
Complete Genome and Phylogeny of Puumala Hantavirus Isolates Circulating in France

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Rodent Trapping and Dissection
2.2. Serological and Molecular Analysis
3. Results
3.1. Seroprevalence

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Geographic Area: City (Department) | Date | NE Status | N° of Tested Bank Voles | N° of PUUV Seropositives | Seroprevalence (%) | N° of RT-PCR + | N° of Nested + |
|---|---|---|---|---|---|---|---|
| Charleville (Ardennes) | October 2008 | Endemic | 511 | 82 | 16.0 | 6 (1%) | 42 (8%) |
| May–June 2009 | |||||||
| June 2010 | |||||||
| November 2011 | |||||||
| Troyes (Aube) | May 2008 | Endemic | 34 | 5 | 14.7 | 0 | 1 (3%) |
| October 2008 | |||||||
| September 2009 | |||||||
| Dole (Jura) | Jun 2010 | Endemic | 229 | 68 | 29.7 | 9 (3.9%) | 33 (14%) |
| Senart (Essonne) | May 2008 | Endemic | 106 | 4 | 3.8 | 0 | 1 (0.9%) |
| October 2008 | |||||||
| September 2009 | |||||||
| Orléans (Loiret) | June 2008 | Free | 145 | 25 | 17.2 | 1 (0.7%) | 17 (12%) |
| October 2008 | |||||||
| April 2009 | |||||||
| June–July 2010 | |||||||
| Fontenay-le Comte (Vendée) | Jun 2009 | Free | 19 | 0 | 0 | ||
| Meillers (Allier) | Jun 2008 | Free | 227 | 0 | 0 | ||
| Sept 2008 | |||||||
| Apr 2009 | |||||||
| May 2009 | |||||||
| Bourg en Bresse (Ain) | Mar 2008 | Free | 31 | 0 | 0 | ||
| May 2008 | |||||||
| Sept 2009 | |||||||
| TOTAL | 1302 | 184 | 14.1 | 16 (1.2%) | 94 (7.2%) |
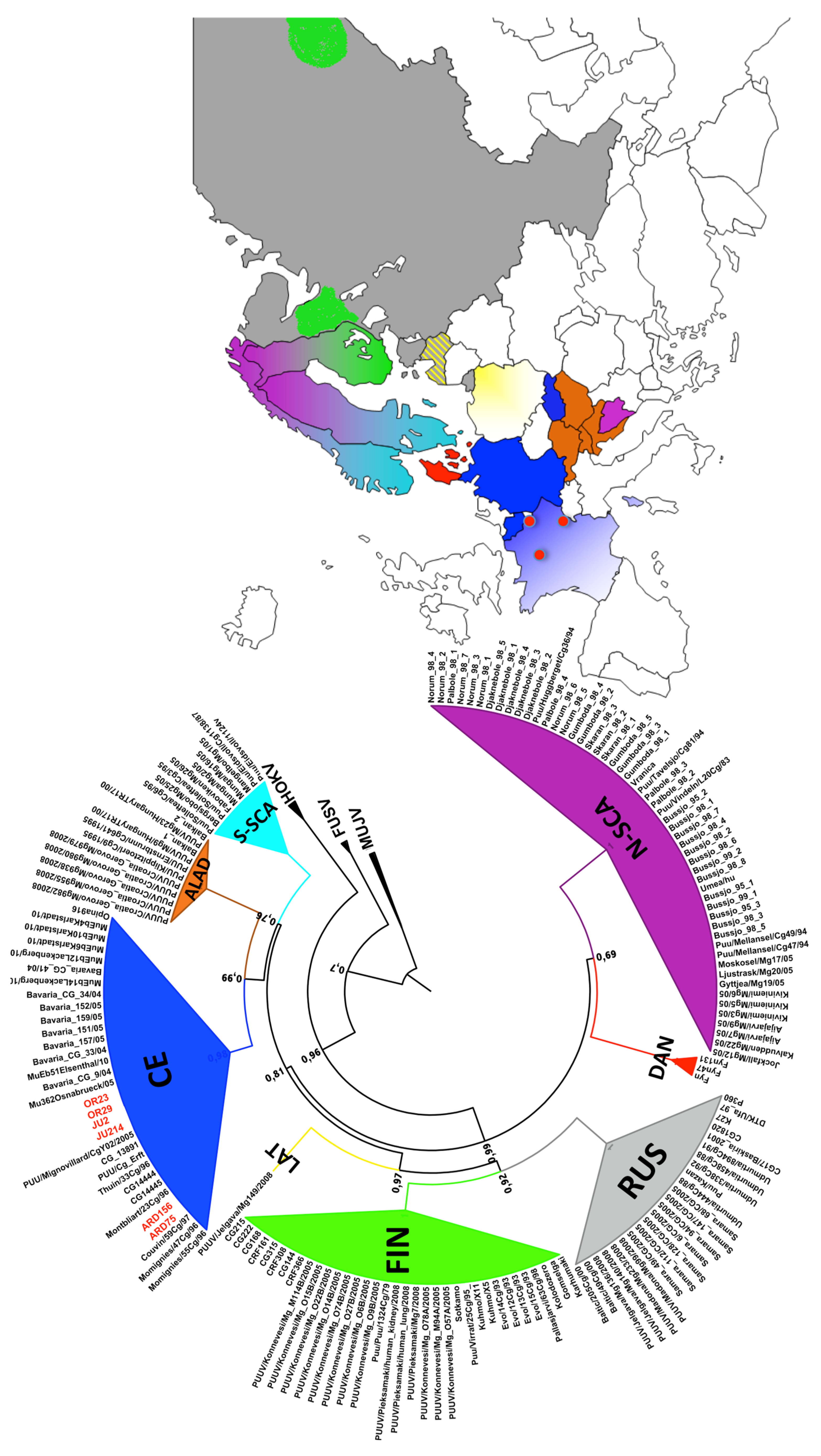
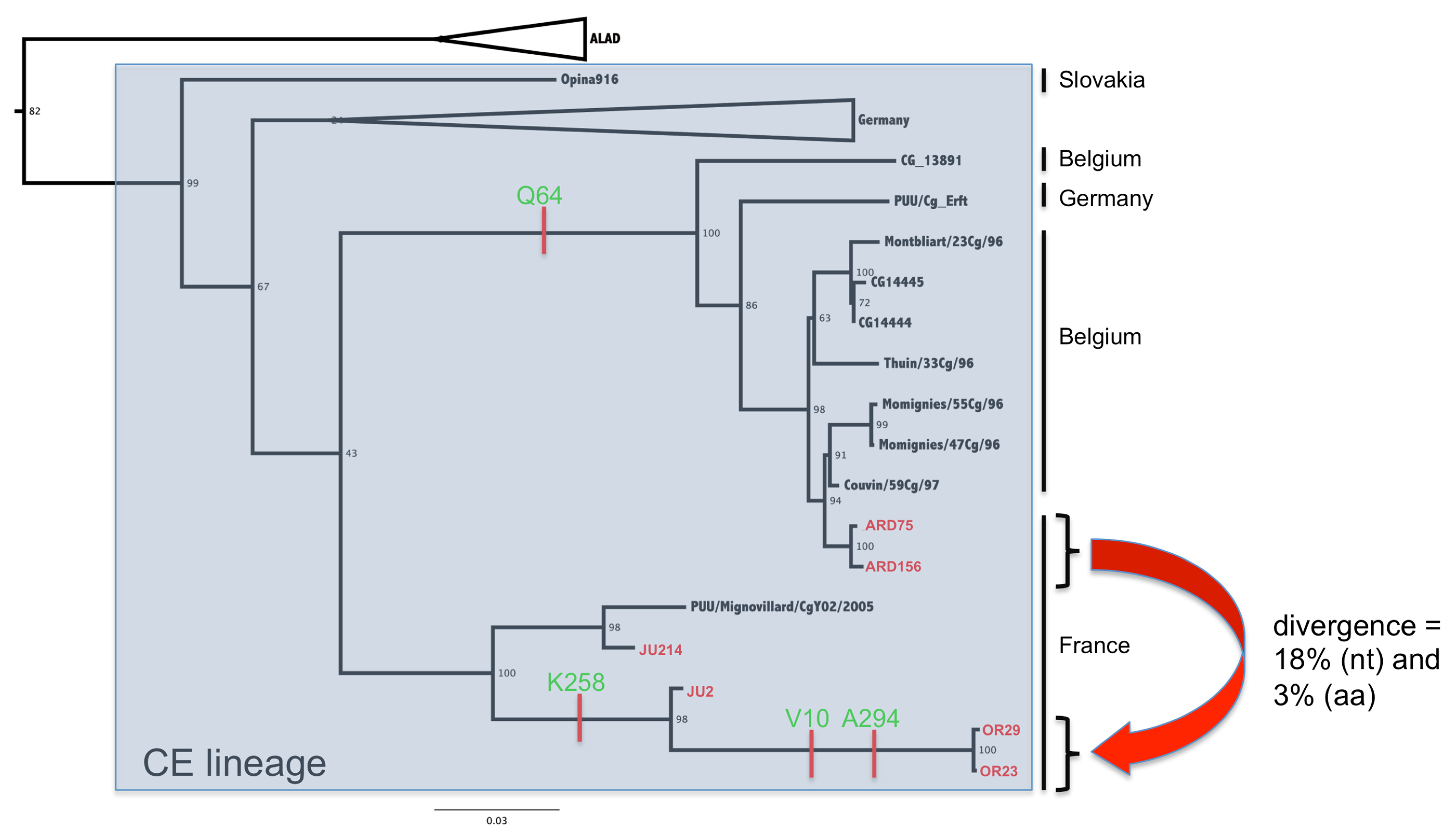
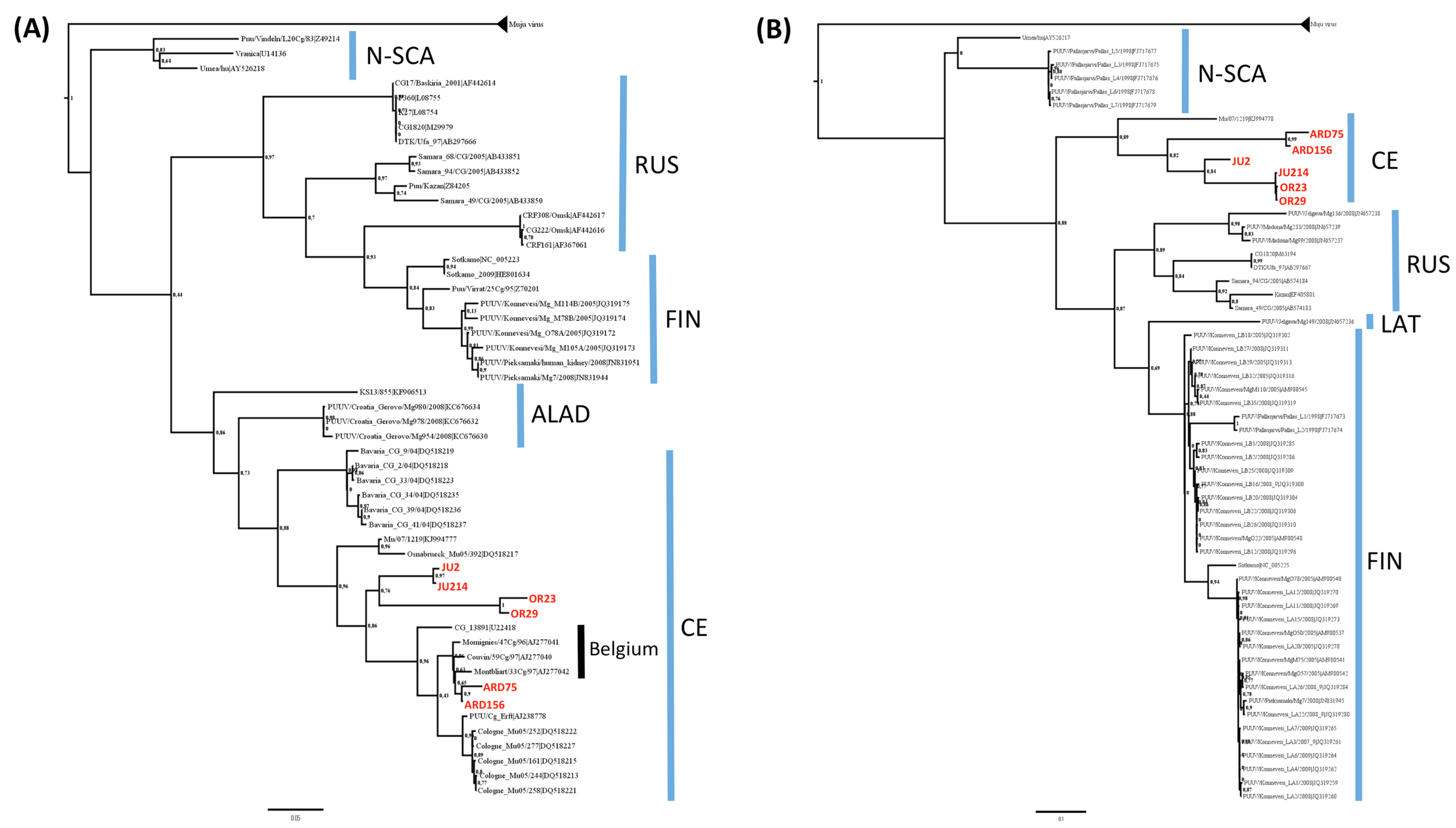
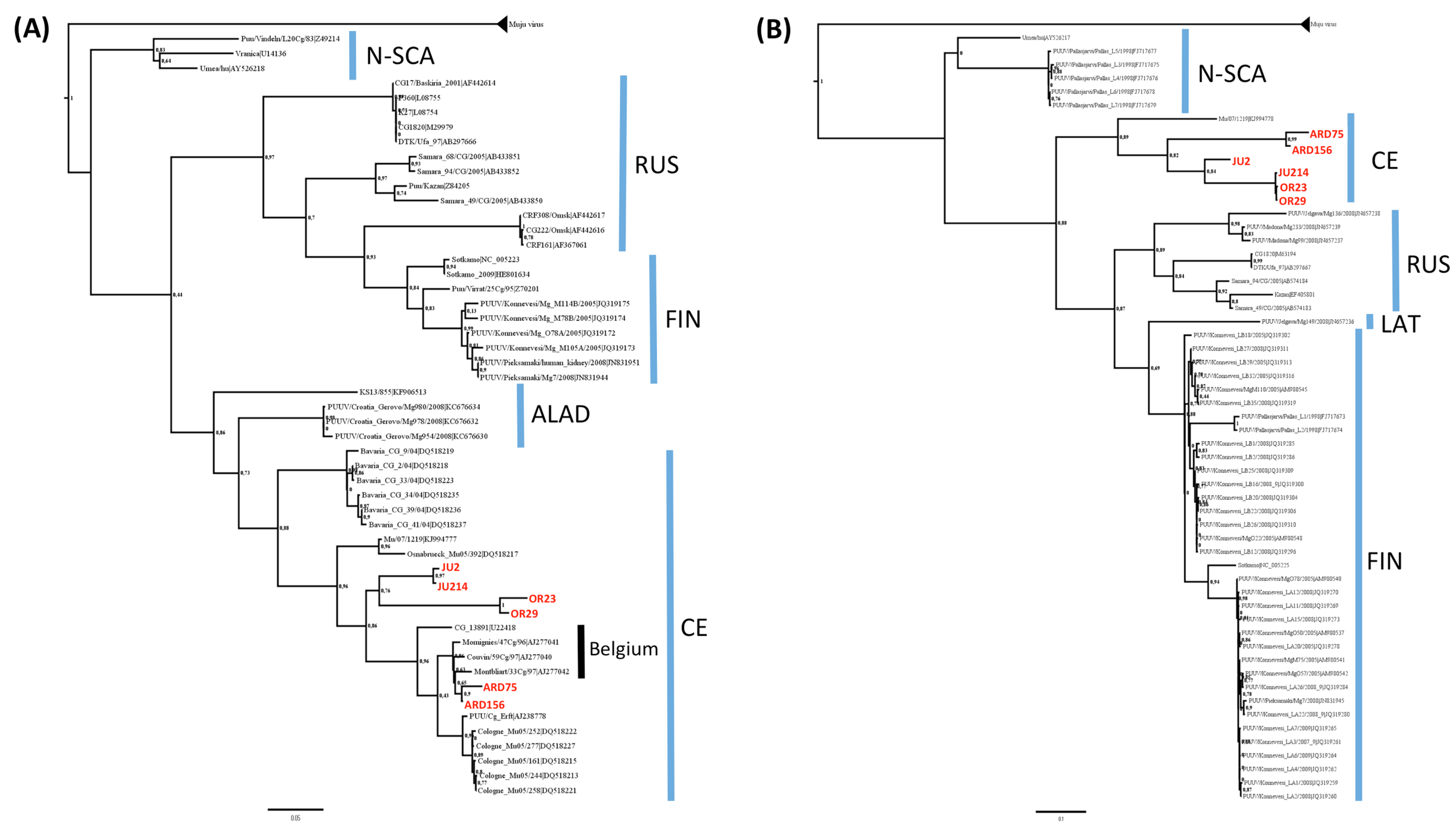
3.2. Phylogenetic Analyses
3.3. Genetic Diversity of French Isolates


| (A) | Nucleotide | |||||||||||||||||
| S segment | M segment | L segment | ||||||||||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 1 | 2 | 3 | 4 | 5 | 6 | 1 | 2 | 3 | 4 | 5 | 6 | |
| (1) Ard156 | * | * | * | |||||||||||||||
| (2) Ard75 | 0 | * | 1 | * | 2 | * | ||||||||||||
| (3) OR23 | 17 | 17 | * | 19 | 18 | * | 14 | 15 | * | |||||||||
| (4) OR29 | 18 | 17 | 0 | * | 18 | 17 | 2 | * | 14 | 15 | 1 | * | ||||||
| (5) JU2 | 16 | 15 | 7 | 7 | * | 14 | 14 | 17 | 16 | * | 12 | 12 | 10 | 10 | * | |||
| (6) JU214 | 16 | 15 | 15 | 15 | 7 | * | 14 | 14 | 17 | 15 | 1 | * | 12 | 11 | 9 | 9 | 3 | * |
| (B) | Amino acid | |||||||||||||||||
| S segment | M segment | L segment | ||||||||||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 1 | 2 | 3 | 4 | 5 | 6 | 1 | 2 | 3 | 4 | 5 | 6 | |
| (1) Ard156 | * | * | * | |||||||||||||||
| (2) Ard75 | 0 | * | 0 | * | 1 | * | ||||||||||||
| (3) OR23 | 3 | 3 | * | 3 | 3 | * | 3 | 3 | * | |||||||||
| (4) OR29 | 3 | 3 | 0 | * | 3 | 3 | 1 | * | 2 | 3 | 0 | * | ||||||
| (5) JU2 | 2 | 2 | 2 | 2 | * | 2 | 2 | 4 | 3 | * | 2 | 2 | 2 | 2 | * | |||
| (6) JU214 | 1 | 1 | 2 | 2 | 1 | 2 | 3 | 4 | 3 | 0 | * | 2 | 3 | 2 | 2 | 1 | * | |
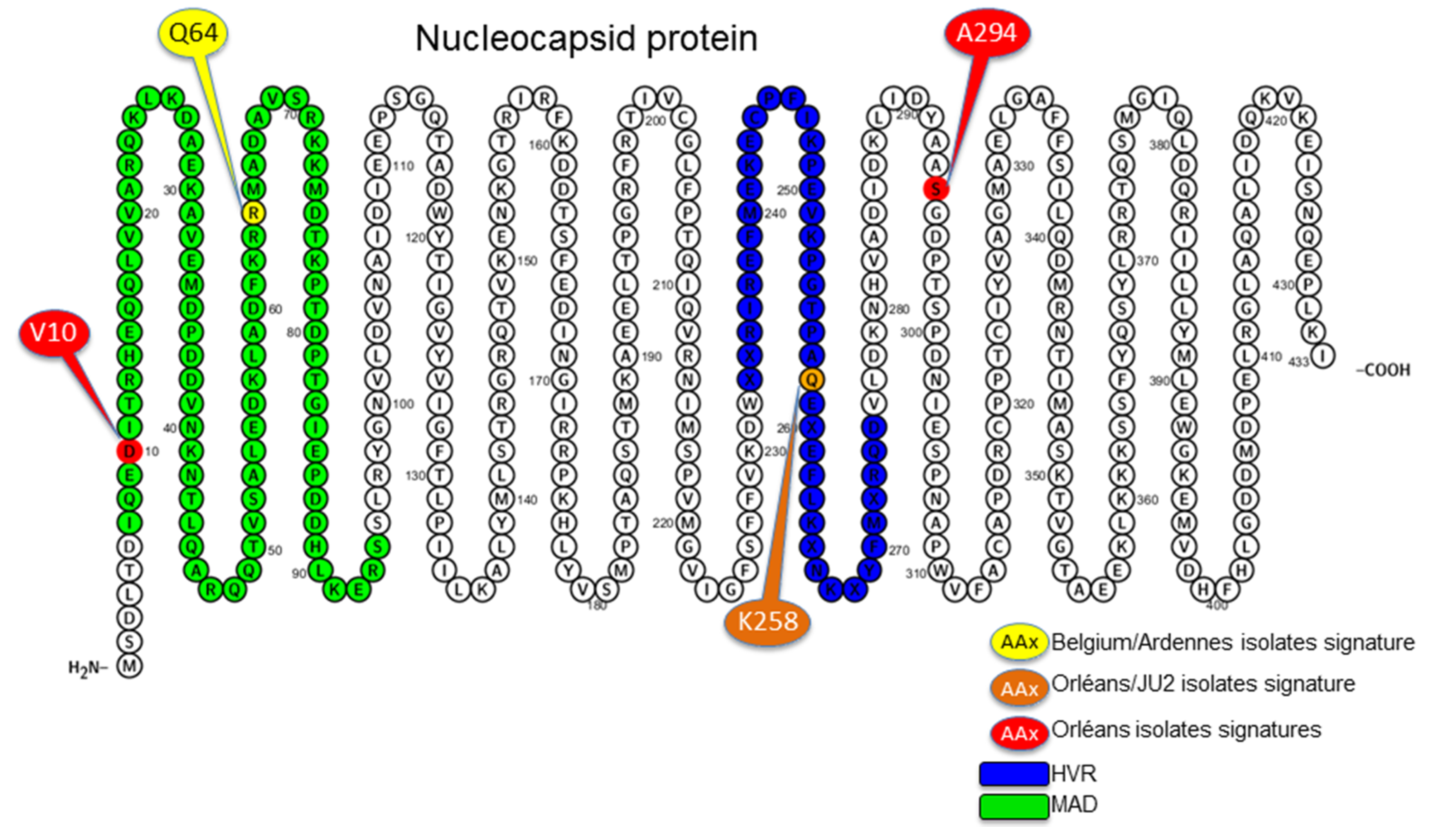
3.4. Amino Acid Specific Signatures
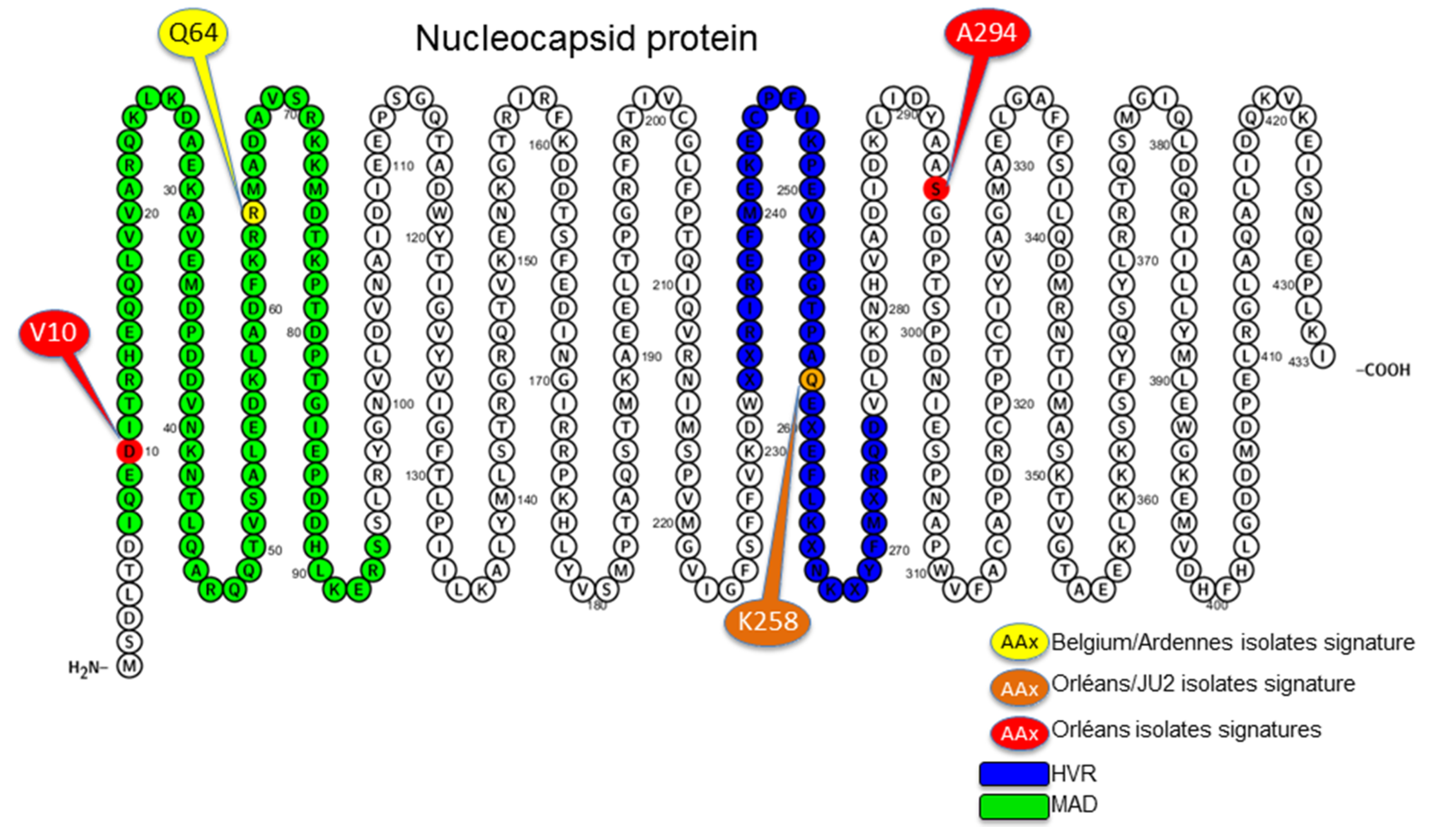
4. Discussion
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Plyusnin, A.; Sironen, T. Evolution of hantaviruses: Co-speciation with reservoir hosts for more than 100 MYR. Virus Res. 2014, 187, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Vaheri, A.; Strandin, T.; Hepojoki, J.; Sironen, T.; Henttonen, H.; Mäkelä, S.; Mustonen, J. Uncovering the mysteries of hantavirus infections. Nat. Rev. Microbiol. 2013, 11, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Z. Discovery of hantaviruses in bats and insectivores and the evolution of the genus Hantavirus. Virus Res. 2014, 187, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Jaaskelainen, K.M.; Kaukinen, P.; Minskaya, E.S.; Plyusnina, A.; Vapalahti, O.; Elliott, R.M.; Weber, F.; Vaheri, A.; Plyusnin, A. Tula and Puumala hantavirus NSs ORFs are functional and the products inhibit activation of the interferon-beta promoter. J. Med. Virol. 2007, 79, 1527–1536. [Google Scholar] [CrossRef] [PubMed]
- Plyusnin, A. Genetics of hantaviruses: Implications to taxonomy. Arch Virol. 2002, 147, 665–682. [Google Scholar] [CrossRef] [PubMed]
- Vaheri, A.; Henttonen, H.; Voutilainen, L.; Mustonen, J.; Sironen, T.; Vapalahti, O. Hantavirus infections in Europe and their impact on public health. Rev. Med. Virol. 2013, 23, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Olsson, G.E.; Leirs, H.; Henttonen, H. Hantaviruses and their hosts in Europe: Reservoirs here and there, but not everywhere? Vector Borne Zoonot. Dis. 2010, 10, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Plyusnin, A.; Vapalahti, O.; Ulfves, K.; Lehväslaiho, H.; Apekina, N.; Gavrilovskaya, I.; Blinov, V.; Vaheri, A. Sequences of wild Puumala virus genes show a correlation of genetic variation with geographic origin of the strains. J. Gen. Virol. 1994, 75, 405–409. [Google Scholar] [CrossRef] [PubMed]
- Sironen, T.; Vaheri, A.; Plyusnin, A. Molecular evolution of Puumala hantavirus. J. Virol. 2001, 75, 11803–11810. [Google Scholar] [CrossRef] [PubMed]
- Heiske, A.; Anheier, B.; Pilaski, J.; Volchkov, V.E.; Feldmann, H. A new Clethrionomys-derived hantavirus from Germany: Evidence for distinct genetic sublineages of Puumala viruses in Western Europe. Virus Res. 1999, 61, 101–112. [Google Scholar] [CrossRef]
- Escutenaire, S.; Chalon, P.; Heyman, P.; van der Auwera, G.; van der Groen, G.; Verhagen, R.; Thomas, I.; Karelle-Bui, L.; Vaheri, A.; Pastoret, P.P.; et al. Genetic characterization of Puumala hantavirus strains from Belgium: Evidence for a distinct phylogenetic lineage. Virus Res. 2001, 74, 1–15. [Google Scholar] [CrossRef]
- Plyusnina, A.; Aberle, S.W.; Aberle, J.H.; Plyusnin, A. Genetic analysis of Puumala hantavirus strains from Austria. Scand. J. Infect. Dis. 2006, 38, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Razzauti, M.; Plyusnina, A.; Niemimaa, J.; Henttonen, H.; Plyusnin, A. Co-circulation of two Puumala hantavirus lineages in Latvia: A Russian lineage described previously and a novel Latvian lineage. J. Med. Virol. 2012, 84, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Asikainen, K.; Hänninen, T.; Henttonen, H.; Niemimaa, J.; Laakkonen, J.; Andersen, H.K.; Bille, N.; Leirs, H.; Vaheri, A.; Plyusnin, A. Molecular evolution of puumala hantavirus in Fennoscandia: Phylogenetic analysis of strains from two recolonization routes, Karelia and Denmark. J. Gen. Virol. 2000, 81, 2833–2841. [Google Scholar] [CrossRef] [PubMed]
- Horling, J.; Lundkvist, A.; Jaarola, M.; Plyusnin, A.; Tegelström, H.; Persson, K.; Lehväslaiho, H.; Hörnfeldt, B.; Vaheri, A.; Niklasson, B. Distribution and genetic heterogeneity of Puumala virus in Sweden. J. Gen. Virol. 1996, 77, 2555–2562. [Google Scholar] [CrossRef] [PubMed]
- Johansson, P.; Olsson, G.E.; Low, H.T.; Bucht, G.; Ahlm, C.; Juto, P.; Elgh, F. Puumala hantavirus genetic variability in an endemic region (Northern Sweden). Infect. Genet. Evol. 2008, 8, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Sikes, R.S.; Gannon, W.L. Guidelines of the American Society of Mammalogists for the use of wild mammals in research. J. Mammal. 2011, 92, 235–253. [Google Scholar] [CrossRef]
- Billecocq, A.; Coudrier, D.; Boué, F.; Combes, B.; Zeller, H.; Artois, M.; Bouloy, M. Expression of the nucleoprotein of the Puumala virus from the recombinant Semliki Forest virus replicon: Characterization and use as a potential diagnostic tool. Clin. Diagn. Lab. Immunol. 2003, 10, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Plyusnina, A.; Razzauti, M.; Sironen, T.; Niemimaa, J.; Vapalahti, O.; Vaheri, A.; Henttonen, H.; Plyusnin, A. Analysis of complete Puumala virus genome, Finland. Emerg. Infect. Dis. 2012, 18, 2070–2072. [Google Scholar] [CrossRef] [PubMed]
- Plyusnin, A.; Beaty, B.J.; Elliott, R.M.; Goldbach, R.; Kormelink, R.; Lundkvist, A.; Schmaljohn, C.S.; Tesh, R.B. Family—Bunyaviridae, in Virus Taxonomy; King, A.M.Q., Ed.; Elsevier: San Diego, CA, USA, 2012; pp. 725–741. [Google Scholar]
- Pickett, B.E.; Greer, D.S.; Zhang, Y.; Stewart, L.; Zhou, L.; Sun, G.; Gu, Z.; Kumar, S.; Zaremba, S.; Larsen, C.N.; et al. Virus pathogen database and analysis resource (ViPR): A comprehensive bioinformatics database and analysis resource for the coronavirus research community. Viruses 2012, 4, 3209–3226. [Google Scholar] [CrossRef] [PubMed]
- Pickett, B.E.; Sadat, E.L.; Zhang, Y.; Noronha, J.M.; Squires, R.B.; Hunt, V.; Liu, M.; Kumar, S.; Zaremba, S.; Gu, Z.; et al. ViPR: An open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012, 40, D593–D598. [Google Scholar] [CrossRef] [PubMed]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Bowen, M.D.; Gelbmann, W.; Ksiazek, T.G.; Nichol, S.T.; Nowotny, N. Puumala virus and two genetic variants of Tula virus are present in Austrian rodents. J. Med. Virol. 1997, 53, 174–181. [Google Scholar] [CrossRef]
- Plyusnina, A.; Deter, J.; Charbonnel, N.; Cosson, J.F.; Plyusnin, A. Puumala and Tula hantaviruses in France. Virus Res. 2007, 129, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Razzauti, M.; Plyusnina, A.; Henttonen, H.; Plyusnin, A. Accumulation of point mutations and reassortment of genomic RNA segments are involved in the microevolution of Puumala hantavirus in a bank vole (Myodes glareolus) population. J. Gen. Virol. 2008, 89, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Razzauti, M.; Plyusnina, A.; Henttonen, H.; Plyusnin, A. Microevolution of Puumala hantavirus during a complete population cycle of its host, the bank vole (Myodes glareolus). PLoS ONE 2013, 8, e64447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castel, G.; Razzauti, M.; Jousselin, E.; Kergoat, G.J.; Cosson, J.F. Changes in diversification patterns and signatures of selection during the evolution of murinae-associated hantaviruses. Viruses 2014, 6, 1112–1134. [Google Scholar] [CrossRef] [PubMed]
- Omasits, U.; Ahrens, C.H.; Müller, S.; Wollscheid, B. Protter: Interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics 2014, 30, 884–886. [Google Scholar] [CrossRef] [PubMed]
- Gott, P.; Zöller, L.; Darai, G.; Bautz, E.K.F. A major antigenic domain of hantaviruses is located on the aminoproximal site of the viral nucleocapsid protein. Virus Genes 1997, 14, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Elgh, F.; Lundkvist, A.; Alexeyev, O.A.; Wadell, G.; Juto, P. A major antigenic domain for the human humoral response to Puumala virus nucleocapsid protein is located at the amino-terminus. J. Virol. Methods 1996, 59, 161–172. [Google Scholar] [CrossRef]
- Lundkvist, A.; Kallio-Kokko, H.; Sjölander, K.B.; Lankinen, H.; Niklasson, B.; Vaheri, A.; Vapalahti, O. Characterization of Puumala virus nucleocapsid protein: Identification of B-cell epitopes and domains involved in protective immunity. Virology 1996, 216, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Katze, M.G.; Fornek, J.L.; Palermo, R.E.; Walters, K.A.; Korth, M.J. Innate immune modulation by RNA viruses: Emerging insights from functional genomics. Nat. Rev. Immunol. 2008, 8, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, M.; Eckerle, I.; Daniel, V.; Burkhardt, U.; Opelz, G.; Schnitzler, P. Cytokine expression during early and late phase of acute Puumala hantavirus infection. BMC Immunol. 2011, 12, 65. [Google Scholar] [CrossRef] [PubMed]
- Charbonnel, N.; Pagès, M.; Sironen, T.; Henttonen, H.; Vapalahti, O.; Mustonen, J.; Vaheri, A. Immunogenetic factors affecting susceptibility of humans and rodents to hantaviruses and the clinical course of hantaviral disease in humans. Viruses 2014, 6, 2214–2241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauvage, F.; Penalba, C.; Vuillaume, P.; Boue, F.; Coudrier, D.; Pontier, D.; Artois, M. Puumala hantavirus infection in humans and in the reservoir host, Ardennes region, France. Emerg. Infect. Dis. 2002, 8, 1509–1511. [Google Scholar] [CrossRef] [PubMed]
- Sauvage, F.; Langlais, M.; Pontier, D. Predicting the emergence of human hantavirus disease using a combination of viral dynamics and rodent demographic patterns. Epidemiol. Infect. 2007, 135, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Reynes, J.M.; Dutrop, C.M.; Carli, D.; Levast, M.; Fontaine, N.; Denoyel, G.A.; Philit, J.B. Puumala hantavirus infection in Isere: Geographic extension of this zoonosis in France. Med. Mal. Infect. 2015, 45, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.M.; Duan, D.L. Insufficient geographical sampling could severely influence phylogeographic interpretations. Mar. Biol. 2013, 160, 1521–1522. [Google Scholar] [CrossRef]
- Gutierrez-Garcia, T.A.; Vazquez-Dominguez, E. Comparative Phylogeography: Designing Studies while Surviving the Process. Bioscience 2011, 61, 857–868. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castel, G.; Couteaudier, M.; Sauvage, F.; Pons, J.-B.; Murri, S.; Plyusnina, A.; Pontier, D.; Cosson, J.-F.; Plyusnin, A.; Marianneau, P.; et al. Complete Genome and Phylogeny of Puumala Hantavirus Isolates Circulating in France. Viruses 2015, 7, 5476-5488. https://doi.org/10.3390/v7102884
Castel G, Couteaudier M, Sauvage F, Pons J-B, Murri S, Plyusnina A, Pontier D, Cosson J-F, Plyusnin A, Marianneau P, et al. Complete Genome and Phylogeny of Puumala Hantavirus Isolates Circulating in France. Viruses. 2015; 7(10):5476-5488. https://doi.org/10.3390/v7102884
Chicago/Turabian StyleCastel, Guillaume, Mathilde Couteaudier, Frank Sauvage, Jean-Baptiste Pons, Séverine Murri, Angelina Plyusnina, Dominique Pontier, Jean-François Cosson, Alexander Plyusnin, Philippe Marianneau, and et al. 2015. "Complete Genome and Phylogeny of Puumala Hantavirus Isolates Circulating in France" Viruses 7, no. 10: 5476-5488. https://doi.org/10.3390/v7102884