
Composite Sequence–Structure Stability Models as Screening Tools for Identifying Vulnerable Targets for HIV Drug and Vaccine Development


Abstract
:1. Introduction
2. Results
2.1. Initial Explorations of Different Mutation Modeling Methods and Protein Scoring Functions
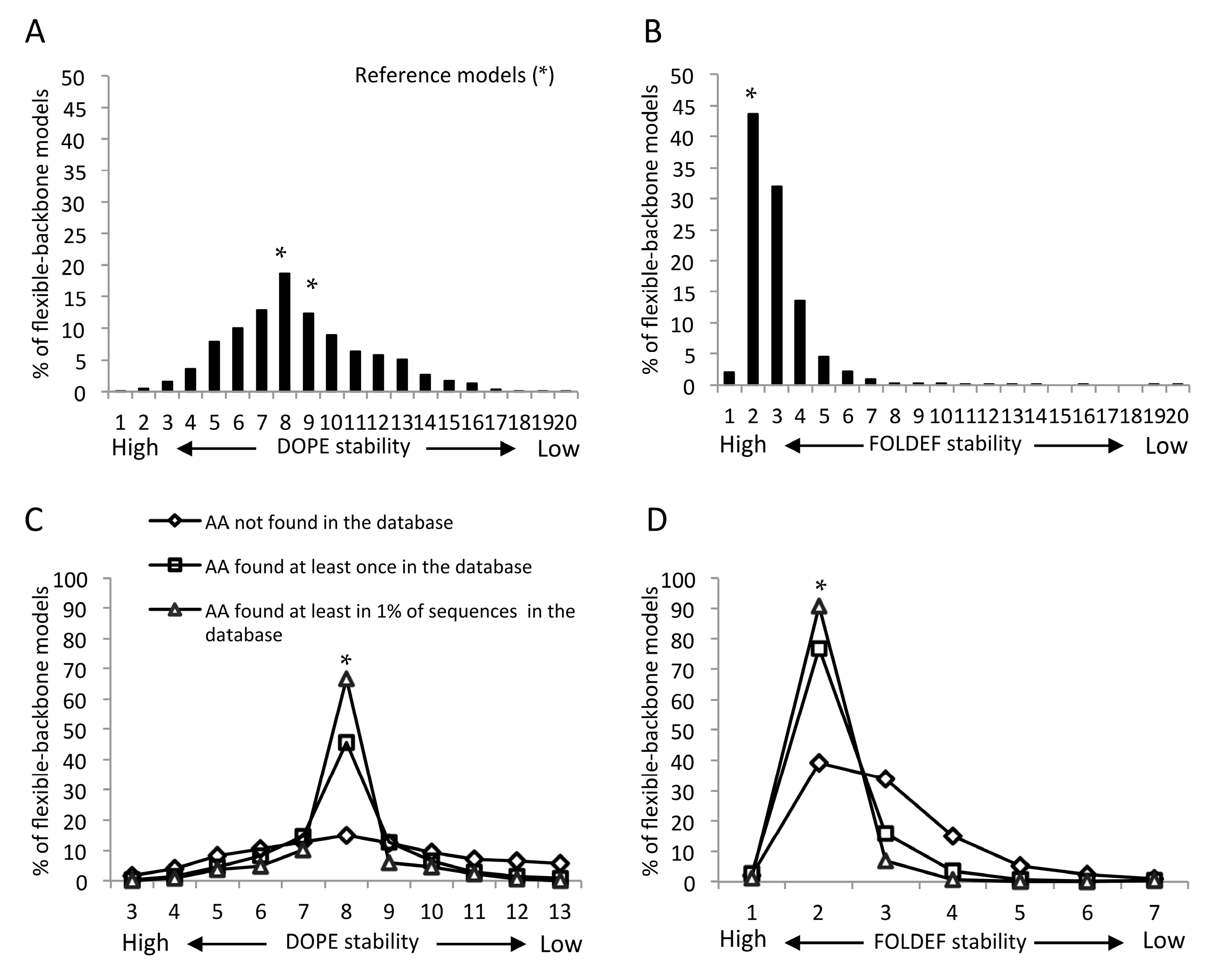
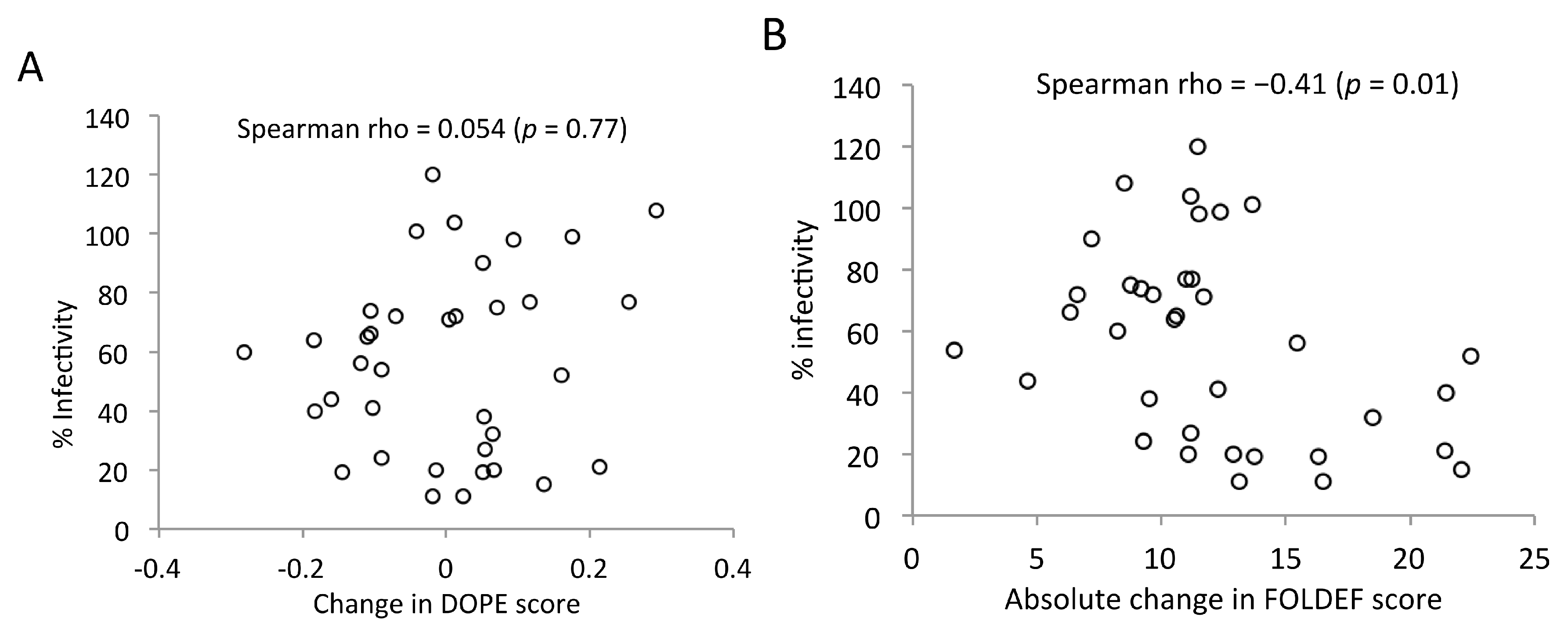
2.2. Statistical- and Empirical-Based Scoring Functions Showed Different Patterns of Predicted Stabilities
2.3. Influence of Input Templates on Predicted Stabilities
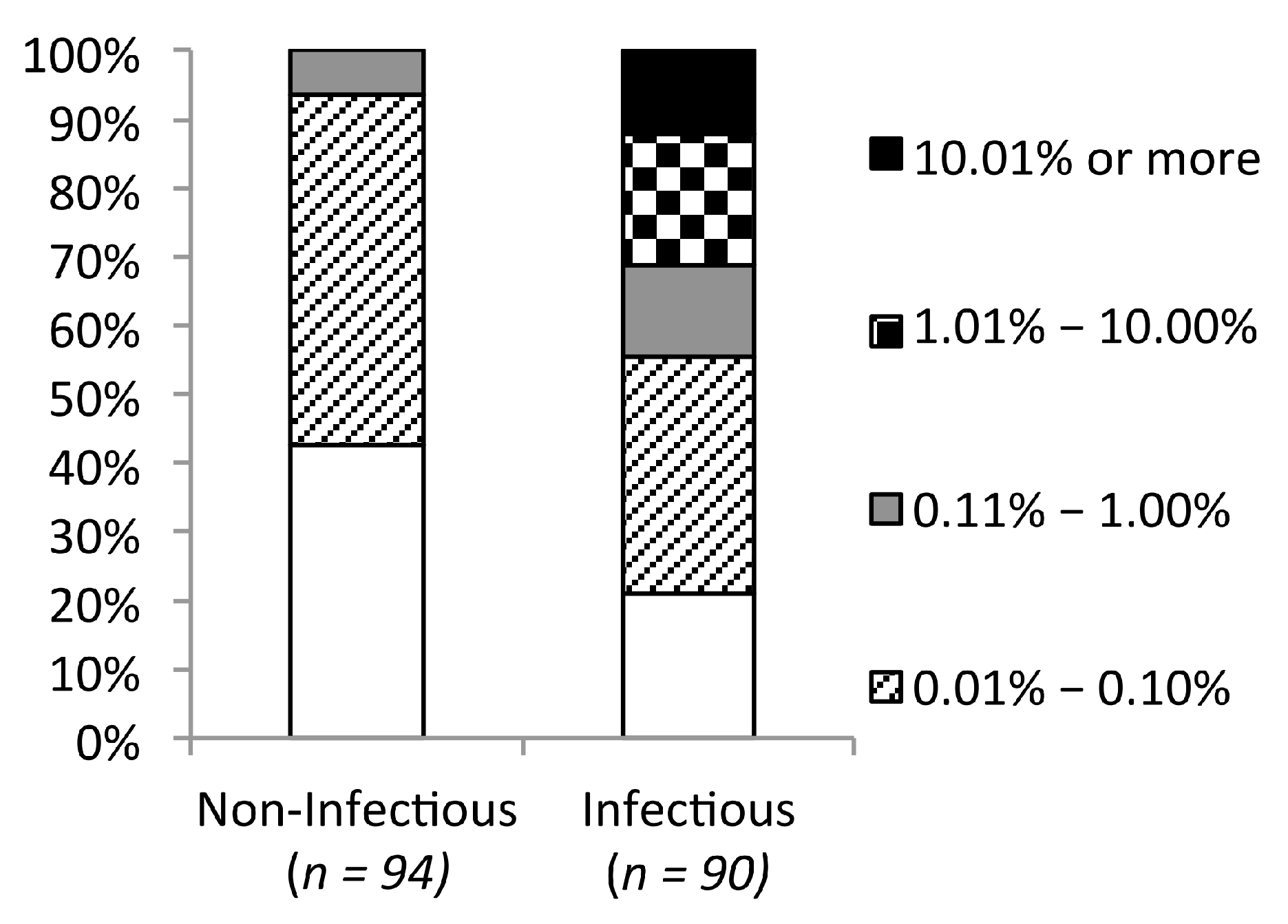
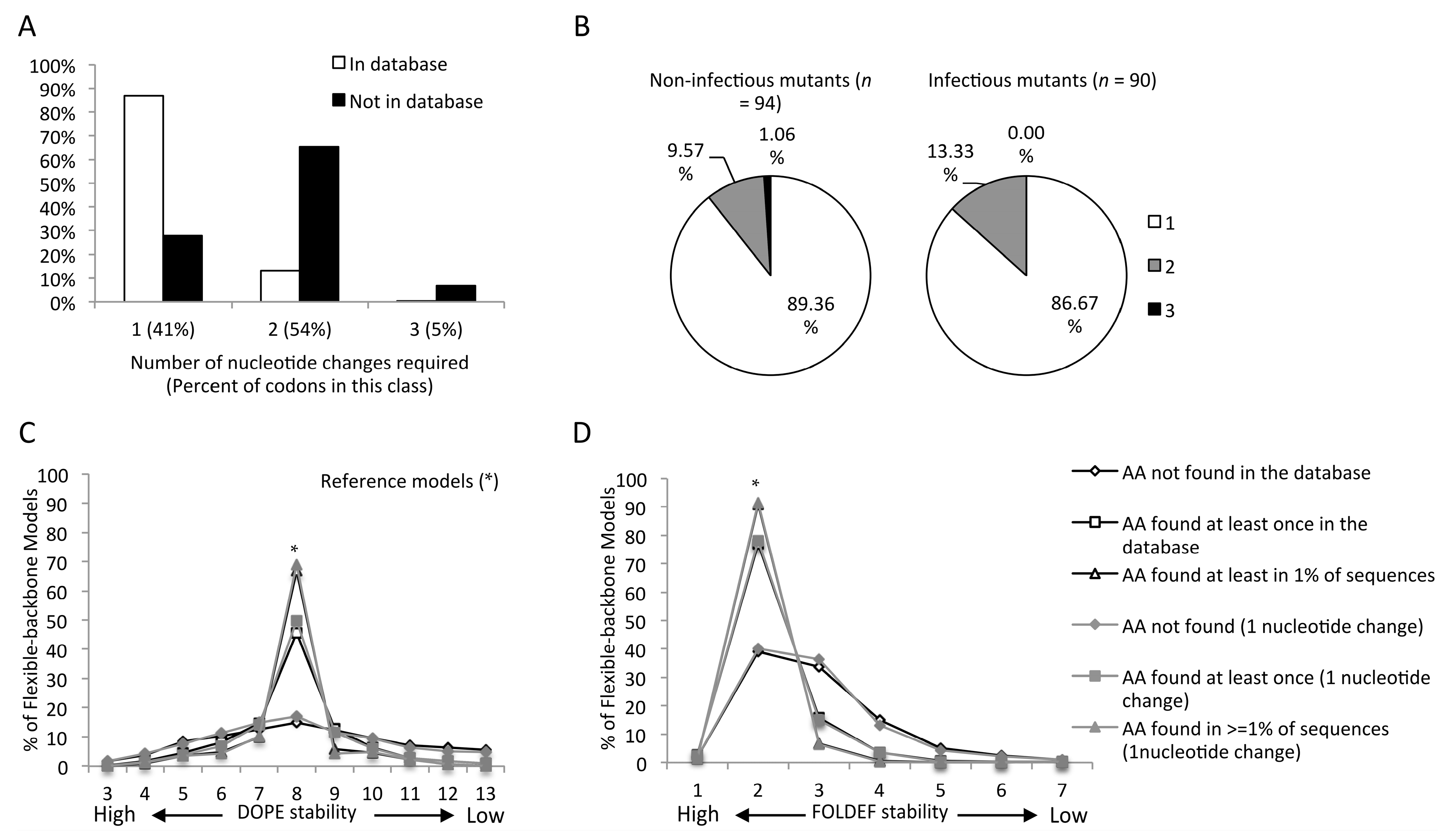
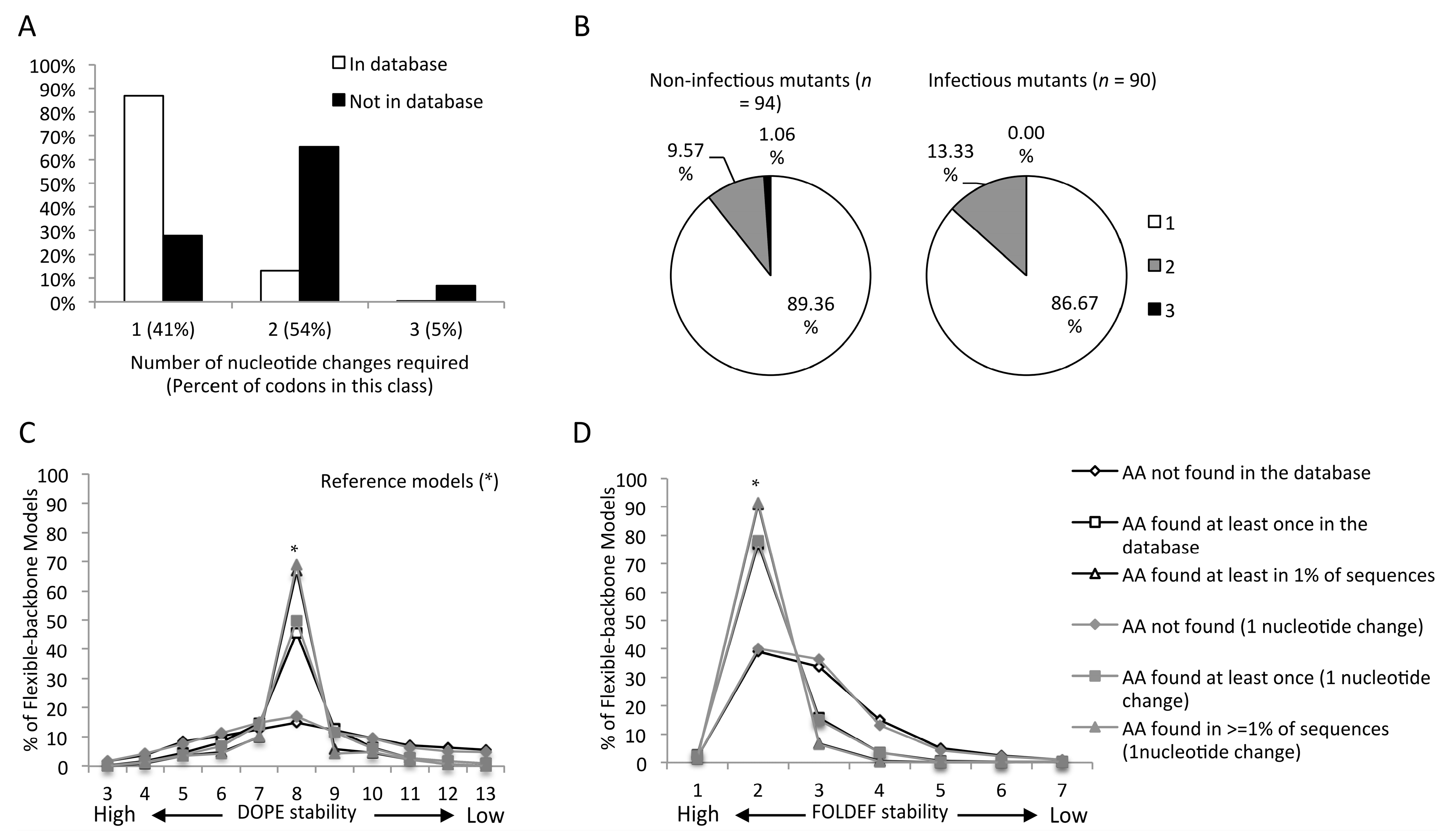
2.4. Mutants Observed in the HIVDB Were Predicted to Have Stabilities Close to the Reference Models by Both Scoring Functions
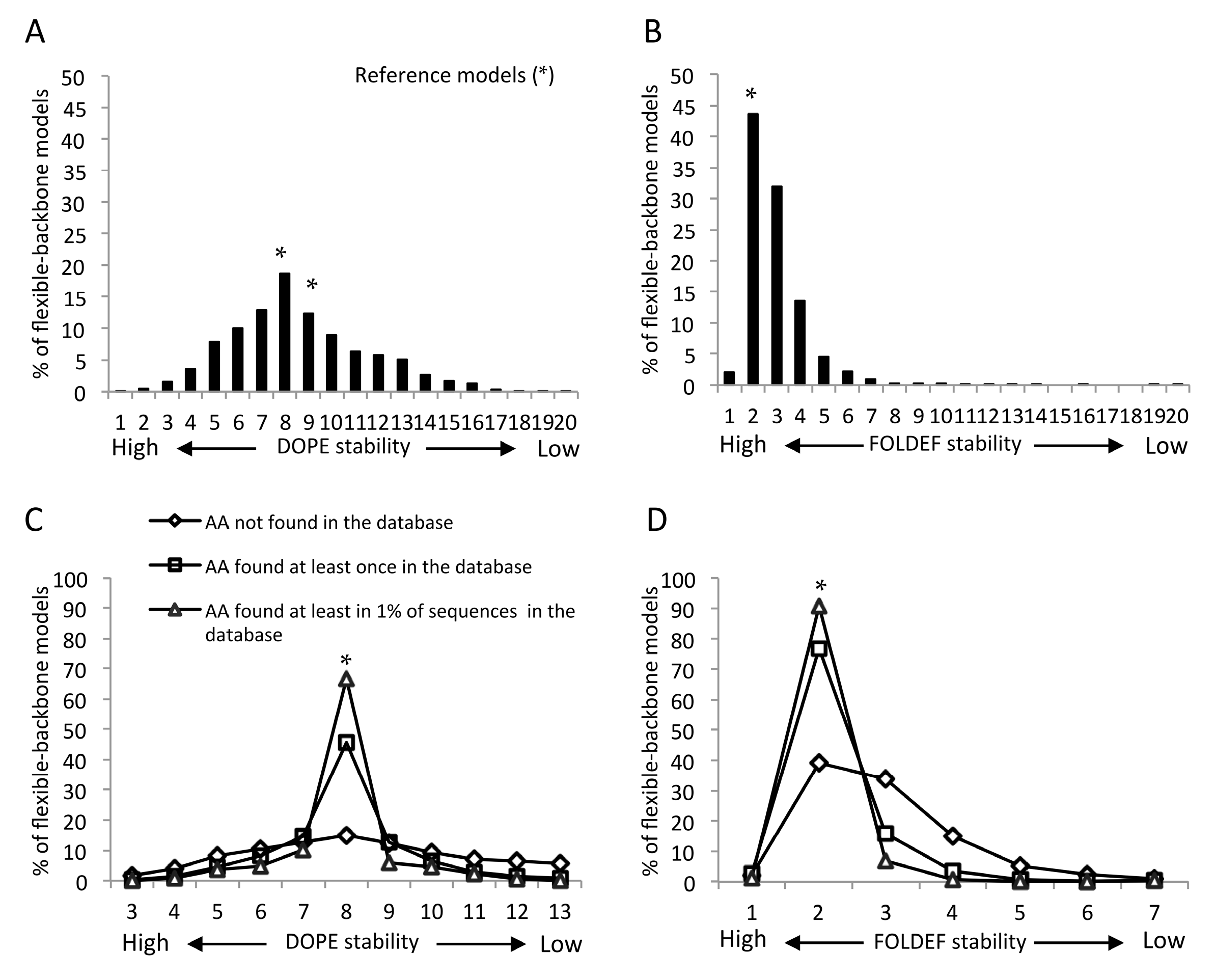
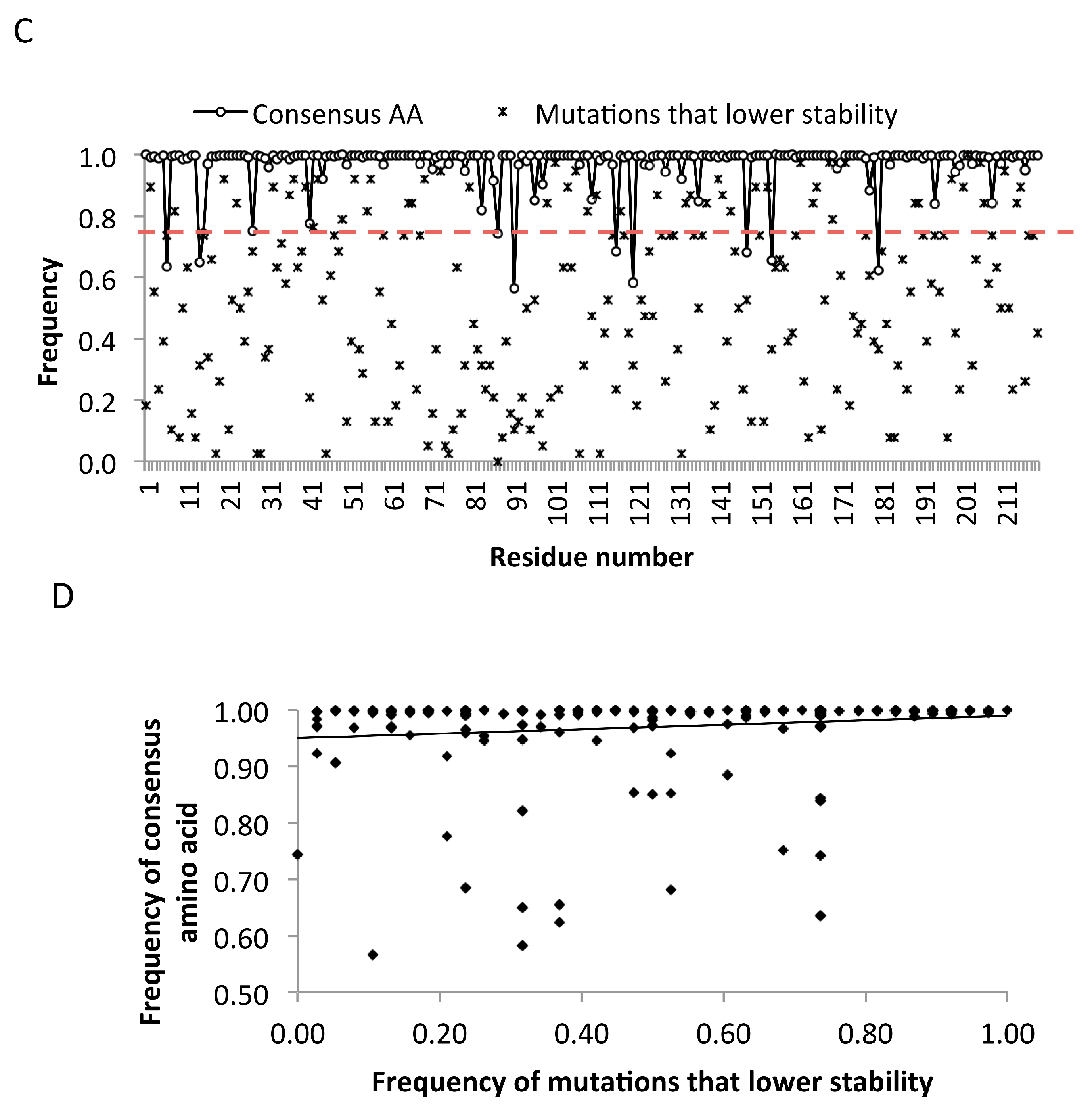
2.5. Frequency Threshold of Tolerated Mutations
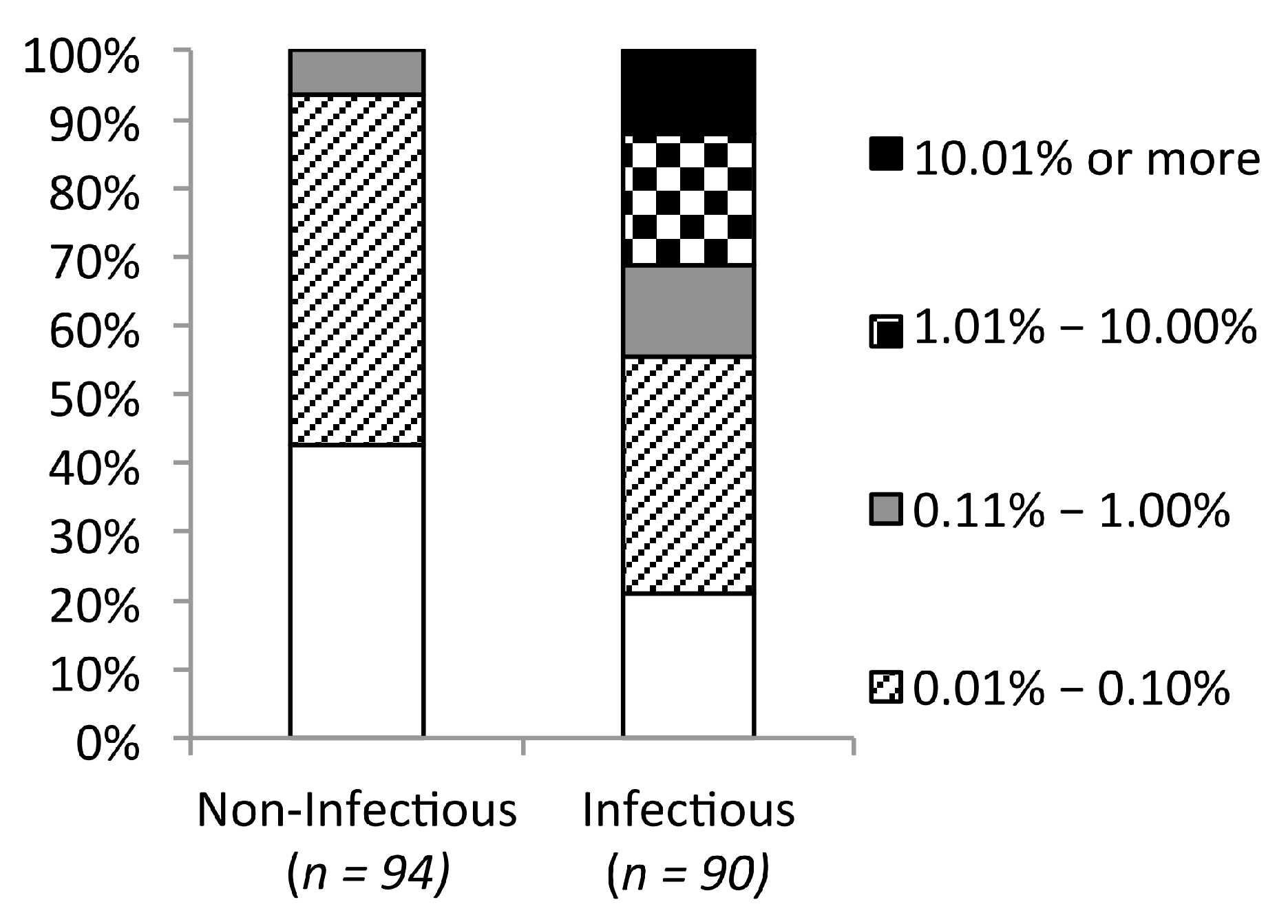

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Predictor | Sensitivity a | Specificity b | Precision c | Accuracy d |
|---|---|---|---|---|
| Mutation Frequency e | 59.5% | 70.0% | 67.10% | 64.10% |
| Stability of Reference Models f | 73.33% | 78.72% | 76.74% | 76.12% |
| Composite Score g | 80.00% | 79.57% | 78.16% | 79.78% |
2.6. Genetic Barrier Influences the Emergence but Not Outcomes of Amino Acid Changes
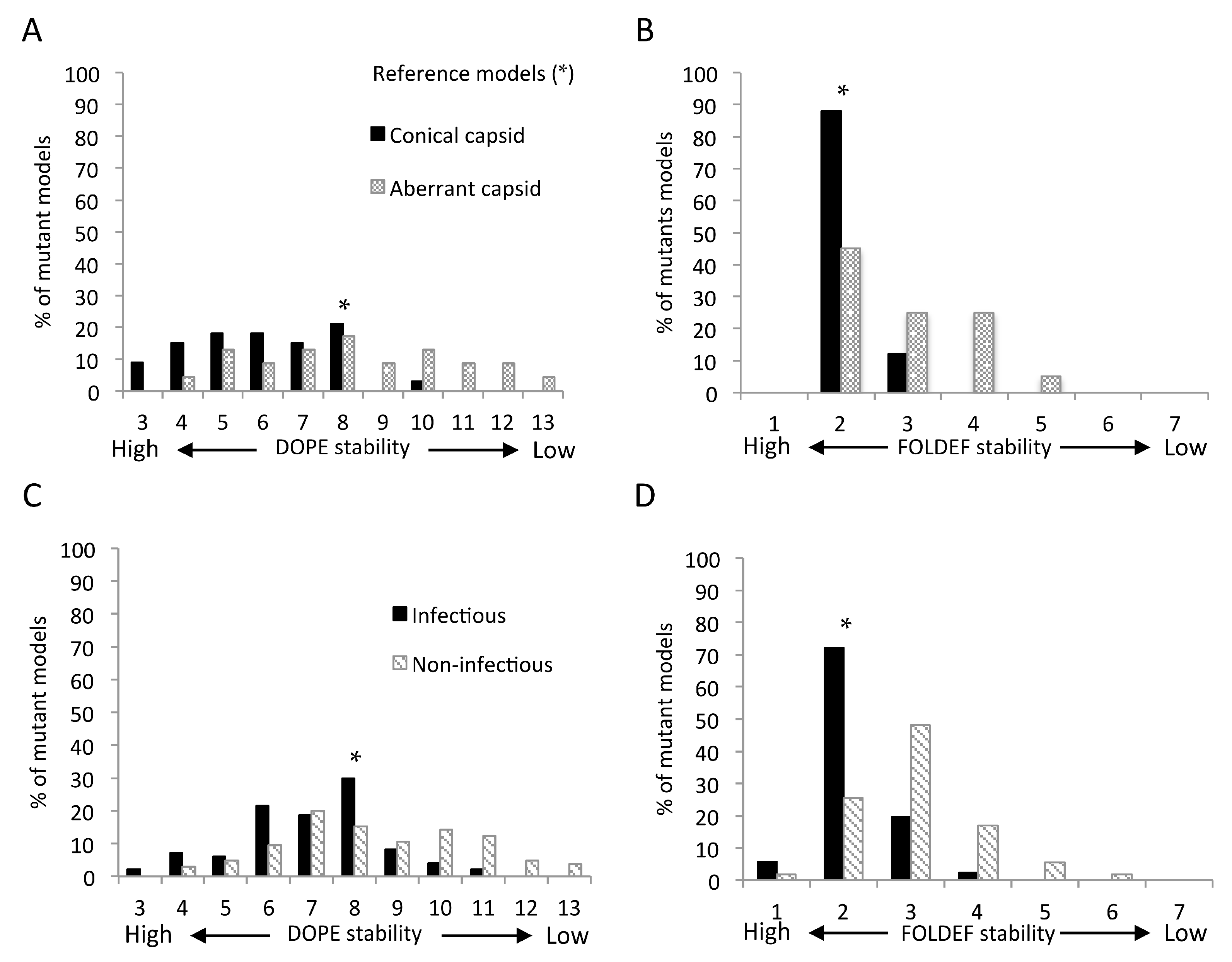
2.7. Predicted Stabilities in Relation to the Impact of Mutations on Mature Capsid Morphology and Viral Infectivity
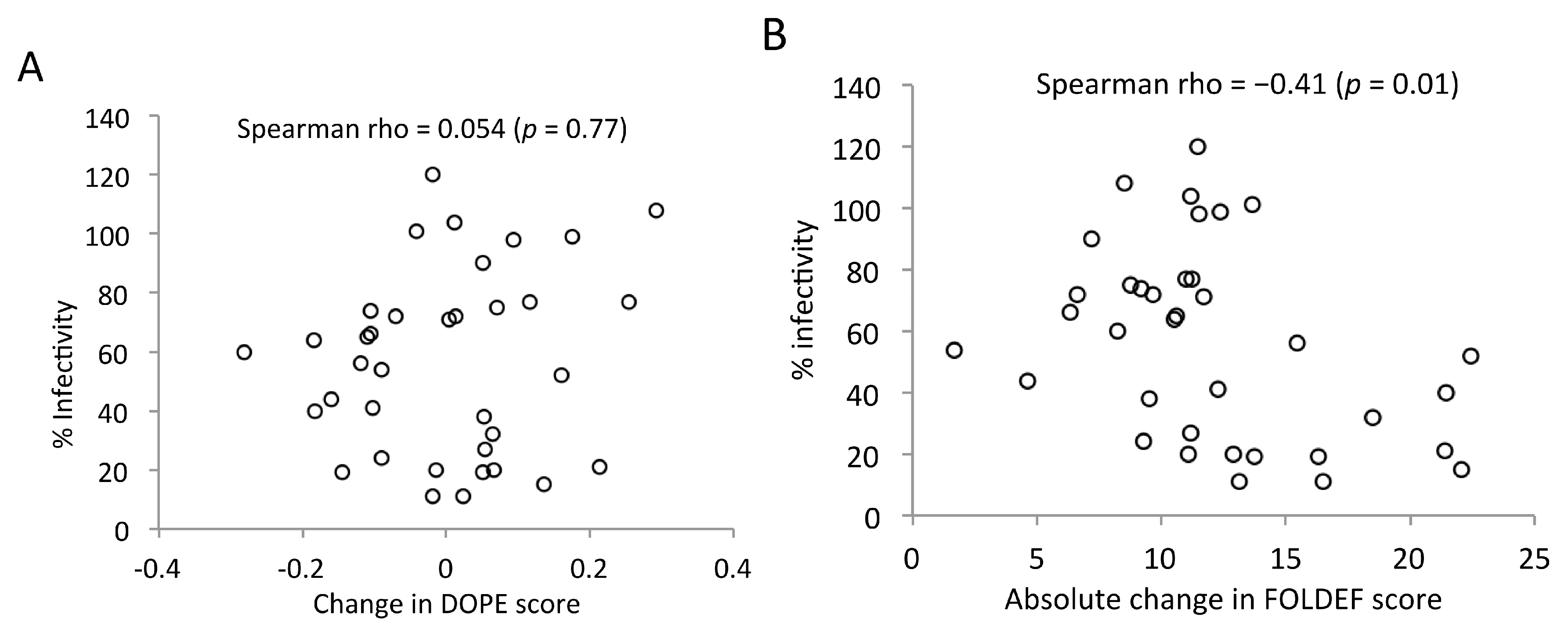
2.8. More than One-Fifth of HIV-1 CA Is Prone to Destabilizing Mutations
2.9. Clusters of Sites Prone to Destabilizing Mutations Significantly Overlap with Peptides Shown to Induce Immune Response and Associated with Viral Control

2.10. Composite Sequence–Structure Score for Predicting the Impact of Mutations
3. Discussion
4. Materials and Methods
4.1. In Silico Mutagenesis
4.2. Proteins Stabilities
4.3. Sequence Dataset and Amino acid Database Frequencies
4.4. Composite Sequence–Dtructure Stability Score
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Altfeld, M.; Allen, T.M. Hitting HIV where it hurts: An alternative approach to HIV vaccine design. Trends Immunol. 2006, 27, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Letourneau, S.; Im, E.J.; Mashishi, T.; Brereton, C.; Bridgeman, A.; Yang, H.; Dorrell, L.; Dong, T.; Korber, B.; McMichael, A.J.; et al. Design and pre-clinical evaluation of a universal HIV-1 vaccine. PLoS ONE 2007, 2, e984. [Google Scholar] [CrossRef] [PubMed]
- Rolland, M.; Nickle, D.C.; Mullins, J.I. HIV-1 group M conserved elements vaccine. PLoS Pathog. 2007, 3, e157. [Google Scholar] [CrossRef] [PubMed]
- Yang, O.O. Candidate vaccine sequences to represent intra- and inter-clade HIV-1 variation. PLoS ONE 2009, 4, e7388. [Google Scholar] [CrossRef] [PubMed]
- Kunwar, P.; Hawkins, N.; Dinges, W.L.; Liu, Y.; Gabriel, E.E.; Swan, D.A.; Stevens, C.E.; Maenza, J.; Collier, A.C.; Mullins, J.I.; et al. Superior control of HIV-1 replication by CD8+ T cells targeting conserved epitopes: Implications for HIV vaccine design. PLoS ONE 2013, 8, e64405. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Holland, J.J. Rna virus mutations and fitness for survival. Annu. Rev. Microbiol. 1997, 51, 151–178. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E. Mechanisms of viral emergence. Vet. Res. 2010, 41. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Picado, J.; Prado, J.G.; Fry, E.E.; Pfafferott, K.; Leslie, A.; Chetty, S.; Thobakgale, C.; Honeyborne, I.; Crawford, H.; Matthews, P.; et al. Fitness cost of escape mutations in p24 Gag in association with control of human immunodeficiency virus type 1. J. Virol. 2006, 80, 3617–3623. [Google Scholar] [CrossRef] [PubMed]
- Schneidewind, A.; Brockman, M.A.; Yang, R.; Adam, R.I.; Li, B.; Le Gall, S.; Rinaldo, C.R.; Craggs, S.L.; Allgaier, R.L.; Power, K.A.; et al. Escape from the dominant HLA-B27-restricted cytotoxic T-lymphocyte response in Gag is associated with a dramatic reduction in human immunodeficiency virus type 1 replication. J. Virol. 2007, 81, 12382–12393. [Google Scholar] [CrossRef] [PubMed]
- Arrive, E.; Chaix, M.L.; Nerrienet, E.; Blanche, S.; Rouzioux, C.; Coffie, P.A.; Kruy Leang, S.; McIntyre, J.; Avit, D.; Srey, V.H.; et al. Tolerance and viral resistance after single-dose nevirapine with tenofovir and emtricitabine to prevent vertical transmission of HIV-1. AIDS 2009, 23, 825–833. [Google Scholar] [PubMed]
- Crawford, H.; Prado, J.G.; Leslie, A.; Hue, S.; Honeyborne, I.; Reddy, S.; van der Stok, M.; Mncube, Z.; Brander, C.; Rousseau, C.; et al. Compensatory mutation partially restores fitness and delays reversion of escape mutation within the immunodominant HLA-B*5703-restricted Gag epitope in chronic HIV-1 infection. J. Virol. 2007, 81, 8346–8351. [Google Scholar] [CrossRef] [PubMed]
- Boutwell, C.L.; Rowley, C.F.; Essex, M. Reduced viral replication capacity of human immunodeficiency virus type 1 subtype C caused by cytotoxic-T-lymphocyte escape mutations in HLA-B57 epitopes of capsid protein. J. Virol. 2009, 83, 2460–2468. [Google Scholar] [CrossRef]
- Boutwell, C.L.; Schneidewind, A.; Brumme, Z.; Brockman, M.; Streeck, H.; Brumme, C.; Dudek, T.; Kane, K.; Kemper, M.; Walker, B.; et al. CTL escape mutations in gag epitopes restricted by protective HLA class I alleles cause substantial reductions in viral replication capacity. Retrovirology 2009, 6, 399–399. [Google Scholar] [CrossRef]
- Rolland, M.; Manocheewa, S.; Swain, J.V.; Lanxon-Cookson, E.C.; Kim, M.; Westfall, D.H.; Larsen, B.B.; Gilbert, P.B.; Mullins, J.I. HIV-1 conserved-element vaccines: Relationship between sequence conservation and replicative capacity. J. Virol. 2013, 87, 5461–5467. [Google Scholar] [CrossRef] [PubMed]
- Manocheewa, S.; Swain, J.V.; Lanxon-Cookson, E.; Rolland, M.; Mullins, J.I. Fitness costs of mutations at the HIV-1 capsid hexamerization interface. PLoS ONE 2013, 8, e66065. [Google Scholar] [CrossRef] [PubMed]
- Rihn, S.J.; Wilson, S.J.; Loman, N.J.; Alim, M.; Bakker, S.E.; Bhella, D.; Gifford, R.J.; Rixon, F.J.; Bieniasz, P.D. Extreme genetic fragility of the HIV-1 capsid. PLoS Pathog. 2013, 9, e1003461. [Google Scholar] [CrossRef] [PubMed]
- Tieu, H.-V.; Rolland, M.; Hammer, S.M.; Sobieszczyk, M.E. Translational research insights from completed HIV vaccine efficacy trials. J. Acquir. Immune Defic. Syndr. 2013, 63, S150–S154. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, Y.; Pfafferott, K.; Frater, J.; Matthews, P.; Payne, R.; Addo, M.; Gatanaga, H.; Fujiwara, M.; Hachiya, A.; Koizumi, H.; et al. Adaptation of HIV-1 to human leukocyte antigen class I. Nature 2009, 458, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, C.M.; Lockhart, D.W.; Listgarten, J.; Maley, S.N.; Kadie, C.; Learn, G.H.; Nickle, D.C.; Heckerman, D.E.; Deng, W.; Brander, C.; et al. Rare hla drive additional HIV evolution compared to more frequent alleles. AIDS Res. Hum. Retroviruses 2009, 25, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Fassati, A. Multiple roles of the capsid protein in the early steps of HIV-1 infection. Virus Res. 2012, 170, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Sundquist, W.I.; Krausslich, H.G. HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef]
- Bocanegra, R.; Rodriguez-Huete, A.; Fuertes, M.A.; del Alamo, M.; Mateu, M.G. Molecular recognition in the human immunodeficiency virus capsid and antiviral design. Virus Res. 2012, 169, 388–410. [Google Scholar] [CrossRef] [PubMed]
- Kiepiela, P.; Ngumbela, K.; Thobakgale, C.; Ramduth, D.; Honeyborne, I.; Moodley, E.; Reddy, S.; de Pierres, C.; Mncube, Z.; Mkhwanazi, N.; et al. Cd8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat. Med. 2007, 13, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, K.E.; Li, H.; Walker, B.D.; Michael, N.L.; Barouch, D.H. Gag-specific cellular immunity determines in vitro viral inhibition and in vivo virologic control following simian immunodeficiency virus challenges of vaccinated rhesus monkeys. J. Virol. 2012, 86, 9583–9589. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, N.; Takahashi, N.; Seki, S.; Nomura, T.; Yamamoto, H.; Inoue, M.; Shu, T.; Naruse, T.K.; Kimura, A.; Matano, T. Control of simian immunodeficiency virus replication by vaccine-induced Gag- and Vif-specific CD8+ T cells. J. Virol. 2014, 88, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.; Robertson, D.L.; Lovell, S.C. Constraints on HIV-1 diversity from protein structure. J. Virol. 2010, 84, 12995–13003. [Google Scholar] [CrossRef] [PubMed]
- Boutwell, C.L.; Carlson, J.M.; Lin, T.H.; Seese, A.; Power, K.A.; Peng, J.; Tang, Y.; Brumme, Z.L.; Heckerman, D.; Schneidewind, A.; et al. Frequent and variable cytotoxic-T-lymphocyte escape-associated fitness costs in the human immunodeficiency virus type 1 subtype B Gag proteins. J. Virol. 2013, 87, 3952–3965. [Google Scholar] [CrossRef] [PubMed]
- Pornillos, O.; Ganser-Pornillos, B.K.; Kelly, B.N.; Hua, Y.; Whitby, F.G.; Stout, C.D.; Sundquist, W.I.; Hill, C.P.; Yeager, M. X-ray structures of the hexameric building block of the HIV capsid. Cell 2009, 137, 1282–1292. [Google Scholar] [CrossRef] [PubMed]
- Pornillos, O.; Ganser-Pornillos, B.K.; Yeager, M. Atomic-level modelling of the HIV capsid. Nature 2011, 469, 424–427. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Perilla, J.R.; Yufenyuy, E.L.; Meng, X.; Chen, B.; Ning, J.; Ahn, J.; Gronenborn, A.M.; Schulten, K.; Aiken, C.; et al. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 2013, 497, 643–646. [Google Scholar] [CrossRef] [PubMed]
- HIV sequence database. Available online: http://www.HIV.lanl.gov/content/sequence/HIV/HIVTools.html (accessed on 1 July 2015).
- Ganser-Pornillos, B.K.; von Schwedler, U.K.; Stray, K.M.; Aiken, C.; Sundquist, W.I. Assembly properties of the human immunodeficiency virus type 1 CA protein. J. Virol. 2004, 78, 2545–2552. [Google Scholar] [CrossRef] [PubMed]
- Schur, F.K.; Hagen, W.J.; Rumlova, M.; Ruml, T.; Muller, B.; Krausslich, H.G.; Briggs, J.A. Structure of the immature HIV-1 capsid in intact virus particles at 8.8 Å resolution. Nature 2015, 517, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Worthylake, D.K.; Wang, H.; Yoo, S.; Sundquist, W.I.; Hill, C.P. Structures of the HIV-1 capsid protein dimerization domain at 2.6 a resolution. Acta Crystallogr. D Biol. Crystallogr. 1999, 55, 85–92. [Google Scholar] [CrossRef] [PubMed]
- von Schwedler, U.K.; Stray, K.M.; Garrus, J.E.; Sundquist, W.I. Functional surfaces of the human immunodeficiency virus type 1 capsid protein. J. Virol. 2003, 77, 5439–5450. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Murakami, T.; Agresta, B.E.; Campbell, S.; Freed, E.O.; Levin, J.G. Human immunodeficiency virus type 1 N-terminal capsid mutants that exhibit aberrant core morphology and are blocked in initiation of reverse transcription in infected cells. J. Virol. 2001, 75, 9357–9366. [Google Scholar] [CrossRef] [PubMed]
- Forshey, B.M.; von Schwedler, U.; Sundquist, W.I.; Aiken, C. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J. Virol. 2002, 76, 5667–5677. [Google Scholar] [CrossRef] [PubMed]
- Bartonova, V.; Igonet, S.; Sticht, J.; Glass, B.; Habermann, A.; Vaney, M.C.; Sehr, P.; Lewis, J.; Rey, F.A.; Krausslich, H.G. Residues in the HIV-1 capsid assembly inhibitor binding site are essential for maintaining the assembly-competent quaternary structure of the capsid protein. J. Biol. Chem. 2008, 283, 32024–32033. [Google Scholar] [CrossRef] [PubMed]
- Brockman, M.A.; Schneidewind, A.; Lahaie, M.; Schmidt, A.; Miura, T.; Desouza, I.; Ryvkin, F.; Derdeyn, C.A.; Allen, S.; Hunter, E.; et al. Escape and compensation from early HLA-B57-mediated cytotoxic T-lymphocyte pressure on human immunodeficiency virus type 1 Gag alter capsid interactions with cyclophilin A. J. Virol. 2007, 81, 12608–12618. [Google Scholar] [CrossRef] [PubMed]
- Llano, A.; Williams, A.; Olvera, A.; Silva-Arrieta, S.; Brander, C. Best-characterized HIV-1 CTL epitopes: The 2013 update. HIV Mol. Immunol. 2013 2013, 3–25. [Google Scholar]
- Kulkarni, V.; Rosati, M.; Valentin, A.; Ganneru, B.; Singh, A.K.; Yan, J.; Rolland, M.; Alicea, C.; Beach, R.K.; Zhang, G.M.; et al. HIV-1 p24(Gag) derived conserved element DNA vaccine increases the breadth of immune response in mice. PLoS ONE 2013, 8, e60245. [Google Scholar] [CrossRef] [PubMed]
- Mothe, B.; Llano, A.; Ibarrondo, J.; Daniels, M.; Miranda, C.; Zamarreno, J.; Bach, V.; Zuniga, R.; Perez-Alvarez, S.; Berger, C.T.; et al. Definition of the viral targets of protective HIV-1-specific T cell responses. J. Transl. Med. 2011, 9, 208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, V.; Valentin, A.; Rosati, M.; Alicea, C.; Singh, A.K.; Jalah, R.; Broderick, K.E.; Sardesai, N.Y.; Le Gall, S.; Mothe, B.; et al. Altered response hierarchy and increased T-cell breadth upon HIV-1 conserved element DNA vaccination in macaques. PLoS ONE 2014, 9, e86254. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, V.; Valentin, A.; Rosati, M.; Rolland, M.; Mullins, J.I.; Pavlakis, G.N.; Felber, B.K. HIV-1 conserved elements p24CE DNA vaccine induces humoral immune responses with broad epitope recognition in macaques. PLoS ONE 2014, 9, e111085. [Google Scholar] [CrossRef] [PubMed]
- Humphris-Narayanan, E.; Akiva, E.; Varela, R.; Conchúir, S.Ó.; Kortemme, T. Prediction of mutational tolerance in HIV-1 protease and reverse transcriptase using flexible backbone protein design. PLoS Comput. Biol. 2012, 8, e1002639. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.G.; Madan, R.; Norris, M.G.; Archer, J.; Mizuguchi, K.; Robertson, D.L.; Lovell, S.C. Using knowledge of protein structural constraints to predict the evolution of HIV-1. J. Mol. Biol. 2011, 410, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.K.; Jordan, M.R.; Sultan, B.J.; Hill, A.; Davis, D.H.; Gregson, J.; Sawyer, A.W.; Hamers, R.L.; Ndembi, N.; Pillay, D.; et al. Global trends in antiretroviral resistance in treatment-naive individuals with HIV after rollout of antiretroviral treatment in resource-limited settings: A global collaborative study and meta-regression analysis. Lancet 2012, 380, 1250–1258. [Google Scholar] [CrossRef]
- Schneidewind, A.; Brumme, Z.L.; Brumme, C.J.; Power, K.A.; Reyor, L.L.; O’Sullivan, K.; Gladden, A.; Hempel, U.; Kuntzen, T.; Wang, Y.E.; et al. Transmission and long-term stability of compensated CD8 escape mutations. J. Virol. 2008. [Google Scholar] [CrossRef] [PubMed]
- Yeh, W.W.; Cale, E.M.; Jaru-Ampornpan, P.; Lord, C.I.; Peyerl, F.W.; Letvin, N.L. Compensatory substitutions restore normal core assembly in simian immunodeficiency virus isolates with Gag epitope cytotoxic T-lymphocyte escape mutations. J. Virol. 2006, 80, 8168–8177. [Google Scholar] [CrossRef] [PubMed]
- Nijhuis, M.; Schuurman, R.; de Jong, D.; Erickson, J.; Gustchina, E.; Albert, J.; Schipper, P.; Gulnik, S.; Boucher, C.A. Increased fitness of drug resistant HIV-1 protease as a result of acquisition of compensatory mutations during suboptimal therapy. AIDS 1999, 13, 2349–2359. [Google Scholar] [CrossRef] [PubMed]
- Goudreau, N.; Coulombe, R.; Faucher, A.M.; Grand-Maitre, C.; Lacoste, J.E.; Lemke, C.T.; Malenfant, E.; Bousquet, Y.; Fader, L.; Simoneau, B.; et al. Monitoring binding of HIV-1 capsid assembly inhibitors using (19)F ligand-and (15)N protein-based NMR and X-ray crystallography: Early hit validation of a benzodiazepine series. Chem. Med. Chem. 2013, 8, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Curreli, F.; Zhang, X.; Bhattacharya, S.; Waheed, A.A.; Cooper, A.; Cowburn, D.; Freed, E.O.; Debnath, A.K. Antiviral activity of α-helical stapled peptides designed from the HIV-1 capsid dimerization domain. Retrovirology 2011, 8. [Google Scholar] [CrossRef] [PubMed]
- Blair, W.S.; Pickford, C.; Irving, S.L.; Brown, D.G.; Anderson, M.; Bazin, R.; Cao, J.; Ciaramella, G.; Isaacson, J.; Jackson, L.; et al. Hiv capsid is a tractable target for small molecule therapeutic intervention. PLoS Pathog 2010, 6, e1001220. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhou, J.; Halambage, U.D.; Shah, V.B.; Burse, M.J.; Wu, H.; Blair, W.S.; Butler, S.L.; Aiken, C. Compensatory substitutions in the HIV-1 capsid reduce the fitness cost associated with resistance to a capsid-targeting small-molecule inhibitor. J. Virol. 2015, 89, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Janes, H.; Friedrich, D.P.; Krambrink, A.; Smith, R.J.; Kallas, E.G.; Horton, H.; Casimiro, D.R.; Carrington, M.; Geraghty, D.E.; Gilbert, P.B.; et al. Vaccine-induced Gag-specific T cells are associated with reduced viremia after HIV-1 infection. J. Infect. Dis. 2013, 208, 1231–1239. [Google Scholar] [CrossRef]
- Zuniga, R.; Lucchetti, A.; Galvan, P.; Sanchez, S.; Sanchez, C.; Hernandez, A.; Sanchez, H.; Frahm, N.; Linde, C.H.; Hewitt, H.S.; et al. Relative dominance of Gag p24-specific cytotoxic T lymphocytes is associated with human immunodeficiency virus control. J. Virol. 2006, 80, 3122–3125. [Google Scholar] [CrossRef] [PubMed]
- Masemola, A.; Mashishi, T.; Khoury, G.; Mohube, P.; Mokgotho, P.; Vardas, E.; Colvin, M.; Zijenah, L.; Katzenstein, D.; Musonda, R.; et al. Hierarchical targeting of subtype C human immunodeficiency virus type 1 proteins by CD8+ T cells: Correlation with viral load. J. Virol. 2004, 78, 3233–3243. [Google Scholar] [CrossRef] [PubMed]
- Hancock, G.; Yang, H.; Yorke, E.; Wainwright, E.; Bourne, V.; Frisbee, A.; Payne, T.L.; Berrong, M.; Ferrari, G.; Chopera, D.; et al. Identification of effective subdominant anti-HIV-1 CD8+ T cells within entire post-infection and post-vaccination immune responses. PLoS Pathog. 2015, 11, e1004658. [Google Scholar] [CrossRef] [PubMed]
- Prince, J.L.; Claiborne, D.T.; Carlson, J.M.; Schaefer, M.; Yu, T.; Lahki, S.; Prentice, H.A.; Yue, L.; Vishwanathan, S.A.; Kilembe, W.; et al. Role of transmitted Gag CTL polymorphisms in defining replicative capacity and early HIV-1 pathogenesis. PLoS Pathog. 2012, 8, e1003041. [Google Scholar] [CrossRef] [PubMed]
- Yue, L.; Pfafferott, K.J.; Baalwa, J.; Conrod, K.; Dong, C.C.; Chui, C.; Rong, R.; Claiborne, D.T.; Prince, J.L.; Tang, J.; et al. Transmitted virus fitness and host T cell responses collectively define divergent infection outcomes in two HIV-1 recipients. PLoS Pathog. 2015, 11, e1004565. [Google Scholar] [CrossRef] [PubMed]
- Briggs, J.A.; Riches, J.D.; Glass, B.; Bartonova, V.; Zanetti, G.; Krausslich, H.G. Structure and assembly of immature HIV. Proc. Natl. Acad. Sci. USA 2009, 106, 11090–11095. [Google Scholar] [CrossRef] [PubMed]
- Bharat, T.A.; Castillo Menendez, L.R.; Hagen, W.J.; Lux, V.; Igonet, S.; Schorb, M.; Schur, F.K.; Krausslich, H.G.; Briggs, J.A. Cryo-electron microscopy of tubular arrays of HIV-1 Gag resolves structures essential for immature virus assembly. Proc. Natl. Acad. Sci. USA 2014, 111, 8233–8238. [Google Scholar] [CrossRef] [PubMed]
- Christensen, N.J.; Kepp, K.P. Accurate stabilities of laccase mutants predicted with a modified foldx protocol. J. Chem. Inf. Model. 2012, 52, 3028–3042. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.W.; Torbett, B.E. Accessory mutations maintain stability in drug-resistant HIV-1 protease. J. Mol. Biol. 2011, 410, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.I.; Suchard, M.A.; Bloom, J.D. Stability-mediated epistasis constrains the evolution of an influenza protein. eLife 2013, 2, e00631. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Rao, U.; McClure, J.; Konopa, P.; Manocheewa, S.; Kim, M.; Chen, L.; Troyer, R.M.; Tebit, D.M.; Holte, S.; et al. Impact of mutations in highly conserved amino acids of the HIV-1 Gag-p24 and Env-gp120 proteins on viral replication in different genetic backgrounds. PLoS ONE 2014, 9, e94240. [Google Scholar] [CrossRef] [PubMed]
- Krivov, G.G.; Shapovalov, M.V.; Dunbrack, R.L., Jr. Improved prediction of protein side-chain conformations with SCWRl4. Proteins 2009, 77, 778–795. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The foldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.Y.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [PubMed]
- Guerois, R.; Nielsen, J.E.; Serrano, L. Predicting changes in the stability of proteins and protein complexes: A study of more than 1000 mutations. J. Mol. Biol. 2002, 320, 369–387. [Google Scholar] [CrossRef]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.S.; Eramian, D.; Shen, M.Y.; Pieper, U.; Sali, A. Comparative protein structure modeling using modeller. Curr. Protoc. bioinformatics 2006, 5. [Google Scholar] [CrossRef]
- Rose, P.P.; Korber, B.T. Detecting hypermutations in viral sequences with an emphasis on G → A hypermutation. Bioinformatics 2000, 16, 400–401. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Muscle: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 2004, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesquite: A modular system for evolutionary analysis. Available online: https://mesquiteproject.wikispaces.com/home (accessed on 1 July 2015).
- CountAAFreq. Available online: http://indra.mullins.microbiol.washington.edu/perlscript/docs/CountAAFreq.html (accessed on 1 July 2015).
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manocheewa, S.; Mittler, J.E.; Samudrala, R.; Mullins, J.I. Composite Sequence–Structure Stability Models as Screening Tools for Identifying Vulnerable Targets for HIV Drug and Vaccine Development. Viruses 2015, 7, 5718-5735. https://doi.org/10.3390/v7112901
Manocheewa S, Mittler JE, Samudrala R, Mullins JI. Composite Sequence–Structure Stability Models as Screening Tools for Identifying Vulnerable Targets for HIV Drug and Vaccine Development. Viruses. 2015; 7(11):5718-5735. https://doi.org/10.3390/v7112901
Chicago/Turabian StyleManocheewa, Siriphan, John E Mittler, Ram Samudrala, and James I Mullins. 2015. "Composite Sequence–Structure Stability Models as Screening Tools for Identifying Vulnerable Targets for HIV Drug and Vaccine Development" Viruses 7, no. 11: 5718-5735. https://doi.org/10.3390/v7112901