Exploiting Herpes Simplex Virus Entry for Novel Therapeutics



{kind=link}
Abstract
:1. Introduction
2. Entry
2.1. Attachment and Surfing
2.2. Fusion
2.2.1. Phase I
2.2.2. Phase II
2.3. Endocytosis
3. Therapeutics
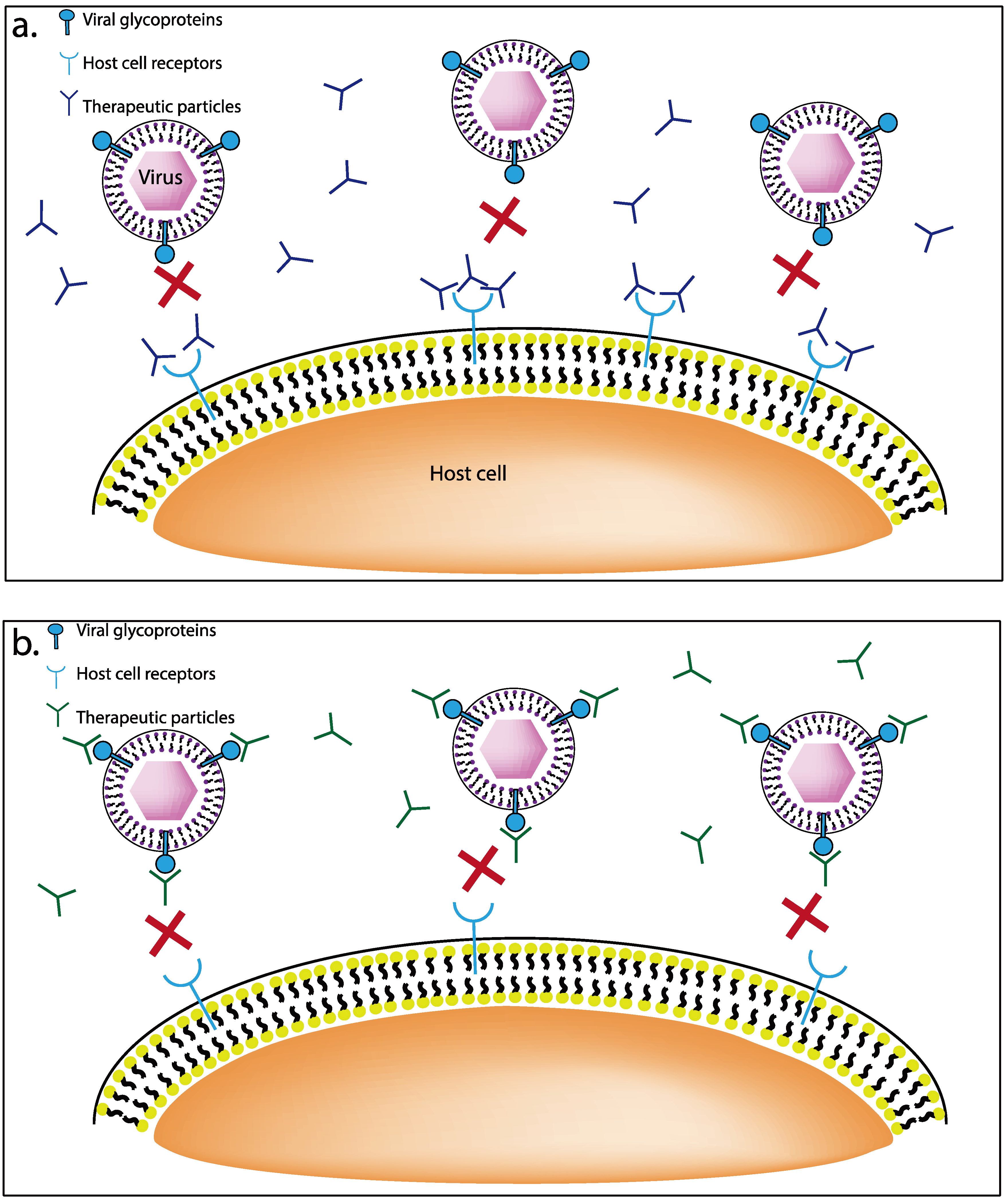
3.1. Receptor Targeting Therapeutics
3.1.1. G1 and G2 Anti-Heparan Sulfate Peptides
3.1.2. Apolipoprotein E
3.1.3. AC-8
3.1.4. Aptamers
3.1.5. Dermaseptins
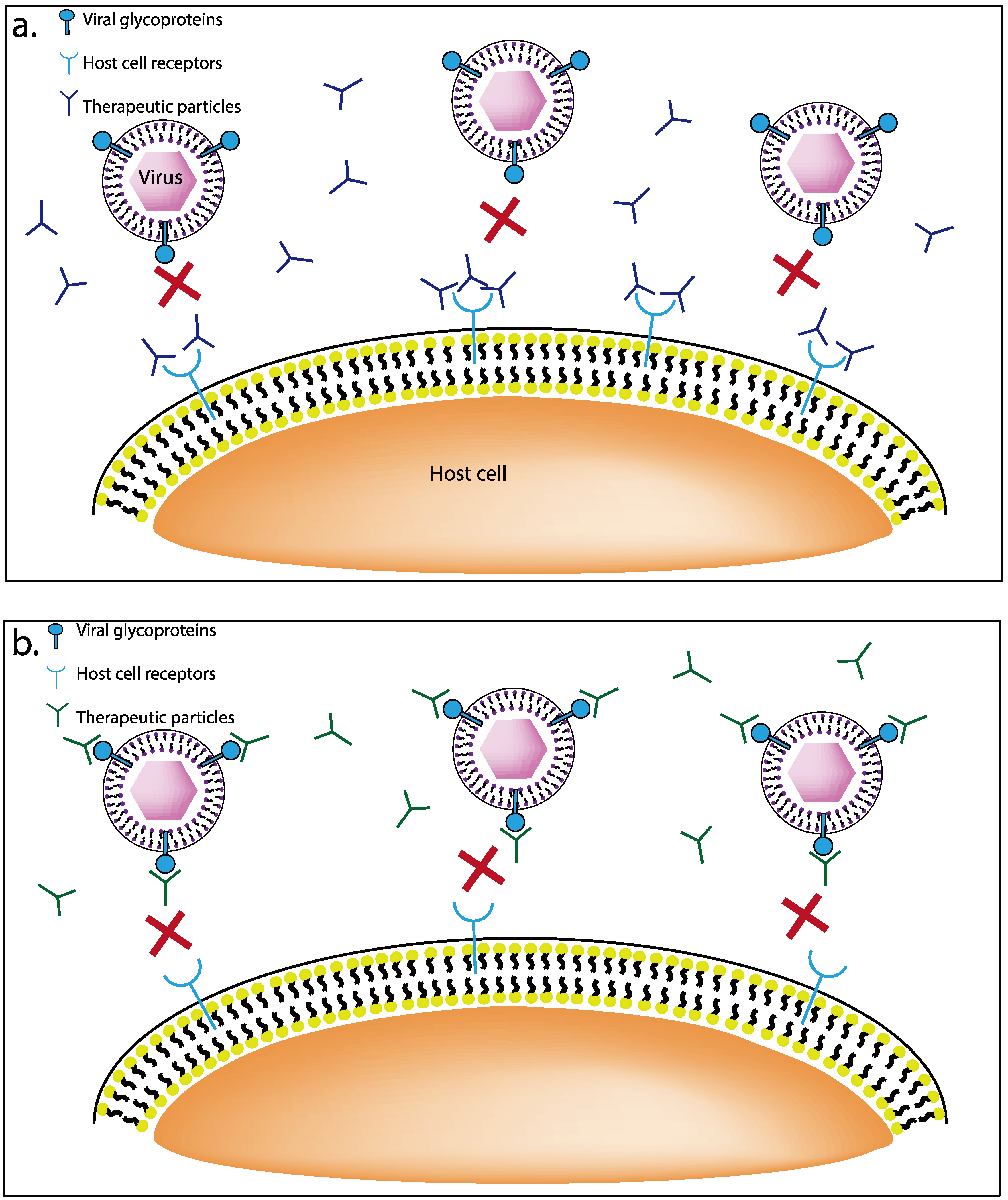
3.2. Viral Glycoprotein Targeting Therapeutics
3.2.1. Nanoparticles
3.2.2. K-5 Compounds
3.2.3. Compound SP-510-50
3.2.4. Dendrimers
3.3. Targeting Downstream Signaling Mechanism
4. Conclusion
Acknowledgments
Conflict of Interest
References and Notes
- Whitley, R.J. Herpesviruses. In Medical Microbiology, 4th; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996; Chapter 68. [Google Scholar]
- Ball, M.J. Limbic predilection in Alzheimer dementia: Is reactivated herpesvirus involved? Can. J. Neurol. Sci. 1982, 9, 303–306. [Google Scholar]
- Nahmias, A.J.; Lee, F.K.; Beckman-Nahmias, S. Sero-epidemiological and -sociological patterns of herpes simplex virus infection in the world. Scand. J. Infect. Dis. Suppl. 1990, 69, 19–36. [Google Scholar]
- Heldwein, E.E.; Krummenacher, C. Entry of herpesviruses into mammalian cells. Cell Mol. Life Sci. 2008, 65, 1653–1668. [Google Scholar] [CrossRef]
- Willis, S.H.; Rux, A.H.; Peng, C.; Whitbeck, J.C.; Nicola, A.V.; Lou, H.; Hou, W.; Salvador, L.; Eisenberg, R.J.; Cohen, G.H. Examination of the kinetics of herpes simplex virus glycoprotein D binding to the herpesvirus entry mediator, using surface plasmon resonance. J. Virol. 1998, 72, 5937–5947. [Google Scholar]
- Razonable, R.R. Antiviral drugs for viruses other than human immunodeficiency virus. Mayo Clin. Proc. 2011, 86, 1009–1026. [Google Scholar] [CrossRef]
- Gerber, S.I.; Belval, B.J.; Herold, B.C. Differences in the role of glycoprotein C of HSV-1 and HSV-2 in viral binding may contribute to serotype differences in cell tropism. Virology 1995, 214, 29–39. [Google Scholar] [CrossRef]
- Cheshenko, N.; Herold, B.C. Glycoprotein B plays a predominant role in mediating herpes simplex virus type 2 attachment and is required for entry and cell-to-cell spread. J. Gen. Virol. 2002, 83, 2247–2255. [Google Scholar]
- Herold, B.C.; Gerber, S.I.; Belval, B.J.; Siston, A.M.; Shulman, N. Differences in the susceptibility of herpes simplex virus types 1 and 2 to modified heparin compounds suggest serotype differences in viral entry. J. Virol. 1996, 70, 3461–3469. [Google Scholar]
- Herold, B.C.; Visalli, R.J.; Susmarski, N.; Brandt, C.R.; Spear, P.G. Glycoprotein C-independent binding of herpes simplex virus to cells requires cell surface heparan sulphate and glycoprotein B. J. Gen. Virol. 1994, 75, 1211–1222. [Google Scholar] [CrossRef]
- Herold, B.C.; WuDunn, D.; Soltys, N.; Spear, P.G. Glycoprotein C of herpes simplex virus type 1 plays a principal role in the adsorption of virus to cells and in infectivity. J. Virol. 1991, 65, 1090–1098. [Google Scholar]
- Spear, P.G.; Longnecker, R. Herpesvirus entry: An update. J. Virol. 2003, 77, 10179–10185. [Google Scholar] [CrossRef]
- Svitkina, T.M.; Bulanova, E.A.; Chaga, O.Y.; Vignjevic, D.M.; Kojima, S.; Vasiliev, J.M.; Borisy, G.G. Mechanism of filopodia initiation by reorganization of a dendritic network. J. Cell Biol. 2003, 160, 409–421. [Google Scholar] [CrossRef]
- Oh, M.J.; Akhtar, J.; Desai, P.; Shukla, D. A role for heparan sulfate in viral surfing. Biochem. Biophys. Res. Commun. 2010, 391, 176–181. [Google Scholar] [CrossRef]
- Nobes, C.D.; Hall, A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 1995, 81, 53–62. [Google Scholar] [CrossRef]
- Connolly, S.A.; Jackson, J.O.; Jardetzky, T.S.; Longnecker, R. Fusing structure and function: A structural view of the herpesvirus entry machinery. Nat. Rev. Microbiol. 2011, 9, 369–381. [Google Scholar]
- Karasneh, G.A.; Shukla, D. Herpes simplex virus infects most cell types in vitro: Clues to its success. Virol. J. 2011, 8, 481. [Google Scholar] [CrossRef]
- Gianni, T.; Amasio, M.; Campadelli-Fiume, G. Herpes simplex virus gD forms distinct complexes with fusion executors gB and gH/gL in part through the C-terminal profusion domain. J. Biol. Chem. 2009, 284, 17370–17382. [Google Scholar] [CrossRef]
- Atanasiu, D.; Whitbeck, J.C.; Cairns, T.M.; Reilly, B.; Cohen, G.H.; Eisenberg, R.J. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc. Natl. Acad. Sci. USA 2007, 104, 18718–18723. [Google Scholar]
- Campadelli-Fiume, G.; Cocchi, F.; Menotti, L.; Lopez, M. The novel receptors that mediate the entry of herpes simplex viruses and animal alphaherpesviruses into cells. Rev. Med. Virol. 2000, 10, 305–319. [Google Scholar] [CrossRef]
- Spear, P.G.; Eisenberg, R.J.; Cohen, G.H. Three classes of cell surface receptors for alphaherpesvirus entry. Virology 2000, 275, 1–8. [Google Scholar] [CrossRef]
- Krummenacher, C.; Supekar, V.M.; Whitbeck, J.C.; Lazear, E.; Connolly, S.A.; Eisenberg, R.J.; Cohen, G.H.; Wiley, D.C.; Carfi, A. Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. EMBO J. 2005, 24, 4144–4153. [Google Scholar] [CrossRef]
- Fusco, D.; Forghieri, C.; Campadelli-Fiume, G. The pro-fusion domain of herpes simplex virus glycoprotein D (gD) interacts with the gD N terminus and is displaced by soluble forms of viral receptors. Proc. Natl. Acad. Sci. USA 2005, 102, 9323–9328. [Google Scholar] [CrossRef]
- Lazear, E.; Carfi, A.; Whitbeck, J.C.; Cairns, T.M.; Krummenacher, C.; Cohen, G.H.; Eisenberg, R.J. Engineered disulfide bonds in herpes simplex virus type 1 gD separate receptor binding from fusion initiation and viral entry. J. Virol. 2008, 82, 700–709. [Google Scholar] [CrossRef]
- Hutchinson, L.; Browne, H.; Wargent, V.; Davis-Poynter, N.; Primorac, S.; Goldsmith, K.; Minson, A.C.; Johnson, D.C. A novel herpes simplex virus glycoprotein, gL, forms a complex with glycoprotein H (gH) and affects normal folding and surface expression of gH. J. Virol. 1992, 66, 2240–2250. [Google Scholar]
- Cairns, T.M.; Landsburg, D.J.; Whitbeck, J.C.; Eisenberg, R.J.; Cohen, G.H. Contribution of cysteine residues to the structure and function of herpes simplex virus gH/gL. Virology 2005, 332, 550–562. [Google Scholar] [CrossRef]
- Peng, T.; Ponce-de-Leon, M.; Jiang, H.; Dubin, G.; Lubinski, J.M.; Eisenberg, R.J.; Cohen, G.H. The gH-gL complex of herpes simplex virus (HSV) stimulates neutralizing antibody and protects mice against HSV type 1 challenge. J. Virol. 1998, 72, 65–72. [Google Scholar]
- Avitabile, E.; Forghieri, C.; Campadelli-Fiume, G. Cross talk among the glycoproteins involved in herpes simplex virus entry and fusion: The interaction between gB and gH/gL does not necessarily require gD. J. Virol. 2009, 83, 10752–10760. [Google Scholar] [CrossRef]
- Subramanian, R.P.; Geraghty, R.J. Herpes simplex virus type 1 mediates fusion through a hemifusion intermediate by sequential activity of glycoproteins D, H, L, and B. Proc. Natl. Acad. Sci. USA 2007, 104, 2903–2908. [Google Scholar] [CrossRef]
- Jackson, J.O.; Longnecker, R. Reevaluating herpes simplex virus hemifusion. J. Virol. 2010, 84, 11814–11821. [Google Scholar] [CrossRef]
- Jackson, J.O.; Lin, E.; Spear, P.G.; Longnecker, R. Insertion mutations in herpes simplex virus 1 glycoprotein H reduce cell surface expression, slow the rate of cell fusion, or abrogate functions in cell fusion and viral entry. J. Virol. 2010, 84, 2038–2046. [Google Scholar] [CrossRef]
- Gianni, T.; Cerretani, A.; Dubois, R.; Salvioli, S.; Blystone, S.S.; Rey, F.; Campadelli-Fiume, G. Herpes simplex virus glycoproteins H/L bind to cells independently of {alpha}V{beta}3 integrin and inhibit virus entry, and their constitutive expression restricts infection. J. Virol. 2010, 84, 4013–4025. [Google Scholar]
- Gianni, T.; Gatta, V.; Campadelli-Fiume, G. {alpha}V{beta}3-integrin routes herpes simplex virus to an entry pathway dependent on cholesterol-rich lipid rafts and dynamin2. Proc. Natl. Acad. Sci. USA 2010, 107, 22260–22265. [Google Scholar]
- Atanasiu, D.; Saw, W.T.; Cohen, G.H.; Eisenberg, R.J. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J. Virol. 2010, 84, 12292–12299. [Google Scholar] [CrossRef]
- Bzik, D.J.; Fox, B.A.; DeLuca, N.A.; Person, S. Nucleotide sequence of a region of the herpes simplex virus type 1 gB glycoprotein gene: Mutations affecting rate of virus entry and cell fusion. Virology 1984, 137, 185–190. [Google Scholar] [CrossRef]
- Heldwein, E.E.; Lou, H.; Bender, F.C.; Cohen, G.H.; Eisenberg, R.J.; Harrison, S.C. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 2006, 313, 217–220. [Google Scholar] [CrossRef]
- Atanasiu, D.; Whitbeck, J.C.; de Leon, M.P.; Lou, H.; Hannah, B.P.; Cohen, G.H.; Eisenberg, R.J. Bimolecular complementation defines functional regions of Herpes simplex virus gB that are involved with gH/gL as a necessary step leading to cell fusion. J. Virol. 2010, 84, 3825–3834. [Google Scholar]
- Smith, A.E.; Helenius, A. How viruses enter animal cells. Science 2004, 304, 237–242. [Google Scholar] [CrossRef]
- Milne, R.S.; Nicola, A.V.; Whitbeck, J.C.; Eisenberg, R.J.; Cohen, G.H. Glycoprotein D receptor-dependent, low-pH-independent endocytic entry of herpes simplex virus type 1. J. Virol. 2005, 79, 6655–6663. [Google Scholar] [CrossRef]
- Nicola, A.V.; McEvoy, A.M.; Straus, S.E. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J. Virol. 2003, 77, 5324–5332. [Google Scholar] [CrossRef]
- Nicola, A.V.; Straus, S.E. Cellular and viral requirements for rapid endocytic entry of herpes simplex virus. J. Virol. 2004, 78, 7508–7517. [Google Scholar] [CrossRef]
- Clement, C.; Tiwari, V.; Scanlan, P.M.; Valyi-Nagy, T.; Yue, B.Y.; Shukla, D. A novel role for phagocytosis-like uptake in herpes simplex virus entry. J. Cell Biol. 2006, 174, 1009–1021. [Google Scholar] [CrossRef]
- Marsh, M.; Bron, R. SFV infection in CHO cells: Cell-type specific restrictions to productive virus entry at the cell surface. J. Cell Sci. 1997, 110, 95–103. [Google Scholar]
- Wittels, M.; Spear, P.G. Penetration of cells by herpes simplex virus does not require a low pH-dependent endocytic pathway. Virus Res. 1991, 18, 271–290. [Google Scholar] [CrossRef]
- Treister, N.S.; Woo, S.B. Topical n-docosanol for management of recurrent herpes labialis. Expert Opin. Pharmacother. 2010, 11, 853–860. [Google Scholar] [CrossRef]
- Pope, L.E.; Marcelletti, J.F.; Katz, L.R.; Lin, J.Y.; Katz, D.H.; Parish, M.L.; Spear, P.G. The anti-herpes simplex virus activity of n-docosanol includes inhibition of the viral entry process. Antivir. Res. 1998, 40, 85–94. [Google Scholar] [CrossRef]
- Shukla, D.; Spear, P.G. Herpesviruses and heparan sulfate: An intimate relationship in aid of viral entry. J. Clin. Invest. 2001, 108, 503–510. [Google Scholar]
- Dobson, C.B.; Sales, S.D.; Hoggard, P.; Wozniak, M.A.; Crutcher, K.A. The receptor-binding region of human apolipoprotein E has direct anti-infective activity. J. Infect. Dis 2006, 193, 442–450. [Google Scholar] [CrossRef]
- Berdugo, M.; Larsen, I.V.; Abadie, C.; Deloche, C.; Kowalczuk, L.; Touchard, E.; Dubielzig, R.; Brandt, C.R.; Behar-Cohen, F.; Combette, J.M. Ocular distribution, spectrum of activity, and in vivo viral neutralization of a fully humanized anti-herpes simplex virus IgG Fab fragment following topical application. Antimicrob. Agents Chemother. 2012, 56, 1390–1402. [Google Scholar]
- Lass, J.H.; Langston, R.H.; Foster, C.S.; Pavan-Langston, D. Antiviral medications and corneal wound healing. Antivir. Res. 1984, 4, 143–157. [Google Scholar]
- Bunka, D.H.; Platonova, O.; Stockley, P.G. Development of aptamer therapeutics. Curr. Opin. Pharmacol. 2010, 10, 557–562. [Google Scholar]
- James, W. Nucleic acid and polypeptide aptamers: A powerful approach to ligand discovery. Curr. Opin. Pharmacol. 2001, 1, 540–546. [Google Scholar]
- Khati, M.; Schuman, M.; Ibrahim, J.; Sattentau, Q.; Gordon, S.; James, W. Neutralization of infectivity of diverse R5 clinical isolates of human immunodeficiency virus type 1 by gp120-binding 2'F-RNA aptamers. J. Virol. 2003, 77, 12692–12698. [Google Scholar] [CrossRef]
- Moore, M.D.; Cookson, J.; Coventry, V.K.; Sproat, B.; Rabe, L.; Cranston, R.D.; McGowan, I.; James, W. Protection of HIV neutralizing aptamers against rectal and vaginal nucleases: Implications for RNA-based therapeutics. J. Biol. Chem. 2011, 286, 2526–2535. [Google Scholar] [CrossRef]
- Wang, J.; Jiang, H.; Liu, F. In vitro selection of novel RNA ligands that bind human cytomegalovirus and block viral infection. RNA 2000, 6, 571–583. [Google Scholar] [CrossRef]
- Gopinath, S.C.; Hayashi, K.; Kumar, P.K. Aptamer that binds to the gD protein of herpes simplex virus 1 and efficiently inhibits viral entry. J. Virol. 2012, 86, 6732–6744. [Google Scholar] [CrossRef]
- Moore, M.D.; Bunka, D.H.; Forzan, M.; Spear, P.G.; Stockley, P.G.; McGowan, I.; James, W. Generation of neutralizing aptamers against herpes simplex virus type 2: Potential components of multivalent microbicides. J. Gen. Virol. 2011, 92, 1493–1499. [Google Scholar] [CrossRef]
- Para, M.F.; Parish, M.L.; Noble, A.G.; Spear, P.G. Potent neutralizing activity associated with anti-glycoprotein D specificity among monoclonal antibodies selected for binding to herpes simplex virions. J. Virol. 1985, 55, 483–488. [Google Scholar]
- Mor, A.; Nguyen, V.H.; Delfour, A.; Migliore-Samour, D.; Nicolas, P. Isolation, amino acid sequence, and synthesis of dermaseptin, a novel antimicrobial peptide of amphibian skin. Biochemistry 1991, 30, 8824–8830. [Google Scholar] [CrossRef]
- Amiche, M.; Ladram, A.; Nicolas, P. A consistent nomenclature of antimicrobial peptides isolated from frogs of the subfamily Phyllomedusinae. Peptides 2008, 29, 2074–2082. [Google Scholar] [CrossRef]
- Nicolas, P.; El Amri, C. The dermaseptin superfamily: A gene-based combinatorial library of antimicrobial peptides. Biochim. Biophys. Acta 2009, 1788, 1537–1550. [Google Scholar] [CrossRef]
- Belaid, A.; Aouni, M.; Khelifa, R.; Trabelsi, A.; Jemmali, M.; Hani, K. In vitro antiviral activity of dermaseptins against herpes simplex virus type 1. J. Med. Virol. 2002, 66, 229–234. [Google Scholar] [CrossRef]
- Bergaoui, I.; Zairi, A.; Tangy, F.; Aouni, M.; Selmi, B.; Hani, K. In vitro antiviral activity of dermaseptin S(4) and derivatives from amphibian skin against herpes simplex virus type 2. J. Med. Virol. 2013, 85, 272–281. [Google Scholar] [CrossRef]
- Yaron, S.; Rydlo, T.; Shachar, D.; Mor, A. Activity of dermaseptin K4-S4 against foodborne pathogens. Peptides 2003, 24, 1815–1821. [Google Scholar]
- Albiol Matanic, V.C.; Castilla, V. Antiviral activity of antimicrobial cationic peptides against Junin virus and herpes simplex virus. Int. J. Antimicrob. Agents 2004, 23, 382–389. [Google Scholar] [CrossRef]
- Daher, K.A.; Selsted, M.E.; Lehrer, R.I. Direct inactivation of viruses by human granulocyte defensins. J. Virol. 1986, 60, 1068–1074. [Google Scholar]
- Wachinger, M.; Kleinschmidt, A.; Winder, D.; von Pechmann, N.; Ludvigsen, A.; Neumann, M.; Holle, R.; Salmons, B.; Erfle, V.; Brack-Werner, R. Antimicrobial peptides melittin and cecropin inhibit replication of human immunodeficiency virus 1 by suppressing viral gene expression. J. Gen. Virol. 1998, 79, 731–740. [Google Scholar]
- Hancock, R.E.; Chapple, D.S. Peptide antibiotics. Antimicrob. Agents Chemother. 1999, 43, 1317–1323. [Google Scholar]
- Cheshenko, N.; Keller, M.J.; MasCasullo, V.; Jarvis, G.A.; Cheng, H.; John, M.; Li, J.H.; Hogarty, K.; Anderson, R.A.; Waller, D.P.; et al. Candidate topical microbicides bind herpes simplex virus glycoprotein B and prevent viral entry and cell-to-cell spread. Antimicrob. Agents Chemother. 2004, 48, 2025–2036. [Google Scholar] [CrossRef]
- Jones, L.S.; Yazzie, B.; Middaugh, C.R. Polyanions and the proteome. Mol. Cell Proteomics 2004, 3, 746–769. [Google Scholar]
- WuDunn, D.; Spear, P.G. Initial interaction of herpes simplex virus with cells is binding to heparan sulfate. J. Virol. 1989, 63, 52–58. [Google Scholar]
- Antoine, T.E.; Mishra, Y.K.; Trigilio, J.; Tiwari, V.; Adelung, R.; Shukla, D. Prophylactic, therapeutic and neutralizing effects of zinc oxide tetrapod structures against herpes simplex virus type-2 infection. Antivir. Res. 2012, 96, 363–375. [Google Scholar] [CrossRef]
- Trigilio, J.; Antoine, T.E.; Paulowicz, I.; Mishra, Y.K.; Adelung, R.; Shukla, D. Tin oxide nanowires suppress herpes simplex virus-1 entry and cell-to-cell membrane fusion. PLoS One 2012, 7, e48147. [Google Scholar]
- Baram-Pinto, D.; Shukla, S.; Richman, M.; Gedanken, A.; Rahimipour, S.; Sarid, R. Surface-modified protein nanospheres as potential antiviral agents. Chem. Commun. (Camb.) 2012, 48, 8359–8361. [Google Scholar] [CrossRef]
- Baram-Pinto, D.; Shukla, S.; Gedanken, A.; Sarid, R. Inhibition of HSV-1 attachment, entry, and cell-to-cell spread by functionalized multivalent gold nanoparticles. Small 2010, 6, 1044–1050. [Google Scholar]
- Freeman, E.E.; Weiss, H.A.; Glynn, J.R.; Cross, P.L.; Whitworth, J.A.; Hayes, R.J. Herpes simplex virus 2 infection increases HIV acquisition in men and women: Systematic review and meta-analysis of longitudinal studies. AIDS 2006, 20, 73–83. [Google Scholar] [CrossRef]
- Vicenzi, E.; Gatti, A.; Ghezzi, S.; Oreste, P.; Zoppetti, G.; Poli, G. Broad spectrum inhibition of HIV-1 infection by sulfated K5 Escherichia coli polysaccharide derivatives. AIDS 2003, 17, 177–181. [Google Scholar]
- Lindahl, U.; Li, J.P.; Kusche-Gullberg, M.; Salmivirta, M.; Alaranta, S.; Veromaa, T.; Emeis, J.; Roberts, I.; Taylor, C.; Oreste, P.; et al. Generation of "neoheparin" from E. coli K5 capsular polysaccharide. J. Med. Chem. 2005, 48, 349–352. [Google Scholar] [CrossRef]
- Pinna, D.; Oreste, P.; Coradin, T.; Kajaste-Rudnitski, A.; Ghezzi, S.; Zoppetti, G.; Rotola, A.; Argnani, R.; Poli, G.; Manservigi, R.; et al. Inhibition of herpes simplex virus types 1 and 2 in vitro infection by sulfated derivatives of Escherichia coli K5 polysaccharide. Antimicrob. Agents Chemother. 2008, 52, 3078–3084. [Google Scholar]
- Epstein, S.P.; Nurozler, M.; Smetana, C.R.; Asbell, P.A. Efficacy of polyclonal antibodies for treatment of ocular herpes simplex infection. Cornea 2001, 20, 495–500. [Google Scholar]
- Heegaard, P.M.; Boas, U. Dendrimer based anti-infective and anti-inflammatory drugs. Recent Pat. Antiinfect. Drug Discov. 2006, 1, 331–351. [Google Scholar]
- Rosa Borges, A.; Schengrund, C.L. Dendrimers and antivirals: A review. Curr. Drug Targets Infect. Disord. 2005, 5, 247–254. [Google Scholar]
- Menjoge, A.R.; Kannan, R.M.; Tomalia, D.A. Dendrimer-based drug and imaging conjugates: Design considerations for nanomedical applications. Drug Discov. Today 2010, 15, 171–185. [Google Scholar]
- Gong, E.; Matthews, B.; McCarthy, T.; Chu, J.; Holan, G.; Raff, J.; Sacks, S. Evaluation of dendrimer SPL7013, a lead microbicide candidate against herpes simplex viruses. Antivir. Res. 2005, 68, 139–146. [Google Scholar] [CrossRef]
- Bernstein, D.I.; Stanberry, L.R.; Sacks, S.; Ayisi, N.K.; Gong, Y.H.; Ireland, J.; Mumper, R.J.; Holan, G.; Matthews, B.; McCarthy, T.; et al. Evaluations of unformulated and formulated dendrimer-based microbicide candidates in mouse and guinea pig models of genital herpes. Antimicrob. Agents Chemother. 2003, 47, 3784–3788. [Google Scholar] [CrossRef]
- McGowan, I.; Gomez, K.; Bruder, K.; Febo, I.; Chen, B.A.; Richardson, B.A.; Husnik, M.; Livant, E.; Price, C.; Jacobson, C.; et al. Phase 1 randomized trial of the vaginal safety and acceptability of SPL7013 gel (VivaGel) in sexually active young women (MTN-004). AIDS 2011, 25, 1057–1064. [Google Scholar] [CrossRef]
- O'Loughlin, J.; Millwood, I.Y.; McDonald, H.M.; Price, C.F.; Kaldor, J.M.; Paull, J.R. Safety, tolerability, and pharmacokinetics of SPL7013 gel (VivaGel): A dose ranging, phase I study. Sex. Transm. Dis. 2010, 37, 100–104. [Google Scholar]
- Chen, M.Y.; Millwood, I.Y.; Wand, H.; Poynten, M.; Law, M.; Kaldor, J.M.; Wesselingh, S.; Price, C.F.; Clark, L.J.; Paull, J.R.; et al. A randomized controlled trial of the safety of candidate microbicide SPL7013 gel when applied to the penis. J. Acquir. Immune Defic. Syndr. 2009, 50, 375–380. [Google Scholar]
- Tarallo, R.; Carberry, T.P.; Falanga, A.; Vitiello, M.; Galdiero, S.; Galdiero, M.; Weck, M. Dendrimers functionalized with membrane-interacting peptides for viral inhibition. Int. J. Nanomedicine 2013, 8, 521–534. [Google Scholar] [CrossRef]
- Luganini, A.; Nicoletto, S.F.; Pizzuto, L.; Pirri, G.; Giuliani, A.; Landolfo, S.; Gribaudo, G. Inhibition of herpes simplex virus type 1 and type 2 infections by peptide-derivatized dendrimers. Antimicrob. Agents Chemother. 2011, 55, 3231–3239. [Google Scholar]
- Vo, T.S.; Ngo, D.H.; Ta, Q.V.; Kim, S.K. Marine organisms as a therapeutic source against herpes simplex virus infection. Eur. J. Pharm. Sci. 2011, 44, 11–20. [Google Scholar]
- Tiwari, V.; Darmani, N.A.; Yue, B.Y.; Shukla, D. In vitro antiviral activity of neem (Azardirachta indica L.) bark extract against herpes simplex virus type-1 infection. Phytother. Res. 2010, 24, 1132–1140. [Google Scholar]
- Tiwari, V.; Shukla, S.Y.; Shukla, D. A sugar binding protein cyanovirin-N blocks herpes simplex virus type-1 entry and cell fusion. Antivir. Res. 2009, 84, 67–75. [Google Scholar] [CrossRef]
- Radtke, K.; Dohner, K.; Sodeik, B. Viral interactions with the cytoskeleton: A hitchhiker's guide to the cell. Cell Microbiol. 2006, 8, 387–400. [Google Scholar] [CrossRef]
- Lehmann, M.J.; Sherer, N.M.; Marks, C.B.; Pypaert, M.; Mothes, W. Actin- and myosin-driven movement of viruses along filopodia precedes their entry into cells. J. Cell Biol. 2005, 170, 317–325. [Google Scholar] [CrossRef]
- Sherer, N.M.; Lehmann, M.J.; Jimenez-Soto, L.F.; Horensavitz, C.; Pypaert, M.; Mothes, W. Retroviruses can establish filopodial bridges for efficient cell-to-cell transmission. Nat. Cell Biol. 2007, 9, 310–315. [Google Scholar] [CrossRef]
- Coffer, P.J.; Jin, J.; Woodgett, J.R. Protein kinase B (c-Akt): A multifunctional mediator of phosphatidylinositol 3-kinase activation. Biochem. J. 1998, 335, 1–13. [Google Scholar]
- Tiwari, V.; Shukla, D. Phosphoinositide 3 kinase signalling may affect multiple steps during herpes simplex virus type-1 entry. J. Gen. Virol. 2010, 91, 3002–3009. [Google Scholar] [CrossRef]
- Lairson, D.R.; Begley, C.E.; Reynolds, T.F.; Wilhelmus, K.R. Prevention of herpes simplex virus eye disease: A cost-effectiveness analysis. Arch. Ophthalmol. 2003, 121, 108–112. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hadigal, S.; Shukla, D. Exploiting Herpes Simplex Virus Entry for Novel Therapeutics. Viruses 2013, 5, 1447-1465. https://doi.org/10.3390/v5061447
Hadigal S, Shukla D. Exploiting Herpes Simplex Virus Entry for Novel Therapeutics. Viruses. 2013; 5(6):1447-1465. https://doi.org/10.3390/v5061447
Chicago/Turabian StyleHadigal, Satvik, and Deepak Shukla. 2013. "Exploiting Herpes Simplex Virus Entry for Novel Therapeutics" Viruses 5, no. 6: 1447-1465. https://doi.org/10.3390/v5061447
APA StyleHadigal, S., & Shukla, D. (2013). Exploiting Herpes Simplex Virus Entry for Novel Therapeutics. Viruses, 5(6), 1447-1465. https://doi.org/10.3390/v5061447