
Molecular Biology of KSHV Lytic Reactivation


Department of Microbiology and Immunology, University of Nevada, Reno, School of Medicine, 1664 N Virginia Street, MS 320, Reno, NV 89557, USA
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Viruses 2015, 7(1), 116-153; https://doi.org/10.3390/v7010116
Submission received: 31 October 2014
/
Accepted: 5 January 2015
/
Published: 14 January 2015
(This article belongs to the Special Issue Kaposi's Sarcoma-Associated Herpesvirus)
Abstract
:Kaposi’s sarcoma-associated herpesvirus (KSHV) primarily persists as a latent episome in infected cells. During latent infection, only a limited number of viral genes are expressed that help to maintain the viral episome and prevent lytic reactivation. The latent KSHV genome persists as a highly ordered chromatin structure with bivalent chromatin marks at the promoter-regulatory region of the major immediate-early gene promoter. Various stimuli can induce chromatin modifications to an active euchromatic epigenetic mark, leading to the expression of genes required for the transition from the latent to the lytic phase of KSHV life cycle. Enhanced replication and transcription activator (RTA) gene expression triggers a cascade of events, resulting in the modulation of various cellular pathways to support viral DNA synthesis. RTA also binds to the origin of lytic DNA replication to recruit viral, as well as cellular, proteins for the initiation of the lytic DNA replication of KSHV. In this review we will discuss some of the pivotal genetic and epigenetic factors that control KSHV reactivation from the transcriptionally restricted latent program.
1. Introduction
Kaposi’s sarcoma-associated herpesvirus (KSHV) or human herpesvirus 8 (HHV 8) is one of the seven human oncogenic viruses, and is the etiologic agent of Kaposi’s sarcoma (KS, a multifocal, angiogenic and inflammatory malignancy of endothelial cell origin), as well as certain B-cell lymphomas, including primary effusion lymphoma (PEL) and multicentric Castleman’s disease (MCD) [1,2,3]. KSHV has been consistently detected in all four clinical forms of KS, including: classical KS, endemic KS in Africa, epidemic AIDS-related KS, and iatrogenic/organ-transplant KS. Lately, a newly characterized KSHV-associated condition, abbreviated as KICS (KSHV Inflammatory Cytokine Syndrome) has been reported in patients with HIV and KSHV co-infection, displaying elevated levels of interleukin-6 (IL-6) production [4]. In healthy seropositive individuals, KSHV causes persistent infection by establishing latency in CD19+ peripheral B-lymphocytes [5].
Since its initial discovery, there has been intense research on understanding the molecular biology of KSHV-mediated tumorigenesis [6]. KSHV is a γ2-lymphotropic-oncogenic-herpesvirus and is genetically linked to the Epstein-Barr virus (EBV), murine γ-herpesvirus-68 (MHV-68), and herpesvirus saimiri (HVS) (reviewed in [7]). Similar to the other members of herpesvirus family, KSHV enters the host cell as a linear double-stranded DNA genome (160–175 kb), encapsidated in an icosahedral protein capsid that is surrounded by a tegument layer and an outer lipid bilayer envelope containing glycoproteins (reviewed in [8]). Upon infection, viral DNA is delivered to the nucleus, where it circularizes to a functional circular minichromosome and persists as a non-integrated episome for the lifetime of the host (reviewed in [9]).
Inside the host cell, KSHV exhibits a biphasic life cycle consisting of a life-long reversible latent phase and a transient lytic reactivation phase, which are distinguished by their virtually distinct gene expression profiles [10]. During latent infection, KSHV genome persists as a circular episome in the infected cell with a restricted latent gene expression without the production of progeny virions. The limited region within the KSHV genome, which is transcriptionally active during latency, encodes for four major open reading frames (ORFs), consisting of ORF73/Latency-associated nuclear antigen (LANA), ORF72/viral-cyclin (v-Cyc), ORF71/viral FLICE-inhibitory protein (v-FLIP), and ORFK12/Kaposins, along with 18 mature miRNAs (at last count) and viral interferon regulatory factor-3 (vIRF3) (reviewed in [11]). KSHV has a propensity to cause latent infection that is tightly regulated by the host immune responses and has been reported to play a significant role in the development of KSHV-associated malignancies. Since the KSHV genome does not encode any viral components required for latent DNA replication, Latency Associated Nuclear Antigen (LANA), the multifarious latent protein, is considered necessary (and sufficient) for latent viral episomal DNA replication and segregation, ensuring equal distribution of replicated episomes to each daughter cell during mitosis. To achieve this, LANA binds to the LANA-binding sequences within the terminal repeat (TR) region of the KSHV genome and tethers it to the host mitotic chromosomes via interaction, with several cellular chromatin-binding proteins, followed by replication of the viral genome using a cis-acting sequence in the TR region as a replication origin [12].
Global analysis of the viral gene expression of KS tumor cells indicated that KSHV predominantly expresses viral latent transcripts with only a few percent of cells being lytically active at any specific given time [13]. The latent phase of the viral life cycle is reversible and can be reactivated to enter into the second, well-ordered program of viral gene expression, i.e., lytic reactivation. This phase predominantly consists of: (1) KSHV Reactivation from latency, followed by (2) Lytic DNA Replication and virion production. Upon reactivation from latency, a full repertoire of lytic viral genes are activated in a temporally regulated manner, leading to the transcriptional activation of three classes of lytic genes, namely, immediate early (IE), early (E), and late (L) genes [14,15,16]. The cellular machinery is switched on for an extensive viral DNA replication and gene expression, resulting in the assembly and release of infectious mature virion particles that egress out of the cell on disruption of the host-cell membrane. KSHV reactivation and lytic replication are not only important for viral propagation but also critical for KSHV-induced tumorigenesis.
Members of all three classes of lytic viral genes encode for proteins that assist in the formation of infectious virions [17]. The IE-lytic genes primarily govern the transition of KSHV genome from latent-to-lytic phase and consist of ORF50/RTA, ORF45, K8α, K8.2, K4.2, K4.1, K4, ORF48, ORF29b, K3, and ORF70. These genes are expressed within 10 h of induction and encode viral proteins that are directly involved in gene transcription and cellular modifications for viral replication. A series of studies have established that a single major IE-lytic protein-RTA acts as the quintessential latent-lytic switch that redirects KSHV to enter the productive transcriptional program required for viral spread and KS tumorigenesis. RTA protein (691 aa and 110 kDa) is the only viral lytic protein, both necessary and sufficient to disrupt latency and promote complete lytic cascade [18]. The RTA gene is reported to auto-activate its own promoter and transactivate the expression of multiple downstream lytic genes, including K8, K5, K2, K12, ORF6, ORF57, ORF74, K9, ORF59, K3, ORF37, K1, K8.1A, ORF21, vIL-6, PAN RNA, vIRF1, K1, and ORF65, either by itself (through RTA-responsive element, RRE) or in accord with other viral regulatory genes [19]. These E-lytic genes are expressed between 10–24 h post-induction and encode viral proteins primarily required for DNA replication and gene expression. The l-lytic genes that appear after 48 h post infection consist of viral structural proteins, including membrane glycoproteins (gB and K8.1), and a small viral capsid antigen required for assembly and maturation of the virions [20].
RTA plays an important role as both an initiator and a controller of KSHV lytic DNA replication [21]. Unlike latent DNA replication, lytic DNA replication: (1) depends on KSHV-encoded replication proteins; (2) initiates from a different origin (ori-Lyt); (3) replicates via a rolling-circle mechanism; and (4) leads to a multifold amplification of the viral DNA. The lytic origin of replication (ori-Lyt) consists of a specific origin binding protein (OBP) that plays a significant role in recruiting the core replication machinery to the site of replication. The two ori-Lyt domains, namely left ori-Lyt (ori-Lyt-L) and right ori-Lyt (ori-Lyt-R), are located between K4.2 and K5 and between ORF69 and ORF71, respectively, in the KSHV genome [22,23]. The ori-Lyts contain regions for various transcription factor-binding sites and RRE element that is essential for RTA-binding and ori-Lyt dependent DNA replication [22,23].
Despite the induction of lytic cycle following KSHV infection, there is a rapid inhibition of RTA promoter that further decelerates the full-blown KSHV reactivation [24]. The mechanisms that regulate the temporally ordered activation and genome-wide repression of lytic genes during primary infection are beginning to be resolved [25]. As both phases of KSHV life cycle are important for the development of KS and associated disorders, further understanding of the underlying mechanisms that coordinate regulation of gene expression may advance our knowledge of KSHV virology and assist in designing preventive therapeutic agents against KSHV lytic replication and associated tumorigenesis.
KSHV reactivation is an extremely complex process that involves a combination of both viral and cellular factors including but not limited to, temporary or permanent immune suppression, oxidative stress, inflammatory cytokines, hypoxia, viral co-infection and treatment with chromatin modifying agents. Thus far, a number of factors have been reported to stimulate or inhibit major viral proteins, however, the physiological relevance of these stimuli or repressors is far from being fully elucidated. In the following sections of this review, we will summarize recent studies that highlight the activation of KSHV lytic cycle and replication and will primarily focus on the relevant physiological, environmental, cellular, and viral regulatory factors involved in the regulation of KSHV’s biphasic life cycle, gene expression, and viral infection.
2. LANA and KSHV Reactivation
The two major KSHV proteins-LANA and RTA are shown to interact with each other and control the switch between latency and lytic reactivation [26]. Studies from multiple research groups reported a tremendous increase in the expression of several IE-lytic genes including RTA, MTA, vIL-6, ORF59, and K8.1 in 293T cells following deletion of LANA, indicating LANA-associated repression of basal level of RTA promoter as well as other RTA-responsive promoters [27,28]. LANA is shown to interact with RTA promoter and inhibit RTA gene expression via functional interaction with a recombination signal binding protein for immunoglobulin κ J region (RBP-Jκ protein), which is a major transcriptional repressor of the Notch signaling pathway [29]. LANA-mediated repression of RTA promoter and RTA auto-activation depends on RBP-Jκ binding sites. LANA recruits RBP-Jκ protein to repress the expression of RTA gene and down-regulates RTA self-activation by competing with RTA in RBP-Jκ-binding. In addition, RTA protein itself contributes to the establishment of KSHV latency by activating LANA protein expression following de novo infection. Therefore, the molecular transition between latency and lytic reactivation is controlled by the interplay between LANA and RTA proteins in KSHV-infected cells.
DNA methylation or CpG dinucleotide methylation, associated with the transcriptional silencing, also plays a key role in the induction of KSHV lytic cycle as the treatment of PEL-derived cell lines with DNA methyltransferase inhibitor, 12-o-tetradecanoylphorbol-13-acetate (TPA) or 5-Azacytidine (5-AzaC) caused demethylation of lytic promoters and induced KSHV lytic phase in vitro [30]. Bisulfite sequencing of latently infected BCBL-1 cell lines revealed hypermethylation of functionally conserved RTA gene of KSHV genome by de novo methyltransferases DNMT3a/DNMT3b and establishment of methylation marks exclusively on RTA promoter, leading to gene silencing during latency [30,31,32]. Recent studies by Grundhoff’s group reported a comprehensive tempo-spatial analysis of DNA methylation in several tumor-derived cell lines, as well as de novo infected endothelial cells using high resolution tiling microarrays together with immunoprecipitation of methylated DNA (MeDIP) [32]. These studies revealed that the KSHV genome is indeed subjected to hypermethylation at CpG dinucleotides, leading to the distinct, genome-wide DNA methylation patterns that include extensive methylation of lytic promoters followed by a poised state of repression during latency.
Interestingly, post-translational modifications of LANA, such as arginine methylation, phosphorylation and SUMOylation, have been shown to down-regulate the expression of lytic genes during the establishment of latency [28,33,34,35]. Treatment of BCBL-1 cells with histone deacetylase inhibitors, including sodium butyrate (NaB) and trichostatin A (TSA), caused a rapid dissociation of LANA from the RTA promoter and initiated transcription activation of RTA gene [28]. Furthermore, reports on phosphorylation of LANA by several kinases including glycogen synthase kinase (GSK-3β), DNA-PK/Ku and Pim-family kinase members, Pim-1 and Pim-3, have been reported to promote viral reactivation by negative modulation of LANA function [36,37,38]. LANA is also identified as a substrate for protein arginine methyltransferase 1 (PRMT1) and methylation at R20 site is found to influence strong binding of LANA to the KSHV genome and repression of lytic genes [39]. LANA is proposed to enhance histones (H2A and H2B) SUMOylation on the local chromatin by recruiting SUMO-Ubc9 complexes through SUMO-binding, resulting in a condensed chromatin and silencing of the KSHV genome (reviewed in [40]).
The early stage of KSHV infection is defined by the constitutive expression of latent genes, as well as temporally ordered expression of viral lytic genes. Recent genome-wide ChIP-seq studies described the epigenetic map of KSHV episomes during latency and indicated that chromatin of the KSHV genome is enriched with both active (H3ac or H3K4me3) and repressive histone marks (H3K9me3 and H3K27me3) [32,41]. Based on studies reported by several independent groups, KSHV-encoded latent genes are found to be associated with activating H3ac/H3K4me3-histone marks, whereas KSHV-encoded IE and E-lytic genes are found to possess either a H3ac/H3K4me3-rich euchromatin or a H3ac/H3K4me3 and H3K27me3-rich bivalent chromatin, and L-lytic genes are found to have increased levels of heterochromatin-associated repressive H3K9me3 and H3K27me3-histone marks. In addition, H3K9 histone demethylase JMJD2A, and H3K27 histone methyltransferase EZH2 of the Polycomb Repressive Complex 2 (PRC2), predominantly bind to the KSHV genome and their recruitment by LANA is shown to maintain H3K27me3-associated silencing marks on lytic genes and repress their expression during latency (reviewed in [9]). Decrease of H3K27me3 marks, by either transient expression of UTX/JMJD3, or by blocking with EZH2 of PRC2 complex, disrupts latency and induces lytic reactivation [32,41]. As LANA is continuously expressed following de novo infection/during latency and interacts with several transcriptional repressors (heterochromatin protein HP1α, methyl-CpG-binding protein MeCP2, histone deacetylase co-repressor mSin3 and DNA methyltransferases) and chromatin-remodeling proteins (H3K9me3 histone methyltransferase SUV39H1 and hSET1 complexes, H3K9 demethylase KDM3A, histone acetyltransferase CBP, histone deacetylase mSin3 and chromatin transcription complex FACT), it is evident that LANA helps to silence lytic gene expression and promotes KSHV latency through epigenetic control [42,43,44,45,46,47].
3. Stimulus Triggering KSHV Reactivation
Thus far, several PEL-infected cells, endothelial cells, CV-1, human fibroblasts and HEK cells are known to maintain KSHV in the latent form that can be induced to enter the complete productive cycle of KSHV, following treatment of cells with the broad-spectrum protein kinase C-activator (TPA) or histone deacetylase inhibitor (NaB) (reviewed in [48]). As a result, these cell lines serve as an authentic tumor model to study KSHV life cycle, providing several insights into the numerous cellular pathways that control viral reactivation. As these chemicals target numerous cellular and viral pathways, it appears that more than one mechanism is necessary to reactivate KSHV. More recently, KSHV was found to efficiently infect, immortalize, and transform, rat embryonic metanephric mesenchymal precursor (MM) cells [49]. KSHV-transformed MM cells (KMM) support the growth of KSHV-induced tumors, hence, providing a novel animal model to study the intrinsic oncogenic pathways underlying KSHV latency and reactivation.
3.1. Viral Co-Infection
While KSHV infection appears to be necessary for the development of KS, the immunodeficiency appears to be another significant factor, as the immunosuppressed patients are often susceptible to many other infectious agents [50]. Several viral proteins, including HIV-1 trans-activating protein (HIV-1 tat) [51], HIV-1 negative factor protein (HIV-1 Nef) [52], herpes simplex virus type 1 (HSV-1) [53], herpes simplex virus type 2 (HSV-2) [54], human cytomegalovirus (HCMV) [50], human herpesvirus-6 (HHV-6), herpes simplex virus type 2 (HSV-2) [54], human cytomegalovirus (HCMV) [50], human herpesvirus-6 (HHV-6) [55], human herpesvirus-7 (HHV-7) [56], and papillomavirus [57] are proven to be potent cofactors that can activate KSHV lytic replication and influence KSHV pathogenesis. In addition, it has been demonstrated that inflammatory cytokines, such as oncostatin M (OSM), hepatocyte growth factor (HGF), interferon-γ (IFN-γ) [58], and toll-like receptors 7 and 8 (TLR7/8), when stimulated by viral infections, can trigger KSHV reactivation (reviewed in [59]).
3.2. Hypoxia
As an important co-factor, hypoxia (low tissue oxygen concentration) is physiologically linked with the initiation and progression of KSHV-associated cancers and known to induce the accumulation of hypoxia-inducible factors (HIF-1α/2α) (reviewed in [59]). Hypoxic stress in PEL cells is shown to stimulate KSHV lytic reactivation through accumulation of HIF-1α within the hypoxia-responsive elements (HRE, 5'-RCGTCG-3') region of RTA promoter, and accumulation of HIF-1α/2α within the HRE2 regions of ORF34-37 promoters. Hypoxia also triggers the activation of plasma cell-differentiation factor X-box binding protein 1 (XBP-1) that trans-activates the KSHV RTA promoter with HIF-1α, leading to the expression of RTA protein and reactivation from latency [60]. Splicing of XBP-1 mRNA, an event that occurs during B-cell differentiation, is also critical for disrupting latency and promoting KSHV reactivation, with the possibility of integration of latter into the host cell differentiation program. In addition, under hypoxic conditions, LANA is reported to interact with HIF-1α bound to HREs within the RTA promoter to upregulate its gene expression [61]. Recent studies by Cai et al. have demonstrated that LANA interacts with a new host nuclear protein and hypoxia-sensitive chromatin remodeler, KAP1 (KRAB-associated protein 1), through its SUMO-2 interacting motif (LANASIM), and recruits it to the lytic promoter region of the KSHV genome for transcriptional repression [62]. Inhibition of KAP1 in KSHV-infected PEL cells enhanced the hypoxia-induced lytic reactivation through association of RBP-Jκ with HIF-1α within the RTA promoter region [63]. In KSHV-harboring cells, shRNA knockdown of KAP-1 resulted in the induction of lytic genes and a five-fold increase of RTA-mediated lytic reactivation.
3.3. Oxidative Stress and Reactive Oxygen Species (ROS)
As all forms of KS are characterized by increased levels of inflammation and oxidative stress, it is postulated that reactive oxygen species (ROS), such as hydrogen peroxide (H2O2), mediate KSHV reactivation from latency (reviewed in [59]). A recent report showed that hypoxia and pro-inflammatory cytokines-mediated spontaneous KSHV reactivation and lytic replication are supported by hydrogen peroxide (H2O2) through both autocrine and paracrine signaling [64]. H2O2 is sufficient for inducing and mediating KSHV lytic replication in KS tumors by activating ERK1/2, JNK, and p38 mitogen-activated protein kinase (p38 MAPK) pathways [65]. Significantly, treatment with antioxidant/H2O2 scavengers; N-acetyl-l-cysteine (NAC), catalase and glutathione peroxidase inhibits KSHV lytic replication and tumor progression in vivo and slows down the development of KSHV-induced lymphoma in a mouse xenograft model [64]. Another study reported that, in infected PEL cell lines BC-3 and BCBL-1, increased levels of reactive oxygen species (ROS) may induce oxidative stress that can trigger transcriptional activation of KSHV lytic cycle and promote cell death [66]. Additionally, ROS levels can be upregulated by NF-kB inhibition and treatment of infected cells with an increased amount of NF-kB inhibitor than used for inducing KSHV reactivation,,can further elevate ROS levels and induce apoptosis [66]. In addition, p38 signaling and anti-cancer drugs (cisplatin and arsenic trioxide) are found to induce KSHV lytic cycle and host cell death in an ROS-dependent manner [66]. These results directly relate KSHV reactivation to oxidative stress and inflammation, suggesting that the antioxidants and anti-inflammation drugs could be potential drugs for effectively targeting KSHV lytic replication and KSHV-associated tumorigenesis.
3.4. Histone Deacetylases and Histone Deacetylase Inhibitors (HDACs and HDACi)
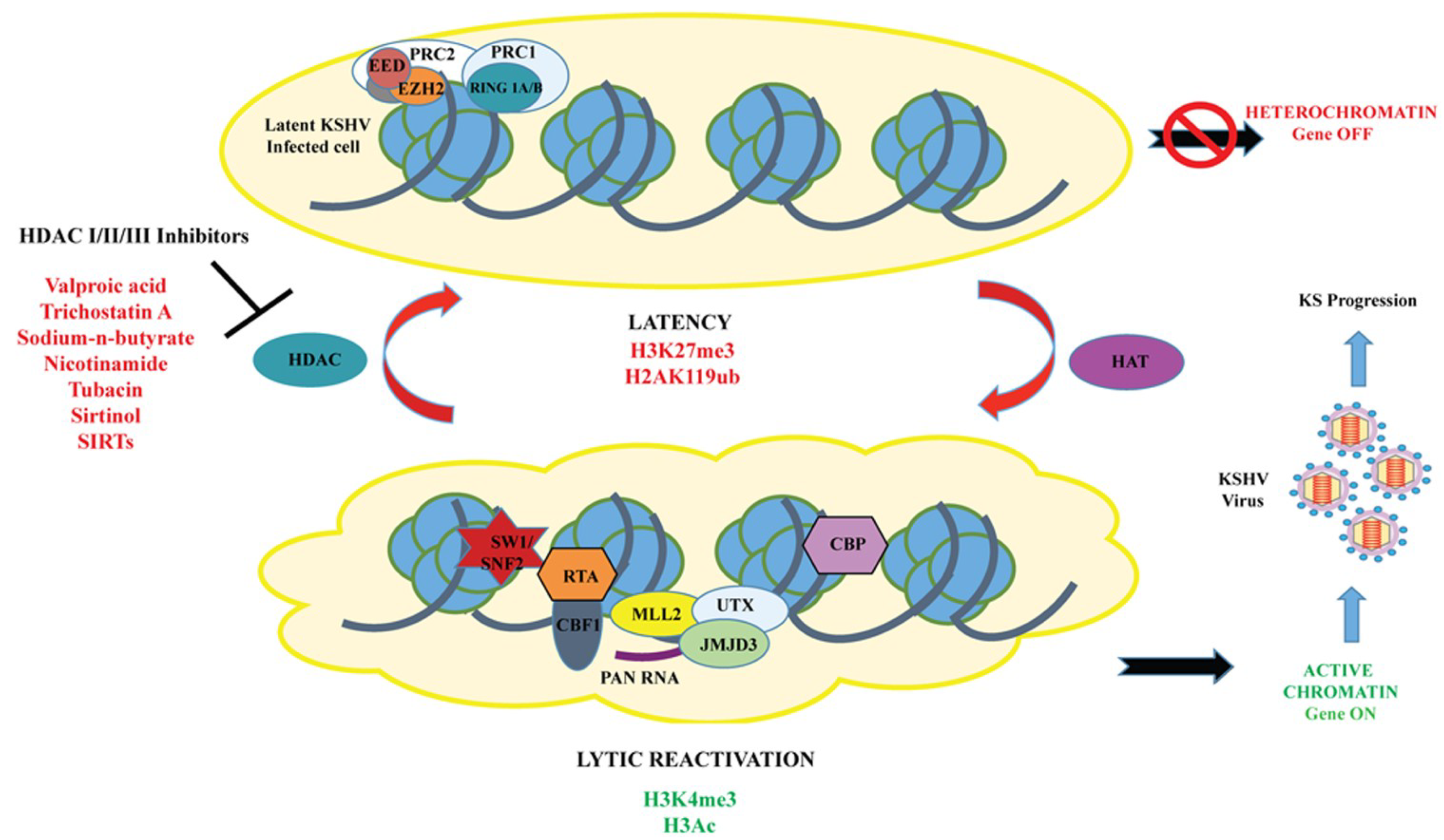
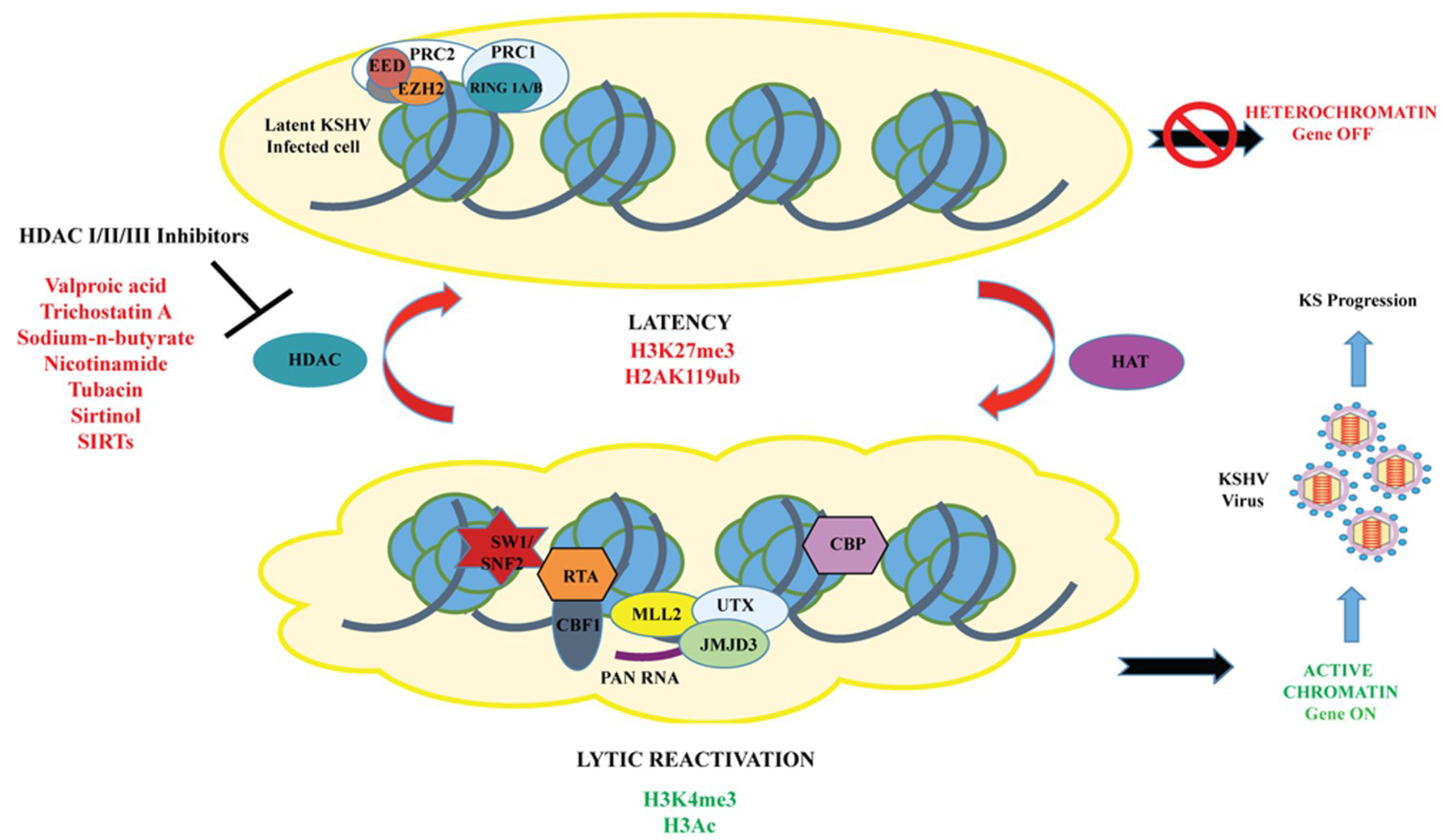
Several research groups have reported that HDAC Class I, II, and III can regulate KSHV reactivation, and activation of lytic gene expression can be triggered by treatment of KSHV latent cells with HDAC inhibitors [9,67,68]. HDACs are a group of enzymes that remove acetyl groups from ε-N-acetyl lysine amino acids in histones/proteins and play an important role in the regulation of gene expression. As mentioned earlier, during latency, IE and E-lytic genes possess bivalent chromatin associated with both repressive (H3K9me3 and H3K27me3) and activating (H3K4me3, H3ac and H3K9/K14-ac)-histone marks. In addition, previous studies showed that demethylation of H3K27me3 using UTX or dissociation of the histone methyltransferase EZH2, counteracts PRC2 repression of the RTA promoter [32,41]. In order to determine which HDAC classes (Class I and II) regulate KSHV latency and reactivation, five latently infected Vero- and PEL-cell lines were treated with a series of HDACi, including Valproic acid (VPA), trichostatin A (TSA), nicotinamide, sirtinol, tubacin, and NaB (Figure 1) [67]. The results indicated that HDAC class I inhibitors of were sufficient enough to induce KSHV virus and lytic gene expression with varied reactivation potential. Out of all the HDACi tested, VPA was found to be the most effective inducer of lytic cycle gene expression, followed by TSA. The data suggested that inhibition of HDAC class I molecules, alone, is sufficient to reactivate KSHV but the inhibition of class I and IIa molecules, together, is optimal for reactivation [67].
Additionally, Gao’s research group recently determined the role of Class III HDACs inhibitors or sirtuins (SIRTs) on the KSHV life cycle and reactivation by treatment of KSHV-positive PEL cell lines (BCP-1, BC-3 and BCBL-1) with three distinct HDACi, namely-nicotinamide (NAM), sirtinol and NaB [68]. The studies revealed that both NAM and sirtinol could efficiently reactivate KSHV from latency. In addition, it was shown that SIRT1 is involved in the control of latency and can prevent the expression of several downstream genes due to its interaction with the RTA promoter [68].
Figure 1.
A model for the chromatin landscape of RTA promoter during KSHV latency and lytic reactivation. During latency, the chromatin of RTA promoter is enriched in both activating (H3ac/H3K4me3) and repressive (H3K27me3)-histone marks, as well as the transcription repressors (Polycomb Repressive Complex 2 and HDACs), hence, the RTA promoter is transcriptionally silent. Following reactivation, the bivalent chromatin of RTA promoter is remodeled into transcriptionally active euchromatin by histone modifying enzymes, such as histone acetylases (HAT/CBP), H3K27me3 demethylase (UTX/JMJD3), H3K4 methyltransferase (MLL complex), and inhibitors of HDACs (Valproic acid, trichostatin A, NaB, nicotinamide, sirtinol, tubacin, and SIRTs, leading to the production of infectious KSHV virious and progression of KSHV-induced malignancies.
Figure 1.
A model for the chromatin landscape of RTA promoter during KSHV latency and lytic reactivation. During latency, the chromatin of RTA promoter is enriched in both activating (H3ac/H3K4me3) and repressive (H3K27me3)-histone marks, as well as the transcription repressors (Polycomb Repressive Complex 2 and HDACs), hence, the RTA promoter is transcriptionally silent. Following reactivation, the bivalent chromatin of RTA promoter is remodeled into transcriptionally active euchromatin by histone modifying enzymes, such as histone acetylases (HAT/CBP), H3K27me3 demethylase (UTX/JMJD3), H3K4 methyltransferase (MLL complex), and inhibitors of HDACs (Valproic acid, trichostatin A, NaB, nicotinamide, sirtinol, tubacin, and SIRTs, leading to the production of infectious KSHV virious and progression of KSHV-induced malignancies.
3.5. Dietary Supplements
A recent report determined that Resveratrol (Rev), an important dietary supplement, inhibits KSHV reactivation by altering the interactions between early growth response-1 (Egr-1) and the RTA promoter [69]. Electrophoretic mobility shift assays (EMSA) and chromatin immunoprecipitation (ChIP) experiments revealed that Egr-1, a cellular transcription factor known to play a critical role in the replication of several viruses, may potentially bind to the KSHV RTA promoter via at least two different GC-rich binding regions and follow a similar expression profile during de novo KSHV infection. Elevated cellular Egr-1 expression is reported to enhance viral RTA expression in a Raf > MEK > ERK-dependent manner [69]. Further, Rev is found to lower ERK1/2 activity and expression of Egr-1 in KSHV-infected cells, resulting in the suppression of virus reactivation from latency, though the precise mechanism by which Rev regulates KSHV reactivation is still unclear [69].
4. Role of Viral and Cellular Proteins Important for Lytic DNA Replication
KSHV lifecycle undergoes a transition between a dormant, latent phase and an active lytic replication phase [15,59]. KSHV lytic DNA replication requires the expression of at least eight viral genes including: ORF9 (DNA polymerase), ORF6 (single-stranded DNA binding protein), ORF40/41 (primase-associated factor), ORF44 (helicase), ORF56 (primase), ORF59 (processivity factor), ORF50 (replication and transcription activator or RTA), and ORF K8 (K-bZIP) [70,71]. RTA, an immediate early protein, is the most important protein required for the activation of lytic replication, transcription initiation, as well as recruitment of additional factors (reviewed in [48,59,72]). This section will describe several viral, as well as cellular, proteins that are important for lytic reactivation.
4.1. Viral Factors
4.1.1. K-RTA (KSHV Replication and Transcription Activator)
KSHV encoded ORF50/ RTA (replication and transcription activator), is a key regulator for the lytic reactivation from viral latency [15,73]. Expression of RTA is both essential and sufficient for KSHV reactivation [71,73,74]. Genetic mutation of RTA results in impaired reactivation and lytic DNA replication [75]. RTA has been reported to be phosphorylated [76,77], Poly (ADP-ribosyl)ated [77] and ubiquitinated [78]. RTA also autoactivates its own promoter [19] and transactivates other important lytic genes, including vIL-6 [79,80] polyadenylated nuclear RNA (PAN) [81] ORF57 (MTA) [82], ORF59 (PF8) [83,84], K-bZIP [82], vIRF1 (ORF-K9) [85], ORF-K1 [86], small viral capsid protein (ORF65) [87], ORF56 [88], SOX (ORF37) [89], vOX [90], and ORF52 [79]. RTA binds and transactivate many promoters containing K-RTA response element (RRE) [91]. KSHV LANA is also known to repress lytic reactivation, as well as RTA-mediated autoactivation [92]. LANA-mediated suppression of RTA autoactivation is dependent on RBP-Jκ, which competes with RTA for binding to RBP-Jκ [29]. Lytic reactivation results in the acetylation of LANA, leading to the dissociation of LANA from the ORF50 promoter bound to Sp1 [28]. Genome-wide screening revealed a consensus RTA interaction motif, TTCCAGGAT(N)(0–16)TTCCTGGGA [93,94]. In addition, specific amplification of bound sequences in vitro showed a number of RTA direct binding targets [93], such as ORF8, ORFK4.1, ORFK5, PAN, ORF16, ORF29, ORF45, RTA, K-bZIP, ORFK10.1, ORF59, ORFK12, ORF71/72, vOX/vGPCR (ORF74), ORF-K15, the two oriLyts, and the miR cluster [94]. These variations indicate that RTA cooperatively binds to its targets by associating with other regulatory proteins [79,93]. Additionally, RTA activates its own promoter by binding to the Oct-1 transcription factor and RBP-Jκ [19,95]. Furthermore, RTA mediated transactivation of viral lytic promoters, such as MTA and thymidine kinase (TK, ORF21), depends on Sp1, octamer-binding protein-1 (Oct-1), and XBP-1 [96,97,98]. However, direct binding of RTA to its promoter is not critical for its autoactivation [95,99]. Several other recent studies have also shown that RTA is recruited to RREs through interaction with RBP-Jκ [100,101]. Similarly, in a recent study using recombinant viruses with deleted RBP-Jκ sites within RTA promoter showed an increased viral latency and a reduced efficiency for lytic replication [102]. In addition, RTA stimulates the Notch signaling pathway, RTA mediated intracellular-activation of Notch1 is sufficient to reactivate KSHV from latency to the lytic replication cycle [103,104].
RTA transactivation of viral promoters also depends on its interactions with other cellular proteins. RTA recruits CREB binding protein (CBP), the SWI/SNF chromatin-remodeling complex, and the TRAP/mediator coactivator into viral promoters [105]. RTA binding positively regulates Histone acetyl transferase (HAT) activity of CREB [106]. A recent report showed that RTA transactivates cellular Bcl-2 through targeting of CCN9GG-like RTA responsive elements (RREs) for lytic reactivation and enhanced virion production [107]. Furthermore, it has been shown that K-RTA associates with a homologue of the Kruppel-associated box-zinc finger proteins (KRAB-ZFPs), for its transactivation function [108,109,110]. The co-repressor of K-RBP, Kruppel-associated box domain-associated protein-1 (KAP-1), is a cellular transcriptional repressor that regulates chromosomal remodeling, participates in the maintenance of latency by repressing lytic promoters [111]. During latency KAP-1 binds to viral lytic promoters to repress gene expression and depletion of KAP-1 is sufficient to induce KSHV reactivation [62]. Studies show that sumoylation and phosphorylation are required to regulate KAP-1 association with heterochromatin [62,111]. KAP-1 is phosphorylated at Ser 824, during lytic reactivation, resulting in decreased sumoylation and association to the condensed chromatin on viral promoters [111]. A recent study confirmed that KAP1 is targeted by KSHV-encoded latency-associated nuclear antigen (LANA) to repress the transactivation of K-RTA [112]. Additionally, knockdown of KAP1 in KSHV-infected primary effusion lymphoma (PEL) cells reduced viral episome stability and enhanced the efficiency of KSHV lytic reactivation by hypoxia, suggesting that both KAP1 and the cooperative interaction of RBS HRE within the RTA promoter are crucial for KSHV latency and hypoxia-induced lytic reactivation [63]. K-RTA interacts with K-bZIP, and increasing evidence indicates that repression of K-RTA transactivation by K-bZIP, a basic leucine zipper (bZIP) transcription factor encoded by KSHV, is essential for the modulation of lytic DNA replication by a feedback circuit [70,113,114]. RTA also interacts with C/EBPα, and the cooperative interaction of K-bZIP and RTA with C/EBPα is essential for the activation of K-bZIP promoter by binding to a proximal C/EBPα binding site [115]. The promoters of RTA, PAN, and MTA are activated through direct interaction of the C/EBPα and RTA complex [116]. K-RTA is also shown to be associated with viral ORF59, a processivity factor for viral DNA polymerase, and ORF45, a multifunctional tegument protein required for lytic replication [117,118].
Recent studies showed that K-RTA activity is regulated by its association with cellular peptidyl-prolyl cis/trans isomerases (PPIase), Pin1. Pin1 binds specifically to phosphorserine or phosphorthreonine-proline (pS/T-P) motifs in the K-RTA and enhances K-RTA transactivation [119]. Additionally, it has been shown that K-RTA is regulated by a 48aa small peptide, vSP-1, encoded by a polyadenylated RNA of 3.0 kb (T3.0), transcribed from the opposite strand of the KSHV RTA (ORF50) DNA template. vSP-1 associates with RTA at the protein abundance regulatory signal (PARS) motifs, and this interaction prevents RTA from degradation by ubiquitin-proteasome pathways, thus, facilitating KSHV lytic replication [120]. Apart from direct DNA interaction, RTA also cooperates with various host transcriptional factors to transactivate several downstream viral genes. Additionally, K-RTA exhibits an ubiquitin E3 ligase activity, RTA is auto-ubiquitinated and directs several cellular and viral proteins for proteasome-mediated degradation [121]. One of the cellular proteins targeted by RTA is Hey1, which interacts with repressor mSin3A. This, in turns, downregulates the expression of RTA by direct interaction with the RTA promoter [122]. RTA upregulates its own expression through ubiquitin-mediated targeting of Hey1 for degradation. Another cellular protein targeted by RTA mediated degradation is IRF-7, a critical modulator of type I IFN induction [78]. IFN signaling plays a crucial role in repressing KSHV lytic replication, therefore, this finding indicates that RTA might circumvent these cellular innate immune defenses during lytic reactivation. Direct association of RTA to the origin of lytic DNA replication (oriLyt) has been demonstrated [21]; there are two distinct oriLyt regions in the KSHV genome [23]. The left oriLyt (oriLyt-L) lies between ORFK4.2 and K5 and is comprised of a region encoding numerous transcription factor binding sites, an A+T-rich region, and a G+C repeat. Similarly, the right oriLyt (oriLyt-R) is situated between ORF69 and vFLIP and is an inverted duplication of oriLyt-L. Importantly, both oriLyts contain RREs, and [123] a direct interaction of RTA to RREs is critical for oriLyt-dependent DNA replication [23,70,123]. The presence of RREs and a downstream TATA box indicate that this region may serve as an RTA-dependent promoter, and a transcription event may be required for oriLyt-dependent DNA replication [123]. Additionally, recent studies have identified that K-RTA is able to function as a STUbL, which is capable of ubiquitylation of SUMO and SUMO conjugates in vitro and in vivo. Thus, K-RTA is an ubiquitin ligase, preferentially targeting SUMO-containing proteins for ubiquitylation; including sumoylated K-bZIP and promyelocytic leukemia (PML) nuclear bodies [124]. Together, these results suggest that RTA is a master regulator of viral lytic DNA replication.
4.1.2. ORF57-mRNA Transcript Accumulation (MTA)
ORF57 is a viral early protein, which favors viral intron-less transcript accumulation, transports, and enhances splicing of intron-containing viral RNA transcripts [125]. MTA is essential for KSHV lytic replication, moreover, genetic knockout of MTA disrupts KSHV productive lytic replication [125,126]. MTA protein carries domains with putative transcriptional and post-transcriptional functions [127]. MTA directly associate with RTA and both proteins are detected in the RTA promoter during lytic replication. KSHV MTA associates with DNA, which was identified by gel shift and chromatin immunoprecipitation assays [127,128]. In addition, it has been shown that MTA directly associates with K-bZIP protein and binds to promoter as well as transcribed regions of PAN RNA, K4, and K-bZIP [129]. These reports suggest that MTA stimulates RNA export through its association with Aly/REF, a cellular RNA-binding protein acting as an adaptor for the nuclear RNA export receptor NXF1/TAP [130]. Additionally, recent studies suggest that Aly/REF-ORF57 association does not necessarily play any significant role in the ORF57-mediated enhancement of ORF59 expression, as Aly/RE knockdown in host cells did not affect the function of ORF57 [131,132]. MTA enhances the expression of RTA or other lytic genes, most probably by binding to transcription regulatory proteins. Further, MTA cooperates with RTA to modulate the viral gene expression in a cell-line-specific manner [127,128]. It is suggested that a putative A/T hook domain within MTA arbitrates DNA binding and transcriptional initiation [127].
MTA modulates a cascade of viral gene expression and accumulation of specific viral and cellular mRNAs during lytic replication [132]. Physical association of MTA and RTA is essential for the synergistic regulatory effect of MTA. When RTA’s transactivation function is removed, MTA no longer affects the expression of viral genes, indicating that their cooperative effect depends on RTA’s transactivation function [128]. It has been shown that MTA regulates mRNA accumulation. Further, a recent study employing a genome-wide CLIP (cross-linking and immunoprecipitation) approach detected KSHV PAN, a long non-coding polyadenylated nuclear RNA, as an important target of ORF57 [133]. Genetic disruption of ORF57 affects PAN RNA expression. In co-transfection experiments, expression of exogenous ORF57 alone increased PAN RNA expression by 20–30-fold, which was due to the MRE (MTA responsive element) at the 5' PAN RNA, however, not as much on an ENE (expression and nuclear retention element) at the 3' end of PAN RNA. Further studies showed that the major function of the 5' PAN MRE is to increase the half-life of PAN in the presence of ORF57 [133]. Systematic mutational analyses identified a core motif, consisting of nine nucleotides, in MRE-II, which is essential for ORF57 interaction and function. The 9-nt core in MRE-II also interacts with cellular poly (A)-binding protein C1 (PABPC1) [134], but not E1B-AP5, which binds to another region of MRE-II [133]. In the presence of ORF57, PAN RNA is partially exportable, suggesting that ORF57 functions to accumulate a non-coding viral RNA during the course of lytic infection [133,134]. Additionally, MTA is also shown to stabilize RNAs and activates translation of mRNAs that carry internal ribosome entry sites [135]. It has been also shown that KSHV ORF57 specifically binds to ORF59 RNA and associates with cellular RNA export cofactors RBM15 and OTT3 to enhance the expression of ORF59 [136].
4.1.3. KSHV K8-K-bZIP—Lytic Replication-Associated Protein (RAP)
K-bZIP is a basic leucine zipper-containing protein encoded by KSHV K8 [137]. The K-bZIP gene locus consists of two promoters: one early promoter controlling K-bZIP and the second late promoter controlling K8.1 [91,138]. K-bZIP directly binds to K-RTA through K-bZIP’s basic domain and a specific RTA region [139,140]. Further, association of K-bZIP suppresses K-RTA transactivation of the MTA promoter in a dose-dependent manner [109,110]. Recent studies suggest that K-bZIP is not required for lytic reactivation in KSHV BACmid systems [114,141,142], however, it was reported to be crucial for virus production in infected PEL cells [143]. It has been shown that K-bZIP interacts with oriLyt [22,144,145,146] and is critical for oriLyt-dependent DNA replication in a plasmid-based transient expression system [70], but its absence can be complemented by an over-expression of RTA [145]. Similarly, association of cellular transcription factor CCAAT/enhancer-binding protein α (C/EBPα) to K-bZIP has also been shown to increase the expression and stabilization of C/EBPα and p21CIP1 proteins, followed by G0/G1 cell cycle arrest [18,115]. Similarly, KSHV bZIP can also bind to the positive regulatory domain I/III region of the IFNb promoter to block IRF3-mediated IFNb transcription [147,148]. In addition, K-bZIP represses the RTA autoactivation [139] and colocalizes with HDAC1/2 through the leucine zipper domain without the requirement of sumoylation of K-bZIP [149]. K-bZIP is phosphorylated on residues Thr111 and Ser167 by a serine/threonine protein kinase (vPK) encoded by ORF36 [146,150]. However, phosphorylation at T111 has a negative effect on both the extent of sumoylation and the repressive activity of K-bZIP [150]. K-bZIP is sumoylated at residue lysine 158, and this sumoylation is essential for K-bZIP mediated transcription repression [146]. As a SUMO adaptor, KbZIP represses transcription by recruiting Ubc9 to specific viral promoters [146]. In addition, it has been shown that K-bZIP functions as the viral SIM-containing poly-SUMO-specific E3 ligase, with specificity for SUMO-2/3 [35]. Further, K-bZIP catalyzes its auto-sumoylation and the sumoylation of other K-bZIP-interacting proteins, such as p53 and pRB [148].
A genome-wide analysis of K-bZIP’s transcriptional regulation on KSHV gene promoters showed that RTA activated 34 viral promoters whereas K-bZIP alone activated 21 promoters [140]. Nonetheless, when RTA and K-bZIP were combined together, K-bZIP was found to repress three RTA-responsive promoters, suggesting that K-bZIP might also transactivate some viral lytic genes during KSHV reactivation [140]. These data strongly suggest that K-bZIP plays a crucial during lytic gene expression and DNA replication in PEL cells [140,151]. Further, K-bZIP also directly binds to oriLyt, indicating that K-bZIP might be playing a crucial in lytic DNA replication [70]. Further, the interaction of K-bZIP with oriLyt is also modulated by LANA expression [145]. Taken together, these studies show that K-bZIP has dual independent functions in modulating the KSHV life cycle by facilitating lytic DNA replication or repressing the lytic gene expression as a feedback modulator [145]. Together with these results, knockdown of K-bZIP in latently infected BCBL-1 and BC-3 cells showed a significant reduction in the expression of RTA, MTA, and ORF26 transcripts, as well as decreased RTA and ORF-K8.1 protein levels, as well as defective viral DNA replication and virion production [143]. Collectively, these results suggest that K-bZIP regulates its own expression and possibly other RTA-transactivated lytic genes by a feedback loop.
4.1.4. ORF59- Viral Processivity Factor
Kaposi’s sarcoma-associated herpesvirus (KSHV) ORF59 plays a critical role in viral lytic DNA replication as a DNA processivity factor to the viral DNA polymerase (ORF9) [70,83]. ORF59 is highly upregulated during lytic reactivation and de novo primary infection. ORF59 forms a homodimer in the cytoplasm and associates with ORF9 to translocate it to the nucleus during lytic DNA replication [152]. ORF59 associates with C/EBPα binding motifs within oriLyt and this binding is K-RTA dependent, where K-RTA acts as an initiator of lytic replication. Additionally, disruption of the K-RTA–ORF59 interaction by a dominant negative approach impairs oriLyt-dependent DNA replication [84]. This strongly suggests that the K-Rta-ORF59 interaction is crucial for lytic DNA replication. ORF59 is a phosphoprotein and is phosphorylated by KSHV viral Ser/Thr kinase, ORF36 primarily at Ser378, which is essential for ORF59's ability to bind to RTA and the oriLyt [83,84]. In a recent study, it has been shown that lytic infection of KSHV induces severe DNA double-strand breaks (DSBs) and impede non-homologous end joining (NHEJ) in host cells. Further, ORF59 was found to be associated with Ku70 and Ku86 and this association was dependent on DSBs, suggesting that KSHV lytic replication may induce tumorigenesis by causing DNA DSBs and interrupting the DSB repair of mechanism [153].
4.1.5. ORF6-Single Strand Binding Protein
KSHV ORF6, a delayed-early gene encodes for a 126 kDa ssDNA binding protein that has been shown to participate in origin-dependent DNA replication [74,154,155]. The expression of ORF6 is regulated by RTA, which could bind to RBP-Jκ recognition site on the ORF6 promoter via interaction with the RBP-Jκ protein [95,155]. Genetic disruption analysis of the ORF6 gene, using the bacterial artificial chromosome (BAC) system, identified the functional role of ORF6 in lytic DNA replication. The mutant virus showed impaired DNA synthesis and failed to make progeny virions. Additionally, transient expression of ORF6 has rescued both defects, suggesting that ORF6 is critical for KSHV lytic replication [155].
4.2. Cellular Factors
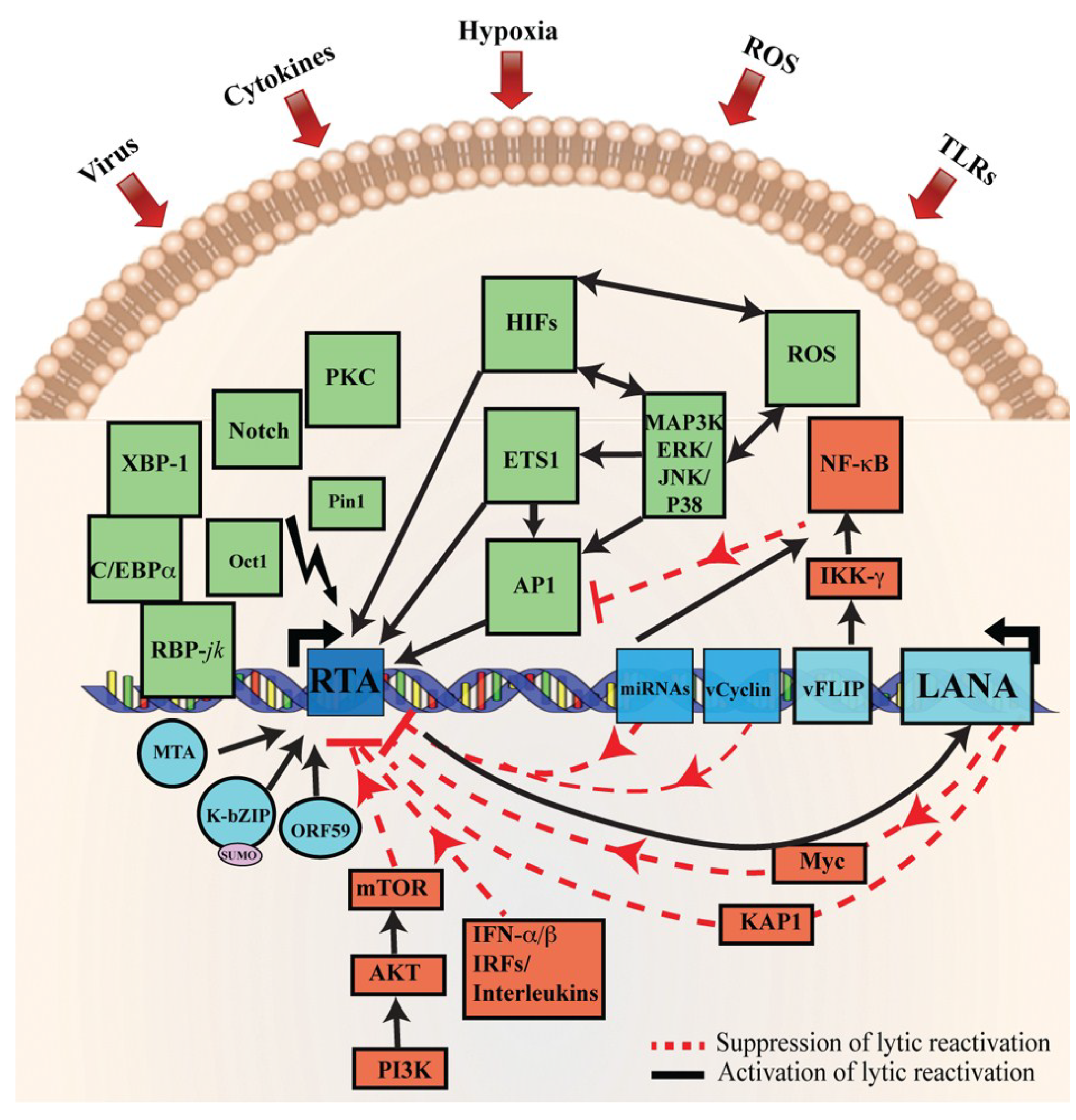
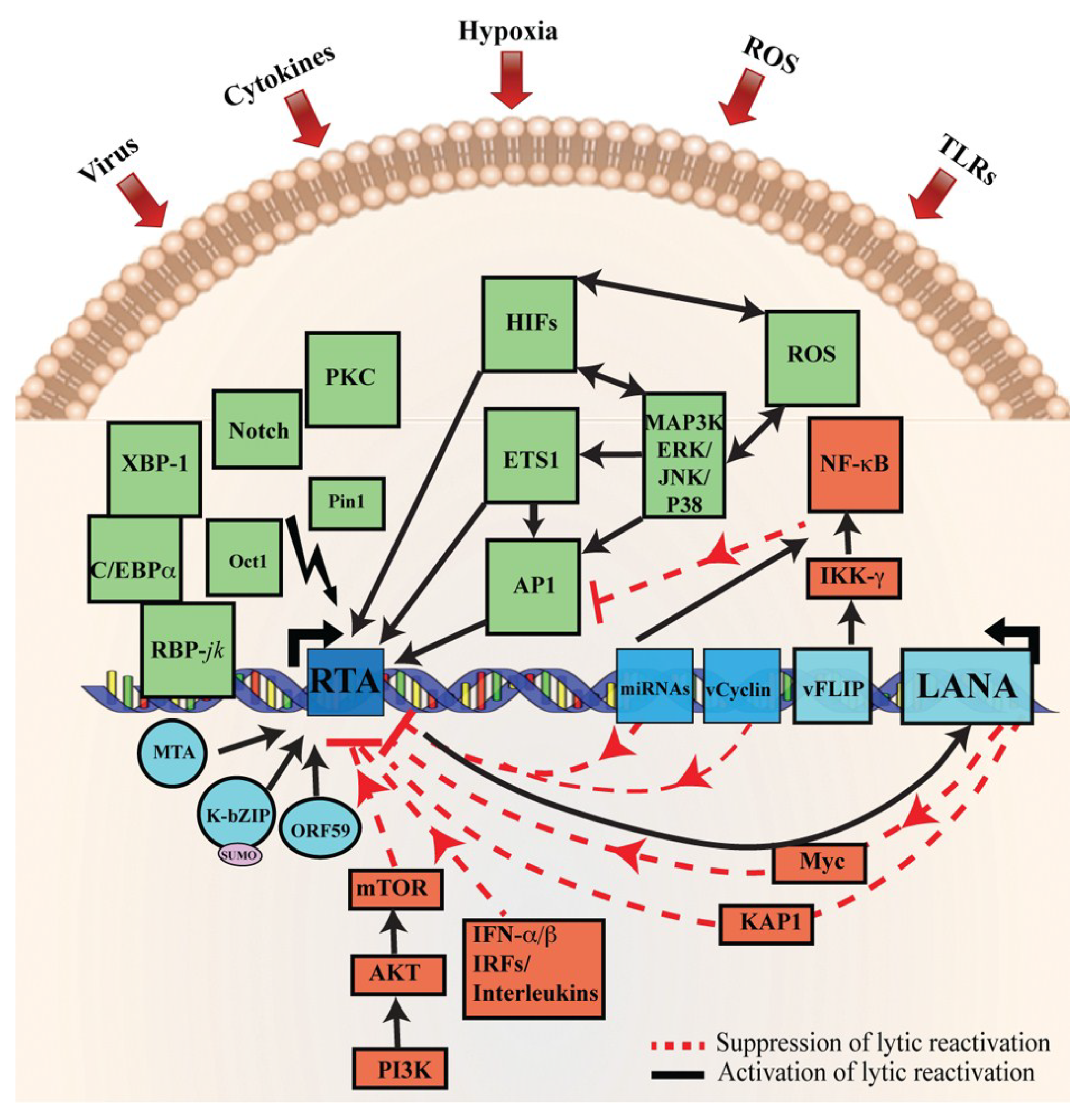
Several cellular signaling pathways are identified to be involved in the reactivation of KSHV from latency, such as PKCd [156], b-Raf/MEK/ERK [157], PKA [104], Notch and RBP-Jκ [95,158], p38 and JNK [159], Pim-1 and Pim-3 [160], PI3K and Akt [161], and TLR7/8 signaling [162]. Apart from these signaling pathways, a number of additional cellular factors also mediate KSHV reactivation [163,164,165,166,167] (Figure 2). It has been shown that intracellular calcium transport activates Ca++ dependent viral reactivation, and inhibition of calcineurin signaling, in turn, blocks KSHV reactivation [168]. Similarly, Protein kinase C delta (PKCdelta) plays a role in KSHV lytic replication [156]. Activation of the MEK/ERK, JNK, and p38 mitogen-activated protein kinase (MAPK) pathways play a central role during KSHV infection. Activation of the MAPK pathway, immediately after infection, enables the establishment of a successful KSHV infection [169,170]. Furthermore, MAPK pathways are induced during lytic reactivation [157,159,163]. Similarly, cellular MAP4K4 is also known to play a crucial role in inflammation, insulin resistance, and the invasiveness of several human malignancies [171,172]. Recently, it has been suggested that MAP4K4 act as a novel mediator of KSHV lytic reactivation from latency [172]. Similarly, yet another essential pathway mediating KSHV reactivation is the Raf/MEK/ERK/Ets-1 pathway [163]. Likewise, promoters of K-RTA, MTA, K-bZIP, and origins of lytic replication (oriLyt) have been shown to carry a functional DNA-binding site for AP-1 and are responsive to AP-1 activation [70,159,173]. During de novo infection, KSHV has been shown to induce MEK/ERK, JNK, and p38 MAPK pathways in human umbilical vascular endothelial cells (HUVEC). This, in turn, regulates AP-1 to facilitate its entry into the target cells and initiate a productive lytic replication at the early acute stage of infection [169,174]. Additionally, in latent KSHV-infected, cells these MAPK pathways modulate both spontaneous and TPA-induced KSHV reactivations and activate the expression of several transcription factors, such as AP-1 and Ets-1 [157,159,175].
KSHV lifecycle is also controlled by the viral protein, K-RTA by altering the Notch signaling pathway through binding with RBP-Jκ [95,101]. Additionally, Notch signaling and the expression of two Notch ligands (JAG1 and DLL4) are upregulated through KSHV genes, expressed during KSHV latent and lytic infection [176,177]. Similarly, Hypoxia-inducible factor (HIF) has been shown to induce numerous genes associated with angiogenesis and tumor growth, and the KSHV infected cells express elevated levels of HIF1α and HIF2α [177,178,179]. Furthermore, both LANA and vIRF3 have been shown to play roles in the stabilization of HIF1α via protein–protein interactions [61,178,180]. Secondary infections by other pathogens, such as HIV and bacteria, have been shown to trigger KSHV reactivation [162,181]. Similarly, short-chain fatty acids (SCFA) from periodontal pathogens suppress histone deacetylases HDAC1, EZH2, and SUV39H1 and downregulates the expression of silent information regulator-1 (SIRT1) to promote KSHV replication [182]. Cytokine-mediated JAK–STAT signaling also regulate various important biological processes, such as immune response, cell growth, and differentiation. KSHV infection has been shown to upregulate gp130 receptor expression, which leads to a constitutive phosphorylation of JAK2/STAT3 activation [183,184]. Further studies have revealed that both LANA and vGPCR play roles in the modulation of JAK2/STAT3 signaling to create angiogenic factors [185,186]. This is further confirmed by the LANA-mediated STAT6 phosphorylation through the inhibition of IL-4 for the maintenance of latency [187].
Figure 2.
Schematic representation of cellular signaling pathways involved in KSHV latency and reactivation. During latency, KSHV latent genes, including LANA, vFLIP, miRNA, and vCyclin activate and maintain various cytokine-mediated cell proliferation and angiogenesis pathways, such as JAK/STAT, PI3K/AKT/mTOR, cMyc, and NF-κB, to suppress KSHV lytic reactivation. The red line represents the inhibitory pathways involved in the maintenance of KSHV latency. Disruption of these signaling pathways by various stimuli, such as secondary infection by bacteria, viruses, hypoxia, inflammatory cytokines, and oxidative stress upregulate RTA expression resulting in KSHV reactivation. The solid black arrows represent signaling pathways that are activated during KSHV lytic reactivation. Moreover, RTA, as well as RTA-induced KSHV genes MTA and K-bZIP, have been shown to interact with XBP-1 and C/EBPα to modulate various cellular signaling pathways. Deregulation of these cellular signaling pathways, such as MAPK, PKCd, b-Raf/MEK/ERK, PKA, Notch, RBP-Jκ, JNK, Pim-1/Pim-3, and TLR7/8 signaling by RTA lead to the reactivation of latently infected KSHV cells to lytic replication. This figure is adopted and modified from a previous review [59].
Figure 2.
Schematic representation of cellular signaling pathways involved in KSHV latency and reactivation. During latency, KSHV latent genes, including LANA, vFLIP, miRNA, and vCyclin activate and maintain various cytokine-mediated cell proliferation and angiogenesis pathways, such as JAK/STAT, PI3K/AKT/mTOR, cMyc, and NF-κB, to suppress KSHV lytic reactivation. The red line represents the inhibitory pathways involved in the maintenance of KSHV latency. Disruption of these signaling pathways by various stimuli, such as secondary infection by bacteria, viruses, hypoxia, inflammatory cytokines, and oxidative stress upregulate RTA expression resulting in KSHV reactivation. The solid black arrows represent signaling pathways that are activated during KSHV lytic reactivation. Moreover, RTA, as well as RTA-induced KSHV genes MTA and K-bZIP, have been shown to interact with XBP-1 and C/EBPα to modulate various cellular signaling pathways. Deregulation of these cellular signaling pathways, such as MAPK, PKCd, b-Raf/MEK/ERK, PKA, Notch, RBP-Jκ, JNK, Pim-1/Pim-3, and TLR7/8 signaling by RTA lead to the reactivation of latently infected KSHV cells to lytic replication. This figure is adopted and modified from a previous review [59].
KSHV has evolved multiple mechanisms to manipulate cellular anti-apoptotic and survival pathways and disruption of these pathways reactivates KSHV [188,189,190]. Apart from AP-1, NF-κB also antagonizes RBP-Jκ to impair the expression and transactivation function of RTA [190]. Furthermore, inhibition of NF-κB pathway in latently infected cells disrupts viral latency and activates viral lytic replication [191]. However, the available data suggest that the role of the NF-κB pathway in the KSHV life cycle is context dependent [192]. It is very likely that the balance of AP-1 and NF-κB pathways decide the fate of virus replication status in a particular cell type [174,193,194]. Consistent with these findings, a recent study showed that inhibition of the pro-survival PI3K-Akt pathway favors KSHV reactivation from latency [195]. Furthermore, inhibition of the Akt pathway reactivates KSHV from latency by increasing the RTA expression [195]. KSHV encoded proteins are also known to modulate the cellular phosphatidyl inositol-3-kinase (PI3K)/AKT/mammalian target of the rapamycin (mTOR) signaling pathway to control cell proliferation. Cellular PI3K/AKT/mTOR signaling is a common to many growth factors and cytokine receptors [196]. However, thus far, only a few KSHV proteins have been shown to regulate PI3K/AKT/mTOR signaling, which include K1 [197,198], (vGPCR) [199,200], vIL-6 [183,201], and ORF45 [170,202].
5. Lytic Proteins in Controlling Immune Regulation and Pathogenesis
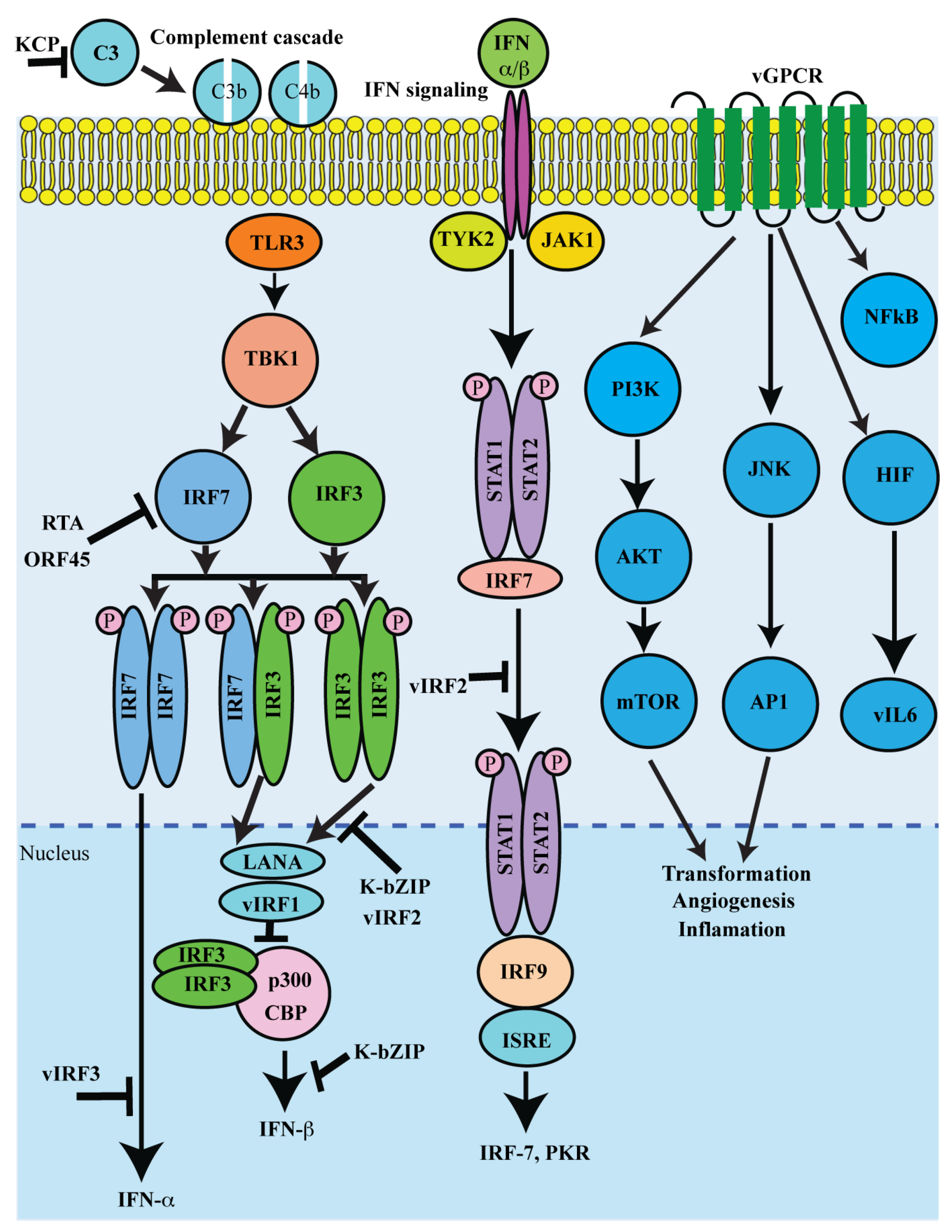
Lytic reactivation results in an expression of several KSHV lytic proteins (Table 1). Many of the proteins encoded by KSHV lytic genes also have pro-growth or transforming abilities. Major functions of KSHV lytic proteins include cellular proliferation and evading the host’s immune response. The immune functions targeted by viral proteins include IFN production, interferon regulatory factor (IRF) activation, complement activation, inflammasome, and chemokine activation (Figure 3).

{kind=link}
{kind=link}
{kind=link}
| KSHV genes | KSHV proteins | Function | References |
|---|---|---|---|
| K1 | Variable ITAM-Containing Protein (VIP) | Type I transmembrane signaling protein containing a functional immunoreceptor tyrosine-based activation motif. Regulate membrane transport in B cells. | [203] |
| K2 | Viral Interleukin-6 (vIL-6) | Homologues of cellular IL-6. Activate JAK/STAT, MAPK, and PI3K/Akt signaling pathways to regulate B-cell proliferation. | [51,204] |
| K3/K5 | Modulator of immune recognition (MIR1/MIR2) | Viral E3 ligases capable of ubiquitinating MHC-I, ICAM-1, B7-2, Tetherin (CD317/BST2), DC-SIGN, and DC-SIGNR. | [205,206] |
| K4/K4.1/K6 | Viral CC-Chemokine Ligands (vCCLs) | Homologues of cellular chemokines: viral CC-chemokine ligand 1 vCCL1 (vMIP1), vCCL2 (vMIP2), and vCCL3 (vMIP3), respectively. Blocks signaling through chemokine receptors. | [207,208] |
| K7 | Viral Inhibitor of Apoptosis (vIAP) | Interact with cellular proteins PLIC1, caspase 3/Bcl-2, CAML, Vps34, and promote cell survival during lytic replication. | [209,210] |
| K9/K10/K11 | KSHV interferon regulatory factors (vIRF-1, vIRF-2, vIRF-3 and vIRF-4) | Homologues of cellular interferon: Inhibitor of IFN1, p53, NFκB RelA, and p300. | [211,212] |
| K14 | vOX2 or vCD200 | Homologues of cellular OX2. A negative regulator of inflammatory signaling and surface glycoproteins. | [213,214] |
| K15 | Viral membrane protein | Regulation of cellular signaling to induce various pro-survival and paracrine-mediated pro- angiogenic cellular cytokines and chemokines, including IL6, IL8, IL-1a/b, CXCL3, and Cox2. | [215,216] |
| ORF4 | KSHV complement Control protein (KCP) | Homologue to cellular RCA. Regulate complement activation by increasing the decay of the classical C3 convertase. | [217,218,219] |
| ORF45 | ORF45 | Inhibit type1 IFN induction by sequestering the cellular interferon regulatory factor-7 to cytoplasm. | [220,221] |
| ORF63 | ORF63 | Homologue to cellular inflammasome complex NLRP1. | [222] |
| ORF64 | Viral deubiquitinase | A non specific deubiquitinase, shown to deubiquitinate RIG-I to suppress RIG-I-mediated activation of the IFNb. | [223] |
| ORF74 | Viral G-protein-coupled receptor (vGPCR) | Homologue of cellular IL-8 receptor. vGPCR induce secretion of proinflammatory cytokines and angiogenic growth factors. | [200,224] |
| ORF75 | ORF75 | A viral effector for the degradation of ND10 proteins. | [225,226] |
| PAN RNA | Polyadenylated Nuclear RNA | Modulator of viral gene expression. | [227,228,229,230] |
KSHV employs diverse mechanisms for controlling both IFN production and signaling as IFN is a potent antiviral defense that is critical for KSHV persistence [231]. The genomic region encompassing ORFs K9 to K11 encodes KSHV vIRFs 1-4 [232]. vIRF1 can bind to and disrupt the transcriptional activities of IRF1, IRF3, and IRF7 [211,212]. Additionally, vIRFs 1, 3, and 4 have been shown to inhibit p53 activity via, either direct binding to the tumor suppressor (vIRF-1 and vIRF-3), or through association with ATM kinase or via stabilization of MDM2, which induces ubiquitination and proteasomal degradation of p53 [233,234]. Similarly, KSHV viral interleukin-6 (vIL6), encoded by ORF K2, shares many functional characteristics with human IL6 and, as a result, the viral cytokine can activate gp130 and downstream signaling pathways, including the JAK/STAT, MAPK, and PI3K/Akt pathways [204,235]. These pathways regulate a variety of transcription factors and response elements (RE), such as the STAT1/3 and STAT5 IL6 RE, C/EBP, and c-jun promoter IL6 RE (JRE-IL-6) [236]. A viral homologue of the cellular angiogenic IL-8 receptor [224], vGPCR has been shown to activate a number of crucial signaling pathways, including PLC, PKC, MAPK, PI3K/Akt/mTOR, and NFκB [237]. Downstream signaling from these pathways activates the AP1, NFAT, NF-kB, HIF-1a, and CREB transcription factors, which, in turn, contribute to vGPCR-mediated production of pro-inflammatory cytokines and chemokines [237].
Figure 3.
Schematic representation of lytic proteins in immune regulation and pathogenesis: The major immune functions targeted by viral lytic proteins include IFN production, interferon regulatory factor (IRF) activation, complement activation, inflammasome and chemokine activation. Regulating both IFN production and signaling is a potent antiviral defense, vIRF can bind and disrupt the transcriptional activities of IRF1, IRF3, and IRF7. Additionally, vGPCR is a constitutively active homologue of the IL8 receptor. vGPCR activates various cell signaling pathways and transcription factors to enhance the production of pro-inflammatory chemokines and cytokines, such as vIL-6. Furthermore, KSHV-encoded KCP regulates complement by increasing the decay of the classical C3 convertase.
Figure 3.
Schematic representation of lytic proteins in immune regulation and pathogenesis: The major immune functions targeted by viral lytic proteins include IFN production, interferon regulatory factor (IRF) activation, complement activation, inflammasome and chemokine activation. Regulating both IFN production and signaling is a potent antiviral defense, vIRF can bind and disrupt the transcriptional activities of IRF1, IRF3, and IRF7. Additionally, vGPCR is a constitutively active homologue of the IL8 receptor. vGPCR activates various cell signaling pathways and transcription factors to enhance the production of pro-inflammatory chemokines and cytokines, such as vIL-6. Furthermore, KSHV-encoded KCP regulates complement by increasing the decay of the classical C3 convertase.
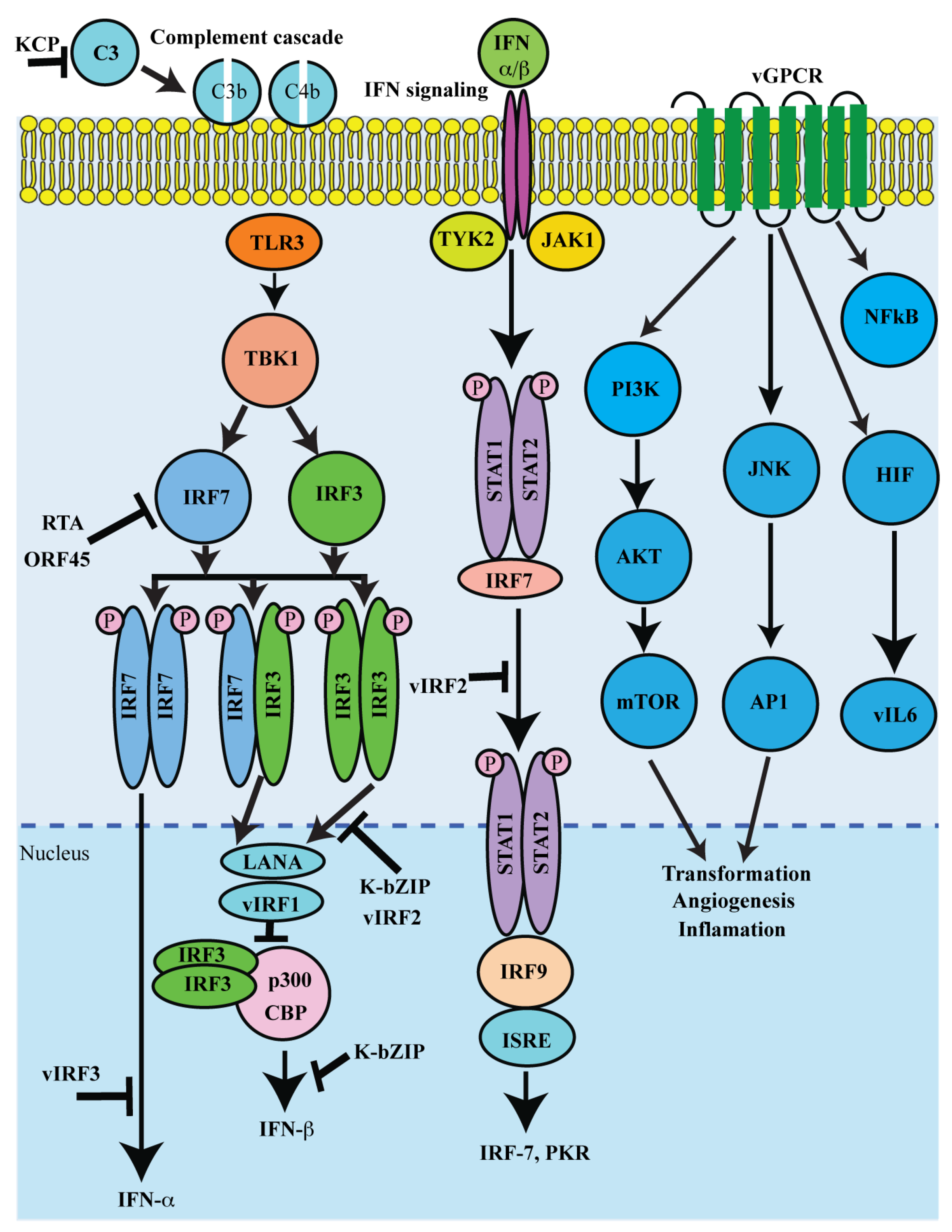
Many of the KSHV K-gene encoded lytic proteins have also been shown to modulate KSHV infection and pathogenesis. Multifunctional transmembrane glycoprotein K1 encoded by the first ORF of KSHV can constitutively activate multiple pro-growth signaling pathways in KSHV-infected cells. [238]. Oligomerization of K1 trigger auto-phosphorylation of ITAM and activate various Src homology 2 (SH2) containing signaling proteins, including PI3K (p85)/Akt, PLCg, Vav, Syk, Lyn, RasGAP, and Grb2 [239,240]. Similarly, it has been shown that K15-activated cellular signaling pathways induce the transcription of a number of cellular cytokines and chemokines, including IL6, IL8, CCL20, CCL2, CXCL3, IL-1a/b, and Cox2 [215,216]. Additionally, KSHV K7 or viral inhibitor of apoptosis (vIAP), is a homologue of cellular Bcl-2 proteins and contains a putative mitochondrial-targeting signal and localizes to mitochondria and ER [210,241]. It has been reported that K7/vIAP inhibits caspase 3 activity by interacting with cellular Bcl-2 via its BIR (baculovirus IAP repeat) [210]. Furthermore, KSHV K3 and K5 (also called modulator of the immune recognition (MIR) 1 and 2, respectively) are viral E3 ligases capable of ubiquitinating the MHC-I cytoplasmic tail to trigger internalization and proteasomal degradation of the MHC-I complex [205,242,243]. K3 and K5 proteins also have been shown to downregulate both C-type lectins, DC-SIGN, and DC-SIGNR by ubiquitin mediated degradation [206]. Similarly, KSHV K6, K4, and K4.1 encode three homologues of cellular chemokines: viral CC-chemokine ligand 1 vCCL1(vMIP1), vCCL2 (vMIP2), and vCCL3 (vMIP3), respectively [154,244,245]. Apart from immune evasion properties, v-chemokines also have been shown to promote angiogenesis through the induction of VEGF [246,247]. KSHV-encoded early lytic protein K14 is another negative regulator of inflammatory signaling and surface glycoprotein (vOX2). K14 shows significant homology with OX2 or CD200, a member of the immunoglobulin superfamily that is broadly distributed on the cell surface [90]. vCD200 promotes the secretion of proinflammatory cytokines on stimulation of monocytes, macrophages, and DCs through a direct interaction with cellular CD200R, inhibiting myeloid cell activation and reducing Th1-cell-associated cytokine production [214,248].
Furthermore, KSHV-ORF4-encoded inhibitor of the complement system, designated as KSHV complement Control Protein (KCP) [217,249,250], regulates complement by increasing the decay of the classical C3 convertase and acting as cofactors for the inactivation of C3b and C4b, components of the C3 and C5 convertases [251,252]. Similarly, KSHV encoded ORF45, an immediate early gene product, plays a crucial role in lytic replication [253]. ORF45 has been shown to inhibit type1 IFN induction upon infection by sequestering the cellular interferon regulatory factor-7 (IRF-7) to the cytoplasm [220,221]. It has been shown that ORF45 can also regulate eIF4B phosphorylation in an mTOR and MAPK independent manner. Additionally, the ORF45 protein is also involved in the transport of the viral capsid-tegument complexes along the microtubule filaments [254].
It has been also been shown that KSHV encoded tegument protein, ORF75 is an essential protein as a new viral effector [255] for the degradation of ND10 proteins, thereby regulating lytic replication and KSHV infection [225]. In addition, the ORF75 also has been shown to induce the degradation of ATRX and relocalization of Daxx, as well as be involved in NF-kB coactivation with KSHV K13/vFLIP [225,256]. Similarly, KSHV encoded ORF64 is a deubiquitinase that non-specifically targets K48 or K63 ubiquitination. It has been shown that KSHV ORF64 is capable of deubiquitinating RIG-I to suppress RIG-I-mediated activation of the IFNb promoter [223]. Studies showed that KSHV ORF63 has homology to parts of cellular inflammasome complex NLRP1 [222,257]. This ORF63 function seems to be critical for supporting viral gene expression and genome replication, as well as suppressing IL-1b production [222,258]. Additionally, KSHV encoded structural PAN RNA has been also shown as a multifunctional transcript that can globally control viral and cellular gene expression during lytic reactivation [259] through direct interaction with chromatin modifying complexes, such as components of PRC2 [228,229]. PAN RNA interacts with demethylases, UTX, and JMJD3 to modify the suppressive H3K27me3 mark within the KSHV genome [260]. Moreover, PAN RNA expression decreased the expression of interferon γ, interleukin 18, interferon α16, and RNase L [229].
6. Conclusions
Kaposi’s sarcoma associated herpesvirus (KSHV) modulates various cellular pathways by which it is able to establish and maintain persistent infection in the host to initiate tumorigenesis. Several of these latent viral and lytic proteins are known to transform host cells, linking KSHV with the development of severe human malignancies. These virus-induced cancers pose a large threat to global public health, specifically in areas that are still struggling with malignancies associated with HIV-AIDS with limited treatment options. Over the years, tremendous progress has been made in elucidating the molecular mechanisms of KSHV latency and lytic replication. Nonetheless, there are still vast aspects of viral infection and transformation that are not well explored. With the help of rapid advancements in modern technology, it is presumed that a thorough knowledge of the KSHV life cycle will be achieved over the next few years. Further understanding of the unique mechanisms that KSHV adopts for the establishment of successful lifetime persistence in the infected host will eventually pave the way for novel therapeutic approaches for the treatment of KSHV diseases.
Acknowledgments
We thank the Verma Lab members for all the constructive comments and helpful discussions. This work was supported by public health grants from the National Institute of Health (grant numbers CA174459 and AI105000) and the Research Scholar Grant (124389-RSG-13-230-01-MPC) from the American Cancer Society.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sturzl, M.; Zietz, C.; Monini, P.; Ensoli, B. Human herpesvirus-8 and Kaposi’s sarcoma: Relationship with the multistep concept of tumorigenesis. Adv. Cancer Res. 2001, 81, 125–159. [Google Scholar] [PubMed]
- Cesarman, E.; Chang, Y.; Moore, P.S.; Said, J.W.; Knowles, D.M. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 1995, 332, 1186–1191. [Google Scholar] [CrossRef] [PubMed]
- Soulier, J.; Grollet, L.; Oksenhendler, E.; Cacoub, P.; Cazals-Hatem, D.; Babinet, P.; d’Agay, M.F.; Clauvel, J.P.; Raphael, M.; Degos, L.; et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood 1995, 86, 1276–1280. [Google Scholar]
- Uldrick, T.S.; Wang, V.; O'Mahony, D.; Aleman, K.; Wyvill, K.M.; Marshall, V.; Steinberg, S.M.; Pittaluga, S.; Maric, I.; Whitby, D.; et al. An interleukin-6-related systemic inflammatory syndrome in patients co-infected with Kaposi sarcoma-associated herpesvirus and HIV but without Multicentric Castleman disease. Clin. Infect. Dis. 2010, 51, 350–358. [Google Scholar] [CrossRef]
- Dittmer, D.; Stoddart, C.; Renne, R.; Linquist-Stepps, V.; Moreno, M.E.; Bare, C.; McCune, J.M.; Ganem, D. Experimental transmission of Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8) to SCID-hu Thy/Liv mice. J. Exp. Med. 1999, 190, 1857–1868. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.C.; Robertson, E.S. Molecular biology and pathogenesis of Kaposi sarcoma-associated herpesvirus. FEMS Microbiol. Lett. 2003, 222, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Veettil, M.V.; Bandyopadhyay, C.; Dutta, D.; Chandran, B. Interaction of KSHV with host cell surface receptors and cell entry. Viruses 2014, 6, 4024–4046. [Google Scholar] [CrossRef] [PubMed]
- Knipe, D.M.; Lieberman, P.M.; Jung, J.U.; McBride, A.A.; Morris, K.V.; Ott, M.; Margolis, D.; Nieto, A.; Nevels, M.; Parks, R.J.; et al. Snapshots: Chromatin control of viral infection. Virology 2013, 435, 141–156. [Google Scholar] [CrossRef]
- Dourmishev, L.A.; Dourmishev, A.L.; Palmeri, D.; Schwartz, R.A.; Lukac, D.M. Molecular genetics of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus-8) epidemiology and pathogenesis. Microbiol. Mol. Biol. Rev. 2003, 67, 175–212. [Google Scholar] [CrossRef] [PubMed]
- Mesri, E.A.; Cesarman, E.; Boshoff, C. Kaposi’s sarcoma and its associated herpesvirus. Nat. Rev. Cancer 2010, 10, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Ballestas, M.E.; Kaye, K.M. The latency-associated nuclear antigen, a multifunctional protein central to Kaposi’s sarcoma-associated herpesvirus latency. Future Microbiol. 2011, 6, 1399–1413. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Lagunoff, M. Establishment and maintenance of Kaposi’s sarcoma-associated herpesvirus latency in B cells. J. Virol. 2005, 79, 14383–14391. [Google Scholar] [CrossRef] [PubMed]
- Renne, R.; Zhong, W.; Herndier, B.; McGrath, M.; Abbey, N.; Kedes, D.; Ganem, D. Lytic growth of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat. Med. 1996, 2, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Lin, S.F.; Staskus, K.; Gradoville, L.; Grogan, E.; Haase, A.; Miller, G. Kinetics of Kaposi’s sarcoma-associated herpesvirus gene expression. J. Virol. 1999, 73, 2232–2242. [Google Scholar] [PubMed]
- Jenner, R.G.; Alba, M.M.; Boshoff, C.; Kellam, P. Kaposi’s sarcoma-associated herpesvirus latent and lytic gene expression as revealed by DNA arrays. J. Virol. 2001, 75, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Verma, S.C.; Lu, J.; Robertson, E.S. Molecular biology of Kaposi’s sarcoma-associated herpesvirus and related oncogenesis. Adv. Virus Res. 2010, 78, 87–142. [Google Scholar] [PubMed]
- Wang, S.E.; Wu, F.Y.; Yu, Y.; Hayward, G.S. CCAAT/enhancer-binding protein-α is induced during the early stages of Kaposi’s sarcoma-associated herpesvirus (KSHV) lytic cycle reactivation and together with the KSHV replication and transcription activator (RTA) cooperatively stimulates the viral RTA, MTA, and PAN promoters. J. Virol. 2003, 77, 9590–9612. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Young, A.; Sun, R. Auto-activation of the rta gene of human herpesvirus-8/Kaposi’s sarcoma-associated herpesvirus. J. Gen. Virol. 2000, 81, 3043–3048. [Google Scholar] [PubMed]
- Song, M.J.; Deng, H.; Sun, R. Comparative study of regulation of RTA-responsive genes in Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8. J. Virol. 2003, 77, 9451–9462. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, Q.; Maul, G.G.; Yuan, Y. Kaposi’s sarcoma-associated herpesvirus ori-Lyt-dependent DNA replication: Dual role of replication and transcription activator. J. Virol. 2006, 80, 12171–12186. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.L.; Li, H.; Wang, Y.; Zhu, F.X.; Kudchodkar, S.; Yuan, Y. Kaposi’s sarcoma-associated herpesvirus lytic origin (ori-Lyt)-dependent DNA replication: Identification of the ori-Lyt and association of K8 bZip protein with the origin. J. Virol. 2003, 77, 5578–5588. [Google Scholar] [CrossRef] [PubMed]
- AuCoin, D.P.; Colletti, K.S.; Xu, Y.; Cei, S.A.; Pari, G.S. Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) contains two functional lytic origins of DNA replication. J. Virol. 2002, 76, 7890–7896. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, H.H.; Naranatt, P.P.; Smith, M.S.; Zeng, L.; Bloomer, C.; Chandran, B. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi’s sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J. Virol. 2004, 78, 3601–3620. [Google Scholar] [CrossRef] [PubMed]
- Toth, Z.; Brulois, K.; Lee, H.R.; Izumiya, Y.; Tepper, C.; Kung, H.J.; Jung, J.U. Biphasic euchromatin-to-heterochromatin transition on the KSHV genome following de novo infection. PLoS Pathog. 2013, 9, e1003813. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Kuppers, D.A.; Verma, S.C.; Sharma, N.; Murakami, M.; Robertson, E.S. Induction of Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen by the lytic transactivator RTA: A novel mechanism for establishment of latency. J. Virol. 2005, 79, 7453–7465. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhou, F.; Ye, F.; Gao, S.J. Genetic disruption of KSHV major latent nuclear antigen LANA enhances viral lytic transcriptional program. Virology 2008, 379, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Day, L.; Gao, S.J.; Lieberman, P.M. Acetylation of the latency-associated nuclear antigen regulates repression of Kaposi’s sarcoma-associated herpesvirus lytic transcription. J. Virol. 2006, 80, 5273–5282. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Kuppers, D.A.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus reactivation is regulated by interaction of latency-associated nuclear antigen with recombination signal sequence-binding protein Jκ, the major downstream effector of the Notch signaling pathway. J. Virol. 2005, 79, 3468–3478. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ueda, K.; Sakakibara, S.; Okuno, T.; Parravicini, C.; Corbellino, M.; Yamanishi, K. Activation of latent Kaposi’s sarcoma-associated herpesvirus by demethylation of the promoter of the lytic transactivator. Proc. Natl. Acad. Sci. USA 2001, 98, 4119–4124. [Google Scholar] [CrossRef] [PubMed]
- Shamay, M.; Krithivas, A.; Zhang, J.; Hayward, S.D. Recruitment of the de novo DNA methyltransferase Dnmt3a by Kaposi’s sarcoma-associated herpesvirus LANA. Proc. Natl. Acad. Sci. USA 2006, 103, 14554–14559. [Google Scholar] [CrossRef] [PubMed]
- Gunther, T.; Grundhoff, A. The epigenetic landscape of latent Kaposi sarcoma-associated herpesvirus genomes. PLoS Pathog. 2010, 6, e1000935. [Google Scholar] [CrossRef] [PubMed]
- Woodard, C.; Shamay, M.; Liao, G.; Zhu, J.; Ng, A.N.; Li, R.; Newman, R.; Rho, H.S.; Hu, J.; Wan, J.; et al. Phosphorylation of the chromatin binding domain of KSHV LANA. PLoS Pathog. 2012, 8, e1002972. [Google Scholar] [CrossRef]
- Chang, P.C.; Cheng, C.Y.; Campbell, M.; Yang, Y.C.; Hsu, H.W.; Chang, T.Y.; Chu, C.H.; Lee, Y.W.; Hung, C.L.; Lai, S.M.; et al. The chromatin modification by SUMO-2/3 but not SUMO-1 prevents the epigenetic activation of key immune-related genes during Kaposi’s sarcoma associated herpesvirus reactivation. BMC Genomics 2013, 14, 824. [Google Scholar] [CrossRef]
- Chang, P.C.; Izumiya, Y.; Wu, C.Y.; Fitzgerald, L.D.; Campbell, M.; Ellison, T.J.; Lam, K.S.; Luciw, P.A.; Kung, H.J. Kaposi’s sarcoma-associated herpesvirus (KSHV) encodes a SUMO E3 ligase that is SIM-dependent and SUMO-2/3-specific. J. Biol. Chem. 2010, 285, 5266–5273. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, B.G.; Verma, S.C.; Lan, K.; Cotter, M.A.; Woodman, Z.L.; Robertson, E.S. KSHV encoded LANA upregulates Pim-1 and is a substrate for its kinase activity. Virology 2006, 351, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.; Lim, C.; Lee, J.Y.; Song, Y.J.; Park, J.; Choe, J.; Seo, T. DNA-PK/Ku complex binds to latency-associated nuclear antigen and negatively regulates Kaposi’s sarcoma-associated herpesvirus latent replication. Biochem. Biophys. Res. Commun. 2010, 394, 934–939. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Martin, H.; Shamay, M.; Woodard, C.; Tang, Q.Q.; Hayward, S.D. Kaposi’s sarcoma-associated herpesvirus LANA protein downregulates nuclear glycogen synthase kinase 3 activity and consequently blocks differentiation. J. Virol. 2007, 81, 4722–4731. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.; Chang, P.C.; Huerta, S.; Izumiya, C.; Davis, R.; Tepper, C.G.; Kim, K.Y.; Shevchenko, B.; Wang, D.H.; Jung, J.U.; et al. Protein arginine methyltransferase 1-directed methylation of Kaposi sarcoma-associated herpesvirus latency-associated nuclear antigen. J. Biol. Chem. 2012, 287, 5806–5818. [Google Scholar]
- Campbell, M.; Izumiya, Y. Post-Translational Modifications of Kaposi’s Sarcoma-Associated Herpesvirus Regulatory Proteins—SUMO and KSHV. Front. Microbiol. 2012, 3, 31. [Google Scholar] [PubMed]
- Toth, Z.; Maglinte, D.T.; Lee, S.H.; Lee, H.R.; Wong, L.Y.; Brulois, K.F.; Lee, S.; Buckley, J.D.; Laird, P.W.; Marquez, V.E.; et al. Epigenetic analysis of KSHV latent and lytic genomes. PLoS Pathog. 2010, 6, e1001013. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.; Lee, D.; Seo, T.; Choi, C.; Choe, J. Latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus functionally interacts with heterochromatin protein 1. J. Biol. Chem. 2003, 278, 7397–7405. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, S.; Persson, L.M.; Wong, L.; Wilson, A.C. The latency-associated nuclear antigen interacts with MeCP2 and nucleosomes through separate domains. J. Virol. 2010, 84, 2318–2330. [Google Scholar] [CrossRef] [PubMed]
- Krithivas, A.; Young, D.B.; Liao, G.; Greene, D.; Hayward, S.D. Human herpesvirus 8 LANA interacts with proteins of the mSin3 corepressor complex and negatively regulates Epstein-Barr virus gene expression in dually infected PEL cells. J. Virol. 2000, 74, 9637–9645. [Google Scholar] [CrossRef] [PubMed]
- Stuber, G.; Mattsson, K.; Flaberg, E.; Kati, E.; Markasz, L.; Sheldon, J.A.; Klein, G.; Schulz, T.F.; Szekely, L. HHV-8 encoded LANA-1 alters the higher organization of the cell nucleus. Mol. Cancer 2007, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.Y.; Huerta, S.B.; Izumiya, C.; Wang, D.H.; Martinez, A.; Shevchenko, B.; Kung, H.J.; Campbell, M.; Izumiya, Y. Kaposi’s sarcoma-associated herpesvirus (KSHV) latency-associated nuclear antigen regulates the KSHV epigenome by association with the histone demethylase KDM3A. J. Virol. 2013, 87, 6782–6793. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Yang, Y.; Turner, P.C.; Jain, V.; McIntyre, L.M.; Renne, R. LANA binds to multiple active viral and cellular promoters and associates with the H3K4methyltransferase hSET1 complex. PLoS Pathog. 2014, 10, e1004240. [Google Scholar] [CrossRef] [PubMed]
- Guito, J.; Lukac, D.M. KSHV Rta Promoter Specification and Viral Reactivation. Front. Microbiol. 2012, 3, 30. [Google Scholar] [CrossRef] [PubMed]
- Jones, T.; Ye, F.; Bedolla, R.; Huang, Y.; Meng, J.; Qian, L.; Pan, H.; Zhou, F.; Moody, R.; Wagner, B.; et al. Direct and efficient cellular transformation of primary rat mesenchymal precursor cells by KSHV. J. Clin. Investig. 2012, 122, 1076–1081. [Google Scholar] [CrossRef] [PubMed]
- Vieira, J.; O’Hearn, P.; Kimball, L.; Chandran, B.; Corey, L. Activation of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) lytic replication by human cytomegalovirus. J. Virol. 2001, 75, 1378–1386. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Xue, M.; Qin, D.; Zhu, X.; Wang, C.; Zhu, J.; Hao, T.; Cheng, L.; Chen, X.; Bai, Z.; et al. HIV-1 Tat promotes Kaposi’s sarcoma-associated herpesvirus (KSHV) vIL-6-induced angiogenesis and tumorigenesis by regulating PI3K/PTEN/AKT/GSK-3beta signaling pathway. PLoS One 2013, 8, e53145. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Guo, Y.; Yao, S.; Yan, Q.; Xue, M.; Hao, T.; Zhou, F.; Zhu, J.; Qin, D.; Lu, C. Synergy between Kaposi’s sarcoma-associated herpesvirus (KSHV) vIL-6 and HIV-1 Nef protein in promotion of angiogenesis and oncogenesis: Role of the AKT signaling pathway. Oncogene 2014, 33, 1986–1996. [Google Scholar] [CrossRef] [PubMed]
- Roupelieva, M.; Griffiths, S.J.; Kremmer, E.; Meisterernst, M.; Viejo-Borbolla, A.; Schulz, T.; Haas, J. Kaposi’s sarcoma-associated herpesvirus Lana-1 is a major activator of the serum response element and mitogen-activated protein kinase pathways via interactions with the Mediator complex. J. Gen. Virol. 2010, 91, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Qin, D.; Lv, Z.; Zhu, X.; Ma, X.; Yan, Q.; Zeng, Y.; Guo, Y.; Feng, N.; Lu, C. Herpes simplex virus type 2 triggers reactivation of Kaposi’s sarcoma-associated herpesvirus from latency and collaborates with HIV-1 Tat. PLoS One 2012, 7, e31652. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Zeng, Y.; Huang, Z.; Huang, L.; Qian, C.; Tang, G.; Qin, D. Human herpesvirus 6 activates lytic cycle replication of Kaposi’s sarcoma-associated herpesvirus. Am. J. Pathol. 2005, 166, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Blauvelt, A. Skin diseases associated with human herpesvirus 6, 7, and 8 infection. J. Investig. Dermatol. Symp. Proc. 2001, 6, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Rady, P.L.; Yen, A.; Rollefson, J.L.; Orengo, I.; Bruce, S.; Hughes, T.K.; Tyring, S.K. Herpesvirus-like DNA sequences in non-Kaposi’s sarcoma skin lesions of transplant patients. Lancet 1995, 345, 1339–1340. [Google Scholar] [CrossRef] [PubMed]
- Mercader, M.; Taddeo, B.; Panella, J.R.; Chandran, B.; Nickoloff, B.J.; Foreman, K.E. Induction of HHV-8 lytic cycle replication by inflammatory cytokines produced by HIV-1-infected T cells. Am. J. Pathol. 2000, 156, 1961–1971. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Lei, X.; Gao, S.J. Mechanisms of Kaposi’s Sarcoma-Associated Herpesvirus Latency and Reactivation. Adv. Virol. 2011, 2011. [Google Scholar] [CrossRef]
- Romero-Ramirez, L.; Cao, H.; Nelson, D.; Hammond, E.; Lee, A.H.; Yoshida, H.; Mori, K.; Glimcher, L.H.; Denko, N.C.; Giaccia, A.J.; et al. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. 2004, 64, 5943–5947. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Lan, K.; Verma, S.C.; Si, H.; Lin, D.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus latent protein LANA interacts with HIF-1α to upregulate RTA expression during hypoxia: Latency control under low oxygen conditions. J. Virol. 2006, 80, 7965–7975. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Cai, S.; Zhu, C.; Verma, S.C.; Choi, J.Y.; Robertson, E.S. A unique SUMO-2-interacting motif within LANA is essential for KSHV latency. PLoS Pathog. 2013, 9, e1003750. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhu, C.; Guo, Y.; Wei, F.; Lu, J.; Qin, J.; Banerjee, S.; Wang, J.; Shang, H.; Verma, S.C.; et al. Inhibition of KAP1 enhances hypoxia-induced Kaposi’s sarcoma-associated herpesvirus reactivation through RBP-Jκ. J. Virol. 2014, 88, 6873–6884. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Zhou, F.; Bedolla, R.G.; Jones, T.; Lei, X.; Kang, T.; Guadalupe, M.; Gao, S.J. Reactive oxygen species hydrogen peroxide mediates Kaposi’s sarcoma-associated herpesvirus reactivation from latency. PLoS Pathog. 2011, 7, e1002054. [Google Scholar] [CrossRef] [PubMed]
- Ruffels, J.; Griffin, M.; Dickenson, J.M. Activation of ERK1/2, JNK and PKB by hydrogen peroxide in human SH-SY5Y neuroblastoma cells: Role of ERK1/2 in H2O2-induced cell death. Eur. J. Pharmacol. 2004, 483, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Feng, J.; Sun, R. Oxidative stress induces reactivation of Kaposi’s sarcoma-associated herpesvirus and death of primary effusion lymphoma cells. J. Virol. 2011, 85, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.J.; DeCotiis, J.; Giron, M.; Palmeri, D.; Lukac, D.M. Histone deacetylase classes I and II regulate Kaposi’s sarcoma-associated herpesvirus reactivation. J. Virol. 2014, 88, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; He, M.; Zhou, F.; Ye, F.; Gao, S.J. Activation of Kaposi’s sarcoma-associated herpesvirus (KSHV) by inhibitors of class III histone deacetylases: Identification of sirtuin 1 as a regulator of the KSHV life cycle. J. Virol. 2014, 88, 6355–6367. [Google Scholar] [CrossRef] [PubMed]
- Dyson, O.F.; Walker, L.R.; Whitehouse, A.; Cook, P.P.; Akula, S.M. Resveratrol inhibits KSHV reactivation by lowering the levels of cellular EGR-1. PLoS One 2012, 7, e33364. [Google Scholar] [CrossRef] [PubMed]
- AuCoin, D.P.; Colletti, K.S.; Cei, S.A.; Papouskova, I.; Tarrant, M.; Pari, G.S. Amplification of the Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 lytic origin of DNA replication is dependent upon a cis-acting AT-rich region and an ORF50 response element and the trans-acting factors ORF50 (K-Rta) and K8 (K-bZIP). Virology 2004, 318, 542–555. [Google Scholar] [CrossRef] [PubMed]
- Gradoville, L.; Gerlach, J.; Grogan, E.; Shedd, D.; Nikiforow, S.; Metroka, C.; Miller, G. Kaposi’s sarcoma-associated herpesvirus open reading frame 50/Rta protein activates the entire viral lytic cycle in the HH-B2 primary effusion lymphoma cell line. J. Virol. 2000, 74, 6207–6212. [Google Scholar] [CrossRef] [PubMed]
- Cousins, E.; Nicholas, J. Molecular biology of human herpesvirus 8: Novel functions and virus-host interactions implicated in viral pathogenesis and replication. Recent Results Cancer Res. 2014, 193, 227–268. [Google Scholar] [PubMed]
- Sun, R.; Lin, S.F.; Gradoville, L.; Yuan, Y.; Zhu, F.; Miller, G. A viral gene that activates lytic cycle expression of Kaposi’s sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 1998, 95, 10866–10871. [Google Scholar] [CrossRef] [PubMed]
- Lukac, D.M.; Renne, R.; Kirshner, J.R.; Ganem, D. Reactivation of Kaposi’s sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 1998, 252, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; AuCoin, D.P.; Huete, A.R.; Cei, S.A.; Hanson, L.J.; Pari, G.S. A Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 ORF50 deletion mutant is defective for reactivation of latent virus and DNA replication. J. Virol. 2005, 79, 3479–3487. [Google Scholar] [CrossRef] [PubMed]
- Lukac, D.M.; Kirshner, J.R.; Ganem, D. Transcriptional activation by the product of open reading frame 50 of Kaposi’s sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J. Virol. 1999, 73, 9348–9361. [Google Scholar] [PubMed]
- Gwack, Y.; Nakamura, H.; Lee, S.H.; Souvlis, J.; Yustein, J.T.; Gygi, S.; Kung, H.J.; Jung, J.U. Poly(ADP-ribose) polymerase 1 and Ste20-like kinase hKFC act as transcriptional repressors for γ2-herpesvirus lytic replication. Mol. Cell Biol. 2003, 23, 8282–8294. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Wang, S.E.; Hayward, G.S. The KSHV immediate-early transcription factor RTA encodes ubiquitin E3 ligase activity that targets IRF7 for proteosome-mediated degradation. Immunity 2005, 22, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Bu, W.; Palmeri, D.; Krishnan, R.; Marin, R.; Aris, V.M.; Soteropoulos, P.; Lukac, D.M. Identification of direct transcriptional targets of the Kaposi’s sarcoma-associated herpesvirus Rta lytic switch protein by conditional nuclear localization. J. Virol. 2008, 82, 10709–10723. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Song, M.J.; Chu, J.T.; Sun, R. Transcriptional regulation of the interleukin-6 gene of human herpesvirus 8 (Kaposi’s sarcoma-associated herpesvirus). J. Virol. 2002, 76, 8252–8264. [Google Scholar] [CrossRef] [PubMed]
- Song, M.J.; Brown, H.J.; Wu, T.T.; Sun, R. Transcription activation of polyadenylated nuclear rna by rta in human herpesvirus 8/Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2001, 75, 3129–3140. [Google Scholar] [CrossRef] [PubMed]
- Byun, H.; Gwack, Y.; Hwang, S.; Choe, J. Kaposi’s sarcoma-associated herpesvirus open reading frame (ORF) 50 transactivates K8 and ORF57 promoters via heterogeneous response elements. Mol. Cells 2002, 14, 185–191. [Google Scholar] [PubMed]
- McDowell, M.E.; Purushothaman, P.; Rossetto, C.C.; Pari, G.S.; Verma, S.C. Phosphorylation of Kaposi’s sarcoma-associated herpesvirus processivity factor ORF59 by a viral kinase modulates its ability to associate with RTA and oriLyt. J. Virol. 2013, 87, 8038–8052. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, C.C.; Susilarini, N.K.; Pari, G.S. Interaction of Kaposi’s sarcoma-associated herpesvirus ORF59 with oriLyt is dependent on binding with K-Rta. J. Virol. 2011, 85, 3833–3841. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Ishikawa, K.; Nishimura, K.; Sakakibara, S.; Do, E.; Yamanishi, K. Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) replication and transcription factor activates the K9 (vIRF) gene through two distinct cis elements by a non-DNA-binding mechanism. J. Virol. 2002, 76, 12044–12054. [Google Scholar] [CrossRef] [PubMed]
- Bowser, B.S.; Morris, S.; Song, M.J.; Sun, R.; Damania, B. Characterization of Kaposi’s sarcoma-associated herpesvirus (KSHV) K1 promoter activation by Rta. Virology 2006, 348, 309–327. [Google Scholar] [CrossRef] [PubMed]
- Sathish, N.; Yuan, Y. Functional characterization of Kaposi’s sarcoma-associated herpesvirus small capsid protein by bacterial artificial chromosome-based mutagenesis. Virology 2010, 407, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Majerciak, V.; Yamanegi, K.; Zheng, Z.M. Gene structure and expression of Kaposi’s sarcoma-associated herpesvirus ORF56, ORF57, ORF58, and ORF59. J. Virol. 2006, 80, 11968–11981. [Google Scholar] [CrossRef] [PubMed]
- Kronstad, L.M.; Brulois, K.F.; Jung, J.U.; Glaunsinger, B.A. Reinitiation after translation of two upstream open reading frames (ORF) governs expression of the ORF35–37 Kaposi’s sarcoma-associated herpesvirus polycistronic mRNA. J. Virol. 2014, 88, 6512–6518. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.H.; Means, R.E.; Choi, J.K.; Lee, B.S.; Jung, J.U. Kaposi's sarcoma-associated herpesvirus OX2 glycoprotein activates myeloid-lineage cells to induce inflammatory cytokine production. J. Virol. 2002, 76, 4688–4698. [Google Scholar] [CrossRef] [PubMed]
- Seaman, W.T.; Quinlivan, E.B. Lytic switch protein (ORF50) response element in the Kaposi’s sarcoma-associated herpesvirus K8 promoter is located within but does not require a palindromic structure. Virology 2003, 310, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Kuppers, D.A.; Verma, S.C.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen inhibits lytic replication by targeting Rta: A potential mechanism for virus-mediated control of latency. J. Virol. 2004, 78, 6585–6594. [Google Scholar] [CrossRef] [PubMed]
- Ziegelbauer, J.; Grundhoff, A.; Ganem, D. Exploring the DNA binding interactions of the Kaposi’s sarcoma-associated herpesvirus lytic switch protein by selective amplification of bound sequences in vitro. J. Virol. 2006, 80, 2958–2967. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ye, F.; Xie, J.; Kuhne, K.; Gao, S.J. Genome-wide identification of binding sites for Kaposi’s sarcoma-associated herpesvirus lytic switch protein, RTA. Virology 2009, 386, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Chang, J.; Lynch, S.J.; Lukac, D.M.; Ganem, D. The lytic switch protein of KSHV activates gene expression via functional interaction with RBP-Jκ (CSL), the target of the Notch signaling pathway. Genes Dev. 2002, 16, 1977–1989. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.J.; Tsao, E.H.; Webb, B.L.; Ye, H.; Dalton-Griffin, L.; Tsantoulas, C.; Gale, C.V.; Du, M.Q.; Whitehouse, A.; Kellam, P. X box binding protein XBP-1s transactivates the Kaposi’s sarcoma-associated herpesvirus (KSHV) ORF50 promoter, linking plasma cell differentiation to KSHV reactivation from latency. J. Virol. 2007, 81, 13578–13586. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Shedd, D.; Miller, G. An Sp1 response element in the Kaposi’s sarcoma-associated herpesvirus open reading frame 50 promoter mediates lytic cycle induction by butyrate. J. Virol. 2005, 79, 1397–1408. [Google Scholar] [CrossRef] [PubMed]
- Carroll, K.D.; Khadim, F.; Spadavecchia, S.; Palmeri, D.; Lukac, D.M. Direct interactions of Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 ORF50/Rta protein with the cellular protein octamer-1 and DNA are critical for specifying transactivation of a delayed-early promoter and stimulating viral reactivation. J. Virol. 2007, 81, 8451–8467. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.J.; Shedd, D.; Miller, G. Two subclasses of Kaposi's sarcoma-associated herpesvirus lytic cycle promoters distinguished by open reading frame 50 mutant proteins that are deficient in binding to DNA. J. Virol. 2005, 79, 8750–8763. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.J.; Boonsiri, J.; Wang, S.S.; Chen, L.Y.; Miller, G. Binding of RBP-Jκ (CSL) protein to the promoter of the Kaposi’s sarcoma-associated herpesvirus ORF47 (gL) gene is a critical but not sufficient determinant of transactivation by ORF50 protein. Virology 2010, 398, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Persson, L.M.; Wilson, A.C. Wide-scale use of Notch signaling factor CSL/RBP-Jκ in RTA-mediated activation of Kaposi’s sarcoma-associated herpesvirus lytic genes. J. Virol. 2010, 84, 1334–1347. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Verma, S.C.; Cai, Q.; Saha, A.; Dzeng, R.K.; Robertson, E.S. The RBP-Jκ binding sites within the RTA promoter regulate KSHV latent infection and cell proliferation. PLoS Pathog. 2012, 8, e1002479. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Murakami, M.; Choudhuri, T.; Kuppers, D.A.; Robertson, E.S. Intracellular-activated Notch1 can reactivate Kaposi’s sarcoma-associated herpesvirus from latency. Virology 2006, 351, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Dittmer, D.P.; Shin, Y.C.; Hong, Y.; Jung, J.U. Role of Notch signal transduction in Kaposi’s sarcoma-associated herpesvirus gene expression. J. Virol. 2005, 79, 14371–14382. [Google Scholar] [CrossRef] [PubMed]
- Gwack, Y.; Baek, H.J.; Nakamura, H.; Lee, S.H.; Meisterernst, M.; Roeder, R.G.; Jung, J.U. Principal role of TRAP/mediator and SWI/SNF complexes in Kaposi’s sarcoma-associated herpesvirus RTA-mediated lytic reactivation. Mol. Cell Biol. 2003, 23, 2055–2067. [Google Scholar] [CrossRef] [PubMed]
- Gwack, Y.; Byun, H.; Hwang, S.; Lim, C.; Choe, J. CREB-binding protein and histone deacetylase regulate the transcriptional activity of Kaposi’s sarcoma-associated herpesvirus open reading frame 50. J. Virol. 2001, 75, 1909–1917. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Cai, Q.; Lu, J.; Jha, H.C.; Robertson, E.S. Upregulation of cellular Bcl-2 by the KSHV encoded RTA promotes virion production. PLoS One 2011, 6, e23892. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, S.; Wu, M.H.; Geng, Y.; Wood, C. Identification of a cellular protein that interacts and synergizes with the RTA (ORF50) protein of Kaposi’s sarcoma-associated herpesvirus in transcriptional activation. J. Virol. 2001, 75, 11961–11973. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wood, C. The transcriptional repressor K-RBP modulates RTA-mediated transactivation and lytic replication of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2007, 81, 6294–6306. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wen, H.J.; Minhas, V.; Wood, C. The zinc finger DNA-binding domain of K-RBP plays an important role in regulating Kaposi’s sarcoma-associated herpesvirus RTA-mediated gene expression. Virology 2009, 391, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.C.; Fitzgerald, L.D.; Van Geelen, A.; Izumiya, Y.; Ellison, T.J.; Wang, D.H.; Ann, D.K.; Luciw, P.A.; Kung, H.J. Kruppel-associated box domain-associated protein-1 as a latency regulator for Kaposi’s sarcoma-associated herpesvirus and its modulation by the viral protein kinase. Cancer Res. 2009, 69, 5681–5689. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Liang, D.; Gao, Y.; Lan, K. Kaposi’s sarcoma-associated herpesvirus-encoded LANA interacts with host KAP1 to facilitate establishment of viral latency. J. Virol. 2014, 88, 7331–7344. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Tang, Y.; Lin, S.F.; Kung, H.J.; Giam, C.Z. K-bZIP of Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 (KSHV/HHV-8) binds KSHV/HHV-8 Rta and represses Rta-mediated transactivation. J. Virol. 2003, 77, 3809–3815. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, C.; Gao, Y.; Yamboliev, I.; Papouskova, I.; Pari, G. Transcriptional repression of K-Rta by Kaposi’s sarcoma-associated herpesvirus K-bZIP is not required for oriLyt-dependent DNA replication. Virology 2007, 369, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.E.; Wu, F.Y.; Fujimuro, M.; Zong, J.; Hayward, S.D.; Hayward, G.S. Role of CCAAT/enhancer-binding protein α (C/EBPα) in activation of the Kaposi’s sarcoma-associated herpesvirus (KSHV) lytic-cycle replication-associated protein (RAP) promoter in cooperation with the KSHV replication and transcription activator (RTA) and RAP. J. Virol. 2003, 77, 600–623. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yuan, Y. Essential role of RBP-Jκ in activation of the K8 delayed-early promoter of Kaposi’s sarcoma-associated herpesvirus by ORF50/RTA. Virology 2007, 359, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cao, Y.; Liang, D.; Gao, Y.; Xia, T.; Robertson, E.S.; Lan, K. Kaposi’s sarcoma-associated herpesvirus RTA activates the processivity factor ORF59 through interaction with RBP-Jκ and a cis-acting RTA responsive element. Virology 2008, 380, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.J.; Wang, S.S.; Chen, L.Y.; Hung, C.H.; Huang, H.Y.; Shih, Y.J.; Yen, J.B.; Liou, J.Y.; Chen, L.W. ORF50-dependent and ORF50-independent activation of the ORF45 gene of Kaposi’s sarcoma-associated herpesvirus. Virology 2013, 442, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Guito, J.; Gavina, A.; Palmeri, D.; Lukac, D.M. The cellular peptidyl-prolyl cis/trans isomerase Pin1 regulates reactivation of Kaposi’s sarcoma-associated herpesvirus from latency. J. Virol. 2014, 88, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Jaber, T.; Yuan, Y. A virally encoded small peptide regulates RTA stability and facilitates Kaposi’s sarcoma-associated herpesvirus lytic replication. J. Virol. 2013, 87, 3461–3470. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yan, Z.; Wood, C. Kaposi’s sarcoma-associated herpesvirus transactivator RTA promotes degradation of the repressors to regulate viral lytic replication. J. Virol. 2008, 82, 3590–3603. [Google Scholar] [CrossRef] [PubMed]
- Gould, F.; Harrison, S.M.; Hewitt, E.W.; Whitehouse, A. Kaposi’s sarcoma-associated herpesvirus RTA promotes degradation of the Hey1 repressor protein through the ubiquitin proteasome pathway. J. Virol. 2009, 83, 6727–6738. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, H.; Chan, M.Y.; Zhu, F.X.; Lukac, D.M.; Yuan, Y. Kaposi’s sarcoma-associated herpesvirus ori-Lyt-dependent DNA replication: Cis-acting requirements for replication and ori-Lyt-associated RNA transcription. J. Virol. 2004, 78, 8615–8629. [Google Scholar] [CrossRef] [PubMed]
- Izumiya, Y.; Kobayashi, K.; Kim, K.Y.; Pochampalli, M.; Izumiya, C.; Shevchenko, B.; Wang, D.H.; Huerta, S.B.; Martinez, A.; Campbell, M.; et al. Kaposi’s sarcoma-associated herpesvirus K-Rta exhibits SUMO-targeting ubiquitin ligase (STUbL) like activity and is essential for viral reactivation. PLoS Pathog. 2013, 9, e1003506. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Swaminathan, S. Kaposi’s sarcoma-associated herpesvirus lytic gene ORF57 is essential for infectious virion production. J. Virol. 2006, 80, 5251–5260. [Google Scholar] [CrossRef] [PubMed]
- Majerciak, V.; Pripuzova, N.; McCoy, J.P.; Gao, S.J.; Zheng, Z.M. Targeted disruption of Kaposi’s sarcoma-associated herpesvirus ORF57 in the viral genome is detrimental for the expression of ORF59, K8α, and K8.1 and the production of infectious virus. J. Virol. 2007, 81, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Palmeri, D.; Spadavecchia, S.; Carroll, K.D.; Lukac, D.M. Promoter- and cell-specific transcriptional transactivation by the Kaposi's sarcoma-associated herpesvirus ORF57/Mta protein. J. Virol. 2007, 81, 13299–13314. [Google Scholar] [CrossRef] [PubMed]
- Malik, P.; Blackbourn, D.J.; Cheng, M.F.; Hayward, G.S.; Clements, J.B. Functional co-operation between the Kaposi’s sarcoma-associated herpesvirus ORF57 and ORF50 regulatory proteins. J. Gen. Virol. 2004, 85, 2155–2166. [Google Scholar] [CrossRef] [PubMed]
- Hunter, O.V.; Sei, E.; Richardson, R.B.; Conrad, N.K. Chromatin immunoprecipitation and microarray analysis suggest functional cooperation between Kaposi’s Sarcoma-associated herpesvirus ORF57 and K-bZIP. J. Virol. 2013, 87, 4005–4016. [Google Scholar] [CrossRef] [PubMed]
- Malik, P.; Blackbourn, D.J.; Clements, J.B. The evolutionarily conserved Kaposi’s sarcoma-associated herpesvirus ORF57 protein interacts with REF protein and acts as an RNA export factor. J. Biol. Chem. 2004, 279, 33001–33011. [Google Scholar] [CrossRef] [PubMed]
- Majerciak, V.; Zheng, Z.M. Kaposi’s sarcoma-associated herpesvirus ORF57 in viral RNA processing. Front. Biosci. (Landmark Ed) 2009, 14, 1516–1528. [Google Scholar] [CrossRef]
- Nekorchuk, M.; Han, Z.; Hsieh, T.T.; Swaminathan, S. Kaposi’s sarcoma-associated herpesvirus ORF57 protein enhances mRNA accumulation independently of effects on nuclear RNA export. J. Virol. 2007, 81, 9990–9998. [Google Scholar] [CrossRef] [PubMed]
- Massimelli, M.J.; Kang, J.G.; Majerciak, V.; Le, S.Y.; Liewehr, D.J.; Steinberg, S.M.; Zheng, Z.M. Stability of a long noncoding viral RNA depends on a 9-nt core element at the RNA 5' end to interact with viral ORF57 and cellular PABPC1. Int. J. Biol. Sci. 2011, 7, 1145–1160. [Google Scholar] [CrossRef] [PubMed]
- Massimelli, M.J.; Majerciak, V.; Kruhlak, M.; Zheng, Z.M. Interplay between polyadenylate-binding protein 1 and Kaposi’s sarcoma-associated herpesvirus ORF57 in accumulation of polyadenylated nuclear RNA, a viral long noncoding RNA. J. Virol. 2013, 87, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Sahin, B.B.; Patel, D.; Conrad, N.K. Kaposi’s sarcoma-associated herpesvirus ORF57 protein binds and protects a nuclear noncoding RNA from cellular RNA decay pathways. PLoS Pathog. 2010, 6, e1000799. [Google Scholar] [CrossRef]
- Majerciak, V.; Uranishi, H.; Kruhlak, M.; Pilkington, G.R.; Massimelli, M.J.; Bear, J.; Pavlakis, G.N.; Felber, B.K.; Zheng, Z.M. Kaposi’s sarcoma-associated herpesvirus ORF57 interacts with cellular RNA export cofactors RBM15 and OTT3 to promote expression of viral ORF59. J. Virol. 2011, 85, 1528–1540. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.F.; Robinson, D.R.; Miller, G.; Kung, H.J. Kaposi’s sarcoma-associated herpesvirus encodes a bZIP protein with homology to BZLF1 of Epstein-Barr virus. J. Virol. 1999, 73, 1909–1917. [Google Scholar] [PubMed]
- Tang, S.; Zheng, Z.M. Kaposi’s sarcoma-associated herpesvirus K8 exon 3 contains three 5'-splice sites and harbors a K8.1 transcription start site. J. Biol. Chem. 2002, 277, 14547–14556. [Google Scholar] [CrossRef] [PubMed]
- Izumiya, Y.; Lin, S.F.; Ellison, T.; Chen, L.Y.; Izumiya, C.; Luciw, P.; Kung, H.J. Kaposi’s sarcoma-associated herpesvirus K-bZIP is a coregulator of K-Rta: Physical association and promoter-dependent transcriptional repression. J. Virol. 2003, 77, 1441–1451. [Google Scholar] [CrossRef] [PubMed]
- Ellison, T.J.; Izumiya, Y.; Izumiya, C.; Luciw, P.A.; Kung, H.J. A comprehensive analysis of recruitment and transactivation potential of K-Rta and K-bZIP during reactivation of Kaposi’s sarcoma-associated herpesvirus. Virology 2009, 387, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Kato-Noah, T.; Xu, Y.; Rossetto, C.C.; Colletti, K.; Papouskova, I.; Pari, G.S. Overexpression of the kaposi’s sarcoma-associated herpesvirus transactivator K-Rta can complement a K-bZIP deletion BACmid and yields an enhanced growth phenotype. J. Virol. 2007, 81, 13519–13532. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sathish, N.; Hollow, C.; Yuan, Y. Functional characterization of Kaposi’s sarcoma-associated herpesvirus open reading frame K8 by bacterial artificial chromosome-based mutagenesis. J. Virol. 2011, 85, 1943–1957. [Google Scholar] [CrossRef] [PubMed]
- Lefort, S.; Flamand, L. Kaposi’s sarcoma-associated herpesvirus K-bZIP protein is necessary for lytic viral gene expression, DNA replication, and virion production in primary effusion lymphoma cell lines. J. Virol. 2009, 83, 5869–5880. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.Y.; Wang, S.E.; Tang, Q.Q.; Fujimuro, M.; Chiou, C.J.; Zheng, Q.; Chen, H.; Hayward, S.D.; Lane, M.D.; Hayward, G.S. Cell cycle arrest by Kaposi’s sarcoma-associated herpesvirus replication-associated protein is mediated at both the transcriptional and posttranslational levels by binding to CCAAT/enhancer-binding protein α and p21(CIP-1). J. Virol. 2003, 77, 8893–8914. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, C.; Yamboliev, I.; Pari, G.S. Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 K-bZIP modulates latency-associated nuclear protein-mediated suppression of lytic origin-dependent DNA synthesis. J. Virol. 2009, 83, 8492–8501. [Google Scholar] [CrossRef] [PubMed]
- Izumiya, Y.; Ellison, T.J.; Yeh, E.T.; Jung, J.U.; Luciw, P.A.; Kung, H.J. Kaposi’s sarcoma-associated herpesvirus K-bZIP represses gene transcription via SUMO modification. J. Virol. 2005, 79, 9912–9925. [Google Scholar] [CrossRef] [PubMed]
- Lefort, S.; Soucy-Faulkner, A.; Grandvaux, N.; Flamand, L. Binding of Kaposi’s sarcoma-associated herpesvirus K-bZIP to interferon-responsive factor 3 elements modulates antiviral gene expression. J. Virol. 2007, 81, 10950–10960. [Google Scholar] [CrossRef] [PubMed]
- Lefort, S.; Gravel, A.; Flamand, L. Repression of interferon-α stimulated genes expression by Kaposi’s sarcoma-associated herpesvirus K-bZIP protein. Virology 2010, 408, 14–30. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.P.; Tang, Q. Leucine zipper domain is required for Kaposi sarcoma-associated herpesvirus (KSHV) K-bZIP protein to interact with histone deacetylase and is important for KSHV replication. J. Biol. Chem. 2012, 287, 15622–15634. [Google Scholar] [CrossRef] [PubMed]
- Izumiya, Y.; Izumiya, C.; van Geelen, A.; Wang, D.H.; Lam, K.S.; Luciw, P.A.; Kung, H.J. Kaposi’s sarcoma-associated herpesvirus-encoded protein kinase and its interaction with K-bZIP. J. Virol. 2007, 81, 1072–1082. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, A.J. bZIP proteins of human γ-herpesviruses. J. Gen. Virol. 2003, 84, 1941–1949. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ciustea, M.; Ricciardi, R.P. Processivity factor of KSHV contains a nuclear localization signal and binding domains for transporting viral DNA polymerase into the nucleus. Virology 2005, 340, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Chen, J.; Liao, Q.; Wu, Y.; Peng, C.; Chen, X. Lytic infection of Kaposi’s sarcoma-associated herpesvirus induces DNA double-strand breaks and impairs non-homologous end joining. J. Gen. Virol. 2013, 94, 1870–1875. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, J.; Ruvolo, V.; Zong, J.; Ciufo, D.; Guo, H.G.; Reitz, M.S.; Hayward, G.S. A single 13-kilobase divergent locus in the Kaposi sarcoma-associated herpesvirus (human herpesvirus 8) genome contains nine open reading frames that are homologous to or related to cellular proteins. J. Virol. 1997, 71, 1963–1974. [Google Scholar] [PubMed]
- Peng, C.; Chen, J.; Tang, W.; Liu, C.; Chen, X. Kaposi’s sarcoma-associated herpesvirus ORF6 gene is essential in viral lytic replication. PLoS One 2014, 9, e99542. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.; Cohen, A.; Kazimirsky, G.; Dovrat, S.; Rubinfeld, H.; Brodie, C.; Sarid, R. Role of protein kinase C delta in reactivation of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2004, 78, 10187–10192. [Google Scholar] [CrossRef] [PubMed]
- Ford, P.W.; Bryan, B.A.; Dyson, O.F.; Weidner, D.A.; Chintalgattu, V.; Akula, S.M. Raf/MEK/ERK signalling triggers reactivation of Kaposi’s sarcoma-associated herpesvirus latency. J. Gen. Virol. 2006, 87, 1139–1144. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.; Choudhuri, T.; Murakami, M.; Kuppers, D.A.; Robertson, E.S. Intracellular activated Notch1 is critical for proliferation of Kaposi’s sarcoma-associated herpesvirus-associated B-lymphoma cell lines in vitro. J. Virol. 2006, 80, 6411–6419. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Ajibade, A.O.; Ye, F.; Kuhne, K.; Gao, S.J. Reactivation of Kaposi’s sarcoma-associated herpesvirus from latency requires MEK/ERK, JNK and p38 multiple mitogen-activated protein kinase pathways. Virology 2008, 371, 139–154. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Weidner-Glunde, M.; Varjosalo, M.; Rainio, E.M.; Lehtonen, A.; Schulz, T.F.; Koskinen, P.J.; Taipale, J.; Ojala, P.M. KSHV reactivation from latency requires Pim-1 and Pim-3 kinases to inactivate the latency-associated nuclear antigen LANA. PLoS Pathog. 2009, 5, e1000324. [Google Scholar] [CrossRef] [PubMed]
- Qin, D.; Feng, N.; Fan, W.; Ma, X.; Yan, Q.; Lv, Z.; Zeng, Y.; Zhu, J.; Lu, C. Activation of PI3K/AKT and ERK MAPK signal pathways is required for the induction of lytic cycle replication of Kaposi’s sarcoma-associated herpesvirus by herpes simplex virus type 1. BMC Microbiol. 2011, 11, 240. [Google Scholar] [CrossRef] [PubMed]
- Gregory, S.M.; West, J.A.; Dillon, P.J.; Hilscher, C.; Dittmer, D.P.; Damania, B. Toll-like receptor signaling controls reactivation of KSHV from latency. Proc. Natl. Acad. Sci. USA 2009, 106, 11725–11730. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Harada, J.N.; Brown, H.J.; Deng, H.; Song, M.J.; Wu, T.T.; Kato-Stankiewicz, J.; Nelson, C.G.; Vieira, J.; Tamanoi, F.; et al. Systematic identification of cellular signals reactivating Kaposi sarcoma-associated herpesvirus. PLoS Pathog. 2007, 3, e44. [Google Scholar] [CrossRef] [PubMed]
- Li, D.J.; Verma, D.; Mosbruger, T.; Swaminathan, S. CTCF and Rad21 act as host cell restriction factors for Kaposi’s sarcoma-associated herpesvirus (KSHV) lytic replication by modulating viral gene transcription. PLoS Pathog. 2014, 10, e1003880. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.R.; Doganay, S.; Chung, B.; Toth, Z.; Brulois, K.; Lee, S.; Kanketayeva, Z.; Feng, P.; Ha, T.; Jung, J.U. Kaposi’s sarcoma-associated herpesvirus viral interferon regulatory factor 4 (vIRF4) targets expression of cellular IRF4 and the Myc gene to facilitate lytic replication. J. Virol. 2014, 88, 2183–2194. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.; Yang, L.; Mancl, M.E.; Barnes, B.J. Modulation of interferon regulatory factor 5 activities by the Kaposi sarcoma-associated herpesvirus-encoded viral interferon regulatory factor 3 contributes to immune evasion and lytic induction. J. Interferon Cytokine Res. 2011, 31, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Kuang, E.; Fu, B.; Liang, Q.; Myoung, J.; Zhu, F. Phosphorylation of eukaryotic translation initiation factor 4B (EIF4B) by open reading frame 45/p90 ribosomal S6 kinase (ORF45/RSK) signaling axis facilitates protein translation during Kaposi sarcoma-associated herpesvirus (KSHV) lytic replication. J. Biol. Chem. 2011, 286, 41171–41182. [Google Scholar] [CrossRef] [PubMed]
- Zoeteweij, J.P.; Moses, A.V.; Rinderknecht, A.S.; Davis, D.A.; Overwijk, W.W.; Yarchoan, R.; Orenstein, J.M.; Blauvelt, A. Targeted inhibition of calcineurin signaling blocks calcium-dependent reactivation of Kaposi sarcoma-associated herpesvirus. Blood 2001, 97, 2374–2380. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Xie, J.; Ye, F.; Gao, S.J. Modulation of Kaposi’s sarcoma-associated herpesvirus infection and replication by MEK/ERK, JNK, and p38 multiple mitogen-activated protein kinase pathways during primary infection. J. Virol. 2006, 80, 5371–5382. [Google Scholar] [CrossRef] [PubMed]
- Sharma-Walia, N.; Krishnan, H.H.; Naranatt, P.P.; Zeng, L.; Smith, M.S.; Chandran, B. ERK1/2 and MEK1/2 induced by Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) early during infection of target cells are essential for expression of viral genes and for establishment of infection. J. Virol. 2005, 79, 10308–10329. [Google Scholar] [CrossRef] [PubMed]
- Hengge, U.R.; Ruzicka, T.; Tyring, S.K.; Stuschke, M.; Roggendorf, M.; Schwartz, R.A.; Seeber, S. Update on Kaposi’s sarcoma and other HHV8 associated diseases. Part 2: Pathogenesis, Castleman’s disease, and pleural effusion lymphoma. Lancet Infect. Dis. 2002, 2, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Haas, D.A.; Bala, K.; Busche, G.; Weidner-Glunde, M.; Santag, S.; Kati, S.; Gramolelli, S.; Damas, M.; Dittrich-Breiholz, O.; Kracht, M.; et al. The inflammatory kinase MAP4K4 promotes reactivation of Kaposi’s sarcoma herpesvirus and enhances the invasiveness of infected endothelial cells. PLoS Pathog. 2013, 9, e1003737. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.E.; Wu, F.Y.; Chen, H.; Shamay, M.; Zheng, Q.; Hayward, G.S. Early activation of the Kaposi’s sarcoma-associated herpesvirus RTA, RAP, and MTA promoters by the tetradecanoyl phorbol acetate-induced AP1 pathway. J. Virol. 2004, 78, 4248–4267. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Pan, H.; Yoo, S.; Gao, S.J. Kaposi’s sarcoma-associated herpesvirus induction of AP-1 and interleukin 6 during primary infection mediated by multiple mitogen-activated protein kinase pathways. J. Virol. 2005, 79, 15027–15037. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.; Brodie, C.; Sarid, R. An essential role of ERK signalling in TPA-induced reactivation of Kaposi’s sarcoma-associated herpesvirus. J. Gen. Virol. 2006, 87, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Emuss, V.; Lagos, D.; Pizzey, A.; Gratrix, F.; Henderson, S.R.; Boshoff, C. KSHV manipulates Notch signaling by DLL4 and JAG1 to alter cell cycle genes in lymphatic endothelia. PLoS Pathog. 2009, 5, e1000616. [Google Scholar] [CrossRef] [PubMed]
- Carroll, K.D.; Bu, W.; Palmeri, D.; Spadavecchia, S.; Lynch, S.J.; Marras, S.A.; Tyagi, S.; Lukac, D.M. Kaposi’s Sarcoma-associated herpesvirus lytic switch protein stimulates DNA binding of RBP-Jk/CSL to activate the Notch pathway. J. Virol. 2006, 80, 9697–9709. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Murakami, M.; Si, H.; Robertson, E.S. A potential α-helix motif in the amino terminus of LANA encoded by Kaposi’s sarcoma-associated herpesvirus is critical for nuclear accumulation of HIF-1α in normoxia. J. Virol. 2007, 81, 10413–10423. [Google Scholar] [CrossRef] [PubMed]
- Carroll, P.A.; Kenerson, H.L.; Yeung, R.S.; Lagunoff, M. Latent Kaposi’s sarcoma-associated herpesvirus infection of endothelial cells activates hypoxia-induced factors. J. Virol. 2006, 80, 10802–10812. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.C.; Joo, C.H.; Gack, M.U.; Lee, H.R.; Jung, J.U. Kaposi’s sarcoma-associated herpesvirus viral IFN regulatory factor 3 stabilizes hypoxia-inducible factor-1α to induce vascular endothelial growth factor expression. Cancer Res. 2008, 68, 1751–1759. [Google Scholar] [CrossRef] [PubMed]
- Gregory, S.M.; Damania, B. KSHV and the toll of innate immune activation. Cell Cycle 2009, 8, 3246–3247. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Shahir, A.M.; Sha, J.; Feng, Z.; Eapen, B.; Nithianantham, S.; Das, B.; Karn, J.; Weinberg, A.; Bissada, N.F.; et al. Short-chain fatty acids from periodontal pathogens suppress histone deacetylases, EZH2, and SUV39H1 to promote Kaposi’s sarcoma-associated herpesvirus replication. J. Virol. 2014, 88, 4466–4479. [Google Scholar] [CrossRef] [PubMed]
- Morris, V.A.; Punjabi, A.S.; Lagunoff, M. Activation of Akt through gp130 receptor signaling is required for Kaposi’s sarcoma-associated herpesvirus-induced lymphatic reprogramming of endothelial cells. J. Virol. 2008, 82, 8771–8779. [Google Scholar] [CrossRef] [PubMed]
- Punjabi, A.S.; Carroll, P.A.; Chen, L.; Lagunoff, M. Persistent activation of STAT3 by latent Kaposi’s sarcoma-associated herpesvirus infection of endothelial cells. J. Virol. 2007, 81, 2449–2458. [Google Scholar] [CrossRef] [PubMed]
- Burger, M.; Hartmann, T.; Burger, J.A.; Schraufstatter, I. KSHV-GPCR and CXCR2 transforming capacity and angiogenic responses are mediated through a JAK2-STAT3-dependent pathway. Oncogene 2005, 24, 2067–2075. [Google Scholar] [CrossRef] [PubMed]
- Muromoto, R.; Okabe, K.; Fujimuro, M.; Sugiyama, K.; Yokosawa, H.; Seya, T.; Matsuda, T. Physical and functional interactions between STAT3 and Kaposi’s sarcoma-associated herpesvirus-encoded LANA. FEBS Lett. 2006, 580, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Verma, S.C.; Choi, J.Y.; Ma, M.; Robertson, E.S. Kaposi’s sarcoma-associated herpesvirus inhibits interleukin-4-mediated STAT6 phosphorylation to regulate apoptosis and maintain latency. J. Virol. 2010, 84, 11134–11144. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.C.; Zhou, F.C.; Xie, J.P.; Kang, T.; Greene, W.; Kuhne, K.; Lei, X.F.; Li, Q.H.; Gao, S.J. Kaposi’s sarcoma-associated herpesvirus latent gene vFLIP inhibits viral lytic replication through NF-κB-mediated suppression of the AP-1 pathway: A novel mechanism of virus control of latency. J. Virol. 2008, 82, 4235–4249. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Bai, Z.; Ye, F.; Xie, J.; Kim, C.G.; Huang, Y.; Gao, S.J. Regulation of NF-κB inhibitor IκBα and viral replication by a KSHV microRNA. Nat. Cell Biol. 2010, 12, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Izumiya, Y.; Izumiya, C.; Hsia, D.; Ellison, T.J.; Luciw, P.A.; Kung, H.J. NF-κB serves as a cellular sensor of Kaposi’s sarcoma-associated herpesvirus latency and negatively regulates K-Rta by antagonizing the RBP-Jκ coactivator. J. Virol. 2009, 83, 4435–4446. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.J.; Song, M.J.; Deng, H.; Wu, T.T.; Cheng, G.; Sun, R. NF-κB inhibits γ-herpesvirus lytic replication. J. Virol. 2003, 77, 8532–8540. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, C.; Ganem, D. Effects of NF-κB activation on KSHV latency and lytic reactivation are complex and context-dependent. Virology 2008, 375, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.J.; Deng, J.H.; Zhou, F.C. Productive lytic replication of a recombinant Kaposi’s sarcoma-associated herpesvirus in efficient primary infection of primary human endothelial cells. J. Virol. 2003, 77, 9738–9749. [Google Scholar] [CrossRef] [PubMed]
- Sadagopan, S.; Sharma-Walia, N.; Veettil, M.V.; Raghu, H.; Sivakumar, R.; Bottero, V.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus induces sustained NF-κB activation during de novo infection of primary human dermal microvascular endothelial cells that is essential for viral gene expression. J. Virol. 2007, 81, 3949–3968. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Wu, T.T.; Tchieu, J.H.; Feng, J.; Brown, H.J.; Feng, J.; Li, X.; Qi, J.; Deng, H.; Vivanco, I.; et al. Inhibition of the phosphatidylinositol 3-kinase-Akt pathway enhances γ2-herpesvirus lytic replication and facilitates reactivation from latency. J. Gen. Virol. 2010, 91, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, A.P.; Damania, B. AKTivation of PI3K/AKT/mTOR signaling pathway by KSHV. Front. Immunol. 2012, 3, 401. [Google Scholar] [PubMed]
- Lagunoff, M.; Majeti, R.; Weiss, A.; Ganem, D. Deregulated signal transduction by the K1 gene product of Kaposi’s sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 1999, 96, 5704–5709. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, C.C.; Damania, B. The K1 protein of Kaposi’s sarcoma-associated herpesvirus activates the Akt signaling pathway. J. Virol. 2004, 78, 1918–1927. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Galisteo, R.; Molinolo, A.A.; Wetzker, R.; Hirsch, E.; Gutkind, J.S. PI3Kγ mediates kaposi’s sarcoma-associated herpesvirus vGPCR-induced sarcomagenesis. Cancer Cell 2011, 19, 805–813. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, A.; Chaisuparat, R.; Hu, J.; Ramsdell, A.K.; Manning, B.D.; Sausville, E.A.; Sawai, E.T.; Molinolo, A.; Gutkind, J.S.; Montaner, S. The TSC2/mTOR pathway drives endothelial cell transformation induced by the Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor. Cancer Cell 2006, 10, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.K.; Foreman, K.; Shin, J.W.; Hirakawa, S.; Curry, C.L.; Sage, D.R.; Libermann, T.; Dezube, B.J.; Fingeroth, J.D.; Detmar, M. Lymphatic reprogramming of blood vascular endothelium by Kaposi sarcoma-associated herpesvirus. Nat. Genet. 2004, 36, 683–685. [Google Scholar] [CrossRef] [PubMed]
- Fu, B.; Kuang, E.; Li, W.; Avey, D.; Li, X.; Turpin, Z.; Valdes, A.; Brulois, K.; Myoung, J.; Zhu, F. Activation of p90 Ribosomal S6 Kinases (RSKs) by ORF45 of Kaposi Sarcoma-Associated Herpesvirus is Critical for Optimal Production of Infectious Viruses. J. Virol. 2015, 89, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, S.; Maeng, H.; Young, D.P.; Prakash, O.; Fayad, L.E.; Younes, A.; Samaniego, F. K1 protein of human herpesvirus 8 suppresses lymphoma cell Fas-mediated apoptosis. Blood 2007, 109, 2174–2182. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Chauhan, D.; Teoh, G.; Raje, N.; Treon, S.P.; Tai, Y.T.; Shima, Y.; Anderson, K.C. Characterization of signaling cascades triggered by human interleukin-6 versus Kaposi’s sarcoma-associated herpes virus-encoded viral interleukin 6. Clin. Cancer Res. 2000, 6, 1180–1189. [Google Scholar] [PubMed]
- Wang, Q.J.; Jenkins, F.J.; Jacobson, L.P.; Meng, Y.X.; Pellett, P.E.; Kingsley, L.A.; Kousoulas, K.G.; Baghian, A.; Rinaldo, C.R., Jr. CD8+ cytotoxic T lymphocyte responses to lytic proteins of human herpes virus 8 in human immunodeficiency virus type 1-infected and -uninfected individuals. J. Infect. Dis. 2000, 182, 928–932. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.M.; Bynoe, M.O.; Karki, R.; Tartell, M.A.; Means, R.E. Kaposi’s sarcoma-associated herpesvirus K3 and K5 proteins down regulate both DC-SIGN and DC-SIGNR. PLoS One 2013, 8, e58056. [Google Scholar] [CrossRef] [PubMed]
- Weber, K.S.; Grone, H.J.; Rocken, M.; Klier, C.; Gu, S.; Wank, R.; Proudfoot, A.E.; Nelson, P.J.; Weber, C. Selective recruitment of Th2-type cells and evasion from a cytotoxic immune response mediated by viral macrophage inhibitory protein-II. Eur. J. Immunol. 2001, 31, 2458–2466. [Google Scholar] [CrossRef] [PubMed]
- Kledal, T.N.; Rosenkilde, M.M.; Coulin, F.; Simmons, G.; Johnsen, A.H.; Alouani, S.; Power, C.A.; Luttichau, H.R.; Gerstoft, J.; Clapham, P.R.; et al. A broad-spectrum chemokine antagonist encoded by Kaposi’s sarcoma-associated herpesvirus. Science 1997, 277, 1656–1659. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Dong, X.; Negaard, A.; Feng, P. Kaposi’s sarcoma-associated herpesvirus K7 induces viral G protein-coupled receptor degradation and reduces its tumorigenicity. PLoS Pathog. 2008, 4, e1000157. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.W.; Sharp, T.V.; Koumi, A.; Koentges, G.; Boshoff, C. Characterization of an anti-apoptotic glycoprotein encoded by Kaposi’s sarcoma-associated herpesvirus which resembles a spliced variant of human survivin. EMBO J. 2002, 21, 2602–2615. [Google Scholar] [CrossRef] [PubMed]
- Burysek, L.; Yeow, W.S.; Lubyova, B.; Kellum, M.; Schafer, S.L.; Huang, Y.Q.; Pitha, P.M. Functional analysis of human herpesvirus 8-encoded viral interferon regulatory factor 1 and its association with cellular interferon regulatory factors and p300. J. Virol. 1999, 73, 7334–7342. [Google Scholar] [PubMed]
- Lin, R.; Genin, P.; Mamane, Y.; Sgarbanti, M.; Battistini, A.; Harrington, W.J., Jr.; Barber, G.N.; Hiscott, J. HHV-8 encoded vIRF-1 represses the interferon antiviral response by blocking IRF-3 recruitment of the CBP/p300 coactivators. Oncogene 2001, 20, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Foster-Cuevas, M.; Wright, G.J.; Puklavec, M.J.; Brown, M.H.; Barclay, A.N. Human herpesvirus 8 K14 protein mimics CD200 in down-regulating macrophage activation through CD200 receptor. J. Virol. 2004, 78, 7667–7676. [Google Scholar] [CrossRef] [PubMed]
- Rezaee, S.A.; Gracie, J.A.; McInnes, I.B.; Blackbourn, D.J. Inhibition of neutrophil function by the Kaposi’s sarcoma-associated herpesvirus vOX2 protein. AIDS 2005, 19, 1907–1910. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, M.M.; Pietrek, M.; Dittrich-Breiholz, O.; Kracht, M.; Schulz, T.F. Modulation of host gene expression by the K15 protein of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2007, 81, 42–58. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Brinkmann, M.M.; Pietrek, M.; Ottinger, M.; Dittrich-Breiholz, O.; Kracht, M.; Schulz, T.F. Functional characterization of the M-type K15-encoded membrane protein of Kaposi’s sarcoma-associated herpesvirus. J. Gen. Virol. 2007, 88, 1698–1707. [Google Scholar] [CrossRef] [PubMed]
- Mullick, J.; Bernet, J.; Singh, A.K.; Lambris, J.D.; Sahu, A. Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) open reading frame 4 protein (kaposica) is a functional homolog of complement control proteins. J. Virol. 2003, 77, 3878–3881. [Google Scholar] [CrossRef] [PubMed]
- Mullick, J.; Singh, A.K.; Panse, Y.; Yadav, V.; Bernet, J.; Sahu, A. Identification of functional domains in kaposica, the complement control protein homolog of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8). J. Virol. 2005, 79, 5850–5856. [Google Scholar] [CrossRef] [PubMed]
- Spiller, O.B.; Robinson, M.; O'Donnell, E.; Milligan, S.; Morgan, B.P.; Davison, A.J.; Blackbourn, D.J. Complement regulation by Kaposi’s sarcoma-associated herpesvirus ORF4 protein. J. Virol. 2003, 77, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.X.; King, S.M.; Smith, E.J.; Levy, D.E.; Yuan, Y. A Kaposi’s sarcoma-associated herpesviral protein inhibits virus-mediated induction of type I interferon by blocking IRF-7 phosphorylation and nuclear accumulation. Proc. Natl. Acad. Sci. USA 2002, 99, 5573–5578. [Google Scholar] [CrossRef] [PubMed]
- Sathish, N.; Zhu, F.X.; Golub, E.E.; Liang, Q.; Yuan, Y. Mechanisms of autoinhibition of IRF-7 and a probable model for inactivation of IRF-7 by Kaposi’s sarcoma-associated herpesvirus protein ORF45. J. Biol. Chem. 2011, 286, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Gregory, S.M.; Damania, B. Inhibition of the inflammasome response by a viral protein that interacts with NLRs. Commun. Integr. Biol. 2011, 4, 416–418. [Google Scholar] [CrossRef] [PubMed]
- Inn, K.S.; Lee, S.H.; Rathbun, J.Y.; Wong, L.Y.; Toth, Z.; Machida, K.; Ou, J.H.; Jung, J.U. Inhibition of RIG-I-mediated signaling by Kaposi’s sarcoma-associated herpesvirus-encoded deubiquitinase ORF64. J. Virol 2011, 85, 10899–10904. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E.; Nador, R.G.; Bai, F.; Bohenzky, R.A.; Russo, J.J.; Moore, P.S.; Chang, Y.; Knowles, D.M. Kaposi's sarcoma-associated herpesvirus contains G protein-coupled receptor and cyclin D homologs which are expressed in Kaposi’s sarcoma and malignant lymphoma. J. Virol. 1996, 70, 8218–8223. [Google Scholar] [PubMed]
- Full, F.; Jungnickl, D.; Reuter, N.; Bogner, E.; Brulois, K.; Scholz, B.; Sturzl, M.; Myoung, J.; Jung, J.U.; Stamminger, T.; et al. Kaposi’s sarcoma associated herpesvirus tegument protein ORF75 is essential for viral lytic replication and plays a critical role in the antagonization of ND10-instituted intrinsic immunity. PLoS Pathog. 2014, 10, e1003863. [Google Scholar] [CrossRef] [PubMed]
- Gunther, T.; Schreiner, S.; Dobner, T.; Tessmer, U.; Grundhoff, A. Influence of ND10 components on epigenetic determinants of early KSHV latency establishment. PLoS Pathog. 2014, 10, e1004274. [Google Scholar] [CrossRef] [PubMed]
- Borah, S.; Darricarrere, N.; Darnell, A.; Myoung, J.; Steitz, J.A. A viral nuclear noncoding RNA binds re-localized poly(A) binding protein and is required for late KSHV gene expression. PLoS Pathog. 2011, 7, e1002300. [Google Scholar] [CrossRef] [PubMed]
- Borah, S.; Nichols, L.A.; Hassman, L.M.; Kedes, D.H.; Steitz, J.A. Tracking expression and subcellular localization of RNA and protein species using high-throughput single cell imaging flow cytometry. RNA 2012, 18, 1573–1579. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, C.C.; Tarrant-Elorza, M.; Verma, S.; Purushothaman, P.; Pari, G.S. Regulation of viral and cellular gene expression by Kaposi’s sarcoma-associated herpesvirus polyadenylated nuclear RNA. J. Virol. 2013, 87, 5540–5553. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, C.C.; Pari, G.S. Kaposi’s sarcoma-associated herpesvirus noncoding polyadenylated nuclear RNA interacts with virus- and host cell-encoded proteins and suppresses expression of genes involved in immune modulation. J. Virol. 2011, 85, 13290–13297. [Google Scholar] [CrossRef] [PubMed]
- Monini, P.; Carlini, F.; Sturzl, M.; Rimessi, P.; Superti, F.; Franco, M.; Melucci-Vigo, G.; Cafaro, A.; Goletti, D.; Sgadari, C.; et al. α-interferon inhibits human herpesvirus 8 (HHV-8) reactivation in primary effusion lymphoma cells and reduces HHV-8 load in cultured peripheral blood mononuclear cells. J. Virol. 1999, 73, 4029–4041. [Google Scholar] [PubMed]
- Jacobs, S.R.; Damania, B. The viral interferon regulatory factors of KSHV: Immunosuppressors or oncogenes? Front. Immunol. 2011, 2, 19. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.R.; Gregory, S.M.; West, J.A.; Wollish, A.C.; Bennett, C.L.; Blackbourn, D.J.; Heise, M.T.; Damania, B. The viral interferon regulatory factors of kaposi’s sarcoma-associated herpesvirus differ in their inhibition of interferon activation mediated by toll-like receptor 3. J. Virol. 2013, 87, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.R.; Choi, W.C.; Lee, S.; Hwang, J.; Hwang, E.; Guchhait, K.; Haas, J.; Toth, Z.; Jeon, Y.H.; Oh, T.K.; et al. Jung Bilateral inhibition of HAUSP deubiquitinase by a viral interferon regulatory factor protein. Nat. Struct. Mol. Biol. 2011, 18, 1336–1344. [Google Scholar] [CrossRef] [PubMed]
- Molden, J.; Chang, Y.; You, Y.; Moore, P.S.; Goldsmith, M.A. A Kaposi’s sarcoma-associated herpesvirus-encoded cytokine homolog (vIL-6) activates signaling through the shared gp130 receptor subunit. J. Biol. Chem. 1997, 272, 19625–19631. [Google Scholar] [CrossRef] [PubMed]
- Arvanitakis, L.; Geras-Raaka, E.; Varma, A.; Gershengorn, M.C.; Cesarman, E. Human herpesvirus KSHV encodes a constitutively active G-protein-coupled receptor linked to cell proliferation. Nature 1997, 385, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Montaner, S. Akt/TSC/mTOR activation by the KSHV G protein-coupled receptor: Emerging insights into the molecular oncogenesis and treatment of Kaposi’s sarcoma. Cell Cycle 2007, 6, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, T.; Itami, M. Seropositivity of human herpesvirus-8 in patients with uveitis. Ocul. Immunol. Inflamm. 2002, 10, 197–199. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.S.; Lee, S.H.; Feng, P.; Chang, H.; Cho, N.H.; Jung, J.U. Characterization of the Kaposi’s sarcoma-associated herpesvirus K1 signalosome. J. Virol. 2005, 79, 12173–12184. [Google Scholar] [CrossRef] [PubMed]
- Prakash, O.; Swamy, O.R.; Peng, X.; Tang, Z.Y.; Li, L.; Larson, J.E.; Cohen, J.C.; Gill, J.; Farr, G.; Wang, S.; et al. Activation of Src kinase Lyn by the Kaposi sarcoma-associated herpesvirus K1 protein: Implications for lymphomagenesis. Blood 2005, 105, 3987–3994. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Park, J.; Lee, B.S.; Lee, S.H.; Bram, R.J.; Jung, J.U. Kaposi’s sarcoma-associated herpesvirus mitochondrial K7 protein targets a cellular calcium-modulating cyclophilin ligand to modulate intracellular calcium concentration and inhibit apoptosis. J. Virol. 2002, 76, 11491–11504. [Google Scholar] [CrossRef] [PubMed]
- Brulois, K.; Jung, J.U. Interplay between Kaposi’s sarcoma-associated herpesvirus and the innate immune system. Cytokine Growth Factor Rev. 2014, 25, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Coscoy, L.; Ganem, D. A viral protein that selectively downregulates ICAM-1 and B7–2 and modulates T cell costimulation. J. Clin. Investig. 2001, 107, 1599–1606. [Google Scholar] [CrossRef] [PubMed]
- Neipel, F.; Albrecht, J.C.; Ensser, A.; Huang, Y.Q.; Li, J.J.; Friedman-Kien, A.E.; Fleckenstein, B. Human herpesvirus 8 encodes a homolog of interleukin-6. J. Virol. 1997, 71, 839–842. [Google Scholar] [PubMed]
- Russo, J.J.; Bohenzky, R.A.; Chien, M.C.; Chen, J.; Yan, M.; Maddalena, D.; Parry, J.P.; Peruzzi, D.; Edelman, I.S.; Chang, Y.; et al. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. USA 1996, 93, 14862–14867. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Okruzhnov, Y.; Li, H.; Nicholas, J. Human herpesvirus 8 (HHV-8)-encoded cytokines induce expression of and autocrine signaling by vascular endothelial growth factor (VEGF) in HHV-8-infected primary-effusion lymphoma cell lines and mediate VEGF-independent antiapoptotic effects. J. Virol. 2001, 75, 10933–10940. [Google Scholar] [CrossRef] [PubMed]
- Stine, J.T.; Wood, C.; Hill, M.; Epp, A.; Raport, C.J.; Schweickart, V.L.; Endo, Y.; Sasaki, T.; Simmons, G.; Boshoff, C.; et al. KSHV-encoded CC chemokine vMIP-III is a CCR4 agonist, stimulates angiogenesis, and selectively chemoattracts TH2 cells. Blood 2000, 95, 1151–1157. [Google Scholar] [PubMed]
- Shiratori, I.; Yamaguchi, M.; Suzukawa, M.; Yamamoto, K.; Lanier, L.L.; Saito, T.; Arase, H. Down-regulation of basophil function by human CD200 and human herpesvirus-8 CD200. J. Immunol. 2005, 175, 4441–4449. [Google Scholar] [CrossRef] [PubMed]
- Mark, L.; Lee, W.H.; Spiller, O.B.; Proctor, D.; Blackbourn, D.J.; Villoutreix, B.O.; Blom, A.M. The Kaposi’s sarcoma-associated herpesvirus complement control protein mimics human molecular mechanisms for inhibition of the complement system. J. Biol. Chem. 2004, 279, 45093–45101. [Google Scholar] [CrossRef] [PubMed]
- Spiller, O.B.; Blackbourn, D.J.; Mark, L.; Proctor, D.G.; Blom, A.M. Functional activity of the complement regulator encoded by Kaposi’s sarcoma-associated herpesvirus. J. Biol. Chem. 2003, 278, 9283–9289. [Google Scholar] [CrossRef] [PubMed]
- Spiller, O.B.; Mark, L.; Blue, C.E.; Proctor, D.G.; Aitken, J.A.; Blom, A.M.; Blackbourn, D.J. Dissecting the regions of virion-associated Kaposi’s sarcoma-associated herpesvirus complement control protein required for complement regulation and cell binding. J. Virol. 2006, 80, 4068–4078. [Google Scholar] [CrossRef] [PubMed]
- Mark, L.; Proctor, D.G.; Blackbourn, D.J.; Blom, A.M.; Spiller, O.B. Separation of decay-accelerating and cofactor functional activities of Kaposi’s sarcoma-associated herpesvirus complement control protein using monoclonal antibodies. Immunology 2008, 123, 228–238. [Google Scholar] [PubMed]
- Zhu, F.X.; Cusano, T.; Yuan, Y. Identification of the immediate-early transcripts of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 1999, 73, 5556–5567. [Google Scholar] [PubMed]
- Sathish, N.; Zhu, F.X.; Yuan, Y. Kaposi’s sarcoma-associated herpesvirus ORF45 interacts with kinesin-2 transporting viral capsid-tegument complexes along microtubules. PLoS Pathog. 2009, 5, e1000332. [Google Scholar] [CrossRef] [PubMed]
- Brousset, P.; Meggetto, F.; Laharrague, P.; Attal, M.; Delsol, G. Kaposi’s sarcoma-associated herpesvirus (KSHV) in bone marrow biopsy from patients with multiple myeloma: PCR amplification of orf26 but not orf72 and orf75 sequences. Br. J. Haematol. 2000, 108, 197–198. [Google Scholar] [CrossRef] [PubMed]
- Konrad, A.; Wies, E.; Thurau, M.; Marquardt, G.; Naschberger, E.; Hentschel, S.; Jochmann, R.; Schulz, T.F.; Erfle, H.; Brors, B.; et al. A systems biology approach to identify the combination effects of human herpesvirus 8 genes on NF-κB activation. J. Virol. 2009, 83, 2563–2574. [Google Scholar] [CrossRef] [PubMed]
- Gregory, S.M.; Davis, B.K.; West, J.A.; Taxman, D.J.; Matsuzawa, S.; Reed, J.C.; Ting, J.P.; Damania, B. Discovery of a viral NLR homolog that inhibits the inflammasome. Science 2011, 331, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Rozen, R.; Sathish, N.; Li, Y.; Yuan, Y. Virion-wide protein interactions of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2008, 82, 4742–4750. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.; Kim, K.Y.; Chang, P.C.; Huerta, S.; Shevchenko, B.; Wang, D.H.; Izumiya, C.; Kung, H.J.; Izumiya, Y. A lytic viral long noncoding RNA modulates the function of a latent protein. J. Virol. 2014, 88, 1843–1848. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, C.C.; Pari, G. KSHV PAN RNA associates with demethylases UTX and JMJD3 to activate lytic replication through a physical interaction with the virus genome. PLoS Pathog. 2012, 8, e1002680. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Purushothaman, P.; Uppal, T.; Verma, S.C. Molecular Biology of KSHV Lytic Reactivation. Viruses 2015, 7, 116-153. https://doi.org/10.3390/v7010116
AMA Style
Purushothaman P, Uppal T, Verma SC. Molecular Biology of KSHV Lytic Reactivation. Viruses. 2015; 7(1):116-153. https://doi.org/10.3390/v7010116
Chicago/Turabian StylePurushothaman, Pravinkumar, Timsy Uppal, and Subhash C. Verma. 2015. "Molecular Biology of KSHV Lytic Reactivation" Viruses 7, no. 1: 116-153. https://doi.org/10.3390/v7010116