
Validation of Candidate Host Cell Entry Factors for Bovine Herpes Virus Type-1 Based on a Genome-Wide CRISPR Knockout Screen
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Plasmids
2.3. dCas9 Knock-In to the Rosa26 Locus
2.4. Guide RNA Design
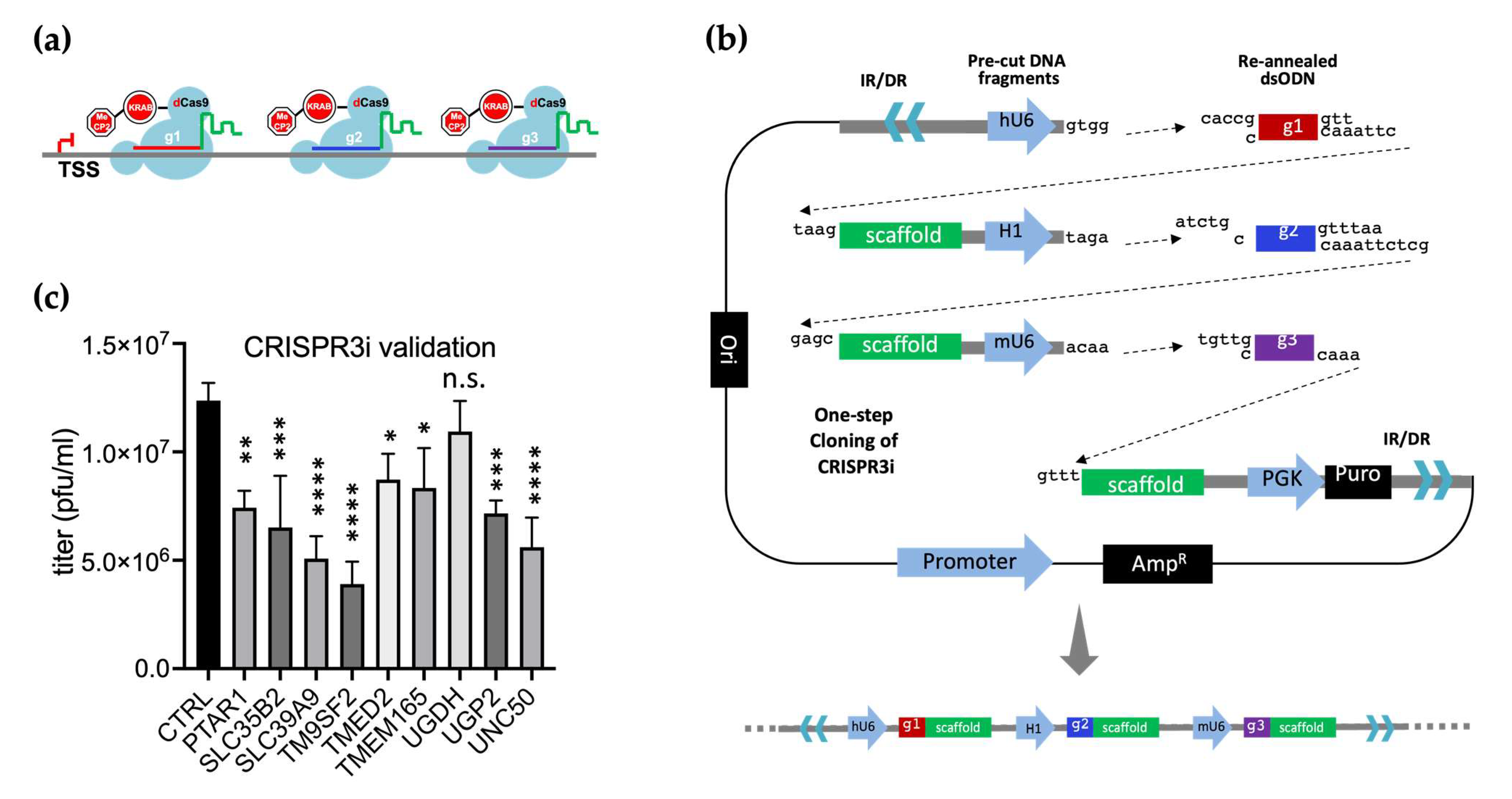
2.5. CRISPR3i Vector Cloning
2.6. Transfections
2.7. Generation of KO Clones and Stable Cells with CRISPRi/3i KD
2.8. Viral Infections and Plaque Assays
2.9. Genome/Pfu and VP26/Pfu Ratio Experiment
2.10. Lectin Staining and Microscopy of KO Cells
2.11. Lectin Staining of Concentrated Viruses
2.12. Statistical Analysis
3. Results
3.1. Genome-Wide CRISPRko Screen Identified Many Host Genes Potentially Important for BoHV-1 Entry into the Cell
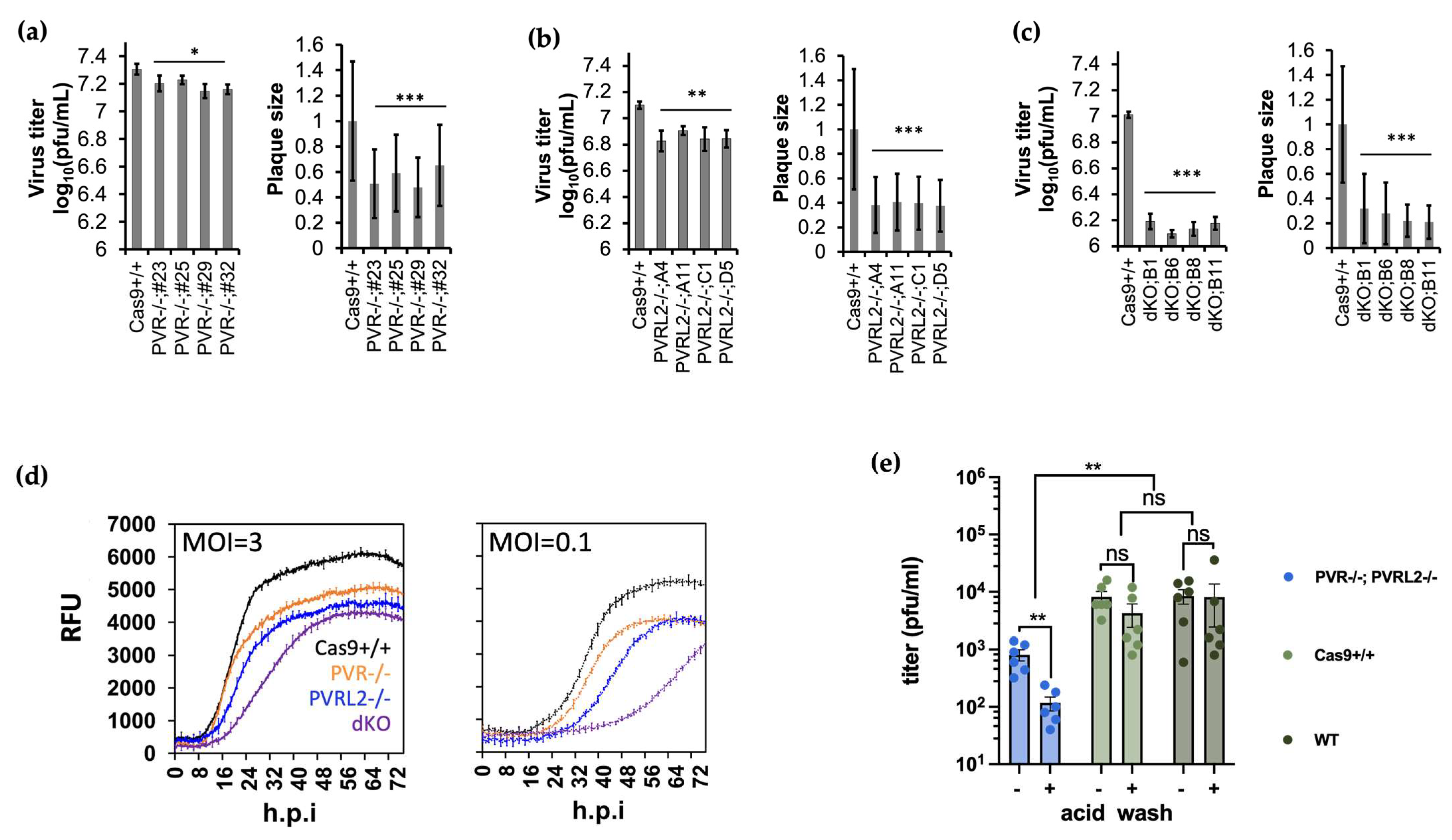
3.2. Knockout of PVR and PVRL2 Impaired BoHV-1 Attachment and Entry into the Host Cell
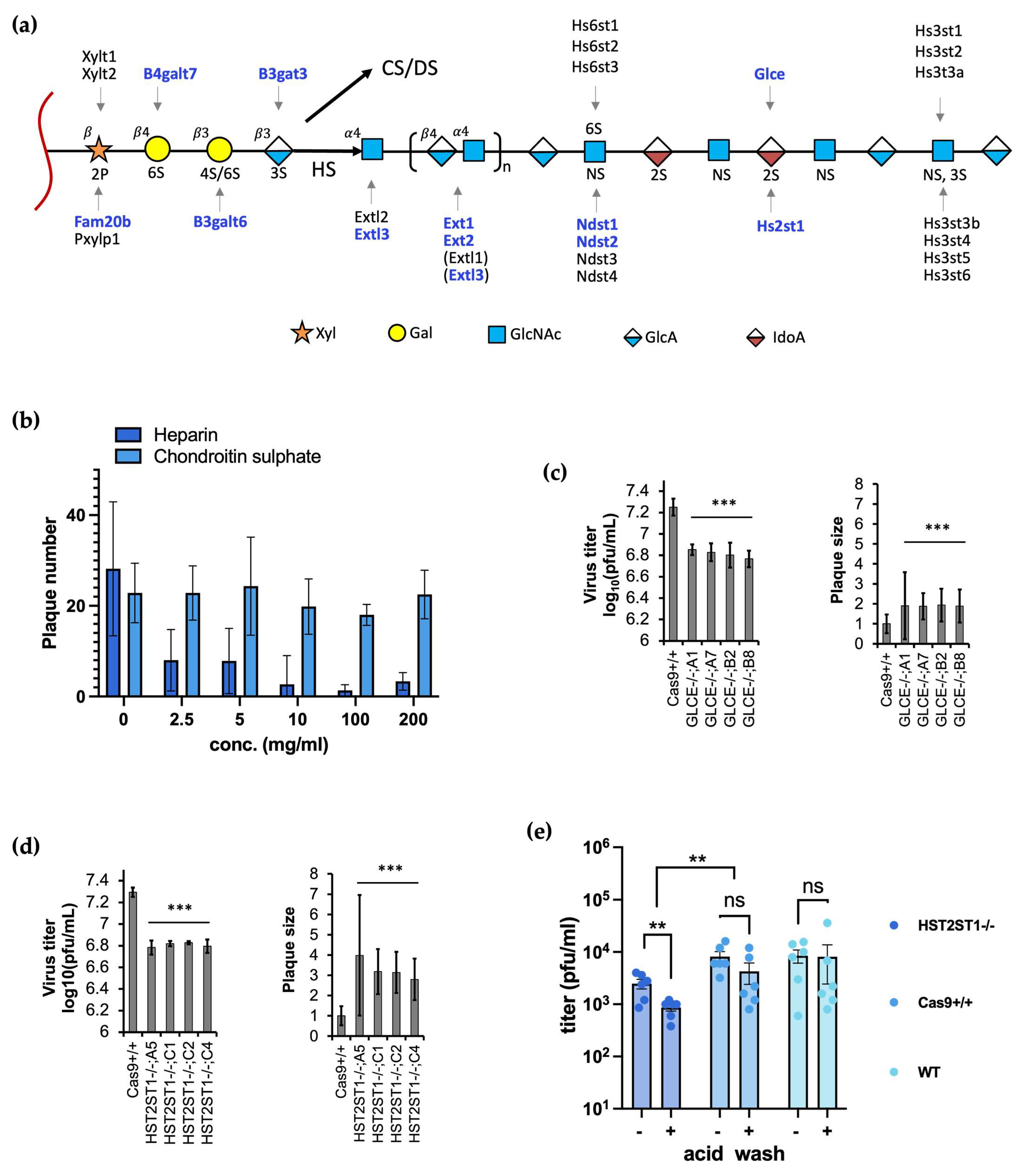
3.3. Heparan Sulfate but Not Chondroitin Sulfate Promotes Efficient BoHV-1 Entry
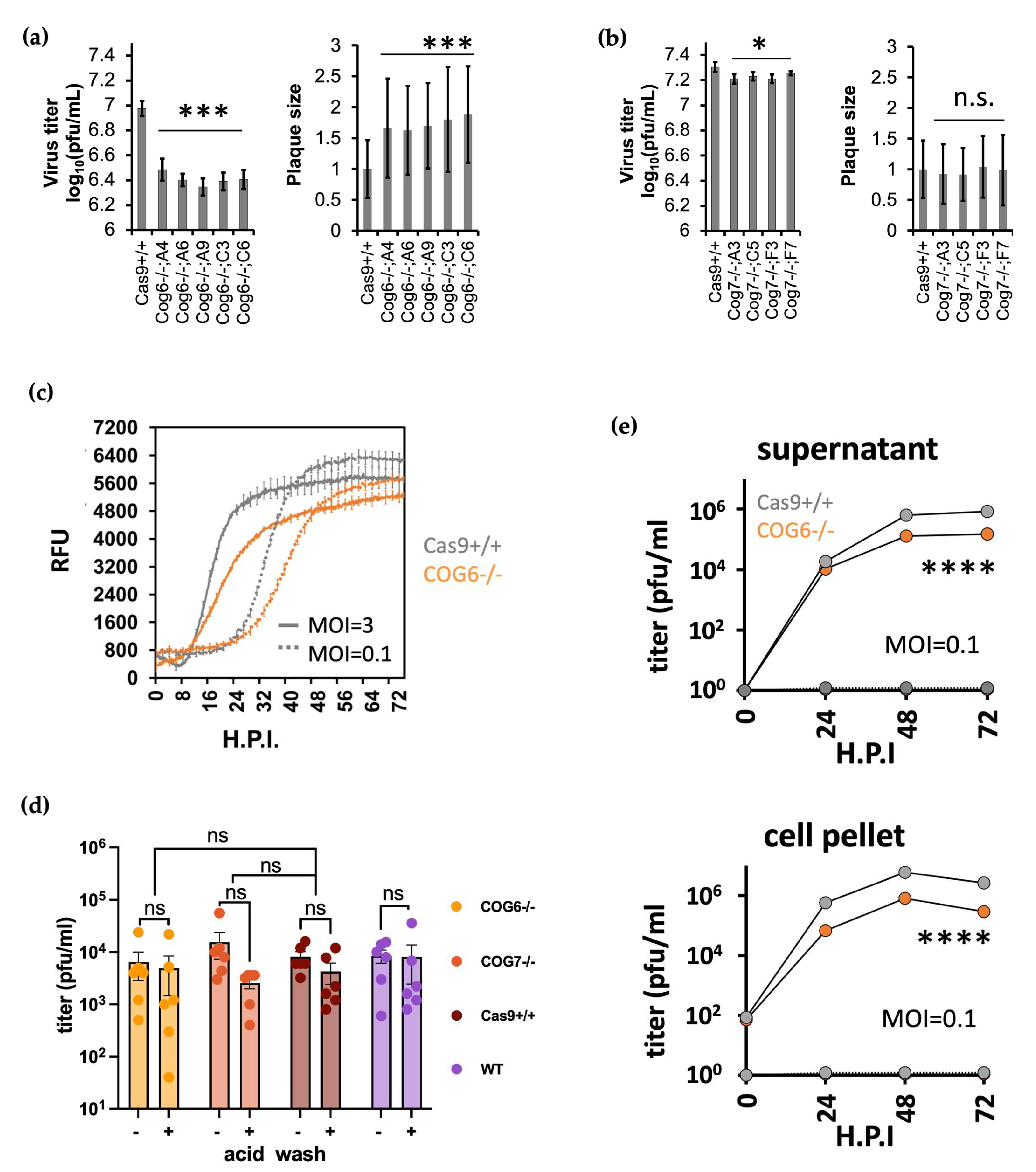
3.4. COG KO Impaired Multi-Cycle but Not Single Cycle BoHV-1 Virus Replication
3.5. COG6-/- Cells Produced Less Infectious Viruses Compared to Those by Wildtype Cells
3.6. COG6-/- Lead to Reduced Glycosylation Patterning of Host and Viral Proteins
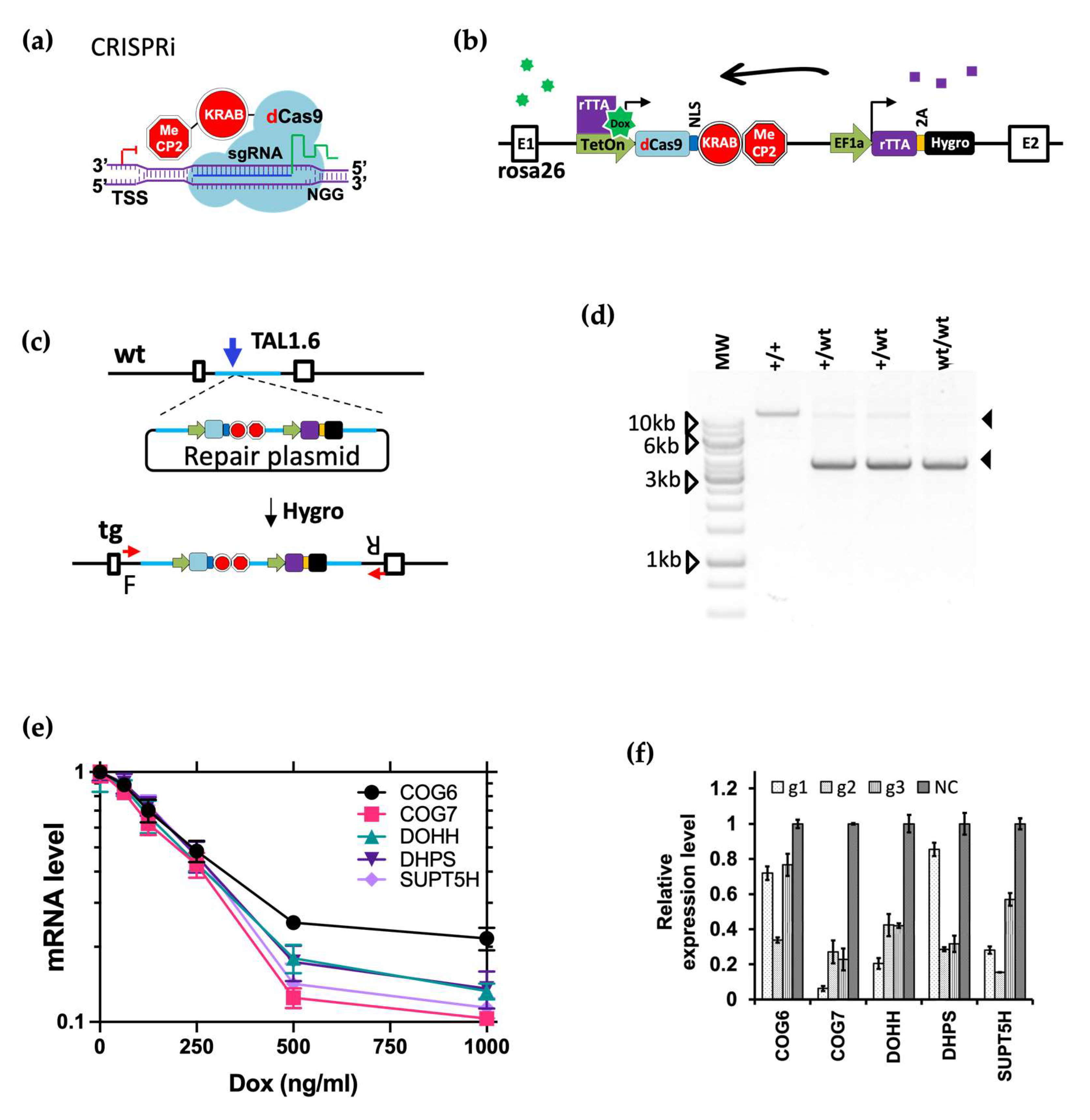
3.7. CRISPRi Efficiently Knocked down Host Gene Transcription
3.8. CRISPR3i Enabled Rapid Validation of Candidate Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jones, C.; Chowdhury, S. A review of the biology of bovine herpesvirus type 1 (BHV-1), its role as a cofactor in the bovine respiratory disease complex and development of improved vaccines. Anim. Health Res. Rev. 1996, 8, 187–205. [Google Scholar] [CrossRef]
- Babiuk, L.A.; van Drunen Littel-van den Hurk, S.; Tikoo, S.K. Immunology of bovine herpesvirus 1 infection. Veter. Microbiol. 1996, 53, 31–42. [Google Scholar] [CrossRef]
- Srikumaran, S.; Kelling, C.L.; Ambagala, A. Immune evasion by pathogens of bovine respiratory disease complex. Anim. Health Res. Rev. 1996, 8, 215–229. [Google Scholar] [CrossRef]
- Pastenkos, G.; Lee, B.; Pritchard, S.M.; Nicola, A.V. Bovine Herpesvirus 1 Entry by a Low-pH Endosomal Pathway. J. Virol. 2018, 92, e00839-18. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.P.; Babiuk, L.A.; van Drunen Littel-van den Hurk, S.; Fitzpatrick, D.R.; Zamb, T.J. Bovine herpesvirus 1 attachment to permissive cells is mediated by its major glycoproteins gI, gIII, and gIV. J. Virol. 1991, 65, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; van Drunen Littel-van den Hurk, S.; Babiuk, L.A.; Liang, X. Characterization of cell-binding properties of bovine herpesvirus 1 glycoproteins B, C, and D: Identification of a dual cell-binding function of gB. J. Virol. 1995, 69, 4758–4768. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Babiuk, L.A.; Zamb, T.J. An in vivo study of a glycoprotein gIII-negative bovine herpesvirus 1 (BHV-1) mutant expressing β-galactosidase: Evaluation of the role of gIII in virus infectivity and its use as a vector for mucosal immunization. Virology 1992, 189, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, K.; Matsuzaki, T.; Sugahara, Y.; Okada, J.; Hasebe, M.; Iwamura, Y.; Ohnishi, M.; Kanno, T.; Shimizu, M.; Honda, E.; et al. BHV-1 Adsorption is mediated by the interaction of glycoprotein gIII with heparinlike moiety on the cell surface. Virology 1991, 181, 666–670. [Google Scholar] [CrossRef]
- Keil, G.M.; Höhle, C.; Giesow, K.; König, P.; Krachmarov, C.; Pinter, A.; Honnen, W.J.; Gorny, M.K.; Nyambi, P.N.; Zolla-Pazner, S.; et al. Engineering Glycoprotein B of Bovine Herpesvirus 1 To Function as Transporter for Secreted Proteins: A New Protein Expression Approach. J. Virol. 2005, 79, 791–799. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Van Drunen Littel-Van Den Hurk, S.; Liang, X.; Babiuk, L.A. Functional analysis of the transmembrane anchor region of bovine herpesvirus 1 glycoprotein gB. Virology 1997, 228, 39–54. [Google Scholar] [CrossRef]
- Alves Dummer, L.; Pereira Leivas Leite, F.; Van Drunen Littel-Van Den Hurk, S. Bovine herpesvirus glycoprotein D: A review of its structural characteristics and applications in vaccinology. Vet. Res. 2014, 45, 111. [Google Scholar] [CrossRef]
- Chase, C.C.; Carter-Allen, K.; Lohff, C.; Letchworth, G.J. Bovine cells expressing bovine herpesvirus 1 (BHV-1) glycoprotein IV resist infection by BHV-1, herpes simplex virus, and pseudorabies virus. J. Virol. 1990, 64, 4866–4872. [Google Scholar] [CrossRef]
- Miethke, A.; Keil, G.M.; Weiland, F.; Mettenleiter, T.C. Unidirectional complementation, between glycoprotein B homologues of pseudorabies virus and bovine herpesvirus 1 is determined by the carboxy-terminal part of the molecule. J. Gen. Virol. 1995, 76, 1623–1635. [Google Scholar] [CrossRef]
- Campadelli-Fiume, G.; Cocchi, F.; Menotti, L.; Lopez, M. The novel receptors that mediate the entry of herpes simplex viruses and animal alphaherpesviruses into cells. Rev. Med. Virol. 2000, 10, 305–319. [Google Scholar] [CrossRef]
- Spear, P.G.; Eisenberg, R.J.; Cohen, G.H. Three classes of cell surface receptors for alphaherpesvirus entry. Virology 2000, 275, 1–8. [Google Scholar] [CrossRef]
- Geraghty, R.J.; Krummenacher, C.; Cohen, G.H.; Eisenberg, R.J.; Spear, P.G. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 1998, 280, 1618–1620. [Google Scholar] [CrossRef]
- Arii, J.; Uema, M.; Morimoto, T.; Sagara, H.; Akashi, H.; Ono, E.; Arase, H.; Kawaguchi, Y. Entry of Herpes Simplex Virus 1 and Other Alphaherpesviruses via the Paired Immunoglobulin-Like Type 2 Receptor α. J. Virol. 2009, 83, 4520–4527. [Google Scholar] [CrossRef] [PubMed]
- Van Drunen Littel-van Den Hurk, S.; Khattar, S.; Tikoo, S.K.; Babiuk, L.A.; Baranowski, E.; Plainchamp, D.; Thiry, E. Glycoprotein H (gII/gp108) and glycoprotein L form a functional complex which plays a role in penetration, but not in attachment, of bovine herpesvirus 1. J. Gen. Virol. 1996, 77, 1515–1520. [Google Scholar] [CrossRef] [PubMed]
- Fuller, A.O.; Santos, R.E.; Spear, P.G. Neutralizing antibodies specific for glycoprotein H of herpes simplex virus permit viral attachment to cells but prevent penetration. J. Virol. 1989, 63, 3435–3443. [Google Scholar] [CrossRef] [PubMed]
- Meyer, G.; Hanon, E.; Georlette, D.; Pastoret, P.P.; Thiry, E. Bovine herpesvirus type 1 glycoprotein H is essential for penetration and propagation in cell culture. J. Gen. Virol. 1998, 79, 1983–1987. [Google Scholar] [CrossRef] [PubMed]
- Khattar, S.K.; Van Drunen Littel-Van Den Hurk, S.; Attah-Poku, S.K.; Babiuk, L.A.; Tikoo, S.K. Identification and characterization of a bovine herpesvirus-1 (BHV-1) glycoprotein gL which is required for proper antigenicity, processing, and transport of BHV-1 glycoprotein gH. Virology 1996, 219, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Schröder, C.; Keil, G.M. Bovine herpesvirus 1 requires glycoprotein H for infectivity and direct spreading and glycoproteins gHW450 and gB for glycoprotein D-independent cell-to-cell spread. J. Gen. Virol. 1999, 80, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Warner, M.S.; Geraghty, R.J.; Martinez, W.M.; Montgomery, R.I.; Whitbeck, J.C.; Xu, R.; Eisenberg, R.J.; Cohen, G.H.; Spear, P.G. A cell surface protein with herpesvirus entry activity (Hveb) confers susceptibility to infection by mutants of herpes simplex virus type 1, herpes simplex virus type 2, and pseudorabies virus. Virology 1998, 246, 179–189. [Google Scholar] [CrossRef]
- Struyf, F.; Martinez, W.M.; Spear, P.G. Mutations in the N-Terminal Domains of Nectin-1 and Nectin-2 Reveal Differences in Requirements for Entry of Various Alphaherpesviruses and for Nectin-Nectin Interactions. J. Virol. 2002, 76, 12940–12950. [Google Scholar] [CrossRef]
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan sulfate proteoglycans. Cold Spring Harb. Perspect. Biol. 2011, 3, a004952. [Google Scholar] [CrossRef] [PubMed]
- Mende, M.; Bednarek, C.; Wawryszyn, M.; Sauter, P.; Biskup, M.B.; Schepers, U.; Bräse, S. Chemical Synthesis of Glycosaminoglycans. Chem. Rev. 2016, 116, 8193–8255. [Google Scholar] [CrossRef]
- Cagno, V.; Tseligka, E.D.; Jones, S.T.; Tapparel, C. Heparan sulfate proteoglycans and viral attachment: True receptors or adaptation bias? Viruses 2019, 11, 596. [Google Scholar] [CrossRef]
- Hallak, L.K.; Spillmann, D.; Collins, P.L.; Peeples, M.E. Glycosaminoglycan Sulfation Requirements for Respiratory Syncytial Virus Infection. J. Virol. 2000, 74, 10508–10513. [Google Scholar] [CrossRef]
- Kalia, M.; Chandra, V.; Rahman, S.A.; Sehgal, D.; Jameel, S. Heparan Sulfate Proteoglycans Are Required for Cellular Binding of the Hepatitis E Virus ORF2 Capsid Protein and for Viral Infection. J. Virol. 2009, 83, 12714–12724. [Google Scholar] [CrossRef]
- Shukla, D.; Liu, J.; Blaiklock, P.; Shworak, N.W.; Bai, X.; Esko, J.D.; Cohen, G.H.; Eisenberg, R.J.; Rosenberg, R.D.; Spear, P.G. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 1999, 99, 13–22. [Google Scholar] [CrossRef]
- Jinno, A.; Park, P.W. Role of glycosaminoglycans in infectious disease. In Glycosaminoglycans; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2015; Volume 1229, pp. 567–585. [Google Scholar] [CrossRef]
- Banfield, B.W.; Leduc, Y.; Esford, L.; Visalli, R.J.; Brandt, C.R.; Tufaro, F. Evidence for an Interaction of Herpes Simplex Virus with Chondroitin Sulfate Proteoglycans during Infection. Virology 1995, 208, 531–539. [Google Scholar] [CrossRef]
- Uyama, T.; Ishida, M.; Izumikawa, T.; Trybala, E.; Tufaro, F.; Bergström, T.; Sugahara, K.; Kitagawa, H. Chondroitin 4-O-sulfotransferase-1 regulates E disaccharide expression of chondroitin sulfate required for herpes simplex virus infectivity. J. Biol. Chem. 2006, 281, 38668–38674. [Google Scholar] [CrossRef]
- Hadley, B.; Maggioni, A.; Ashikov, A.; Day, C.J.; Haselhorst, T.; Tiralongo, J. Structure and function of nucleotide sugar transporters: Current progress. Comput. Struct. Biotechnol. J. 2014, 10, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Lind, T.; Tufaro, F.; McCormick, C.; Lindahl, U.; Lidholt, K. The putative tumor suppressors EXT1 and EXT2 are glycosyltransferases required for the biosynthesis of heparan sulfate. J. Biol. Chem. 1998, 273, 26265–26268. [Google Scholar] [CrossRef] [PubMed]
- Fisher, P.; Ungar, D. Bridging the gap between glycosylation and vesicle traffic. Front. Cell Develop. Biol. 2016, 4, 15. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, J.B.; D’Souza, Z.; Lupashin, V.V. Maintaining order: COG complex controls Golgi trafficking, processing, and sorting. FEBS Lett. 2019, 593, 2466–2487. [Google Scholar] [CrossRef] [PubMed]
- Zeevaert, R.; Foulquier, F.; Jaeken, J.; Matthijs, G. Deficiencies in subunits of the Conserved Oligomeric Golgi (COG) complex define a novel group of Congenital Disorders of Glycosylation. Mol. Genet. Metab. 2008, 93, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Steet, R.A.; Bohorov, O.; Bakker, J.; Newell, J.; Krieger, M.; Spaapen, L.; Kornfeld, S.; Freeze, H.H. Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nat. Med. 2004, 10, 518–523. [Google Scholar] [CrossRef] [PubMed]
- Ungar, D.; Oka, T.; Brittle, E.E.; Vasile, E.; Lupashin, V.V.; Chatterton, J.E.; Heuser, J.E.; Krieger, M.; Gerard Waters, M. Characterization of a mammalian Golgi-localized protein complex, COG, that is required for normal Golgi morphology and function. J. Cell Biol. 2002, 157, 405–415. [Google Scholar] [CrossRef]
- Kingsley, D.M.; Kozarsky, K.F.; Segal, M.; Krieger, M. Three types of low density lipoprotein receptor-deficient mutant have pleiotropic defects in the synthesis of N-linked, O-linked, and lipid-linked carbohydrate chains. J. Cell Biol. 1986, 102, 1576–1585. [Google Scholar] [CrossRef]
- Tan, W.; Carlson, D.F.; Lancto, C.A.; Garbe, J.R.; Webster, D.A.; Hackett, P.B.; Fahrenkrug, S.C. Efficient nonmeiotic allele introgression in livestock using custom endonucleases. Proc. Natl. Acad. Sci. USA 2013, 110, 16526–16531. [Google Scholar] [CrossRef]
- Cermak, T.; Doyle, E.L.; Christian, M.; Wang, L.; Zhang, Y.; Schmidt, C.; Baller, J.A.; Somia, N.V.; Bogdanove, A.J.; Voytas, D.F. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011, 39, e82. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Mali, P.; Esvelt, K.M.; Church, G.M. Cas9 as a versatile tool for engineering biology. Nat. Methods 2013, 10, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; East, A.; Cheng, A.; Lin, S.; Ma, E.; Doudna, J. RNA-programmed genome editing in human cells. eLife 2013, 2013, e00471. [Google Scholar] [CrossRef] [PubMed]
- Park, R.J.; Wang, T.; Koundakjian, D.; Hultquist, J.F.; Lamothe-Molina, P.; Monel, B.; Schumann, K.; Yu, H.; Krupzcak, K.M.; Garcia-Beltran, W.; et al. A genome-wide CRISPR screen identifies a restricted set of HIV host dependency factors. Nat. Genet. 2017, 49, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Muffat, J.; Javed, A.O.; Keys, H.R.; Lungjangwa, T.; Bosch, I.; Khan, M.; Virgilio, M.C.; Gehrke, L.; Sabatini, D.M.; et al. Genome-wide CRISPR screen for Zika virus resistance in human neural cells. Proc. Natl. Acad. Sci. USA 2019, 116, 9527–9532. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.S.; Rong, E.; Dry, I.; Lillico, S.G.; Law, A.; Digard, P.; Whitelaw, B.; Dalziel, R.G. GARP and EARP are required for efficient BoHV-1 replication as identified by a genome wide CRISPR knockout screen. PLOS Pathog. 2023, 19, e1011822. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Clohisey, S.M.; Chia, B.S.; Wang, B.; Cui, A.; Eisenhaure, T.; Schweitzer, L.D.; Hoover, P.; Parkinson, N.J.; Nachshon, A.; et al. Genome-wide CRISPR screen identifies host dependency factors for influenza A virus infection. Nat Commun 2020, 11, 164. [Google Scholar] [CrossRef] [PubMed]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Yeo, N.C.; Chavez, A.; Lance-Byrne, A.; Chan, Y.; Menn, D.; Milanova, D.; Kuo, C.C.; Guo, X.; Sharma, S.; Tung, A.; et al. An enhanced CRISPR repressor for targeted mammalian gene regulation. Nat. Methods 2018, 15, 611–616. [Google Scholar] [CrossRef]
- Wild, P.; Engels, M.; Senn, C.; Tobler, K.; Ziegler, U.; Schraner, E.M.; Loepfe, E.; Ackermann, M.; Mueller, M.; Walther, P. Impairment of nuclear pores in bovine herpesvirus 1-infected MDBK cells. J. Virol. 2005, 79, 1071–1083. [Google Scholar] [CrossRef]
- Brinkman, E.K.; Chen, T.; Amendola, M.; Van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014, 42, 168. [Google Scholar] [CrossRef]
- Chen, Y.H.; Narimatsu, Y.; Clausen, T.M.; Gomes, C.; Karlsson, R.; Steentoft, C.; Spliid, C.B.; Gustavsson, T.; Salanti, A.; Persson, A.; et al. The GAGOme: A cell-based library of displayed glycosaminoglycans. Nat. Methods 2018, 15, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Tumkosit, U.; Nakamura, S.; Motooka, D.; Kishishita, N.; Priengprom, T.; Sa-ngasang, A.; Kinoshita, T.; Takeda, N.; Maeda, Y. Genome-Wide Screening Uncovers the Significance of N-Sulfation of Heparan Sulfate as a Host Cell Factor for Chikungunya Virus Infection. J. Virol. 2017, 91, e00432-17. [Google Scholar] [CrossRef] [PubMed]
- Jae, L.T.; Raaben, M.; Riemersma, M.; Van Beusekom, E.; Blomen, V.A.; Velds, A.; Kerkhoven, R.M.; Carette, J.E.; Topaloglu, H.; Meinecke, P.; et al. Deciphering the glycosylome of dystroglycanopathies using haploid screens for Lassa virus entry. Science 2013, 340, 479–483. [Google Scholar] [CrossRef]
- Riblett, A.M.; Blomen, V.A.; Jae, L.T.; Altamura, L.A.; Doms, R.W.; Brummelkamp, T.R.; Wojcechowskyj, J.A. A Haploid Genetic Screen Identifies Heparan Sulfate Proteoglycans Supporting Rift Valley Fever Virus Infection. J. Virol. 2016, 90, 1414–1423. [Google Scholar] [CrossRef] [PubMed]
- Luteijn, R.D.; van Diemen, F.; Blomen, V.A.; Boer, I.G.J.; Manikam Sadasivam, S.; van Kuppevelt, T.H.; Drexler, I.; Brummelkamp, T.R.; Lebbink, R.J.; Wiertz, E.J. A Genome-Wide Haploid Genetic Screen Identifies Heparan Sulfate-Associated Genes and the Macropinocytosis Modulator TMED10 as Factors Supporting Vaccinia Virus Infection. J. Virol. 2019, 93, e02160-18. [Google Scholar] [CrossRef]
- Mahajan, D.; Tie, H.C.; Chen, B.; Lu, L. Dopey1-Mon2 complex binds to dual-lipids and recruits kinesin-1 for membrane trafficking. Nat. Commun. 2019, 10, 3218. [Google Scholar] [CrossRef]
- Kim, B.T.; Kitagawa, H.; Tamura, J.I.; Saito, T.; Kusche-Gullberg, M.; Lindahl, U.; Sugahara, K. Human tumor suppressor EXT gene family members EXTL1 and EXTL3 encode α1,4-N-acetylglucosaminyltransferases that likely are involved in heparan sulfate/heparin biosynthesis. Proc. Natl. Acad. Sci. USA 2001, 98, 7176–7181. [Google Scholar] [CrossRef]
- Pinhal, M.A.S.; Smith, B.; Olson, S.; Aikawa, J.I.; Kimata, K.; Esko, J.D. Enzyme interactions in heparan sulfate biosynthesis: Uronosyl 5-epimerase and 2-O-sulfotransferase interact in vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 12984–12989. [Google Scholar] [CrossRef] [PubMed]
- Mårdberg, K.; Trybala, E.; Tufaro, F.; Bergström, T. Herpes simplex virus type 1 glycoprotein C is necessary for efficient infection of chondroitin sulfate-expressing gro2C cells. J. Gen. Virol. 2002, 83, 291–300. [Google Scholar] [CrossRef]
- Russell, G.C.; Stewart, J.P.; Haig, D.M. Malignant catarrhal fever: A review. Veter. J. 2009, 179, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Pokrovskaya, I.D.; Willett, R.; Smith, R.D.; Morelle, W.; Kudlyk, T.; Lupashin, V.V. Conserved oligomeric Golgi complex specifically regulates the maintenance of Golgi glycosylation machinery. Glycobiology 2011, 21, 1554–1569. [Google Scholar] [CrossRef] [PubMed]
- Burkard, C.; Lillico, S.G.; Reid, E.; Jackson, B.; Mileham, A.J.; Ait-Ali, T.; Whitelaw, C.B.A.; Archibald, A.L. Precision engineering for PRRSV resistance in pigs: Macrophages from genome edited pigs lacking CD163 SRCR5 domain are fully resistant to both PRRSV genotypes while maintaining biological function. PLOS Pathog. 2017, 13, e1006206. [Google Scholar] [CrossRef] [PubMed]
- Whitworth, K.M.; Rowland, R.R.R.; Ewen, C.L.; Trible, B.R.; Kerrigan, M.A.; Cino-Ozuna, A.G.; Samuel, M.S.; Lightner, J.E.; McLaren, D.G.; Mileham, A.J.; et al. Gene-edited pigs are protected from porcine reproductive and respiratory syndrome virus. Nat. Biotechnol. 2016, 34, 20–22. [Google Scholar] [CrossRef] [PubMed]
- Whitworth, K.M.; Rowland, R.R.R.; Petrovan, V.; Sheahan, M.; Cino-Ozuna, A.G.; Fang, Y.; Hesse, R.; Mileham, A.; Samuel, M.S.; Wells, K.D.; et al. Resistance to coronavirus infection in amino peptidase N-deficient pigs. Transgenic Res. 2019, 28, 21–32. [Google Scholar] [CrossRef]
- Hadigal, S.R.; Agelidis, A.M.; Karasneh, G.A.; Antoine, T.E.; Yakoub, A.M.; Ramani, V.C.; Djalilian, A.R.; Sanderson, R.D.; Shukla, D. Heparanase is a host enzyme required for herpes simplex virus-1 release from cells. Nat. Commun. 2015, 6, 6985. [Google Scholar] [CrossRef]
- Hopkins, J.; Yadavalli, T.; Agelidis, A.M.; Shukla, D. Host Enzymes Heparanase and Cathepsin L Promote Herpes Simplex Virus 2 Release from Cells. J. Virol. 2018, 92, e01179-18. [Google Scholar] [CrossRef]
- Blackburn, J.B.; Kudlyk, T.; Pokrovskaya, I.; Lupashin, V.V. More than just sugars: Conserved oligomeric Golgi complex deficiency causes glycosylation-independent cellular defects. Traffic 2018, 19, 463–480. [Google Scholar] [CrossRef]
- Young, R.; Lefevre, L.; Bush, S.J.; Joshi, A.; Singh, S.H.; Jadhav, S.K.; Dhanikachalam, V.; Lisowski, Z.M.; Iamartino, D.; Summers, K.M.; et al. A Gene Expression Atlas of the Domestic Water Buffalo (Bubalus bubalis). Front. Genet. 2019, 10, 668. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, W.S.; Rong, E.; Dry, I.; Lillico, S.; Law, A.; Digard, P.; Whitelaw, B.; Dalziel, R.G. Validation of Candidate Host Cell Entry Factors for Bovine Herpes Virus Type-1 Based on a Genome-Wide CRISPR Knockout Screen. Viruses 2024, 16, 297. https://doi.org/10.3390/v16020297
Tan WS, Rong E, Dry I, Lillico S, Law A, Digard P, Whitelaw B, Dalziel RG. Validation of Candidate Host Cell Entry Factors for Bovine Herpes Virus Type-1 Based on a Genome-Wide CRISPR Knockout Screen. Viruses. 2024; 16(2):297. https://doi.org/10.3390/v16020297
Chicago/Turabian StyleTan, Wenfang Spring, Enguang Rong, Inga Dry, Simon Lillico, Andy Law, Paul Digard, Bruce Whitelaw, and Robert G. Dalziel. 2024. "Validation of Candidate Host Cell Entry Factors for Bovine Herpes Virus Type-1 Based on a Genome-Wide CRISPR Knockout Screen" Viruses 16, no. 2: 297. https://doi.org/10.3390/v16020297