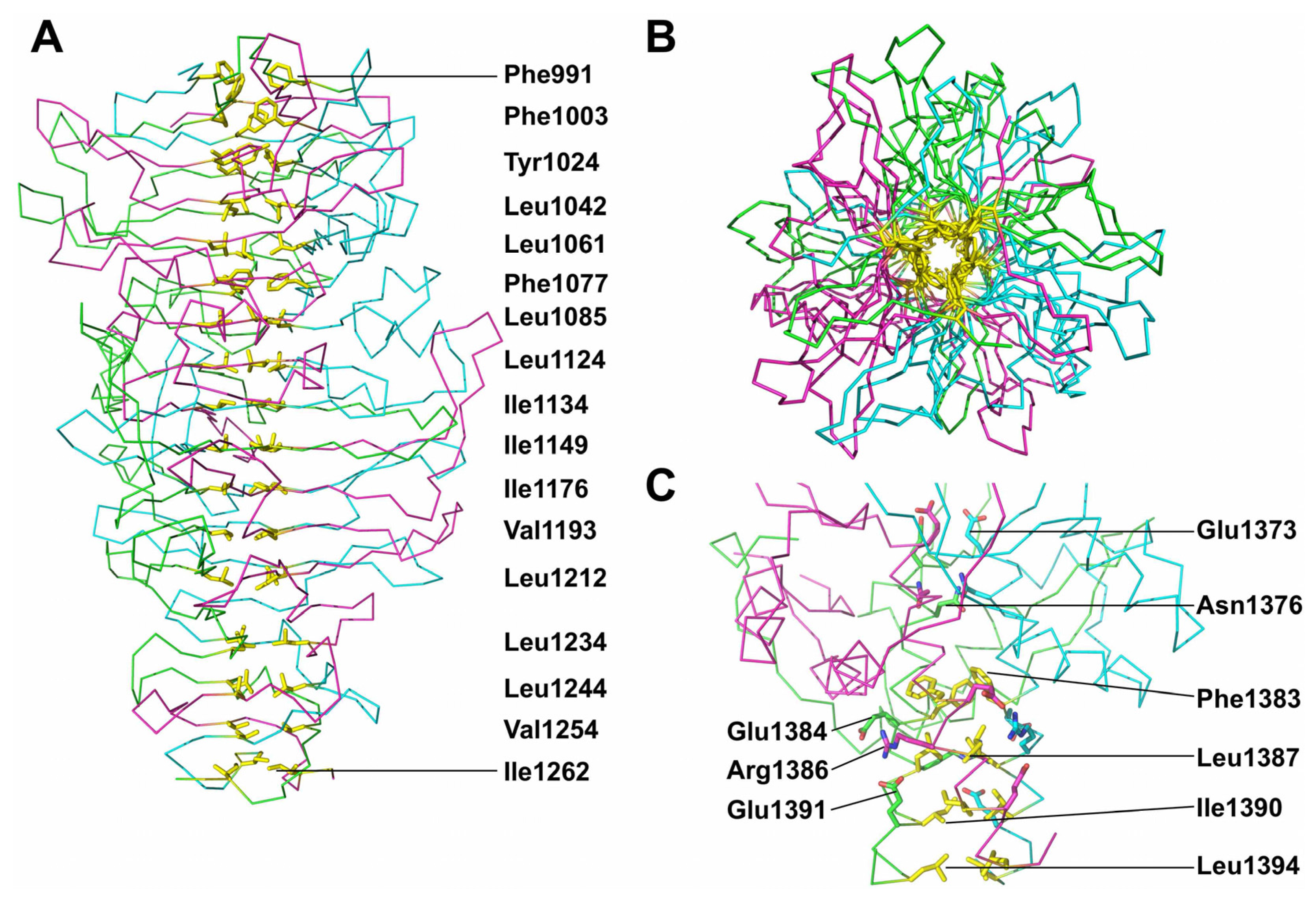
3.6. Trimer Stability
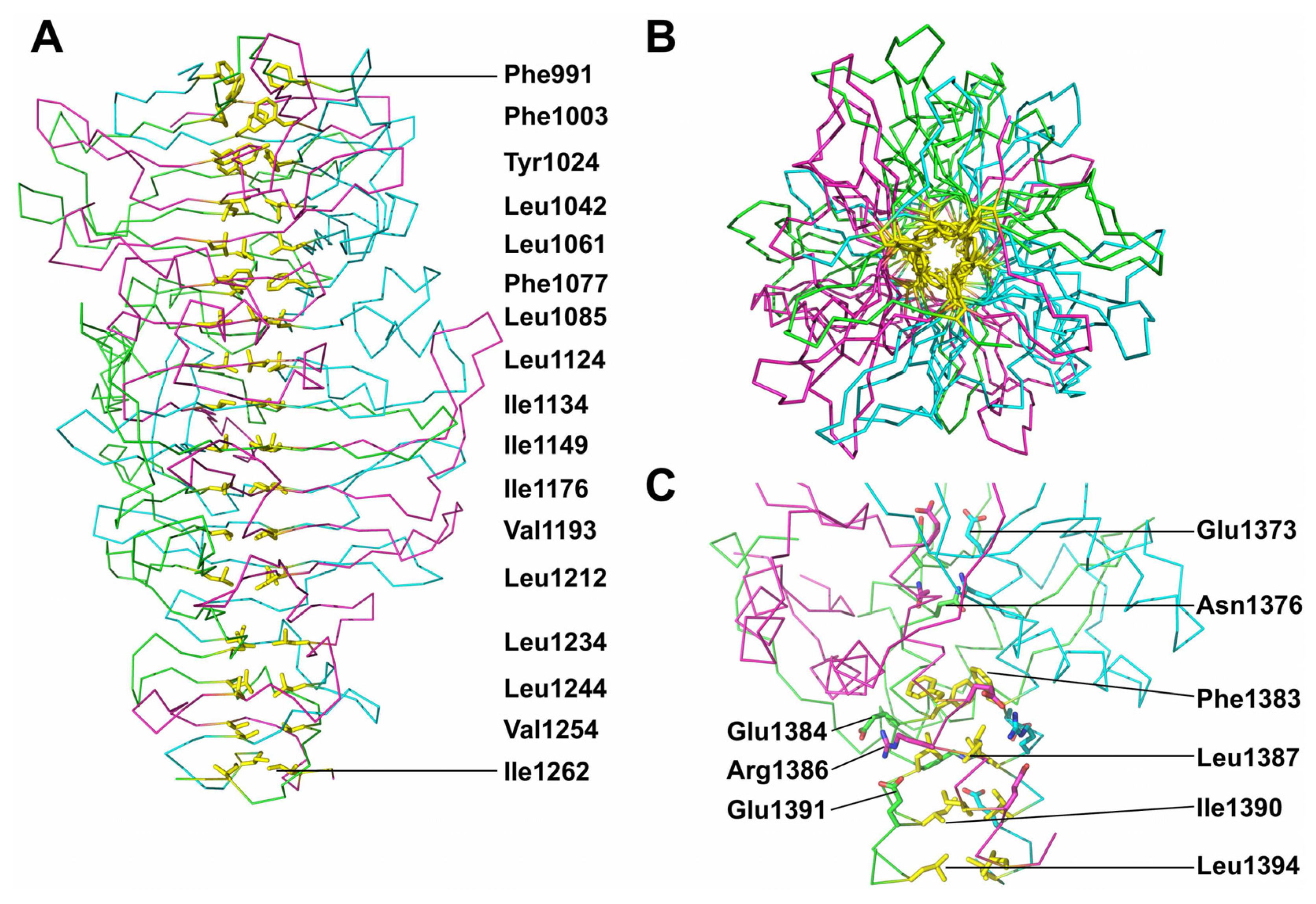
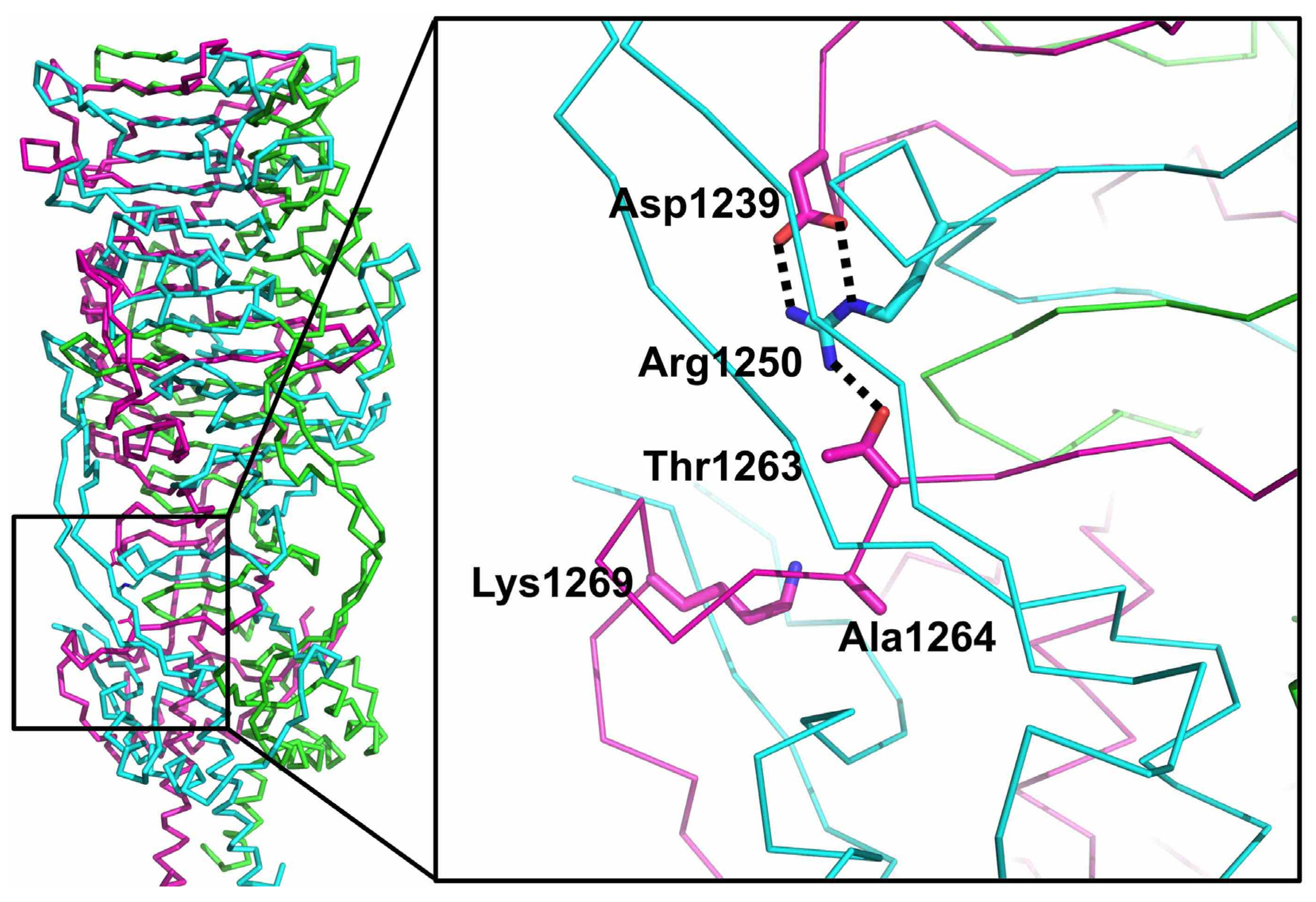
3.7. The Catalytic Site

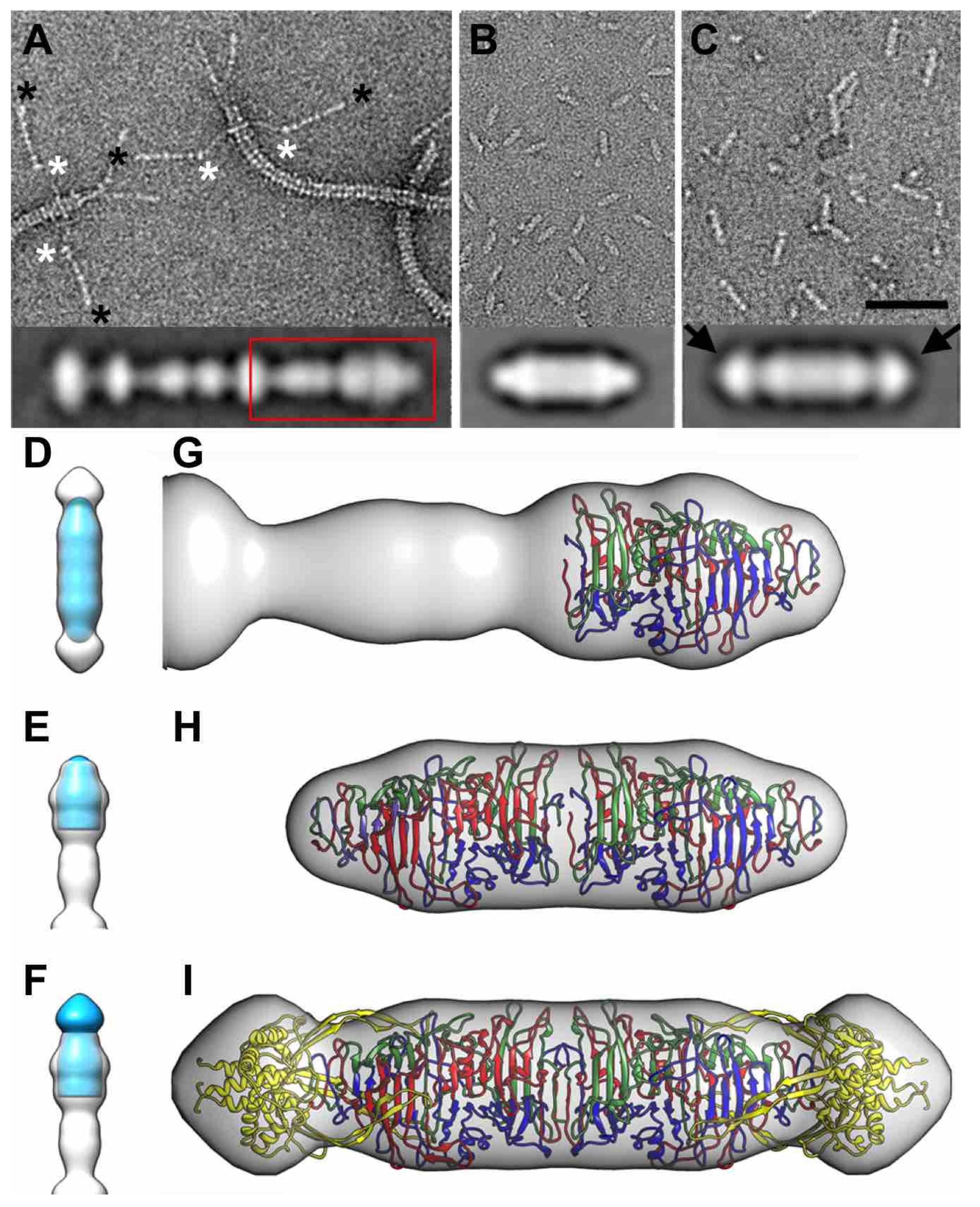
3.8. Electron Microscopy of Phage T5 Tail and Distal Pb1 Assemblies
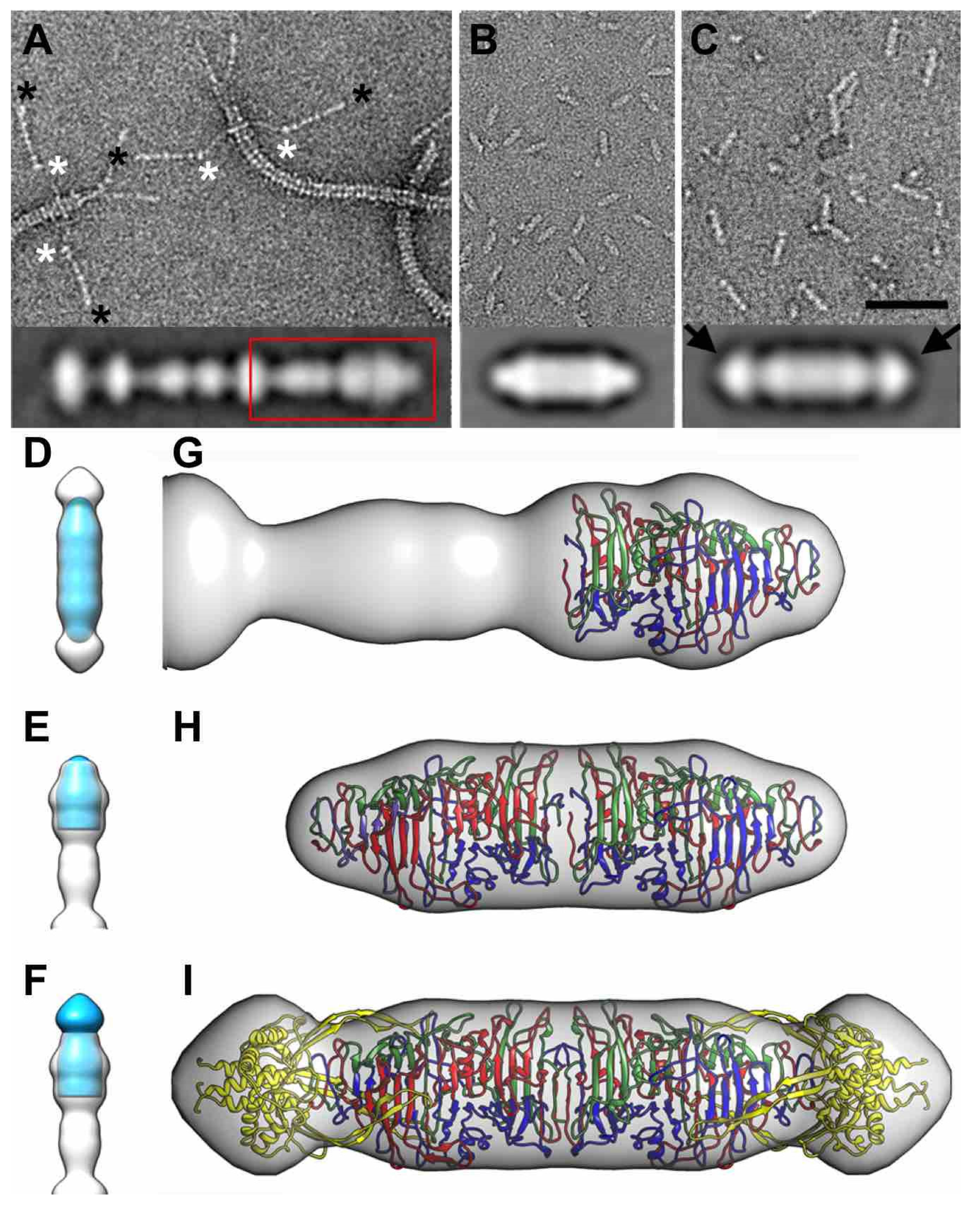
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Marks, T.; Sharp, R. Bacteriophages and biotechnology: A review. J. Chem. Technol. Biotechnol. 2000, 75, 6–17. [Google Scholar] [CrossRef]
- Haq, I.U.; Chaudhry, W.N.; Akhtar, M.N.; Andleeb, S.; Qadri, I. Bacteriophages and their implications on future biotechnology: A review. Virol. J. 2012, 9. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, H.W.; Prangishvili, D. Prokaryote viruses studied by electron microscopy. Arch. Virol. 2012, 157, 1843–1849. [Google Scholar] [CrossRef] [PubMed]
- Heller, K.J.; Krauel, V. Cloning and expression of the ltf gene of bacteriophage T5. J. Bacteriol. 1986, 167, 1071–1073. [Google Scholar] [PubMed]
- Effantin, G.; Boulanger, P.; Neumann, E.; Letellier, L.; Conway, J.F. Bacteriophage T5 structure reveals similarities with HK97 and T4 suggesting evolutionary relationships. J. Mol. Biol. 2006, 361, 993–1002. [Google Scholar] [CrossRef] [PubMed]
- Heller, K.; Braun, V. Polymannose O-antigens of Escherichia coli, the binding sites for the reversible adsorption of bacteriophage T5+ via the L-shaped tail fibers. J. Virol. 1982, 41, 222–227. [Google Scholar] [PubMed]
- Schwarzer, D.; Stummeyer, K.; Gerardy-Schahn, R.; Muhlenhoff, M. Characterization of a novel intramolecular chaperone domain conserved in endosialidases and other bacteriophage tail spike and fiber proteins. J. Biol. Chem. 2007, 282, 2821–2831. [Google Scholar] [CrossRef] [PubMed]
- Reske, K.; Jann, K. The O8 antigen of Escherichia coli. Structure of the polysaccharide chain. Eur. J. Biochem. 1972, 31, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Prehm, P.; Jann, B.; Jann, K. The O9 antigen of Escherichia coli. Structure of the polysaccharide chain. Eur. J. Biochem. 1976, 67, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Heller, K.; Braun, V. Accelerated adsorption of bacteriophage T5 to Escherichia coli F, resulting from reversible tail fiber-lipopolysaccharide binding. J. Bacteriol. 1979, 139, 32–38. [Google Scholar] [PubMed]
- Zivanovic, Y.; Confalonieri, F.; Ponchon, L.; Lurz, R.; Chami, M.; Flayhan, A.; Renouard, M.; Huet, A.; Decottignies, P.; Davidson, A.R.; et al. Insights into bacteriophage T5 structure from analysis of its morphogenesis genes and protein components. J. Virol. 2014, 88, 1162–1174. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, P.; le Maire, M.; Bonhivers, M.; Dubois, S.; Desmadril, M.; Letellier, L. Purification and structural and functional characterization of FhuA, a transporter of the Escherichia coli outer membrane. Biochemistry 1996, 35, 14216–14224. [Google Scholar] [CrossRef] [PubMed]
- Bonhivers, M.; Ghazi, A.; Boulanger, P.; Letellier, L. FhuA, a transporter of the Escherichia coli outer membrane, is converted into a channel upon binding of bacteriophage T5. EMBO J. 1996, 15, 1850–1856. [Google Scholar] [PubMed]
- Garcia-Doval, C.; Luque, D.; Castón, J.R.; Boulanger, P.; van Raaij, M.J. Crystallization of the C-terminal domain of the bacteriophage T5 L-shaped fibre. Acta Cryst. Sect. F 2013, 69, 1363–1367. [Google Scholar] [CrossRef] [PubMed]
- Heller, K.J.; Bryniok, D. O-antigen dependent mutant of bacteriophage T5. J. Virol. 1984, 49, 20–25. [Google Scholar] [PubMed]
- Boulanger, P. Purification of bacteriophages and SDS-PAGE analysis of phage structural proteins from ghost particles. Methods Mol. Biol. 2009, 502, 227–238. [Google Scholar] [PubMed]
- Whitfield, C.; Trent, M.S. Biosynthesis and export of bacterial lipopolysaccharides. Annu. Rev. Biochem. 2014, 83, 99–128. [Google Scholar] [CrossRef] [PubMed]
- Juanhuix, J.; Gil-Ortiz, F.; Cuní, G.; Colldelram, C.; Nicolás, J.; Lidón, J.; Boter, E.; Ruget, C.; Ferrer, S.; Benach, J. Developments in optics and performance at BL13-XALOC, the macromolecular crystallography beamline at the Alba Synchrotron. J. Synchrotron Radiat. 2014, 21, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Battye, T.G.; Kontogiannis, L.; Johnson, O.; Powell, H.R.; Leslie, A.G. iMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM. Acta Cryst. Sect. D 2011, 67, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Cryst. Sect. D 2013, 69, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- Vonrhein, C.; Blanc, E.; Roversi, P.; Bricogne, G. Automated structure solution with autoSHARP. Methods Mol. Biol. 2007, 364, 215–230. [Google Scholar] [PubMed]
- Sheldrick, G. Experimental phasing with SHELXC/D/E: Combining chain tracing with density modification. Acta Cryst. Sect. D 2010, 66, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, J.P.; Leslie, A.G. Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Cryst. Sect. D 1996, 52, 30–42. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Cryst. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Cryst. Sect. D 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Cryst. Sect. D 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Cryst. Sect. D 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Cryst. Sect. D 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.; Rosenstrom, P. Dali server: Conservation mapping in 3D. Nucleic Acids Res. 2010, 38, W545–W549. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Cryst. Sect. D 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System, Version 1.4.1; Schrödinger LLC: New York, NY, USA, 2011.
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera, a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Scheres, S.H.; Núñez-Ramírez, R.; Sorzano, C.O.; Carazo, J.M.; Marabini, R. Image processing for electron microscopy single-particle analysis using XMIPP. Nat. Protoc. 2008, 3, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Mindell, J.A.; Grigorieff, N. Accurate determination of local defocus and specimen tilt in electron microscopy. J. Struct. Biol. 2003, 142, 334–347. [Google Scholar] [CrossRef]
- Scheres, S.H.; Valle, M.; Nuñez, R.; Sorzano, C.O.; Marabini, R.; Herman, G.T.; Carazo, J.M. Maximum-likelihood multi-reference refinement for electron microscopy images. J. Mol. Biol. 2005, 348, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Schulz, E.C.; Dickmanns, A.; Urlaub, H.; Schmitt, A.; Mühlenhoff, M.; Stummeyer, K.; Schwarzer, D.; Gerardy-Schahn, R.; Ficner, R. Crystal structure of an intramolecular chaperone mediating triple-β-helix folding. Nat. Struct. Mol. Biol. 2010, 17, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Steinbacher, S.; Seckler, R.; Miller, S.; Steipe, B.; Huber, R.; Reinemer, P. Crystal structure of P22 tailspike protein: Interdigitated subunits in a thermostable trimer. Science 1994, 265, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Mitraki, A.; Papanikolopoulou, K.; van Raaij, M.J. Natural triple β-stranded fibrous folds. Adv. Prot. Chem. 2006, 73, 97–124. [Google Scholar]
- Xiang, Y.; Leiman, P.G.; Li, L.; Grimes, S.; Anderson, D.L.; Rossmann, M.G. Crystallographic insights into the autocatalytic assembly mechanism of a bacteriophage tail spike. Mol. Cell 2009, 34, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Kreisberg, J.F.; Betts, S.D.; Haase-Pettingell, C.; King, J. The interdigitated β-helix domain of the P22 tailspike protein acts as a molecular clamp in trimer stabilization. Protein Sci. 2002, 11, 820–830. [Google Scholar] [CrossRef] [PubMed]
- Schulz, E.C.; Ficner, R. Knitting and snipping: Chaperones in β-helix folding. Curr. Opin. Struct. Biol. 2011, 21, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Bartual, S.G.; Otero, J.M.; Garcia-Doval, C.; Llamas-Saiz, A.L.; Kahn, R.; Fox, G.C.; van Raaij, M.J. Structure of the bacteriophage T4 long tail fiber receptor-binding tip. Proc. Natl. Acad. Sci. USA 2010, 107, 20287–20292. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Doval, C.; van Raaij, M.J. Structure of the receptor-binding carboxy-terminal domain of bacteriophage T7 tail fibers. Proc. Natl. Acad. Sci. USA 2012, 109, 9390–9395. [Google Scholar] [CrossRef] [PubMed]
- Danner, M.; Fuchs, A.; Miller, S.; Seckler, R. Folding and assembly of phage P22 tailspike endorhamnosidase lacking the N-terminal, head-binding domain. Eur. J. Biochem. 1993, 215, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Doval, C.; van Raaij, M.J. Crystallization of the C-terminal domain of the bacteriophage T7 fibre protein gp17. Acta Cryst. Sect. F 2012, 68, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Griniuviené, B.; Goldberg, E.; Tsugita, A.; Tanaka, N.; Arisaka, F. Isolation and characterization of a molecular chaperone, gp57A, of bacteriophage T4. J. Bacteriol. 1997, 179, 1846–1851. [Google Scholar] [PubMed]
- Van Raaij, M.J.; Schoehn, G.; Jaquinod, M.; Ashman, K.; Burda, M.R.; Miller, S. Identification and crystallisation of a heat- and protease-stable fragment of the bacteriophage T4 short tail fibre. Biol. Chem. 2001, 382, 1049–1055. [Google Scholar] [PubMed]
- Granell, M.; Namura, M.; Alvira, S.; Garcia-Doval, C.; Singh, A.K.; Gutsche, I.; van Raaij, M.J.; Kanamaru, S. Crystallization of the carboxy-terminal region of the bacteriophage T4 proximal long tail fibre protein gp34. Acta Cryst. Sect. F 2014, 70, 970–975. [Google Scholar] [CrossRef] [PubMed]
- Bartual, S.G.; Garcia-Doval, C.; Alonso, J.; Schoehn, G.; van Raaij, M.J. Two-chaperone assisted soluble expression and purification of the bacteriophage T4 long tail fibre protein gp37. Protein Expr. Purif. 2010, 70, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.E.; Pourhossein, M.; Waterhouse, A.; Hudson, T.; Goldrick, M.; Derrick, J.P.; Roberts, I.S. The K5 lyase KflA combines a viral tail spike structure with a bacterial polysaccharide lyase mechanism. J. Biol. Chem. 2010, 285, 23963–23969. [Google Scholar] [CrossRef] [PubMed]
- Takata, T.; Haase-Pettingell, C.; King, J. The C-terminal cysteine annulus participates in auto-chaperone function for Salmonella phage P22 tailspike folding and assembly. Bacteriophage 2012, 2, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Weis, W.I.; Drickamer, K. Structural basis of lectin-carbohydrate recognition. Annu. Rev. Biochem. 1996, 65, 441–473. [Google Scholar] [CrossRef] [PubMed]
- Thomassen, E.; Gielen, G.; Schütz, M.; Schoehn, G.; Abrahams, J.P.; Miller, S.; van Raaij, M.J. The structure of the receptor-binding domain of the bacteriophage T4 short tail fibre reveals a knitted trimeric metal-binding fold. J. Mol. Biol. 2003, 331, 361–373. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia-Doval, C.; Castón, J.R.; Luque, D.; Granell, M.; Otero, J.M.; Llamas-Saiz, A.L.; Renouard, M.; Boulanger, P.; Van Raaij, M.J. Structure of the Receptor-Binding Carboxy-Terminal Domain of the Bacteriophage T5 L-Shaped Tail Fibre with and without Its Intra-Molecular Chaperone. Viruses 2015, 7, 6424-6440. https://doi.org/10.3390/v7122946
Garcia-Doval C, Castón JR, Luque D, Granell M, Otero JM, Llamas-Saiz AL, Renouard M, Boulanger P, Van Raaij MJ. Structure of the Receptor-Binding Carboxy-Terminal Domain of the Bacteriophage T5 L-Shaped Tail Fibre with and without Its Intra-Molecular Chaperone. Viruses. 2015; 7(12):6424-6440. https://doi.org/10.3390/v7122946
Chicago/Turabian StyleGarcia-Doval, Carmela, José R. Castón, Daniel Luque, Meritxell Granell, José M. Otero, Antonio L. Llamas-Saiz, Madalena Renouard, Pascale Boulanger, and Mark J. Van Raaij. 2015. "Structure of the Receptor-Binding Carboxy-Terminal Domain of the Bacteriophage T5 L-Shaped Tail Fibre with and without Its Intra-Molecular Chaperone" Viruses 7, no. 12: 6424-6440. https://doi.org/10.3390/v7122946
APA StyleGarcia-Doval, C., Castón, J. R., Luque, D., Granell, M., Otero, J. M., Llamas-Saiz, A. L., Renouard, M., Boulanger, P., & Van Raaij, M. J. (2015). Structure of the Receptor-Binding Carboxy-Terminal Domain of the Bacteriophage T5 L-Shaped Tail Fibre with and without Its Intra-Molecular Chaperone. Viruses, 7(12), 6424-6440. https://doi.org/10.3390/v7122946