1. Introduction
Begomoviruses are small, circular, single-stranded (ss) DNA viral pathogens (family,
Geminiviridae; genus,
Begomovirus) of plants that infect a wide range of eudicots in the tropics and subtropics. The genome size of this genus ranges in size from 2.8 to 5.2 kb, and is arranged either in two components (bipartite) referred to as DNA-A and DNA-B, each approximately 2.6 kb, or in a single component (monopartite), approximately ~2.8 kb in size [
1]. A number of begomoviruses endemic to the Eastern Hemisphere have a monopartite genome, and are associated with circular, ssDNA alpha- and beta-satellites that are about half the size of the genome of the “helper” begomovirus, at 1.4 kb, upon which they rely for aspects of the infection cycle [
2,
3]. Also, smaller than expected circular, ssDNA satellite molecules of ~0.7 kb in size are frequently detected together with begomoviruses in plant DNA extracts [
4], as well as smaller than unit size (and variable in length) helper genome or satellite, some of which have been shown to function as defective interfering (DI) sequences [
5]. However, the biological functions of DI DNAs with respect to the begomoviral infection cycle, virulence, or evolution are not well understood.
In most instances in which a helper begomovirus has been found to be associated with a beta-type satellite, it has been shown that a satellite is required by the helper virus to systemically infect the plant host, and to contribute to the development of characteristic disease symptoms [
6,
7]. These phenomena are in part made possible by the suppressor activity of the betasatellite-encoded protein, β
C1 that silences the plant host response to viral infection [
8]. Further betasatellites are encapsidated into virions for transmission from plant to plant, along with the helper virus, by the whitefly vector
Bemisia tabaci (Gennadius) sibling species group [
9,
10].
The alpha-type of satellite on the other hand has been shown to be dispensable for systemic infection of the plant when the betasatellite and the helper virus are both present. Most experimental studies have used the plant viral-permissive host,
Nicotiana benthamiana (Domin), instead of naturally infected plant species, resulting in the obfuscation of the possible role of associated satellites in the infection cycle [
11].
Begomoviruses that are associated with DNA satellites represent a pathogen complex. Neither the ssDNA helper virus nor the betasatellite molecule encode a DNA polymerase, and so both must replicate in the plant nucleus using the host polymerase and a double-stranded DNA form by rolling circle replication (RCA) [
12,
13,
14]. Recombination and mutations that occur during the replication cycle are important genetic processes that give rise to genomic and genetic variability presumably due to the ability to adapt to new environments, including host genetic variability [
15,
16,
17]. The contemporary experimental approaches used for detecting and quantifying the extent of genetic variability within begomoviral genomes and the associated satellites employ either polymerase-mediated amplification (as much as 10,000 fold in a few hours) of viral genomic DNA by polymerase chain reaction (PCR) [
18] using sequence-specific (or degenerate) primers, or phi29 DNA polymerase-mediated rolling circle amplification (RCA) through the use of random primers [
19], followed by cloning and capillary DNA sequencing. However, these strategies produce limited information about the presumed predominant genomes, given that the depth of sequencing is low, that precludes assessment of the genetic differentiation and population structure of begomovirus-satellite variants. Indeed, the RCA or PCR amplification methods can be biased to unknown extents during early amplification steps that select for the most abundant viral and satellite DNA variants, and other potential artifacts of the methodologies.
Typically, the complete genome of monopartite or bipartite DNA components of begomoviruses and their associated circular, ssDNA satellites are cloned from the products of RCA [
20,
21] (commercial application available through GE Healthcare, Life Sciences, Piscataway, NJ, USA) or virus-specific PCR [
21]. Also, begomoviral genome characterization has been achieved using a combination of RFLP (restriction fragment length polymorphism) and pyro-sequencing, referred to as “circomics” [
22]. The amplification of begomoviral genomic and associated components by PCR, followed by cloning and capillary DNA sequencing, are limited by the specificity of the primers, and by the number of variants produced by the earliest amplification steps, and then by selection during the molecular cloning step. Typically, RCA produces high molecular weight products as dsDNA concatemers that are digested into unit-length components and cloned, with the inserts verified by capillary sequencing [
19,
23]. This is a time-consuming process, and in addition, a limited number of variants are represented among the resultant clones, based on the expectation that one or a few predominant genotypes are represented in the starting material. However, the innovative approach described here employs the robustness of the bacteriophage phi29 DNA polymerase used in RCA technology, together with deep sequencing using Illumina [
24,
25] and bioinformatics to assess population diversity of begomoviruses and their satellites in naturally infected plants.
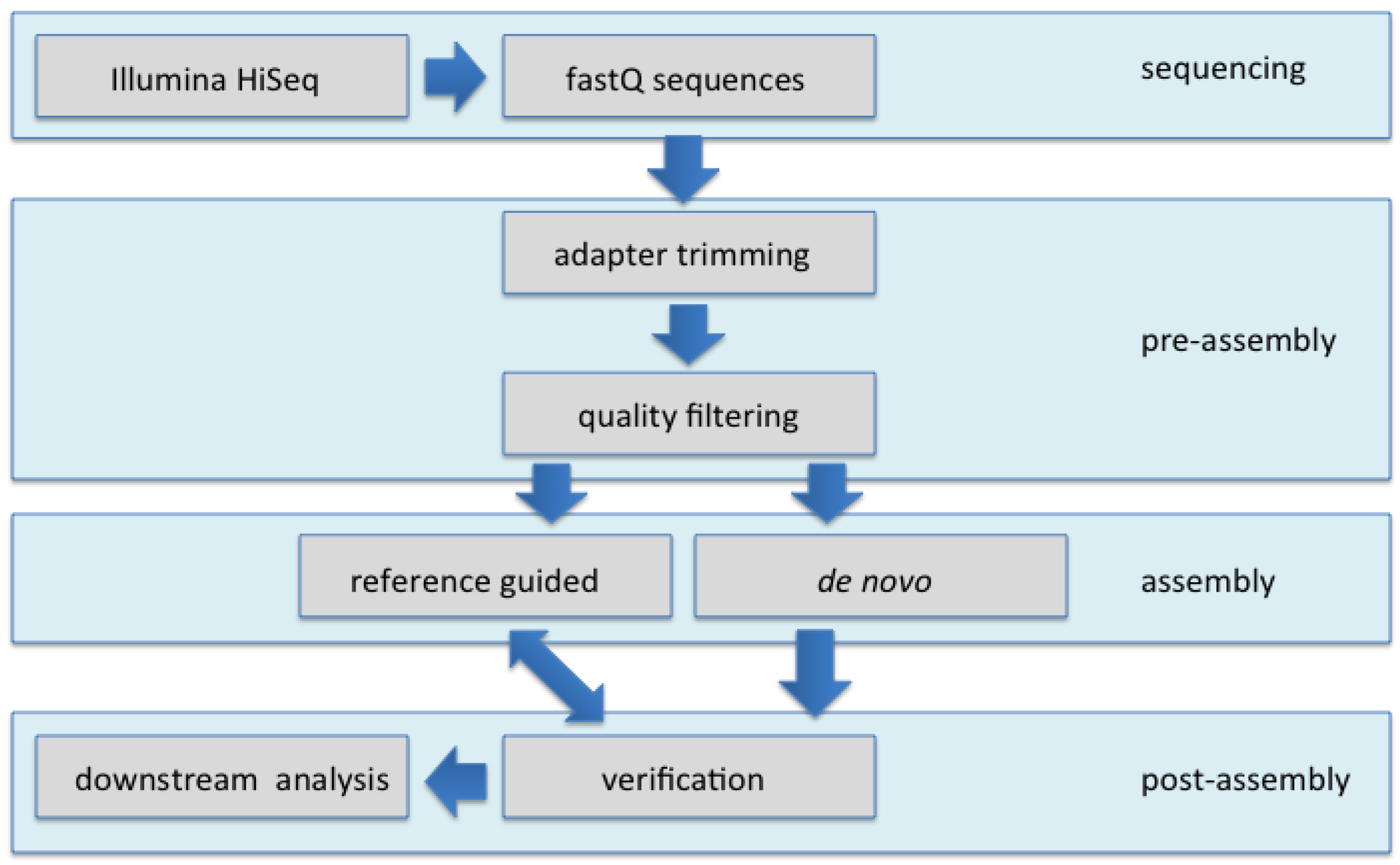
For virus discovery from field samples at the population level, traditional methods such as RCA, or PCR followed by cloning and traditional DNA sequencing, are ineffective, particularly for detecting rare members in a population, or those containing minimal, single nucleotide (nt) polymorphisms in begomoviral populations, where mixtures of isolates, strains, and multiple species prevail. In addition, technical limitations can result in the inability to detect low- abundant begomoviral and/or associated DNA satellite molecules. In this study, circular, single-stranded DNA-containing begomoviruses and their satellites were enriched by RCA from total DNA extracts of naturally infected, symptomatic field plants. The enriched begomoviral genomes and satellites were subjected to Illumina-NGS, assembled, and subjected to analysis to detect polymorphisms.
2. Results and Discussion
The
de novo assembly of the high-throughput Illumina 10 million (10M) reads was carried out using SeqMan NGen3 (DNASTAR Inc., Madison, WI, USA) software, and resulted in a large number of contigs that ranged from 247 and 2244 (
Table 1). The contigs were used to search the NCBI-GenBank database to identify the most closely related begomovirus exemplars. The search result showed that the contigs containing begomoviruses and begomovirus-associated DNA satellite sequences consistently comprised the highest number of assembled reads, indicating that the RCA successfully enriched for these components.
The preliminary
de novo assembly of the resultant high-throughput sequence data (using the default setting) that consisted of 10 million reads, proved to be time-consuming for these relatively short genomic components, and further yielded a higher than necessary depth of coverage, and so was found to be unnecessarily wasteful (
Table 1). This observation was confirmed by reducing the number of reads used in subsequent assemblies of both
de novo and reference-guided assemblies to 100,000 (
Table 1 and
Table 2), from which results were similar to those using the 10M reads, while still achieving a adequate coverage (
Figure 1).
Table 1.
The de novo assembly statistics of enriched begomovirus genomes and satellite molecules.
Table 1.
The de novo assembly statistics of enriched begomovirus genomes and satellite molecules.
| Sample | Number of Contigs | N50 (kb) | Average Read Length (bp) | Average Phred Read Quality | Number of Assembled Viral Components |
|---|
| 10 M | 100 K | 10 M | 100 K | 10 M | 100 K | 10 M | 100 K |
|---|
| KSA27 | 644 | 13 | 3 | 1.993 | 94 | 99 | 33 | 35 | 1 helper virus
2 alphasatellites
1 betasatellite |
| KSA46 | 2244 | 19 | 3 | 3 | 96 | 97 | 34 | 34 | 1 helper virus |
| G11 | 1585 | 7 | 3 | 3 | 94 | 99 | 34 | 34 | 1 helper virus |
| Control | 247 | 5 | 3 | 2 | 94 | 98 | 34 | 34 | 1 helper virus
2 betasatellites |
Table 2.
Reference-guided assembly statistics for the Illumina sequences obtained for the RCA-enriched begomovirus-satellite complexes. Reference genomes were obtained from the de novo assembly. CLCuGV = Cotton leaf curl Gezira virus, ToLCSDV = Tomato leaf curl Sudan virus, TYLCV-OM = Tomato yellow leaf curl virus from Oman, TYLCB = Tomato yellow leaf curl betasatellite, ToLCSDB = Tomato leaf curl Sudan betasatellite, CLCuGB = Cotton leaf curl Gezira betasatellite, DNA1 and DNA2 are alphasatellites.
Table 2.
Reference-guided assembly statistics for the Illumina sequences obtained for the RCA-enriched begomovirus-satellite complexes. Reference genomes were obtained from the de novo assembly. CLCuGV = Cotton leaf curl Gezira virus, ToLCSDV = Tomato leaf curl Sudan virus, TYLCV-OM = Tomato yellow leaf curl virus from Oman, TYLCB = Tomato yellow leaf curl betasatellite, ToLCSDB = Tomato leaf curl Sudan betasatellite, CLCuGB = Cotton leaf curl Gezira betasatellite, DNA1 and DNA2 are alphasatellites.
| Sample | Average Depth of Coverage | N50 (kb) | Identity of the Assembled Viral Components |
|---|
| 10 M | 100 K | 10 M | 100 K |
|---|
| KSA27 | 49,733 | 766 | 1.550 | 1.532 | CLCuGV, CLCuGB, DNA1, DNA2 |
| KSA46 | 11,853 | 125 | 2 | 2 | ToLCSDV |
| G11 | 155,410 | 1480 | 2 | 2 | ToLCSDV |
| Control | 47,135 | 1455 | 1.536 | 1.527 | TYLCV-OM, TYLCB and ToLCuSDB |
The BLASTn results indicated that only one kind of begomoviral genomic component (EMBL accession number HG530539) (
Figure 2) was obtained from the field tomato sample (KSA46) collected in Saudi Arabia. This 2788-nt-long viral genome shared 87%–92% nt identity with several
Tomato leaf curl Sudan virus (ToLCSDV) strains/isolates reported previously from the Nile Basin and neighboring Yemen and Oman [
4,
11]. The genome organization of the assembled ToLCSDV was found to be similar to other strains reported from the region [
11]. However, no begomovirus-associated DNA satellite molecule was identified from this tomato field sample. In contrast, one helper virus, a betasatellite, and two types of alphasatellite (
Figure 3) were found to be present in the field okra sample (KSA27), indicating that single and multiple genomic components for helper viruses and/or satellites were reliably detectable when present in begomovirus-infected field plants.
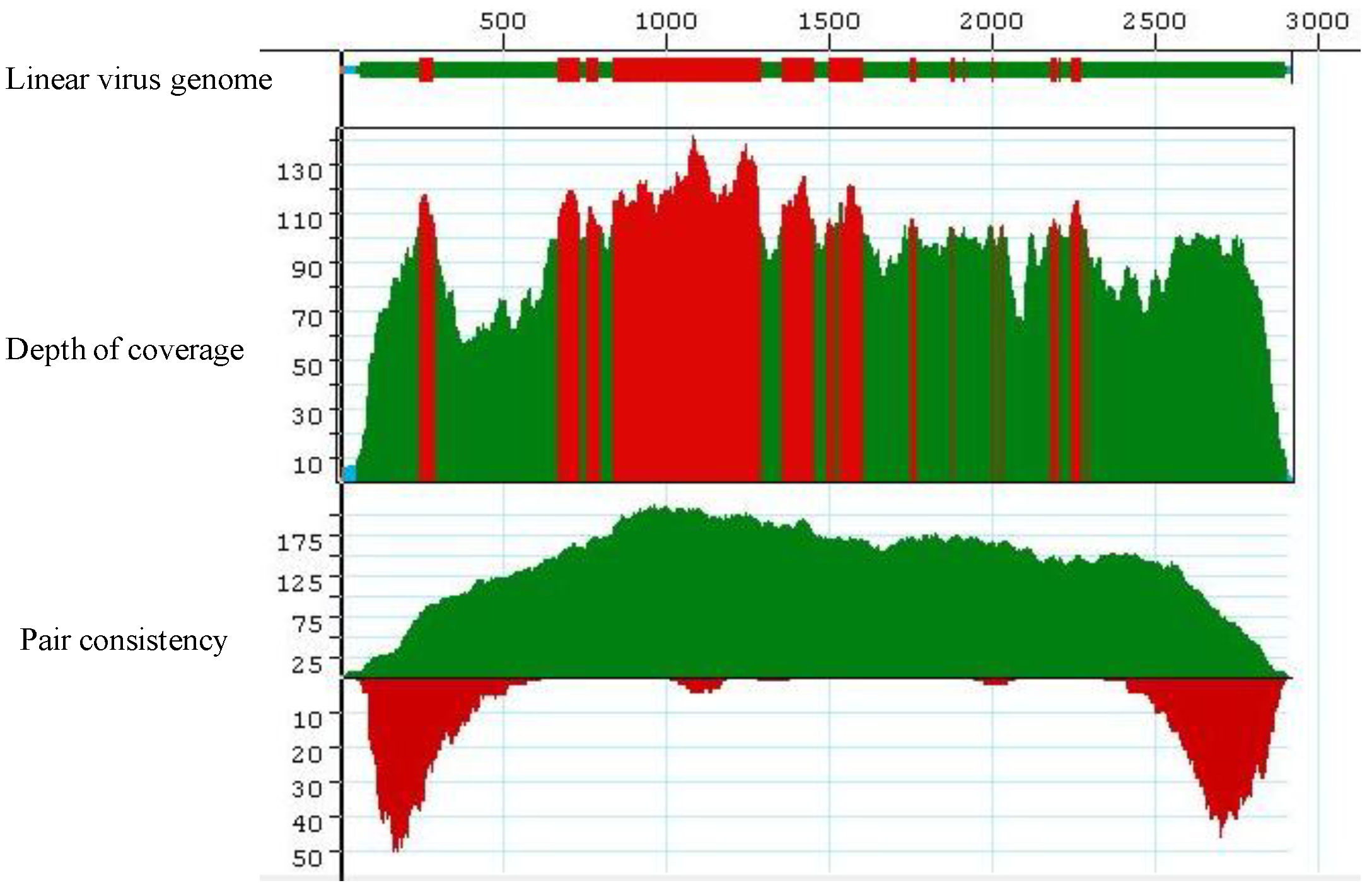
Figure 1.
Complete coverage of the helper virus Cotton leaf curl Gezira virus (field sample KSA27) in reference-guided assembly. 100,000 Illumina short reads were used in this assembly. The red, green and cyan colors in the Coverage Threshold Graph for the linearized (with ori set as coordinate1) virus genome represent various levels of mapping of sequencing reads onto the reference viral genome, Cotton leaf curl Gezira virus. Red color exemplifies regions that exceed the maximum expected coverage level (set for 100 sequences at each position). The color green denotes regions sequenced on both strands that fell above coverage threshold (set for 4 sequences at each position). The color cyan denotes regions sequenced from only one strand. The colors in the Depth of Coverage graph represent the same regions as those in the Coverage Threshold graph. The pair consistency histogram was relatively low at the beginning and the end of the genome, an observation that could reflect the circular nature of the genome.
Figure 1.
Complete coverage of the helper virus Cotton leaf curl Gezira virus (field sample KSA27) in reference-guided assembly. 100,000 Illumina short reads were used in this assembly. The red, green and cyan colors in the Coverage Threshold Graph for the linearized (with ori set as coordinate1) virus genome represent various levels of mapping of sequencing reads onto the reference viral genome, Cotton leaf curl Gezira virus. Red color exemplifies regions that exceed the maximum expected coverage level (set for 100 sequences at each position). The color green denotes regions sequenced on both strands that fell above coverage threshold (set for 4 sequences at each position). The color cyan denotes regions sequenced from only one strand. The colors in the Depth of Coverage graph represent the same regions as those in the Coverage Threshold graph. The pair consistency histogram was relatively low at the beginning and the end of the genome, an observation that could reflect the circular nature of the genome.
![Viruses 06 01219 g001]()
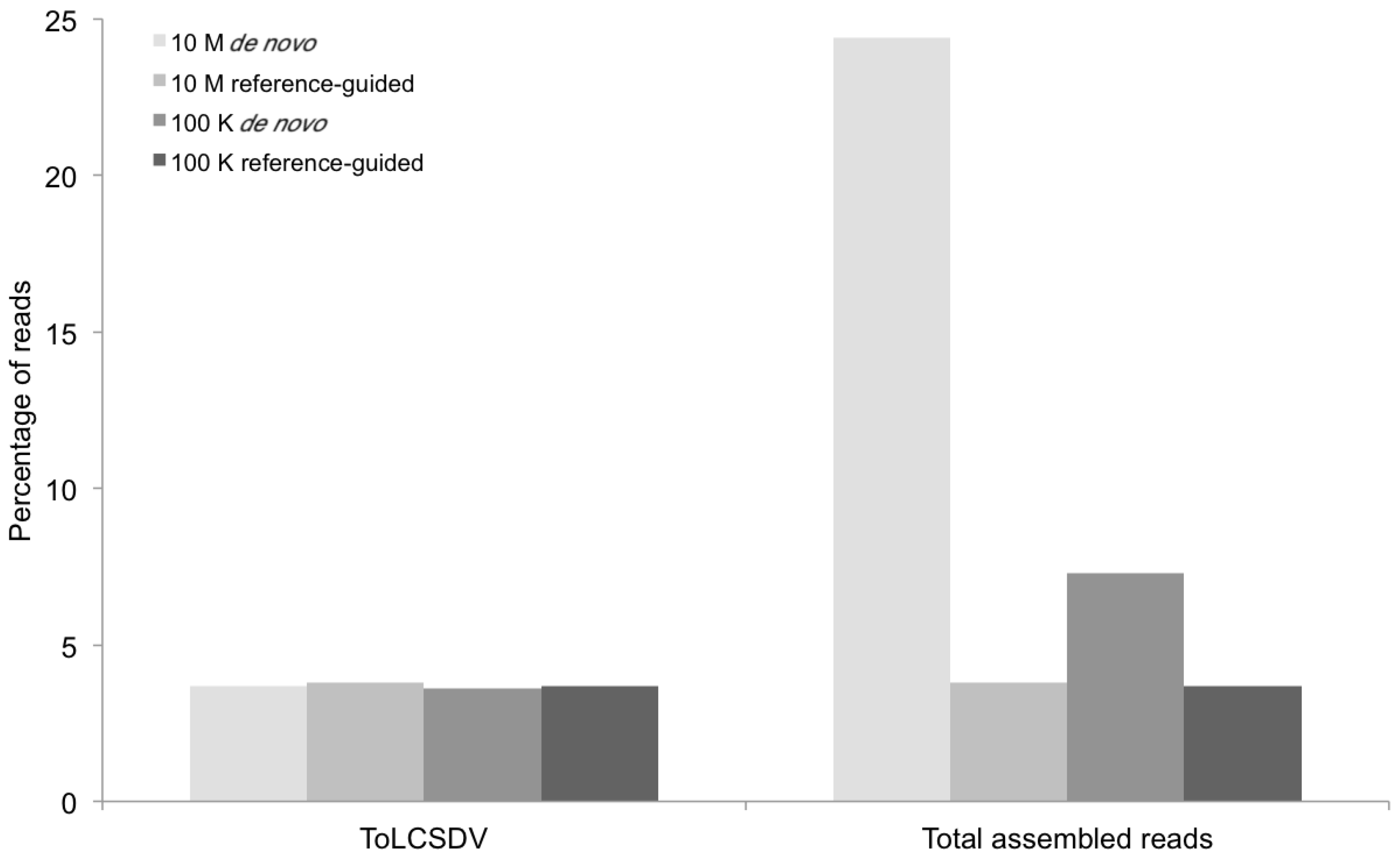
Figure 2.
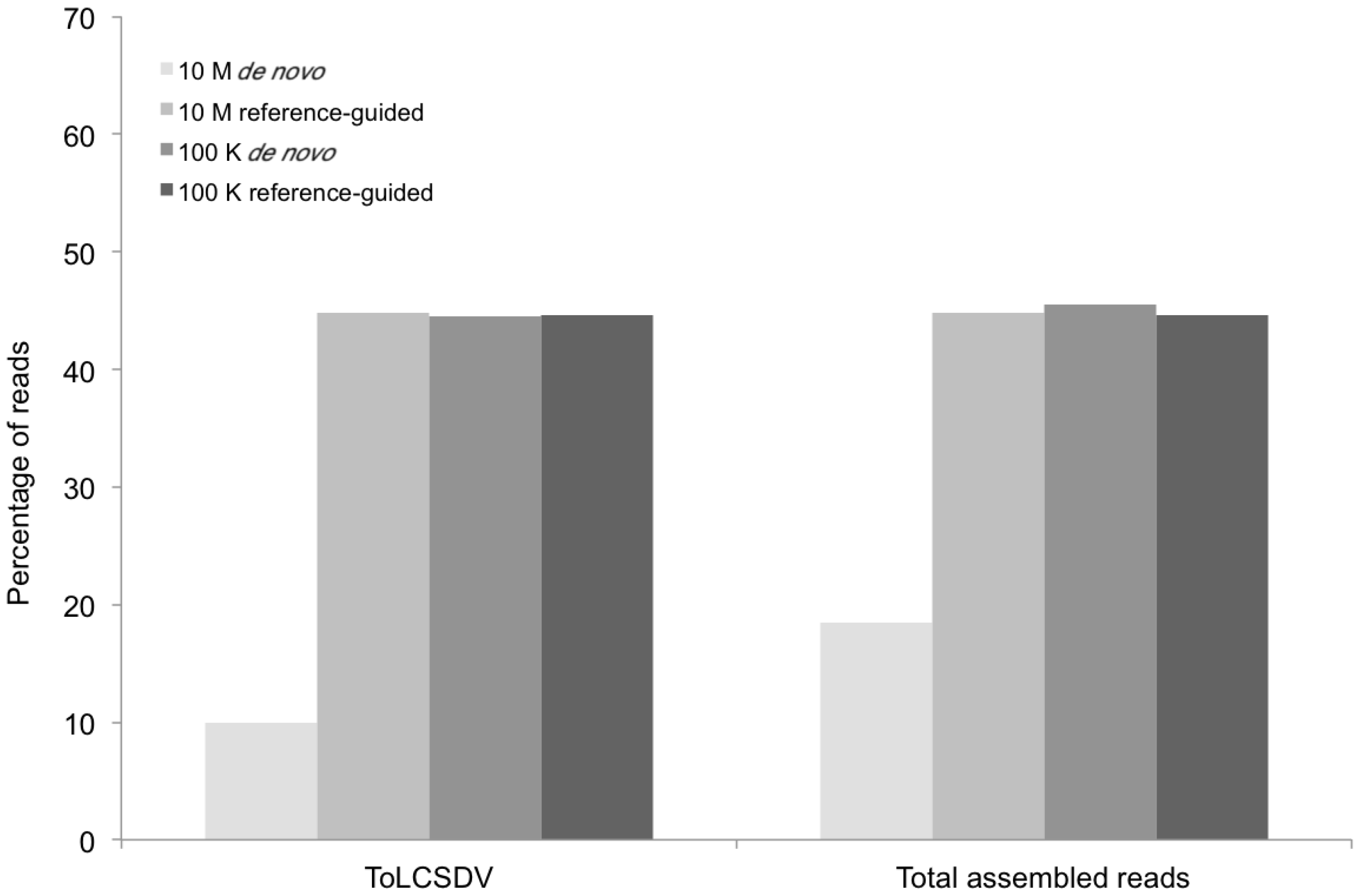
Percentage of DNA sequence reads (Y axis) mapped at different levels of sequence depth that contributed the begomoviral contigs obtained from sample KSA46 (X axis) resulting from de novo and reference-guided assembly. 10 M = 10 million reads, 100 K = 100 thousand reads, ToLCSDV = Tomato leaf curl Sudan virus.
Figure 2.
Percentage of DNA sequence reads (Y axis) mapped at different levels of sequence depth that contributed the begomoviral contigs obtained from sample KSA46 (X axis) resulting from de novo and reference-guided assembly. 10 M = 10 million reads, 100 K = 100 thousand reads, ToLCSDV = Tomato leaf curl Sudan virus.
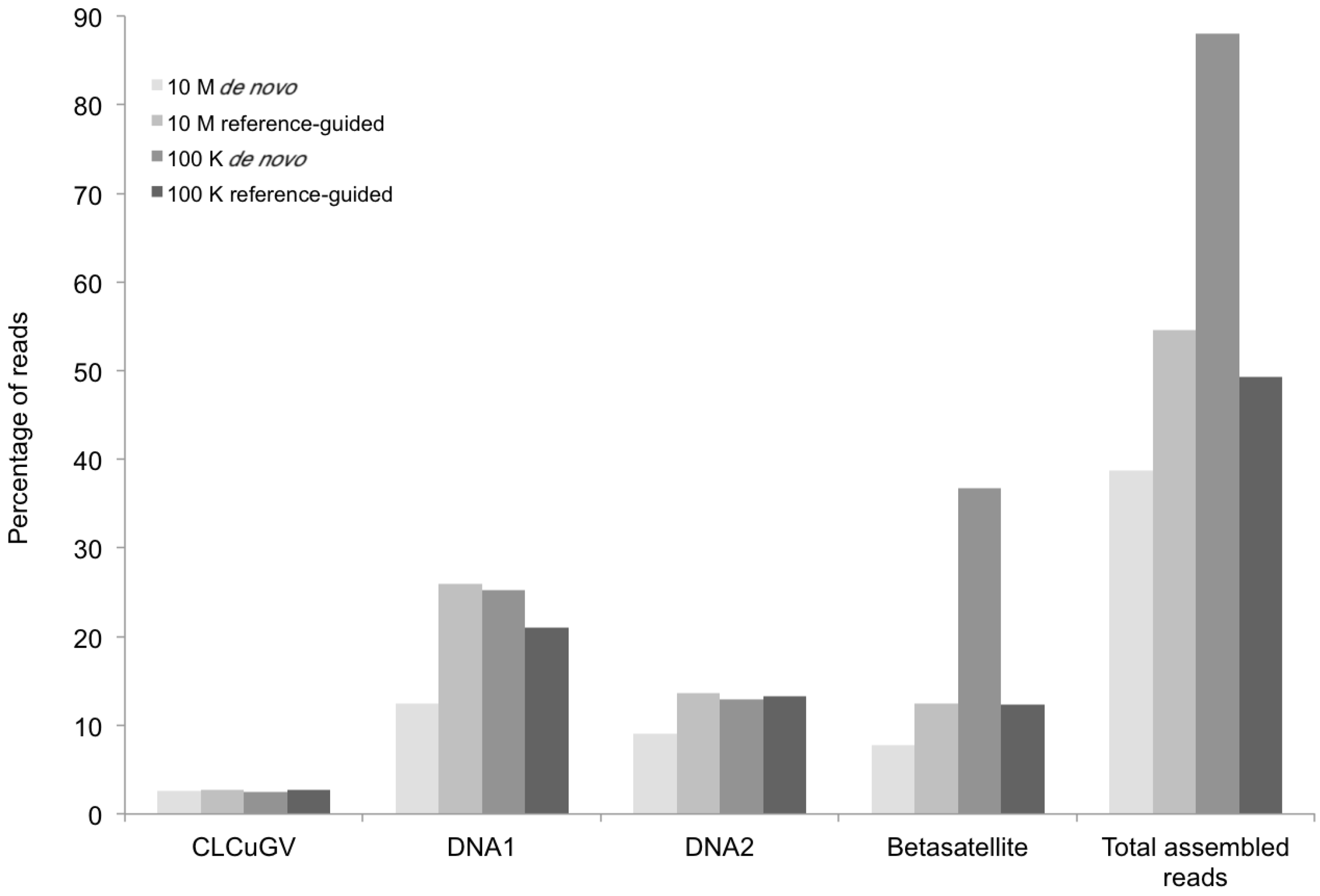
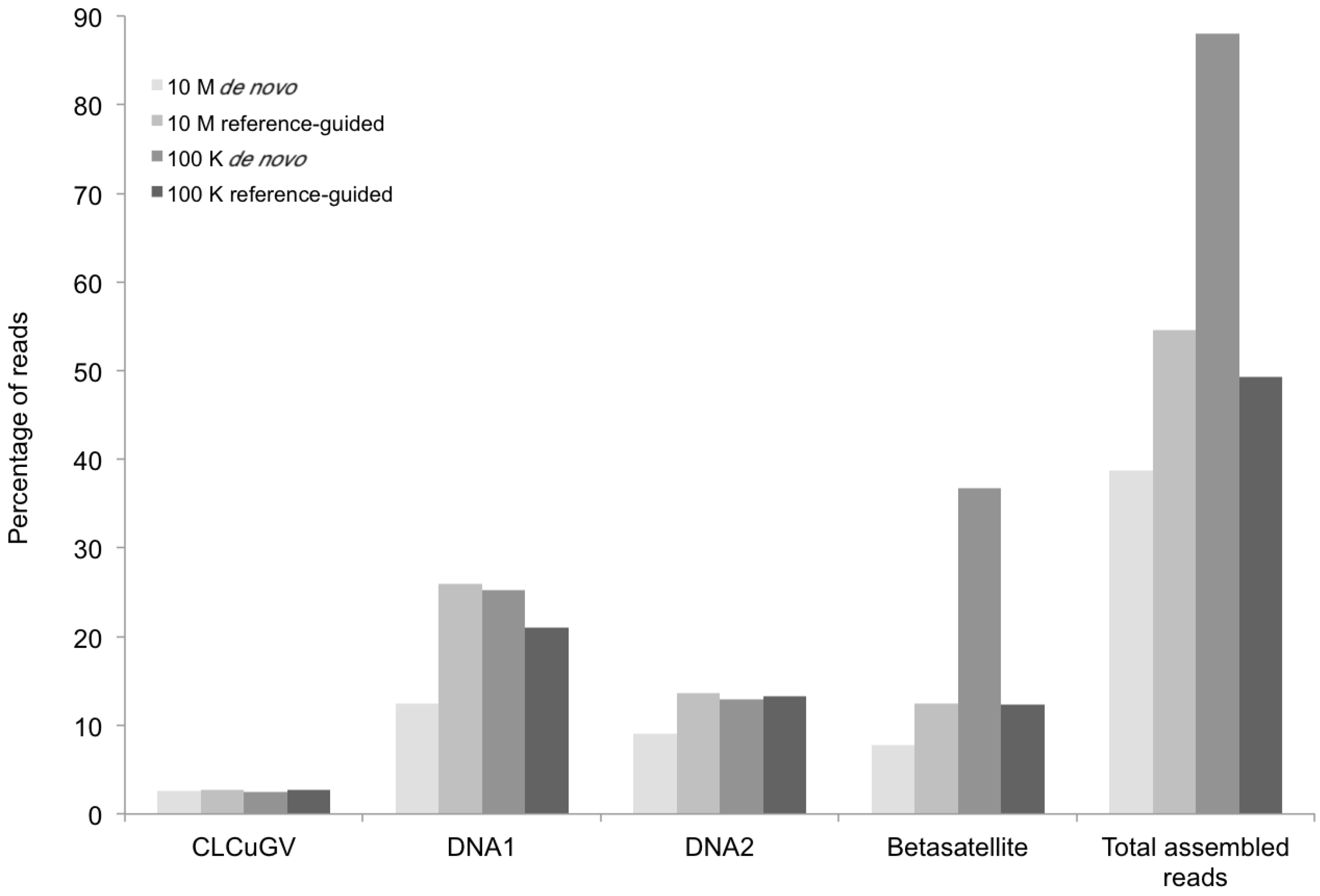
Figure 3.
Percentage of DNA sequence reads (Y axis) mapped at different levels of sequence depth that contributed the begomoviral contigs obtained from sample KSA27 (X axis) resulting from de novo and reference-guided assembly. 10 M = 10 million reads, 100 K = 100 thousand reads, CLCuGV = Cotton leaf curl Gezira virus, DNA1 and DNA2 are new alphasatellites and betasatellites, respectively.
Figure 3.
Percentage of DNA sequence reads (Y axis) mapped at different levels of sequence depth that contributed the begomoviral contigs obtained from sample KSA27 (X axis) resulting from de novo and reference-guided assembly. 10 M = 10 million reads, 100 K = 100 thousand reads, CLCuGV = Cotton leaf curl Gezira virus, DNA1 and DNA2 are new alphasatellites and betasatellites, respectively.
The DNA sequence comparisons indicated that the helper begomovirus, at 2780 nt in size, (EMBL Accession Number HG530540) from okra shared 92% identity with
Cotton leaf curl Gezira virus (CLCuGV), and that the betasatellite (EMBL Accession Number HG530542) also was related to
Cotton leaf curl Gezira betasatellite (CLCuGB) [
7], at 61% shared nt identity. This assembled CLCuGV has genome organization similar to other CLCuGV strains reported from the Nile Basin. However, the 681-nt-long betasatellite identified in okra plant sample (KSA27) was (herein) considered to be “defective”, owing to the absence of the essential β
C1 ORF. Because the
de novo assembled betasatellite was divergent from its closest relatives, and it lacked the β
C1 ORF traditional PCR was employed to amplify the target sequences and verify the identity by cloning and DNA sequencing. PCR betasatellite-specific primers [
26] were used to amplify a defective molecule from total DNA extracted from the field sample, KSA46. Cloning and sequencing of the amplicons confirmed that they were identical to the
de novo assembled molecules (EMBL Accession Number HG530542).
Similarly, DNA sequence analysis of the alphasatellites obtained by
de novo assembly from the field okra sample indicated the presence of four variants of the DNA-1 type alphasatellite at 1382 nt in size (EMBL Accession Number HG530544-HG530547), and one DNA-2 type alphasatellite at 1367 nt (EMBL Accession Number HG530543) (
Figure 4) [
11], are herein referred to as KSA27 DNA1 and KSA27 DNA2, respectively. These two alphasatellites contained the expected conserved ORF (Rep) on the sense strand. These KSA27 DNA1 variants shared 90%–98% identity with each other, and 30%–33% nt identity with KSA27 DNA2 isolated from the same okra plant sample. KSA27 DNA1 shared the highest nt identity with DNA1 from Mali and Gezira at 88%–94% and 86%–89%, respectively, while DNA2 shared its highest nt identity with two other DNA2 alphasatellites, reported from Oman and Singapore, at 64%. To verify the presence of the KSA27 DNA1 and DNA2 alphasatellites in plant extracts (KSA27), PCR specific primers were designed based on the
de novo assembled alphasatellites. Specific primers for KSA27 DNA1 were: 681F-3'-TACACTCGTGGAGGATCTGC-5' and 680R-3'-GAACCAGGTCCCACTTCTGA-5', and for the KSA27 DNA2, primers were 249F-3'-GAGGAAACAACTGGCACTGG-5' and 248R-3'-CGGCGAAGGACTTAACAGAG-5'. The PCR amplicons obtained from the field-infected okra sample KSA27 using these specific primers were cloned and completely sequenced. All of the resultant clones were verified to share 100% nt identity with the genome of the
de novo-assembled alphasatellites. The verification of satellite identities by PCR amplification, cloning, and DNA sequencing, provided robust support for the validity of the approach that involves enrichment, deep sequencing, and
de novo assembly of the circular, ssDNA begomoviral-associated alphasatellites.
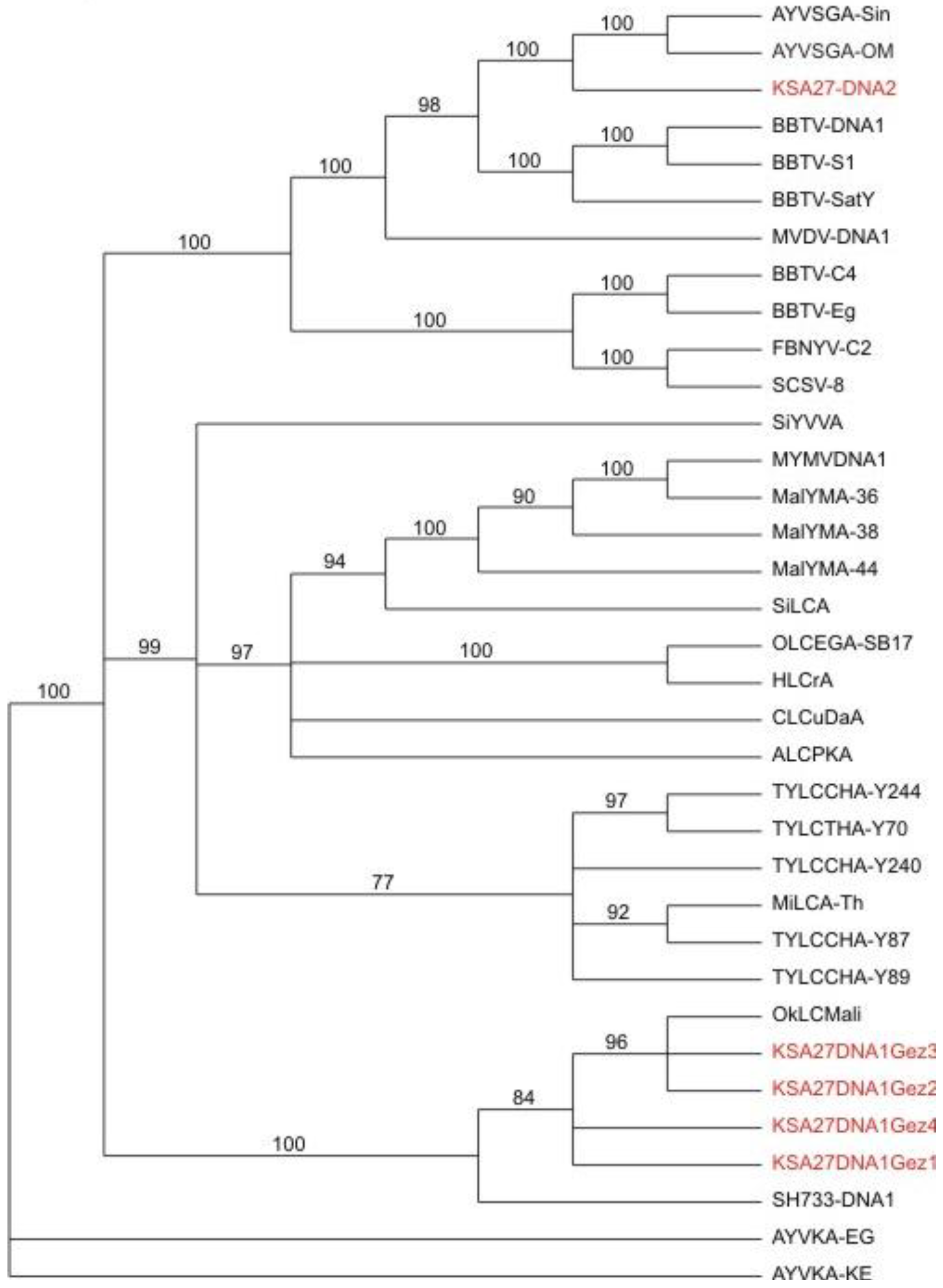
Figure 4.
Phylogenetic relationships of alphasatellite DNA sequences obtained from field samples, and agro-inoculated positive control test plants, obtained by
de novo assembly of the resultant DNA obtained from deep sequencing (in red), in relation to selected, reference alphasatellites. The alphasatellite acronyms and accession numbers are as described by Idris
et al., (2011) [
11].
Figure 4.
Phylogenetic relationships of alphasatellite DNA sequences obtained from field samples, and agro-inoculated positive control test plants, obtained by
de novo assembly of the resultant DNA obtained from deep sequencing (in red), in relation to selected, reference alphasatellites. The alphasatellite acronyms and accession numbers are as described by Idris
et al., (2011) [
11].
A second line of support for the validity of this approach is provided by the DNA sequence analysis of two different (positive) experimental controls. The first experimental control consisted of a field-infected tomato plant sample collected from Gezira (G11) from which the single helper begomovirus, ToLCSDV, had been previously cloned (G11), and the genome sequenced using capillary DNA sequencing (GenBank Accession Number JX483705). DNA extracts from the same plant were then subjected to RCA enrichment, deep sequencing, and
de novo assembly. A rigorous search of the contigs assembled from the deep sequencing experiment resulted in the recovery of the complete ToLCSDV genome (
Figure 5 and
Figure 6) (EMBL Accession Number HG530541) that was identical to the genome sequence obtained using the “traditional” approach (GenBank Accession Number JX483705). The second positive experimental control consisted of
N. benthamiana tobacco plants inoculated (in the seedling stage) with a mixture of three
Agrobacterium (agro)-clones of the helper virus,
Tomato yellow leaf curl virus (TYLCV) (GenBank accession number FJ956703), the Tomato yellow leaf curl betasatellite (TYLCB) (GenBank Accession Number DQ644566), and the Cotton leaf curl Gezira betasatellite (CLCuGB) (GenBank Accession Number AY044143), from previous studies. Total DNA extracted from the agro-inoculated, symptomatic
N. benthamiana seedlings was subjected to RCA enrichment of the begomoviral and satellite genomes present followed by deep sequencing, and
de novo assembly. The assembled viral components and satellite molecules (
Figure 6 and
Figure 7) were found to share 100% nt identity with the sequences for the respective agro-clones that were used for inoculation of the tobacco plants.
Figure 5.
Percentage of mappable DNA sequence reads (Y axis) at different levels of sequence depth that contributed to sample G11 contigs containing the begomovirus genomic sequence (X axis) resulting from the de novo and reference-guided assemblies. 10 M = 10 million reads, 100 K = 100 thousand reads, ToLCSDV = Tomato leaf curl Sudan virus.
Figure 5.
Percentage of mappable DNA sequence reads (Y axis) at different levels of sequence depth that contributed to sample G11 contigs containing the begomovirus genomic sequence (X axis) resulting from the de novo and reference-guided assemblies. 10 M = 10 million reads, 100 K = 100 thousand reads, ToLCSDV = Tomato leaf curl Sudan virus.
Figure 6.
Phylogenetic relationships of helper begomovirus DNA sequences obtained from field samples, and from agro-inoculated positive control test plants, obtained by deep sequencing (in red), in relation to selected begomovirus reference sequences. The
virus acronyms and accession numbers are as described by Idris
et al., (2011) [
11].
Figure 6.
Phylogenetic relationships of helper begomovirus DNA sequences obtained from field samples, and from agro-inoculated positive control test plants, obtained by deep sequencing (in red), in relation to selected begomovirus reference sequences. The
virus acronyms and accession numbers are as described by Idris
et al., (2011) [
11].
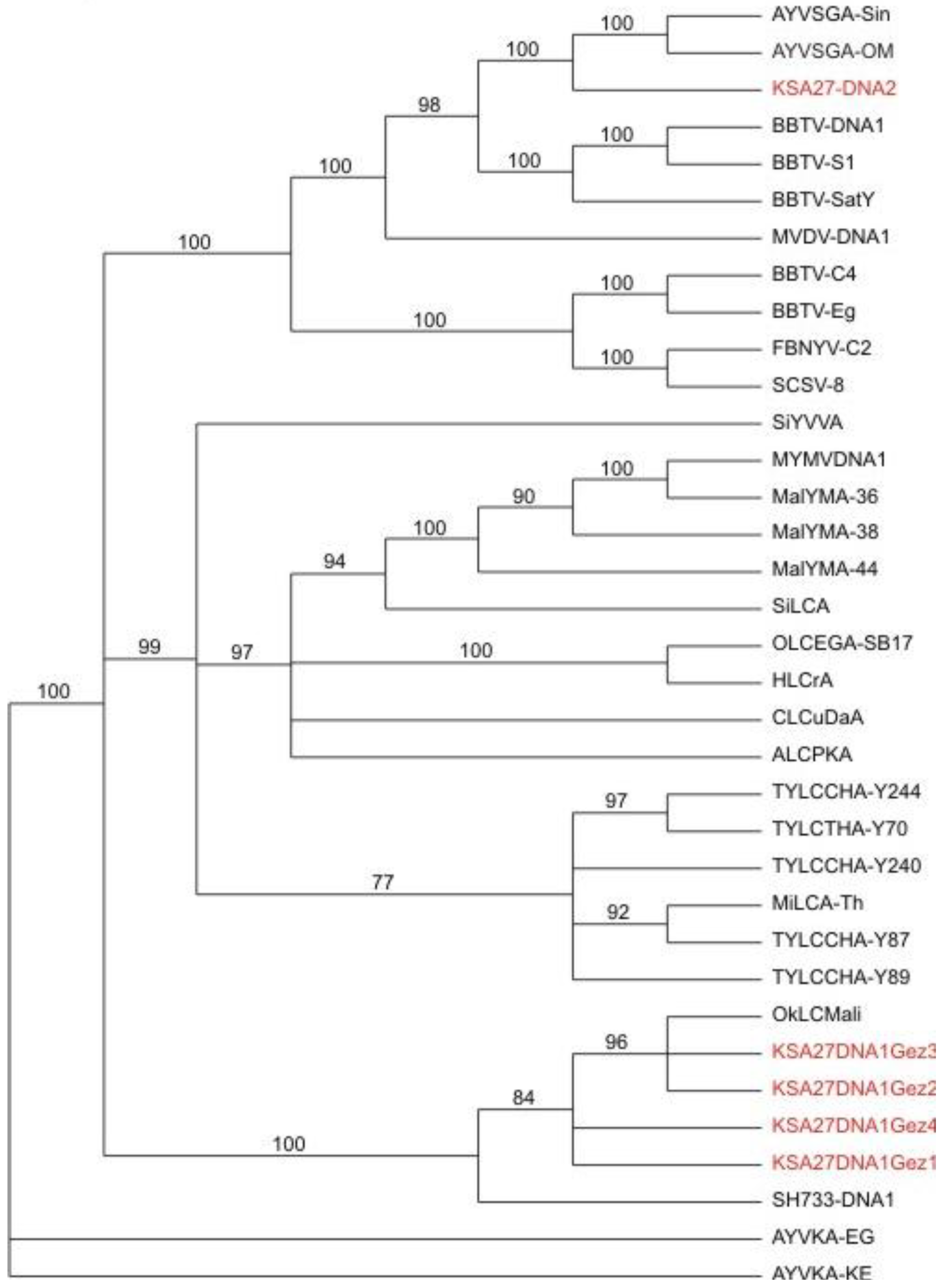
In a third line of support for the deep sequencing approach, the resultant DNA sequences that had been determined for all of the begomoviral and satellite molecules, were subjected to phylogenetic analysis. The results (
Figure 6) confirmed that the helper begomoviruses associated with the KSA46 and G11 satellites, grouped together in the clade that also contained other previously determined genome sequences of ToLCSDV isolates and strains, which also clustered with a basis in known, geographical endemism (
Figure 6). The helper begomovirus (associated with the KSA27 satellites DNA-1 and DNA-2) grouped into the clade containing the CLCuGV sequence. Also, the TYLCV (
Figure 6) isolated from the agro-inoculated tobacco plants grouped with its closest relative, TYLCV-OM, as was expected. The two different alphasatellites obtained from sample KSA27 grouped in different clades, with the DNA-1 type clustered with other DNA1-like alphasatellites, while the second grouped with its closest DNA2 type alphasatellite relatives (
Figure 4).
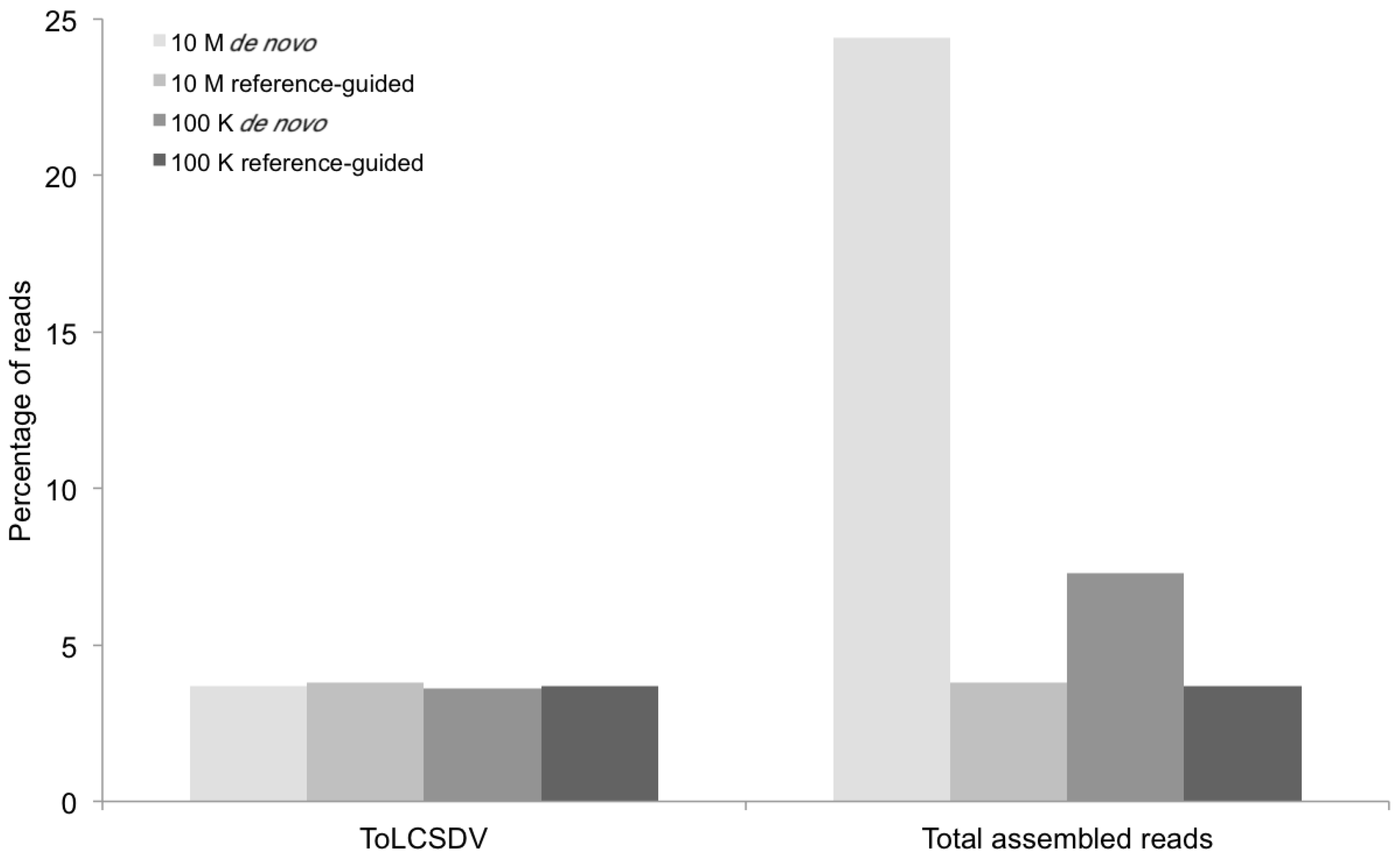
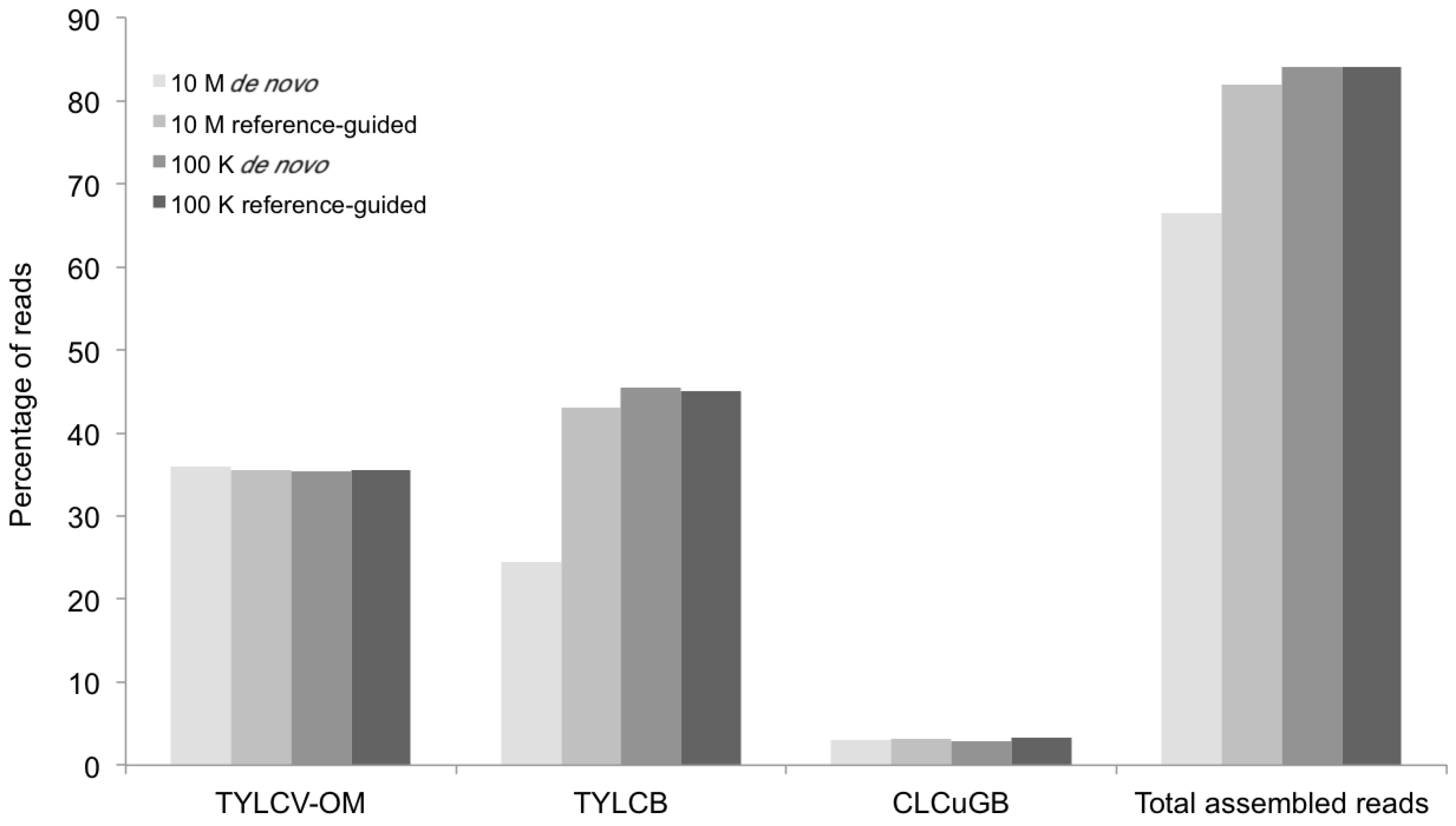
Figure 7.
Percentage of mappable DNA sequence reads (Y axis) at different levels of sequence depth contributing to the agro-begomovirus-betatsatellite clones used to inoculate the positive control plants, and the resultant contigs containing the begomovirus genomic or betasatellite sequences (X axis) produced by the de novo and reference-guided assemblies. 10 M = 10 million reads, 100 K = 100 thousand reads, TYLCV-OM = Tomato yellow leaf curl virus from Oman, TYLCB = Tomato yellow leaf curl betasatellite, CLCuGB = Cotton leaf curl Gezira betasatellite.
Figure 7.
Percentage of mappable DNA sequence reads (Y axis) at different levels of sequence depth contributing to the agro-begomovirus-betatsatellite clones used to inoculate the positive control plants, and the resultant contigs containing the begomovirus genomic or betasatellite sequences (X axis) produced by the de novo and reference-guided assemblies. 10 M = 10 million reads, 100 K = 100 thousand reads, TYLCV-OM = Tomato yellow leaf curl virus from Oman, TYLCB = Tomato yellow leaf curl betasatellite, CLCuGB = Cotton leaf curl Gezira betasatellite.
The contigs resulting from the
de novo assemblies represented primarily concatemeric molecules consisting of repeated sequences that were also present in the respective begomovirus genome, and/or the associated satellite, making it impossible to determine the depth of coverage, and the presence and/or the precise locations of a number of SNPs (
Table 3,
Figure 8). To circumvent this obstacle we reassembled the short, high-throughput Illumina reads using the
de novo assembled begomovirus genomes and satellite DNAs as reference sequences (
Table 4 and
Figure 2,
Figure 3,
Figure 5 and
Figure 7). These results demonstrate a “proof of concept”, for the implementation of deep sequencing as a fast and reliable means of “harvesting” circular, ssDNAs characteristically associated with begomovirus infections in plants, and for exploring the SNPs associated with viral genomes and/or their associated satellites. Further, as few as 100,000 reads was sufficient to assemble up to four (
Table 4) (and probably many more) distinct, begomoviral genomes, making the method feasible for recovering monopartite and bipartite begomoviral genomes and associated smaller circular, ssDNAs (satellites, DI’s, subgenomic molecules, others) from a single plant harboring a mixture of different viral strains or species.
The percentage of total assembled reads was variable among the begomoviruses included in this study (
Figure 2,
Figure 3,
Figure 5 and
Figure 7). This could have resulted from differences in host species, whether plants were grown in greenhouse (
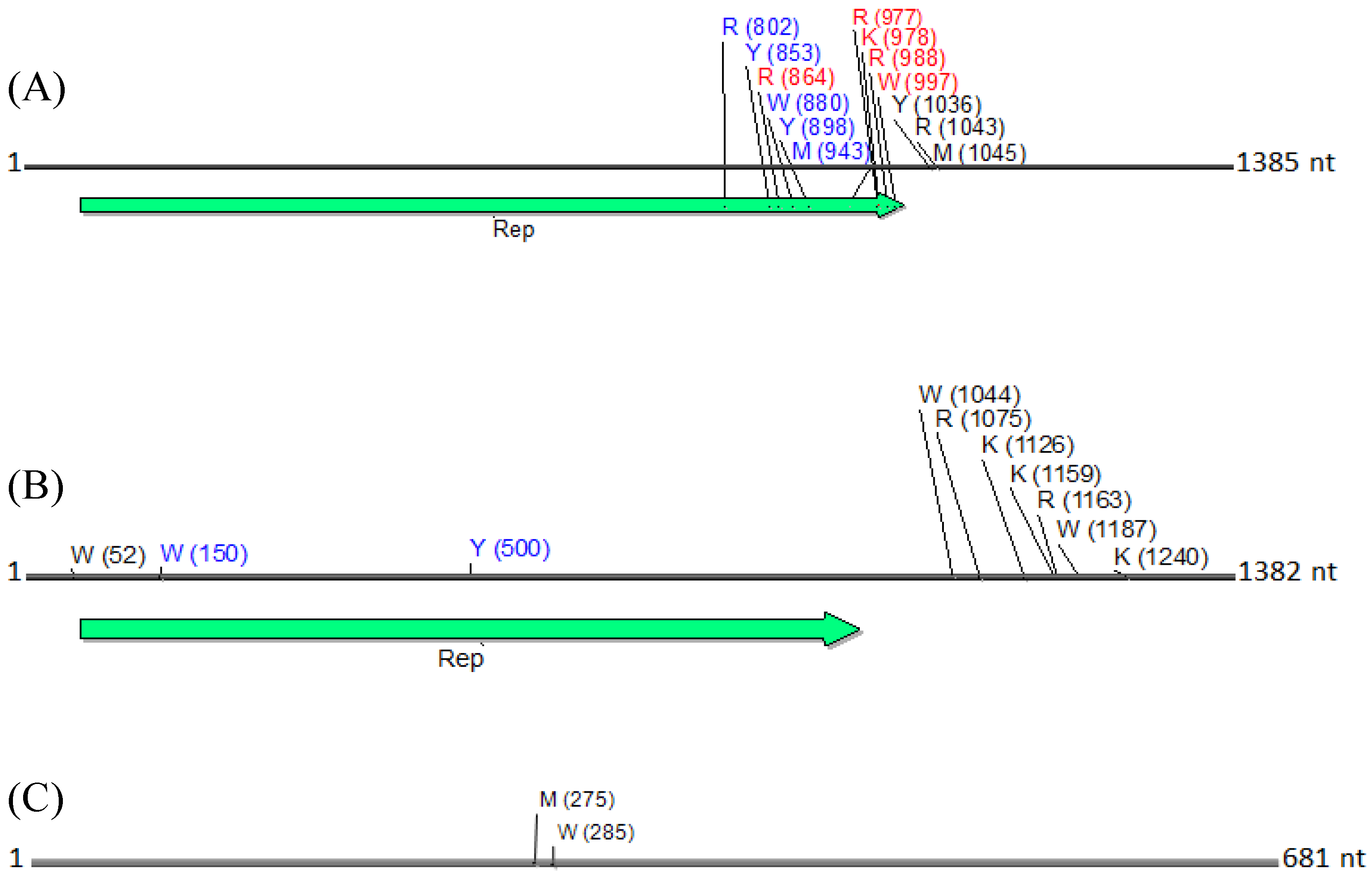
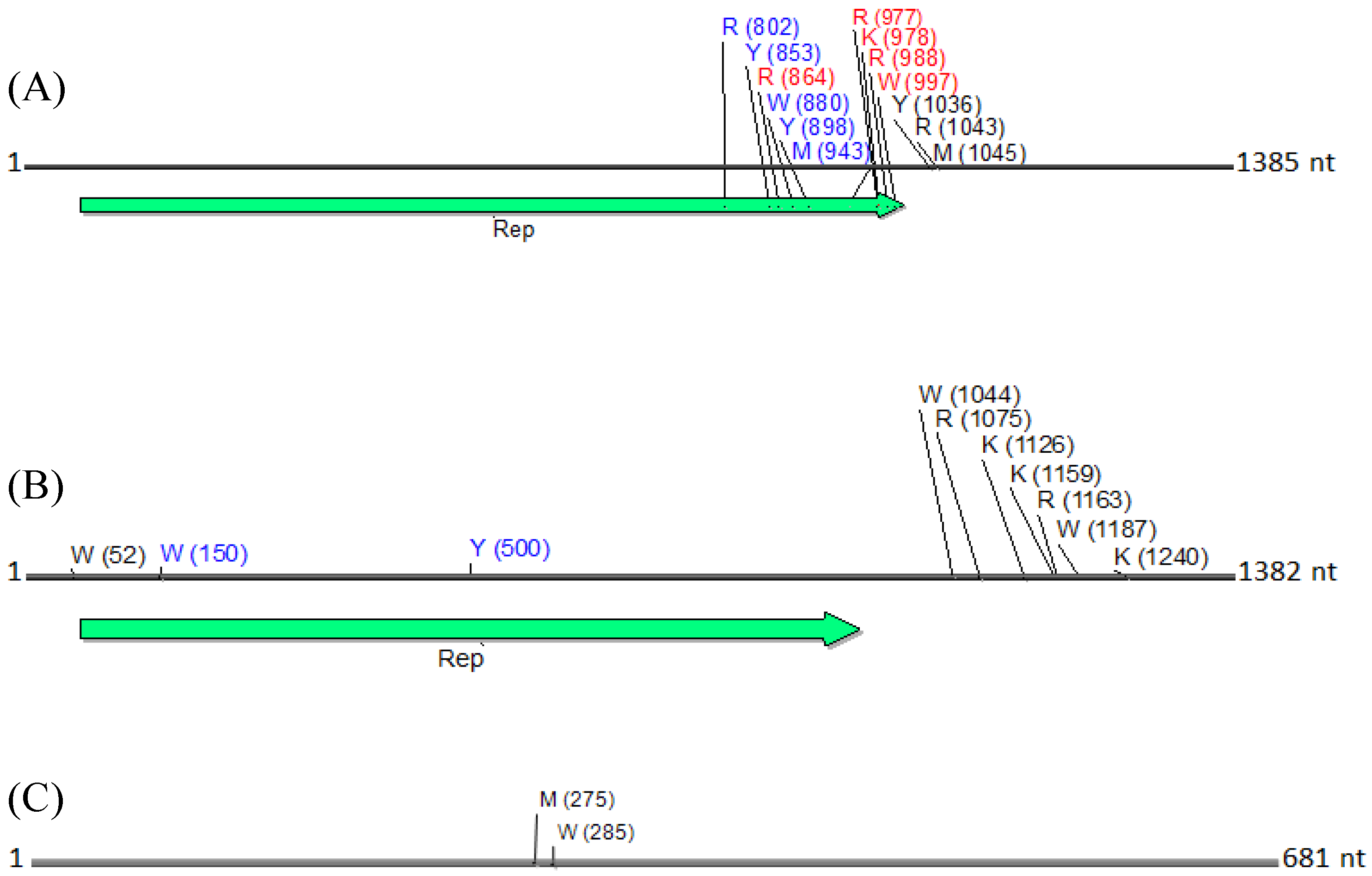
N. benthamiana) or field (okra, tomato), and perhaps also to differences between the plant species with the respect to the extent of amplification (by RCA) of mitochondria and chloroplast organelles, or to circularized chromosomal fragments. The SNPs in DNA 1 (
Figure 8) were mapped to a region that spans the
C-terminus of the satellite Rep protein (the replication associated protein), and the region located immediately downstream of this gene. Of the ten SNPs mapped in the Rep region, five resulted in an amino acid change, as a non-synonymous substitution (
Figure 8). In contrast, two SNPs were present in the Rep gene of DNA2 that would result in no an amino acid change owing to a synonymous substitution (
Figure 8).
Table 3.
Identification of the position and type(s) of single nucleotide polymorphisms (SNPs) in three kinds of satellite DNA molecules assembled from next-generation sequencing reads, obtained from field sample KSA27. The reads were assembled using SeqMan NGen software that identifies SNPs and classifies them based on the International Union of Pure and Applied Chemistry code. The non-synonymous and synonymous SNPs are shown in red and in blue, respectively, in C1 ORF (see
Figure 8). The SNPs in noncoding regions are shown in black. For each nucleotide position the SNPs were confirmed at ≥10 base percentage score.
Table 3.
Identification of the position and type(s) of single nucleotide polymorphisms (SNPs) in three kinds of satellite DNA molecules assembled from next-generation sequencing reads, obtained from field sample KSA27. The reads were assembled using SeqMan NGen software that identifies SNPs and classifies them based on the International Union of Pure and Applied Chemistry code. The non-synonymous and synonymous SNPs are shown in red and in blue, respectively, in C1 ORF (see Figure 8). The SNPs in noncoding regions are shown in black. For each nucleotide position the SNPs were confirmed at ≥10 base percentage score.
| DNA1 | DNA2 | Betasatellite |
|---|
| SNP Position | SNP Type | Base percentage Score | SNP Position | SNP Type | Base Percentage Score | SNP Position | SNP Type | Base Percentage Score |
|---|
| 802 | R | A = 13; G = 87 | 52 | W | A = 58; T = 42 | 275 | M | A = 46; C = 54 |
| 853 | Y | C = 32; T = 68 | 150 | W | A = 56; T = 44 | 285 | W | A = 51; T = 49 |
| 864 | R | A = 64; G = 36 | 500 | Y | C = 62; T = 38 | | | |
| 880 | W | A = 61; T = 39 | 1044 | W | A = 33; T = 67 | | | |
| 898 | Y | C = 63; T = 37 | 1075 | R | A = 77; G = 23 | | | |
| 943 | M | A = 67; G = 33 | 1126 | K | G = 40; T = 60 | | | |
| 977 | R | A = 30; G = 70 | 1159 | Y | C = 50; T = 50 | | | |
| 978 | K | G = 30; T = 70 | 1163 | R | A = 51; G = 49 | | | |
| 988 | R | A = 30; G = 70 | 1187 | W | A = 56; T = 44 | | | |
| 997 | W | A = 69; T = 31 | 1240 | K | G = 45; T = 55 | | | |
| 1036 | Y | C = 69; T = 31 | | | | | | |
| 1043 | R | A = 32; G = 68 | | | | | | |
| 1045 | M | A = 33; C = 67 | | | | | | |
Table 4.
Genomic component size and number of SNPs. SNPs were determined based on ≤90 percent mismatches.
Table 4.
Genomic component size and number of SNPs. SNPs were determined based on ≤90 percent mismatches.
| Sample | Genomic Components | Size (bp) | SNPs |
|---|
| KSA27 | CLCuGV | 2780 | 0 |
| CLCuGB | 681 | 2 |
| DNA1 | 1385 | 13 |
| DNA2 | 1382 | 10 |
| KSA46 | ToLCSDV | 2788 | 0 |
| G11 | ToLCSDV | 2765 | 0 |
| Control | TYLCV-OM | 2767 | 0 |
| TYLCB | 1371 | 0 |
| CLCuGB | 1349 | 0 |
Figure 8.
Physical maps of (
A) DNA1, (
B) DNA2 and (
C) betasatellite, illustrating the locations of SNPs. SNPs are depicted as their respective nucleotide ambiguity code (
Table 3), and their location on each molecule is shown in brackets. The predominant region found to contain SNPs was located in the carboxyl terminus, and immediately rightward of the C1 (Rep) ORF in a non-coding region (
A). Non-synonymous and synonymous substitutions present in the coding region are demarcated in red and in blue, respectively, and those located in the noncoding region are shown in black. The satellite circular maps were presented as linear maps (black horizontal lines) for ease of viewing. The number of nucleotides (nt) in each molecule is indicated.
Figure 8.
Physical maps of (
A) DNA1, (
B) DNA2 and (
C) betasatellite, illustrating the locations of SNPs. SNPs are depicted as their respective nucleotide ambiguity code (
Table 3), and their location on each molecule is shown in brackets. The predominant region found to contain SNPs was located in the carboxyl terminus, and immediately rightward of the C1 (Rep) ORF in a non-coding region (
A). Non-synonymous and synonymous substitutions present in the coding region are demarcated in red and in blue, respectively, and those located in the noncoding region are shown in black. The satellite circular maps were presented as linear maps (black horizontal lines) for ease of viewing. The number of nucleotides (nt) in each molecule is indicated.
Recent advances in deep sequencing technologies, or NGS, has made it possible to utilize multiplexing to explore the composition of begomovirus populations for as many as 96 samples per flow cell lane, and to effectively reduce the cost of deep sequencing such that large numbers of virus samples can be processed simultaneously. However, for organisms with small-sized genomes [
27], such as begomoviruses, the Illumina Hiseq2000/2500 is not an optimal platform because of its massive throughput capacity, unless >96 samples are multiplexed and run in a single lane [
28]. In a successful 2 × 100 bp paired-end, HiSeq2000/2500 run, 30–40 Gb (150–200 million reads) of sequence are generated from one lane of a flow cell, to produce 1.56–2.08 million reads per sample, or about 112,000–149,000× coverage of each 2.8 Kb of begomovirus genome, and requires enormous time and computational capability to assemble such huge amounts of sequence data.
To minimize the sequence throughput and reduce the time devoted to computation and assembly, sequencing of begomoviral genome in an Illumina bench top sequencer, MiSeq, would likely offer a better option than the HiSeq2000/25,000 platform. In both platforms libraries need to be constructed using a high multiplexing barcode [
28] in order to substantially increase the number of samples per lane to make reasonable coverage for this shot genome sizes and reduce the cost per sample. The standard MiSeq run using V3 kits can produce 25 Million reads at 3.75 Gb (2 × 75 bp PE per run) to 15 Gb (2 × 300 bp PE per run). Thus, approximately 260,000 reads can be obtained per sample with coverage of 14,000–56,000× for 96 multiplexed samples.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}