Identification and Characterization of Porcine Kobuvirus Variant Isolated from Suckling Piglet in Gansu Province, China

Abstract
:1. Introduction
2. Results and Discussion
2.1. Prevalence and Complete Genomic Sequences of Porcine Kobuviruses

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region | Length (nt) | Nucleotide identity (%) | Amino acid identity (%) | Predicted N-terminal cleavage site | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| K-11 | K-4 | GS-1 | GS-2 | K-11 | K-4 | GS-1 | GS-2 | K1-1/K-4 | GS-1/GS-2 | S-1-HUN | ||
| 5′UTR | 576 | 97 | 95 | 95 | 95 | - | - | - | - | - | - | - |
| L | 585 | 87 | 87 | 86 | 86 | 93 | 93 | 93 | 93 | - | - | - |
| VP0 | 1,098 | 83 | 87 | 87 | 85 | 90 | 96 | 96 | 93 | Q/G | Q/G | Q/G |
| VP3 | 669 | 87 | 87 | 86 | 77 | 93 | 97 | 95 | 97 | Q/H | Q/H | Q/H |
| VP1 | 762 | 85 | 86 | 82 | 85 | 96 | 96 | 89 | 96 | Q/A | Q/A | Q/A |
| 2A | 408 | 89 | 88 | 88 | 87 | 95 | 93 | 90 | 93 | Q/C | Q/C | Q/C |
| 2B | 585/495 * | 87 | 87 | 76 | 77 | 97 | 93 | 95 | 96 | Q/G | Q/G | Q/G |
| 2C | 1,005 | 92 | 90 | 91 | 91 | 98 | 98 | 98 | 98 | Q/G | Q/G | Q/G |
| 3A | 270 | 87 | 87 | 87 | 88 | 91 | 93 | 93 | 93 | Q/G | Q/G | Q/G |
| 3B | 102 | 96 | 89 | 92 | 91 | 100 | 97 | 100 | 100 | Q/G | Q/G | Q/G |
| 3C | 576 | 86 | 88 | 88 | 88 | 95 | 99 | 97 | 98 | Q/S | Q/C | Q/S |
| 3D | 1,407 | 93 | 93 | 93 | 93 | 98 | 98 | 99 | 99 | Q/S | Q/S | Q/S |
| 3′UTR | 167/168 * | 98 | 97 | 96 | 96 | - | - | - | - | - | - | - |
| Structural (VP0–VP3) | 2,329 | 81 | 86 | 85 | 83 | 93 | 97 | 94 | 95 | - | - | - |
| Non-structural(2A–3D) | 353/4,263 * | 91 | 90 | 89 | 89 | 97 | 97 | 95 | 95 | - | - | - |
| Complete genome | 8,210/8,121 | 86 | 89 | 88 | 87 | 95 | 96 | 94 | 94 | - | - | - |
2.2. Analysis of the UTRs and Coding Regions of the Four Strains
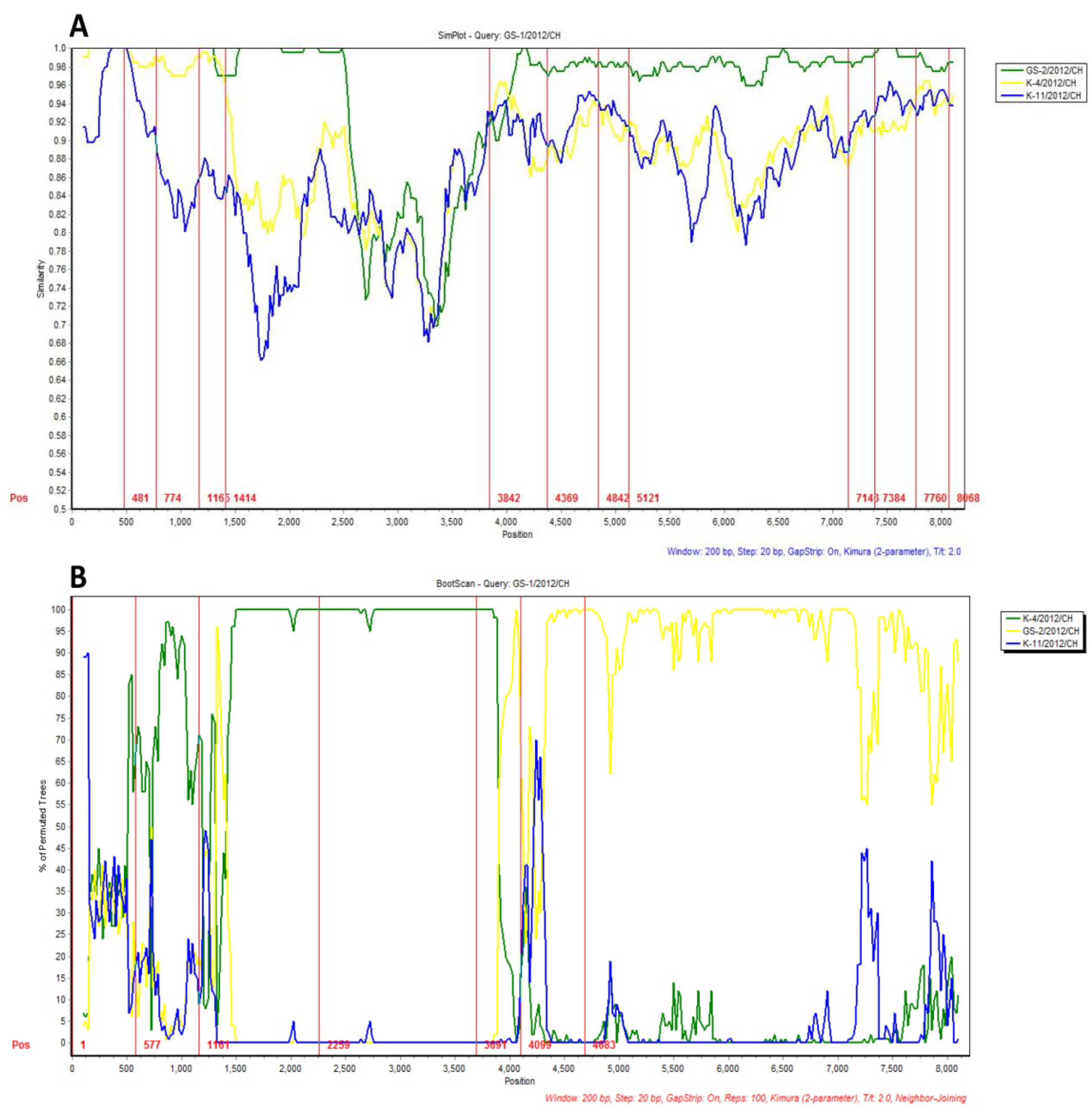
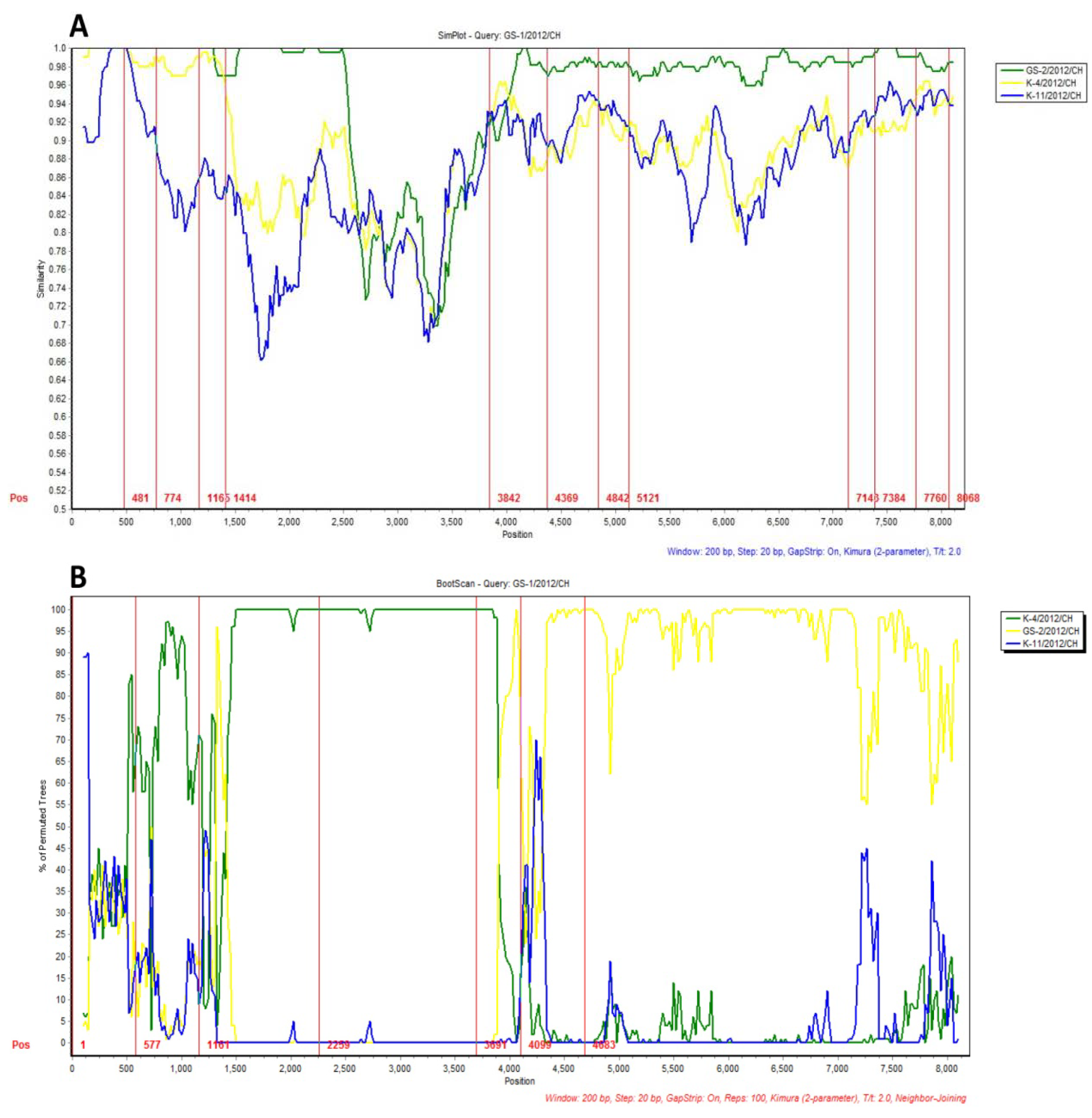
2.3. Recombination Analysis of the Variant Stains
2.4. Analysis of Coding Region Amino Acid Sequences
| Accession No. | Virus strain | Kobuvirus species | Host species | Country reported |
|---|---|---|---|---|
| GQ927711 | D/VI2287/2004 | Aichi virus | Human | Germany |
| JX564249 | kvgh99012632/2010 | Aichi virus | Human | Taiwan |
| DQ028632 | Goiania/GO/03/01/Brazil | Aichi virus | Human | Brazil |
| FJ890523 | Chshc7 | Aichi virus | Human | China |
| AB040749 | A846/88 | Aichi virus | Human | Japan |
| HQ650197 | KB101 | Bovine virus | Cattle | South Korea |
| AB084788 | U-1 | Bovine virus | Cattle | Japan |
| HQ650168 | KB12 | Bovine virus | Cattle | South Korea |
| HQ650180 | KB15 | Bovine virus | Cattle | South Korea |
| JF755427 | M-5/USA/2010 | Mouse virus | Mouse | USA |
| JN387133 | AN211D/USA/2009 | Dog virus | Dog | USA |
| KF006985 | MpKoV38 | Ferret virus | Ferret | Netherlands |
| GU245693 | TB3/HUN/2009 | Sheep virus | Sheep | Hungary |
| HQ400969 | 97DA4 | Porcine virus | Sus scrofa | South Korea |
| HQ400970 | 111DA18 | Porcine virus | Sus scrofa | South Korea |
| HQ400968 | 99DA6 | Porcine virus | Sus scrofa | South Korea |
| HQ400967 | 110DA17 | Porcine virus | Sus scrofa | South Korea |
| HQ400963 | 95DA2 | Porcine virus | Sus scrofa | South Korea |
| HQ400966 | 123DB10 | Porcine virus | Sus scrofa | South Korea |
| HQ400962 | 94DA1 | Porcine virus | Sus scrofa | South Korea |
| HQ400948 | 15OA16 | Porcine virus | Sus scrofa | South Korea |
| JF422792 | D11 | Porcine virus | Pig | China |
| JF422788 | D1 | Porcine virus | Pig | China |
| JX401523 | CH/HNXX-4/2012 | Porcine virus | Piglet | China |
| NC_016769 | SH-W-CHN | Porcine virus | Pig | China |
| JX177612 | WB-1-HUN/2011 | Porcine virus | Sus scrofa | Hungary |
| JQ692069 | WHU1 | Porcine virus | Sus scrofa | China |
| GQ249161 | K-30-HUN/2008 | Porcine virus | Sus scrofa | Hungary |
| EU787450 | S-1-HUN | Porcine virus | Sus scrofa | Hungary |
| JX827598 | CH/HZ/2011 | Porcine virus | Pig | China |
| GU298971 | Ch16/2008/CHN | Porcine virus | Pig | China |
| GU298967 | Ch40/2008/CHN | Porcine virus | Pig | China |
| GU292559 | JY-2010a/CHN | Porcine virus | Pig | China |
| KC414936 | K-11/2012/CH | Porcine virus | Sus scrofa | China |
| KC424638 | K-4/2012/CH | Porcine virus | Sus scrofa | China |
| KC424639 | GS-1/2012/CH | Porcine virus | Sus scrofa | China |
| KC424649 | GS-2/2012/CH | Porcine virus | Sus scrofa | China |
| KC204684 | XX | Porcine virus | Pig | China |
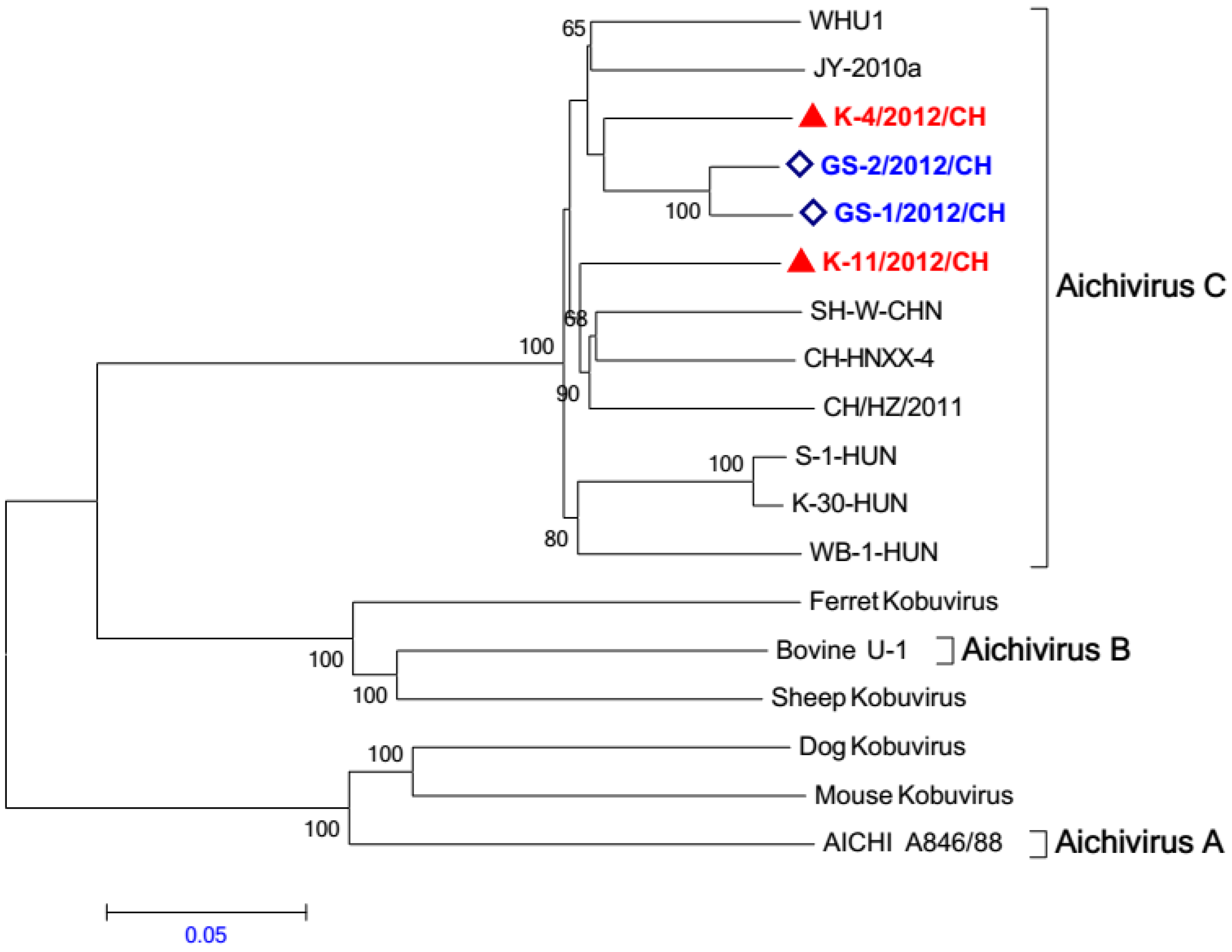
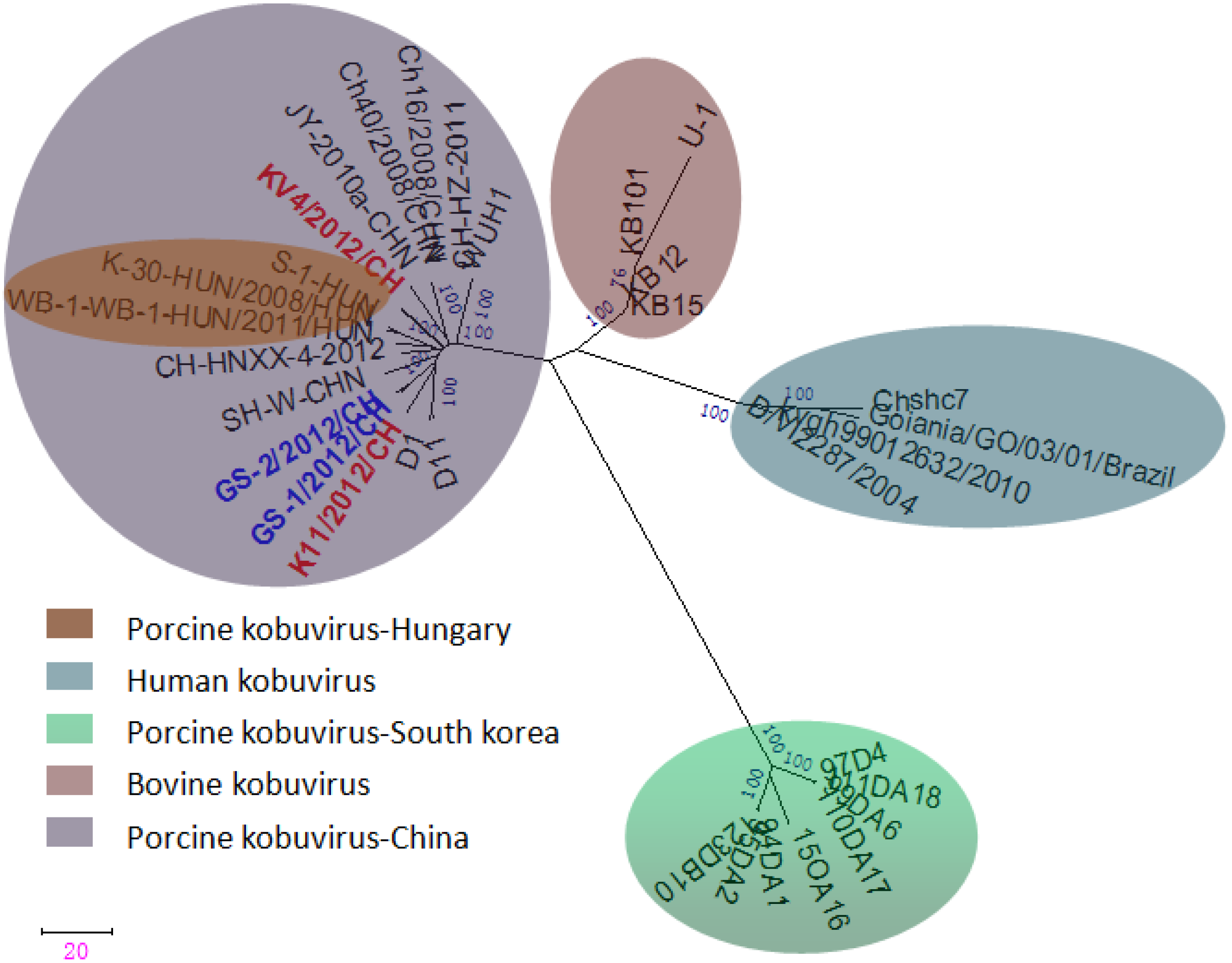
2.5. Phylogenetic Analysis

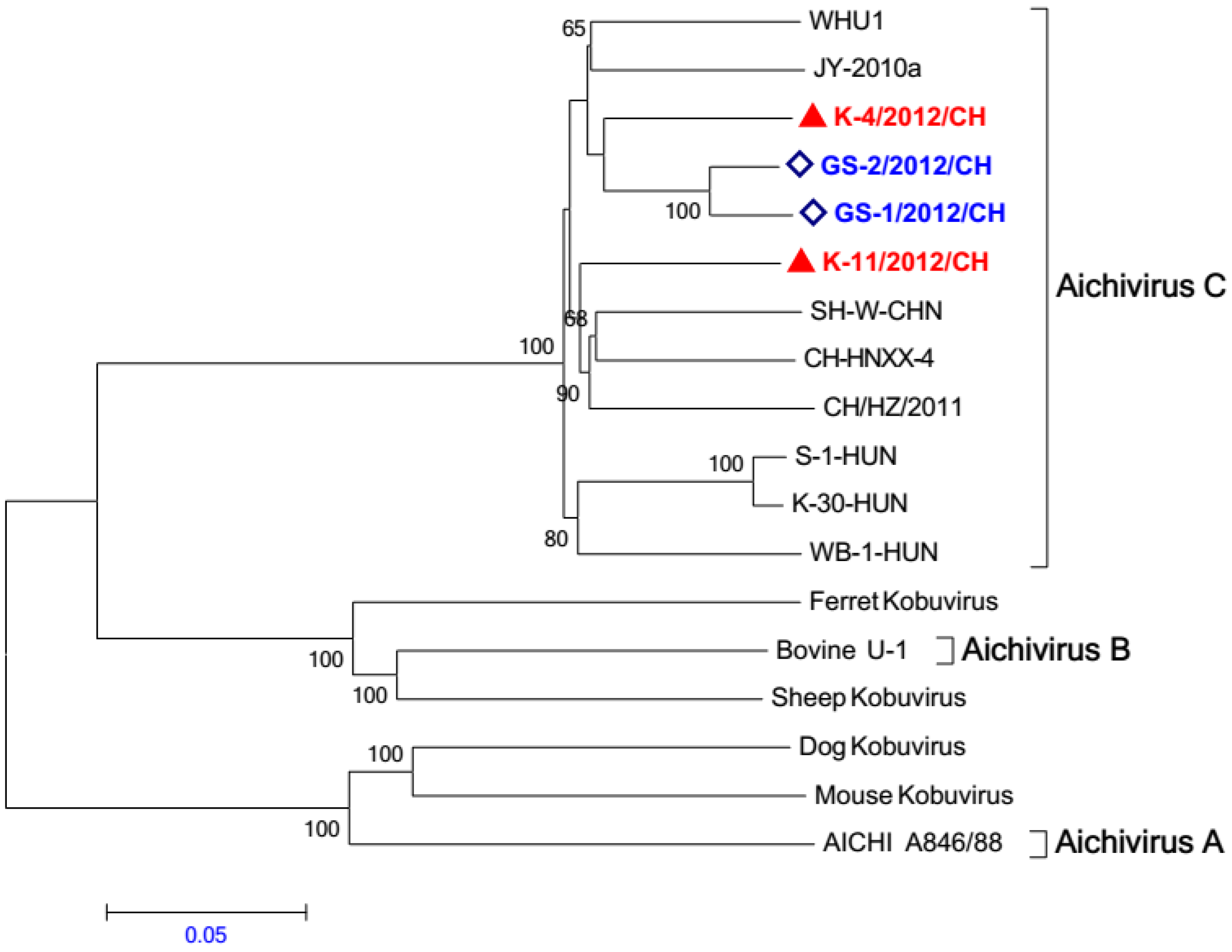

2.6. Viral Culture
3. Experimental
3.1. Fecal and Serum Samples
3.2. RT-PCR, Cloning, and Sequencing
| Primer name | Sequence (5'–3') | Size of PCR product (bp) |
|---|---|---|
| Pokv-30F | CCCTCACCCTCTTTTCCG | 369 |
| Pokv-398R | ACCGCAGTCCATGCTCTA | |
| Pokv-302F | AAACTCCTACCCGACAAA | 966 |
| Pokv-1267R | CAGGRCCATCACCAAGRC | |
| Pokv-1134F | YAMCACTCCTTGCCAGATC | 1048 |
| Pokv-2181R | AGAACCAGTWGGRACAGM | |
| Pokv-2048F | ACCTCTAYCAGGGCAAYA | 955 |
| Pokv-3002R | GGTTCAGGGACWGTAGTAGC | |
| Pokv-2894F | TSCGYGGCATCCAAGCAC | 946 |
| Pokv-3839R | AGACCAAGGCGGGAAAGG | |
| Pokv-3723F | CTGTCCAGACGTGCGGRTYT | 1019 |
| Pokv-4741R | CCATGAGCCACTCGGTGTTC | |
| Pokv-4668F | GACGGTTGAACAYCAAGGTG | 928 |
| Pokv-5595R | GAARGAAGGYTGCCAAAGAG | |
| Pokv-5462F | ARCCCTTCGAYCCTGTGGAG | 925 |
| Pokv-6386R | GGAGGACCAGAAAGAGTAGAAAT | |
| Pokv-6219F | YATCGGTCCRGACACCTTTG | 970 |
| Pokv-7188R | GATAGCGTGKAYGGGAGCAG | |
| Pokv-7036F | TTGCCRMYTCCWGAGTTRGA | 695 |
| Pokv-7730R | AAAGTRTCTGTTTTRTTTGCTG | |
| Pokv-7614F | CTAYGGTGATGATGTGATCTATG | 582 |
| Pokv-8195R | AAGTAAAGGACAGCCAGGGA | |
| Kv-F | TCTGGATGCGTTGGCACTTCCAT | 519 |
| Kv-R | CCAGCGGGTCTGAAGGTAAGAGT |
3.3. Sequencing and Phylogenetic Analysis
3.4. Viral Culture
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- International Committee on Taxonomy of Viruses. ICTV official taxonomy: Updates since the 9th report. Available online: http://talk.ictvonline.org/files/ictv_documents/m/msl/4440.aspx (accessed on 14 January 2012).
- Yamashita, T.; Kobayashi, S.; Sakae, K.; Nakata, S.; Chiba, S.; Ishihara, Y.; Isomura, S. Isolation of cytopathic small round viruses with BS-C-1 cells from patients with gastroenteritis. J. Infect. Dis. 1991, 164, 954–957. [Google Scholar] [CrossRef]
- Yamashita, T.; Ito, M.; Kabashima, Y.; Tsuzuki, H.; Fujiura, A.; Sakae, K. Isolation and characterization of a new species of kobuvirus associated with cattle. J. Gen. Virol. 2003, 84, 3069–3077. [Google Scholar] [CrossRef]
- Reuter, G.; Boldizsar, A.; Kiss, I.; Pankovics, P. Candidate new species of Kobuvirus in porcine hosts. Emerging Infect. Dis. 2008, 14, 1968–1970. [Google Scholar] [CrossRef]
- Reuter, G.; Kecskemeti, S.; Pankovics, P. Evolution of porcine kobuvirus infection, Hungary. Emerging Infect. Dis. 2010, 16, 696–698. [Google Scholar] [CrossRef]
- Li, L.; Victoria, J.G.; Wang, C.; Jones, M.; Fellers, G.M.; Kunz, T.H.; Delwart, E. Bat guano virome: Predominance of dietary viruses from insects and plants plus novel mammalian viruses. J. Virol. 2010, 84, 6955–6965. [Google Scholar]
- Li, L.; Pesavento, P.A.; Shan, T.; Leutenegger, C.M.; Wang, C.; Delwart, E. Viruses in diarrhoeic dogs include novel kobuviruses and sapoviruses. J. Gen. Virol. 2011, 92, 2534–2541. [Google Scholar] [CrossRef]
- Chung, J.Y.; Kim, S.H.; Kim, Y.H.; Lee, M.H.; Lee, K.K.; Oem, J.K. Detection and genetic characterization of feline kobuviruses. Virus Genes 2013. [Google Scholar] [CrossRef]
- Smits, S.L.; Raj, V.S.; Oduber, M.D.; Schapendonk, C.M.; Bodewes, R.; Provacia, L.; Stittelaar, K.J.; Osterhaus, A.D.; Haagmans, B.L. Metagenomic analysis of the ferret fecal viral flora. PLoS One 2013, 8, e71595. [Google Scholar]
- Lee, M.H.; Jeoung, H.Y.; Lim, J.A.; Song, J.Y.; Song, D.S.; An, D.J. Kobuvirus in South Korean black goats. Virus Genes 2012, 45, 186–189. [Google Scholar] [CrossRef]
- Yu, J.M.; Jin, M.; Zhang, Q.; Li, H.Y.; Li, D.D.; Xu, Z.Q.; Li, J.S.; Cui, S.X.; Yang, S.H.; Liu, N.; et al. Candidate porcine Kobuvirus, China. Emerging Infect. Dis. 2009, 15, 823–825. [Google Scholar] [CrossRef]
- Khamrin, P.; Maneekarn, N.; Hidaka, S.; Kishikawa, S.; Ushijima, K.; Okitsu, S.; Ushijima, H. Molecular detection of kobuvirus sequences in stool samples collected from healthy pigs in Japan. Infect. Genet. Evol. 2010, 10, 950–954. [Google Scholar] [CrossRef]
- Khamrin, P.; Maneekarn, N.; Kongkaew, A.; Kongkaew, S.; Okitsu, S.; Ushijima, H. Porcine kobuvirus in piglets, Thailand. Emerging Infect. Dis. 2009, 15, 2075–2076. [Google Scholar] [CrossRef]
- Barry, A.F.; Ribeiro, J.; Alfieri, A.F.; van der Poel, W.H.; Alfieri, A.A. First detection of kobuvirus in farm animals in Brazil and The Netherlands. Infect. Genet. Evol. 2011, 11, 1811–1814. [Google Scholar] [CrossRef]
- Park, S.J.; Kim, H.K.; Moon, H.J.; Song, D.S.; Rho, S.M.; Han, J.Y.; Nguyen, V.G.; Park, B.K. Molecular detection of porcine kobuviruses in pigs in Korea and their association with diarrhea. Arch. Virol. 2010, 155, 1803–1811. [Google Scholar] [CrossRef]
- Reuter, G.; Boros, A.; Pankovics, P. Kobuviruses—A comprehensive review. Rev. Med. Virol. 2011, 21, 32–41. [Google Scholar] [CrossRef]
- Oberste, M.S.; Maher, K.; Pallansch, M.A. Genomic evidence that simian virus 2 and six other simian picornaviruses represent a new genus in Picornaviridae. Virology 2003, 314, 283–293. [Google Scholar] [CrossRef]
- Larkin, E.K.; Morris, N.J.; Li, Y.; Nock, N.L.; Stein, C.M. Comparison of affected sibling-pair linkage methods to identify gene x gene interaction in GAW15 simulated data. BMC Proc. 2007, 1, S66. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar]
- De Jong, A.S.; de Mattia, F.; van Dommelen, M.M.; Lanke, K.; Melchers, W.J.; Willems, P.H.; van Kuppeveld, F.J. Functional analysis of picornavirus 2B proteins: Effects on calcium homeostasis and intracellular protein trafficking. J. Virol. 2008, 82, 3782–3790. [Google Scholar] [CrossRef]
- Okitsu, S.; Khamrin, P.; Thongprachum, A.; Hidaka, S.; Kongkaew, S.; Kongkaew, A.; Maneekarn, N.; Mizuguchi, M.; Hayakawa, S.; Ushijima, H. Sequence analysis of porcine kobuvirus VP1 region detected in pigs in Japan and Thailand. Virus Genes 2012, 44, 253–257. [Google Scholar] [CrossRef]
- Wang, C.; Lan, D.; Cui, L.; Yang, Z.; Yuan, C.; Hua, X. Molecular characterization of a porcine kobuvirus strain in China. Arch. Virol. 2012, 157, 573–578. [Google Scholar] [CrossRef]
- Lukashev, A.N. Recombination among picornaviruses. Rev. Med. Virol. 2010, 20, 327–337. [Google Scholar] [CrossRef]
- Kuiken, T.; Holmes, E.C.; McCauley, J.; Rimmelzwaan, G.F.; Williams, C.S.; Grenfell, B.T. Host species barriers to influenza virus infections. Science 2006, 312, 394–397. [Google Scholar] [CrossRef]
- Hellen, C.U.; de Breyne, S. A distinct group of hepacivirus/pestivirus-like internal ribosomal entry sites in members of diverse picornavirus genera: Evidence for modular exchange of functional noncoding RNA elements by recombination. J. Virol. 2007, 81, 5850–5863. [Google Scholar] [CrossRef]
- Reuter, G.; Boldizsar, A.; Pankovics, P. Complete nucleotide and amino acid sequences and genetic organization of porcine kobuvirus, a member of a new species in the genus Kobuvirus, family Picornavirida. Arch. Virol. 2009, 154, 101–108. [Google Scholar] [CrossRef]
- Yu, Y.; Sweeney, T.R.; Kafasla, P.; Jackson, R.J.; Pestova, T.V.; Hellen, C.U. The mechanism of translation initiation on Aichivirus RNA mediated by a novel type of picornavirus IRES. EMBO J. 2011, 30, 4423–4436. [Google Scholar] [CrossRef]
- Sweeney, T.R.; Dhote, V.; Yu, Y.; Hellen, C.U. A distinct class of internal ribosomal entry site in members of the Kobuvirus and proposed Salivirus and Paraturdivirus genera of the Picornaviridae. J. Virol. 2012, 86, 1468–1486. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fan, S.; Sun, H.; Ying, Y.; Gao, X.; Wang, Z.; Yu, Y.; Li, Y.; Wang, T.; Yu, Z.; Yang, S.; et al. Identification and Characterization of Porcine Kobuvirus Variant Isolated from Suckling Piglet in Gansu Province, China. Viruses 2013, 5, 2548-2560. https://doi.org/10.3390/v5102548
Fan S, Sun H, Ying Y, Gao X, Wang Z, Yu Y, Li Y, Wang T, Yu Z, Yang S, et al. Identification and Characterization of Porcine Kobuvirus Variant Isolated from Suckling Piglet in Gansu Province, China. Viruses. 2013; 5(10):2548-2560. https://doi.org/10.3390/v5102548
Chicago/Turabian StyleFan, Shengtao, Heting Sun, Ying Ying, Xiaolong Gao, Zheng Wang, Yicong Yu, Yuanguo Li, Tiecheng Wang, Zhijun Yu, Songtao Yang, and et al. 2013. "Identification and Characterization of Porcine Kobuvirus Variant Isolated from Suckling Piglet in Gansu Province, China" Viruses 5, no. 10: 2548-2560. https://doi.org/10.3390/v5102548