2.1. Generation of WNV-NY/TAT
To overcome the limitations of instability and reduced viral fitness associated with WN reporter viruses, we sought to generate a recombinant virus in which the reporter was located within the coding region of the viral genome and liberated from the polyprotein via cleavage by the viral protease. Since insertion of
gfp at the 5’ end of the viral coding region is stable [
17], we focused on this region. The WNV NS2B-NS3 protease naturally cleaves the viral polyprotein between Capsid (C) and Capsid Anchor (CA) [
19,
20,
21], making this position an attractive site for insertion of a reporter gene. Because larger inserts are potentially detrimental to viral fitness, we minimized the size of the sequence inserted at this location. To this end, we began by inserting the sequence encoding residues 1-67 of HIV-TAT, which is the minimal portion of TAT required to drive efficient expression from the HIV long-terminal repeat (LTR) [
22]. Although the HIV-TAT protein is not itself a reporter, several cell lines have been developed that express TAT-responsive reporters. For HTSs with WNV, similar TAT-responsive reporters can be stably transfected into cell lines of interest. Thus, TAT(1-67) provided us with the opportunity to probe the flexibility of the WNV C/CA junction using an insert sequence substantially smaller than currently available reporters. In order to ensure that the TAT(1-67) sequence was proteolytically separated from the rest of the WNV polyprotein, we engineered two Gly residues and a Lys-Arg residue at the N- and C-terminus, respectively, of the TAT(1-67) insert (
Figure 1B). Addition of these amino acids preserved the wild-type WNV C/CA cleavage site (P
2-P
2’) at both ends of TAT(1-67) and did not affect the ability of the TAT protein to stimulate HIV LTR-driven expression (
Figure S1). There is substantial natural variation between the P3-P6 positions of the WNV protease, and evidence suggests that substrate specificity is almost exclusively governed by the P2-P2’ residues [
23,
24,
25].
To create the reporter virus, we inserted the modified TAT sequence into a highly virulent strain of WNV, WNV-New York (WNV-NY), which is of interest for high throughput studies. Vero cells were electroporated with
in vitro-transcribed
WNV-NY/TAT RNA. Although no cytopathic effects (CPE) were observed, a relatively high titer viral stock (p0) (2.5 × 10
6 plaque-forming units (pfu)/mL), with a plaque phenotype indistinguishable from that of wild-type WNV-NY [
26], was recovered at 7 days post-electroporation. Passage of this viral stock in Vero cells generated a 4.9 × 10
7 pfu/mL (p1) stock. To assess the integrity of the
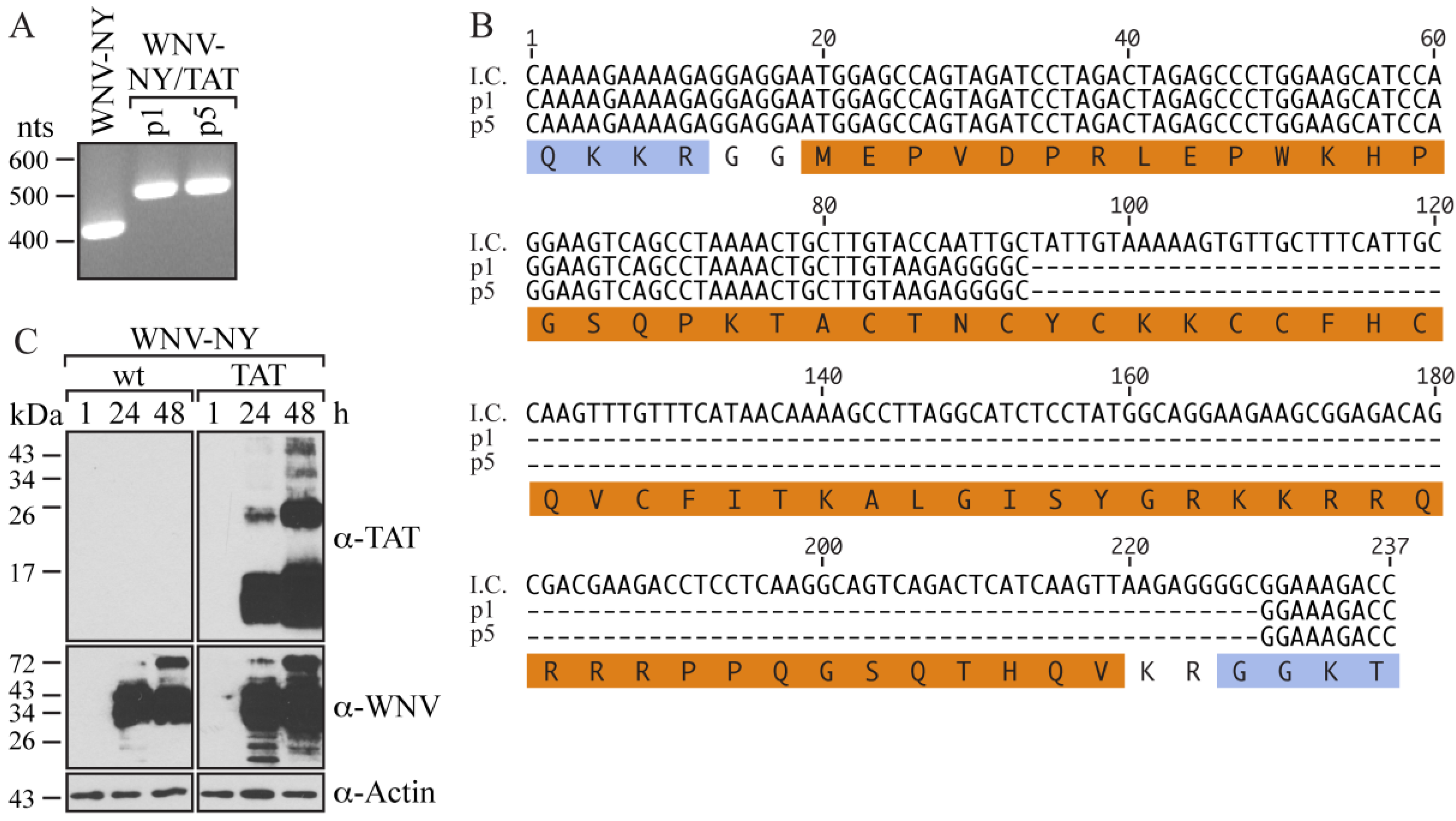
TAT(1-67) gene within the recovered virus, total RNA was extracted from p1 cultures and used as a template for reverse transcription (RT)-PCR with primers spanning the site of insertion. As expected, amplification of wild-type WNV-NY RNA resulted in the production of a ~400 nts band (
Figure 2A). In contrast, the PCR product generated from the amplification of WNV-NY/TAT p1 RNA was 500 nts, which despite being larger than that of wild-type WNV-NY, was approximately 100 nts smaller than expected for a virus containing TAT(1-67). Based on this size difference, we concluded that the recovered virus contained only part of TAT(1-67). Indeed, sequencing indicated that the recovered virus contained only the first 25 residues of TAT (
Figure 2B), suggesting that the TAT(1-67) sequence was unstable in this context. Not surprisingly, we were unable to detect functional TAT protein using the TZM-bl reporter cell line (
Figure S2), which is consistent with TAT’s requirement of residues 1-67 for efficient trans-activation of the HIV LTR [
22].
To evaluate whether the truncated TAT sequence was stable within the WNV-NY genome, we serially passaged the p1 virus in Vero cells four more times. The RT-PCR product generated from passage 5 (p5) RNA was the same size as that generated from p1 RNA (
Figure 2A). Moreover, the sequences of the p1 and p5 RT-PCR products were identical, indicating that the truncated TAT(1-25) sequence was stable within the WNV-NY genome. Thus, WNV can tolerate the addition of coding nucleotides at the C/CA junction, though certain size or sequence restrictions exist.
To assess whether the TAT(1-25) insert was proteolytically separated from the rest of the WNV-NY/TAT polyprotein, we examined TAT expression in Vero cells infected with WNV-NY/TAT (p0). Although TAT-reactive proteins were detected in cultures infected with WNV-NY/TAT p0, most bands were larger than 2.5 kilodaltons (kDa), the size expected for TAT(1-25) (
Figure 2C). Thus, TAT(1-25) was not completely cleaved from the WNV-NY/TAT polyprotein.
Figure 2.
Characterization of WNV-NY/TAT. (A) RT-PCR analysis. RNA extracted from passage 1 (p1) and passage 5 (p5) WNV-NY/TAT-infected cells at 48 h post-infection was used as a template for RT-PCR amplification of the WNV C/CA region (WNV nts 97-500). RNA extracted from cultures infected with wild-type WNV-NY was used as a positive control. DNA molecular size standards (in nts) are indicated on the left; (B) Sequences. The p1 and p5 PCR products (Panel A) were sequenced by GeneWiz. The sequence encompassing the TAT(1-67) insertion in the WNV-NY/TAT infectious clone (I.C.) is shown at the top. The amino acid sequence is shown below with the WNV residues indicated in blue and the TAT(1-67) residues in orange; (C) Immunoblots. Vero monolayers were infected (MOI = 0.05) with WNV-NY or WNV-NY/TAT p0. Cell lysates prepared at the indicated times (h) were subjected to SDS-PAGE and immunoblot analysis with anti-TAT (top), anti-WNV (middle), or anti-actin (bottom) antibodies. Protein molecular size standards (in kilodaltons) are indicated on the left.
Figure 2.
Characterization of WNV-NY/TAT. (A) RT-PCR analysis. RNA extracted from passage 1 (p1) and passage 5 (p5) WNV-NY/TAT-infected cells at 48 h post-infection was used as a template for RT-PCR amplification of the WNV C/CA region (WNV nts 97-500). RNA extracted from cultures infected with wild-type WNV-NY was used as a positive control. DNA molecular size standards (in nts) are indicated on the left; (B) Sequences. The p1 and p5 PCR products (Panel A) were sequenced by GeneWiz. The sequence encompassing the TAT(1-67) insertion in the WNV-NY/TAT infectious clone (I.C.) is shown at the top. The amino acid sequence is shown below with the WNV residues indicated in blue and the TAT(1-67) residues in orange; (C) Immunoblots. Vero monolayers were infected (MOI = 0.05) with WNV-NY or WNV-NY/TAT p0. Cell lysates prepared at the indicated times (h) were subjected to SDS-PAGE and immunoblot analysis with anti-TAT (top), anti-WNV (middle), or anti-actin (bottom) antibodies. Protein molecular size standards (in kilodaltons) are indicated on the left.
![Viruses 06 01637 g002]()
To determine whether portions of the TAT(1-67) sequence were lost during the initial recovery of the virus or during the subsequent (p1) passage, we performed a second recovery of WNV-NY/TAT from Vero cells electroporated with
WNV-TAT(1-67) RNA. Similar to the first recovery, CPE was not observed within the electroporated monolayer. However, the titer of the stock recovered at 7 days post-transfection was substantially lower (35 pfu/mL) than in the initial recovery. Moreover, RT-PCR of total RNA isolated from the electroporated monolayer (p0) yielded a 600 nts band (
Figure 3A), consistent with the presence of the TAT(1-67) insert. Sequencing confirmed the presence of the entire TAT(1-67) coding region as well as coding sequence for the WNV protease cleavage sites (
Figure 3B). Therefore, insertion of the entire TAT cassette did not abrogate WNV-NY infectivity. However, as previously observed, a large portion of the TAT insert was deleted during the first passage of this virus.
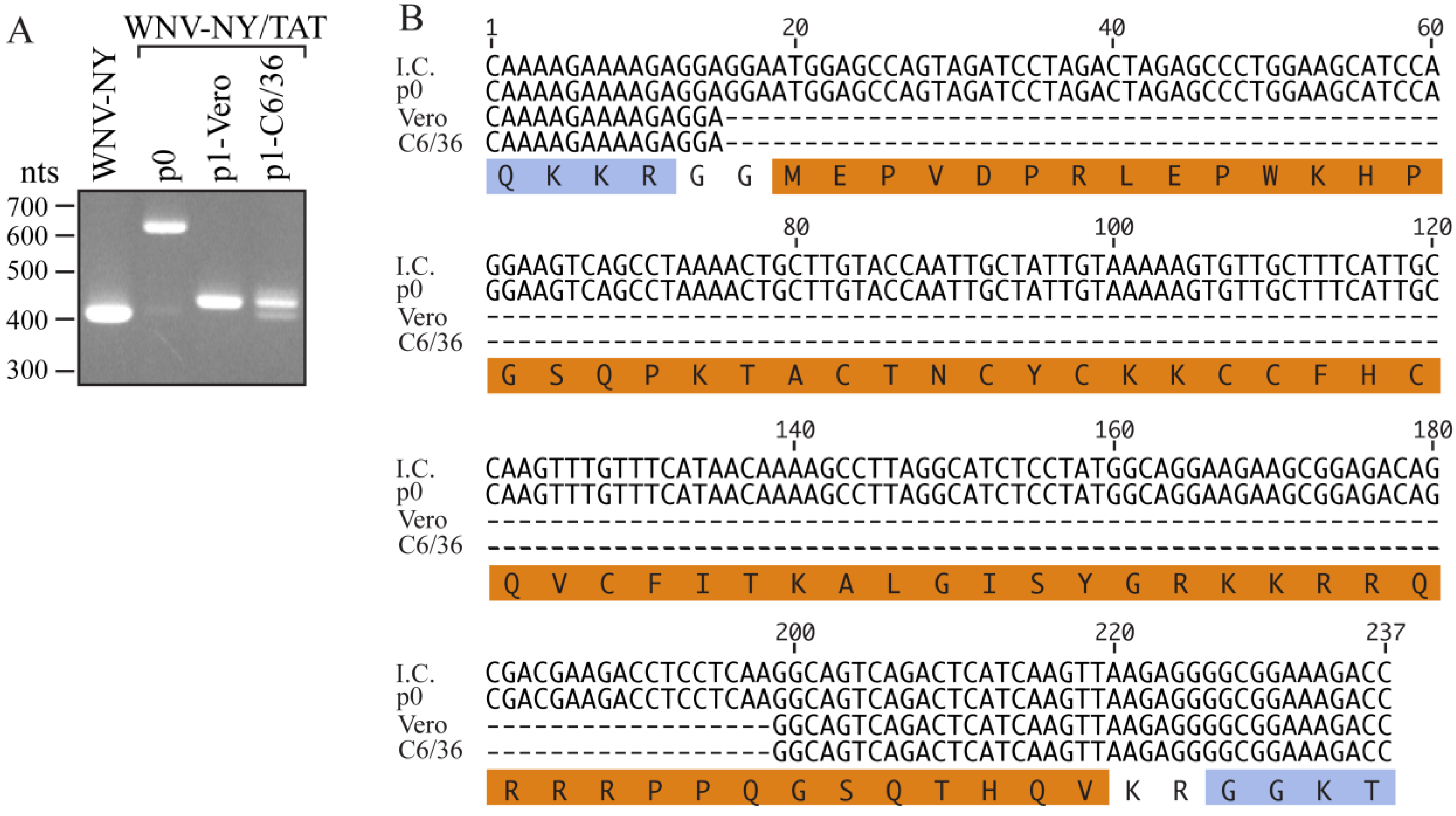
Since extension of the WNV Capsid protein negatively affects WNV replication in Vero cells, but not mosquito cells [
18], we hypothesized that the TAT(1-67) sequence may be more stable in insect cells. Therefore, the recovered WNV-NY/TAT p0 virus was passaged once in Vero or mosquito C6/36 cells. RT-PCR analysis of RNA extracted from the p1 cultures indicated that a substantial portion of the TAT cassette was missing (
Figure 3A). Sequence analyses revealed that the WNV-NY/TAT genomes produced during the Vero and C6/36 p1 amplifications retained only the coding sequence for residues 61-67 of the TAT insert (
Figure 3B), providing additional evidence that the full TAT(1-67) cassette is not well tolerated at the C/CA junction. It is unlikely that the identical deletions in the TAT(1-67) coding sequence arose independently during passage of the virus in two separate cell lines. Rather, the p0 population likely contained undetectable levels of the truncated WNV-NY/TAT(61-67) in addition to full-length WNV-NY/TAT(1-67). The loss of full-length WNV-NY/TAT(1-67) during passage, also suggested that WNV-TAT(61-67) significantly outcompeted WNV-TAT(1-67) and was preferentially amplified. Therefore, the full-length WNV-NY/TAT virus is not only unstable, but also less fit than wild-type WNV-NY.
Figure 3.
Passage of WNV-NY/TAT on insect cells. (A) RT-PCR analysis. RNA was extracted from cells transfected with WNV-NY/TAT RNA (p0) and Vero or C6/36 monolayers infected with the p0 virus (p1). The extracted RNA was used as template for RT-PCR amplification of the WNV C/CA region (WNV nts 97-500). RNA recovered from cultures infected with wild-type WNV-NY was used as a positive control. DNA molecular size standards (in nts) are indicated on the left; (B) Sequences. The p0 and p1 PCR products (Panel A) were sequenced by GeneWiz. The sequence encompassing the TAT(1-67) insertion in the WNV-NY/TAT infectious clone (I.C.) is shown at the top. The amino acid sequence is shown below with the WNV residues indicated in blue and the TAT(1-67) residues in orange.
Figure 3.
Passage of WNV-NY/TAT on insect cells. (A) RT-PCR analysis. RNA was extracted from cells transfected with WNV-NY/TAT RNA (p0) and Vero or C6/36 monolayers infected with the p0 virus (p1). The extracted RNA was used as template for RT-PCR amplification of the WNV C/CA region (WNV nts 97-500). RNA recovered from cultures infected with wild-type WNV-NY was used as a positive control. DNA molecular size standards (in nts) are indicated on the left; (B) Sequences. The p0 and p1 PCR products (Panel A) were sequenced by GeneWiz. The sequence encompassing the TAT(1-67) insertion in the WNV-NY/TAT infectious clone (I.C.) is shown at the top. The amino acid sequence is shown below with the WNV residues indicated in blue and the TAT(1-67) residues in orange.
2.2. Generation of WNV-NY/GLuc
Our findings suggested that the C/CA junction of WNV can tolerate additional sequences. However, the inability to recover stable full-length WNV-NY/TAT(1-67) suggested that the insert was subject to one or more of the following constraints: (1) the size of the full-length insert exceeded that which is tolerated by WNV; (2) specific sequences within the coding regions for TAT residues 26-60 affected WNV replication; or (3) the cellular activities of TAT(1-67) impeded virus replication. To distinguish between these possibilities, we generated a second infectious clone containing Gaussia luciferase (GLuc) [
27,
28] instead of TAT(1-67). A viral (p0) stock of 1.65 × 10
4 pfu/mL was recovered from Vero cells electroporated 7 days earlier with
WNV-NY/GLuc RNA, though no CPE was evident over this time period. The plaque phenotype of the recovered virus was highly variable. Some plaques were indistinguishable from those of wild-type WNV-NY, while others were significantly smaller [
26], suggesting the virus population was mixed. Indeed, when RNA isolated from the infected Vero cells was used as template for RT-PCR, two predominate PCR products were generated (
Figure 4A). The size of the smaller product was similar to that of wild-type WNV-NY, and sequence analysis indicated that it had entirely wild-type WNV-NY sequence within the C/CA region. In contrast, as confirmed by sequencing, the larger PCR product contained the entire GLuc insert. To test whether full-length WNV-NY/GLuc was stable within the virus population, we passaged the WNV-NY/GLuc p0 once in Vero cells for 48 h. Using RT-PCR, we readily detected a band of wild-type WNV-NY size, but did not detect any full-length WNV-NY/GLuc (
Figure 4A). Thus, wild-type WNV-NY outcompeted WNV-NY/GLuc and had a fitness advantage over WNV-NY/GLuc.
In an attempt to isolate a pure WNV-NY/GLuc population, we repeated the electroporation of Vero cells with
WNV-NY/GLuc RNA and collected culture supernatant at 4 and 7 days post-transfection. While only 75 pfu/mL of virus was recovered on day 4, the plaques had a uniformly small plaque phenotype [
26], indicative of a single virus population. In contrast, virus recovered on day 7 displayed both small- and large-plaque phenotypes, similar to those observed in the previous isolation. RT-PCR analysis of RNA extracted on day 7 post-electroporation produced multiple products of various sizes (
Figure 4B). The large, ~1000 nts product contained the full-length GLuc insert, with two nucleotide changes (TG➔CC) at position 26-27 and a three-nucleotide deletion at position 28-30, which together resulted in a two-residue mutation (ALIC➔APC) within the GLuc N-terminus (
Figure 4C). The smaller product at ~400 nts was the same size as wild-type WNV-NY, but retained the first 9 nts of the GLuc gene and lacked the first 15 nts of the CA sequence, which led to a KTG➔MGV mutation within the resulting polyprotein. Thus, the day 7 p0 virus was a mixed population. Moreover, the only virus detected by RT-PCR following a single passage of this population in Vero cells was the smaller KTG➔MGV virus (
Figure 4B), suggesting that the GLuc sequence, like the TAT(1-67) sequence, was unstable. This instability was further confirmed by the observation that a single passage of the day 4 p0 virus, which had produced only small plaques, yielded a virus stock of mixed small- and large-plaque phenotypes [
26] and RT-PCR products of variable lengths (
Figure 4B).
Figure 4.
Characterization of WNV-NY/Gluc. (A) RT-PCR analysis. RNA extracted from cells transfected with WNV-NY/TAT RNA (p0) and cultures infected with the p0 stock (p1) were used as templates for RT-PCR amplification of the WNV C/CA region (WNV nts 97-500). RNA recovered from cultures infected with wild-type WNV-NY was used as a positive control. DNA molecular size standards (in nts) are indicated on the left; (B) RT-PCR analysis of passaged WNV-NY/Gluc. Vero cells were transfected with infectious WNV-NY/GLuc RNA. A portion of the culture supernatant (p0) was collected after 4 days (d4). The remaining supernatant and total RNA was collected at 7 days (d7) post-transfection. Total RNA was extracted from Vero cells infected with d4 or d7 p0 stocks (d4-p1 and d7-p1). Extracted RNA was used as template for RT-PCR as described in Panel A; (C) Sequences. The d7 p0 large (Lg) and small (Sm) PCR products (Panel B) were sequenced by GeneWiz. The sequence encompassing the GLuc-encoding region of the WNV-NY/GLuc infectious clone (I.C.) is shown at the top. The amino acid sequence is shown below with WNV residues indicated in blue and GLuc residues in orange. Bullets represent additional GLuc residues that are not shown but are present in the I.C. and Lg product. The gray box highlights the residues that are different between I.C. and Lg; (D) Plaque purification of WNV-NY/Gluc. The d4 p0 stock (Panel B) was also used to inoculate a confluent monolayer of Vero cells, which were then overlaid with 0.9% agarose/DMEM. After 4 days, two plaques (pp1 and pp2) were picked, incubated with 1 mL DMEM, and used to inoculate Vero cells. After an additional 4 days, culture supernatants were collected. RNA was extracted from the infected Vero cells and used as template for RT-PCR as described in Panel A; (E) RT-PCR analysis of pp1 and pp2 serial passages. The pp1 and pp2 viruses were passaged twice in Vero cells to generate p1 and p2. RNA extracted from the infected Vero cells was used as template for RT-PCR as described in Panel A.
Figure 4.
Characterization of WNV-NY/Gluc. (A) RT-PCR analysis. RNA extracted from cells transfected with WNV-NY/TAT RNA (p0) and cultures infected with the p0 stock (p1) were used as templates for RT-PCR amplification of the WNV C/CA region (WNV nts 97-500). RNA recovered from cultures infected with wild-type WNV-NY was used as a positive control. DNA molecular size standards (in nts) are indicated on the left; (B) RT-PCR analysis of passaged WNV-NY/Gluc. Vero cells were transfected with infectious WNV-NY/GLuc RNA. A portion of the culture supernatant (p0) was collected after 4 days (d4). The remaining supernatant and total RNA was collected at 7 days (d7) post-transfection. Total RNA was extracted from Vero cells infected with d4 or d7 p0 stocks (d4-p1 and d7-p1). Extracted RNA was used as template for RT-PCR as described in Panel A; (C) Sequences. The d7 p0 large (Lg) and small (Sm) PCR products (Panel B) were sequenced by GeneWiz. The sequence encompassing the GLuc-encoding region of the WNV-NY/GLuc infectious clone (I.C.) is shown at the top. The amino acid sequence is shown below with WNV residues indicated in blue and GLuc residues in orange. Bullets represent additional GLuc residues that are not shown but are present in the I.C. and Lg product. The gray box highlights the residues that are different between I.C. and Lg; (D) Plaque purification of WNV-NY/Gluc. The d4 p0 stock (Panel B) was also used to inoculate a confluent monolayer of Vero cells, which were then overlaid with 0.9% agarose/DMEM. After 4 days, two plaques (pp1 and pp2) were picked, incubated with 1 mL DMEM, and used to inoculate Vero cells. After an additional 4 days, culture supernatants were collected. RNA was extracted from the infected Vero cells and used as template for RT-PCR as described in Panel A; (E) RT-PCR analysis of pp1 and pp2 serial passages. The pp1 and pp2 viruses were passaged twice in Vero cells to generate p1 and p2. RNA extracted from the infected Vero cells was used as template for RT-PCR as described in Panel A.
![Viruses 06 01637 g004]()
2.3. Plaque Purification of WNV-NY/GLuc Virus
To further examine the stability of WNV containing the full-length GLuc insert, we plaque purified two clones from the day 4 p0 stock of WNV-NY/GLuc. The two plaques, pp1 and pp2, were amplified for 96 h in Vero cells (p0). RT-PCR of total RNA isolated from p0 cultures yielded a band of ~1000 nts, suggesting that the population consisted predominately of full-length WNV-NY/GLuc (
Figure 4D). Sequence analyses revealed that both pp1 and pp2 had the full-length GLuc(ALIC➔APC) insert. Thus, the ALIC➔APC mutation was introduced prior to 4 days after transfection of the infectious RNA.
To test the stability of the plaque-purified clones, we passaged the pp1 and pp2 stocks twice in Vero cells and extracted total RNA from the infected cells for RT-PCR analysis and sequencing. While WNV-NY/GLuc(ALIC➔APC)-pp1 retained the full-length GLuc(ALIC➔APC) insert through both passages, WNV-NY/GLuc(ALIC➔APC)-pp2 became a mixed population within a single passage (
Figure 4E). Moreover, following two passages, the WNV-NY/GLuc(ALIC➔APC)-pp2 population consisted almost entirely of virus lacking a substantial portion of the GLuc(ALIC➔APC) insert. Since the PCR products were distinctly larger than that of wild-type WNV-NY, the p2 population most likely consisted of virus retaining only a portion of the GLuc(ALIC➔APC) insert. Sequence analysis of the WNV-NY/GLuc(ALIC➔APC)-pp2 p2 PCR product returned a mixed sequence, indicative of the presence of multiple virus species. Thus, the GLuc insertion is not well tolerated at the C/CA junction. Furthermore, the instability of the GLuc(ALIC➔APC) mutant virus (
Figure 4B–E) suggests that the ALIC➔APC mutation does not provide any appreciable fitness advantage, though WNV-NY/GLuc(ALIC➔APC)-pp1 may contain compensatory mutations outside of the C/CA region that offer some stability to the recombinant virus. The inability to recover a WNV-NY/GLuc virus containing an insertion of comparable size to TAT(1-25) (~75 nts) suggests that, in addition to size, the sequence of any additional nucleotides at the C/CA junction influences the stability of resulting viruses. Therefore, this site may tolerate an insertion larger than 75 nts if the sequence is sufficiently inert.
Numerous GLuc-reactive bands were detected in WNV-NY/GLuc(ALIC➔APC)-pp1 or -pp2-infected cells [
26], indicating that the GLuc reporter was translated. However, we did not detect GLuc activity in cell lysates or culture supernatants of Vero cells infected with WNV-NY/GLuc(ALIC➔APC)-pp1 or -pp2 (
Figure S3), suggesting that the virus does not produce functional GLuc. The lack of functional GLuc may be due to the ALIC➔APC mutation altering the protein’s activity. Alternatively, within the context of the virus, GLuc may not be properly folded, or incomplete cleavage of GLuc from the rest of the WNV polyprotein may impede GLuc function.
2.4. Effect of Insertions on WNV Replication
Because the truncated WNV-NY/TAT(1-25) was stable, we hypothesized that the TAT(1-25) insertion had little effect on virus fitness. Therefore, we compared wild-type WNV-NY and WNV-NY/TAT(1-25) replication in Vero cells. Both viruses exhibited similar replication kinetics and reached peak titers at 48 h post infection (
Figure 5A). While we did observe slightly higher peak viral titers for wild-type WNV-NY compared to WNV-NY/TAT(1-25), the difference was not statistically significant. Thus, insertion of the TAT(1-25) sequence did not significantly alter WNV-NY fitness.
Figure 5.
Replication of WNV-NY recombinant viruses. (A) WNV-NY/TAT(1-25). Vero monolayers were infected (MOI = 0.05) with WNV-NY or WNV-NY/TAT(1-25) p0; (B) WNV-NY/GLuc(ALIC➔APC). Vero monolayers were infected (MOI = 0.005) with WNV-NY or WNV-NY-GLuc(ALIC➔APC)-pp1 or -pp2 p0. Culture supernatants were collected at the indicated times after infection and infectious particle production determined by plaque assay on Vero cells. Values represent the average number of pfu per mL of supernatant (± standard deviation) from three independent experiments.
Figure 5.
Replication of WNV-NY recombinant viruses. (A) WNV-NY/TAT(1-25). Vero monolayers were infected (MOI = 0.05) with WNV-NY or WNV-NY/TAT(1-25) p0; (B) WNV-NY/GLuc(ALIC➔APC). Vero monolayers were infected (MOI = 0.005) with WNV-NY or WNV-NY-GLuc(ALIC➔APC)-pp1 or -pp2 p0. Culture supernatants were collected at the indicated times after infection and infectious particle production determined by plaque assay on Vero cells. Values represent the average number of pfu per mL of supernatant (± standard deviation) from three independent experiments.
Unlike the TAT(1-25) insert, the GLuc inserts were unstable in WNV-NY, suggesting that they conferred a fitness disadvantage to the virus. To test this hypothesis, we also compared the replication kinetics of WNV-GLuc(ALIC➔APC)-pp1 and -pp2 with wild-type WNV-NY. WNV-NY replication was readily detected by 24 h after infection and reached peak levels by 48 h (
Figure 5B). In comparison, WNV-NY/GLuc(ALIC➔APC)-pp1 and -pp2 infectious particle production was delayed and peak virus levels, which occurred at 72 h, were significantly reduced. Thus, unlike WNV-NY/TAT(1-25), WNV-NY/GLuc(ALIC➔APC) had a substantial growth disadvantage compared to wild-type virus, indicating that the GLuc(ALIC➔APC) insert reduced viral fitness.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}